 Damian Perez-Mazliah
Damian Perez-Mazliah Jean Langhorne
Jean Langhorne- Division of Parasitology, MRC National Institute for Medical Research, London, UK
CD4+ T-cells have been shown to play a central role in immune control of infection with Plasmodium parasites. At the erythrocytic stage of infection, IFN-γ production by CD4+ T-cells and CD4+ T-cell help for the B-cell response are required for control and elimination of infected red blood cells. CD4+ T-cells are also important for controlling Plasmodium pre-erythrocytic stages through the activation of parasite-specific CD8+ T-cells. However, excessive inflammatory responses triggered by the infection have been shown to drive pathology. Early classical experiments demonstrated a biphasic CD4+ T-cell response against erythrocytic stages in mice, in which T helper (Th)1 and antibody-helper CD4+ T-cells appear sequentially during a primary infection. While IFN-γ-producing Th1 cells do play a role in controlling acute infections, and they contribute to acute erythrocytic-stage pathology, it became apparent that a classical Th2 response producing IL-4 is not a critical feature of the CD4+ T-cell response during the chronic phase of infection. Rather, effective CD4+ T-cell help for B-cells, which can occur in the absence of IL-4, is required to control chronic parasitemia. IL-10, important to counterbalance inflammation and associated with protection from inflammatory-mediated severe malaria in both humans and experimental models, was originally considered be produced by CD4+ Th2 cells during infection. We review the interpretations of CD4+ T-cell responses during Plasmodium infection, proposed under the original Th1/Th2 paradigm, in light of more recent advances, including the identification of multifunctional T-cells such as Th1 cells co-expressing IFN-γ and IL-10, the identification of follicular helper T-cells (Tfh) as the predominant CD4+ T helper subset for B-cells, and the recognition of inherent plasticity in the fates of different CD4+ T-cells.
Introduction
Malaria, caused by infection with Plasmodium transmitted via mosquito bites, represents a major global cause of morbidity and mortality (1). Plasmodium spp. are eukaryotic apicomplexan intracellular parasites with different life-cycle stages within the vertebrate host: an early clinically silent liver stage that can last approximately 7–10 days in humans and 2 days in rodents, followed by an erythrocytic stage, responsible for the pathology of malaria (Figure 1A). Species of Plasmodium that infect humans include P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi. A number of Plasmodium species that infect rodents, but not humans, are available for laboratory research, including P. berghei, P. vinckei, P. chabaudi, and P. yoelii (2), which allow the dissection of immune mechanism of protection and pathology (3).
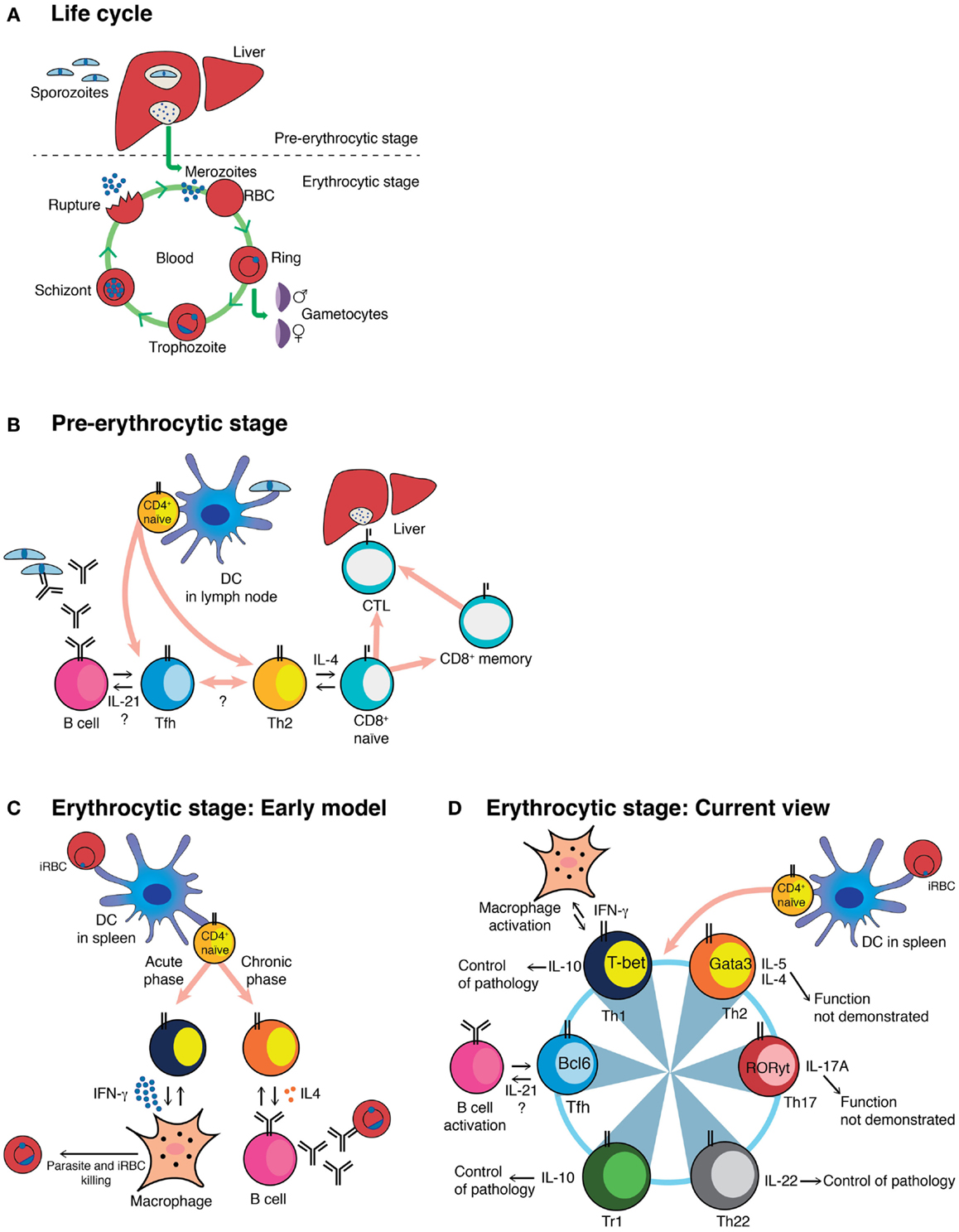
Figure 1. Schematic representation of the Plasmodium life cycle, and different models of CD4+ T-cell activation during Plasmodium infection. (A) Plasmodium life cycle in the mammalian host. (B) The cartoon shows the different subsets known, or proposed to be, activated by the pre-erythrocytic stage of Plasmodium, together with their known or proposed functions. (C) Classical view of the biphasic activation of Th1 and Th2 CD4+ T-cells toward the erythrocytic stage of Plasmodium. (D) Current understanding of the CD4+ T-cell responses to the erythrocytic stage of Plasmodium, together with their known or proposed roles during infection. This model highlights the plastic ability of activated CD4+ T-cells to interconvert into different Th subsets. The master regulator transcription factors known to drive each Th program as well as the cytokines associated to each Th subset are depicted (53). DC, myeloid dendritic cell; iRBC, infected red blood cell; CTL, cytotoxic CD8+ T-cell; Tr1, Foxp3− regulatory T-cell.
In most cases, the host’s immune system can eventually control a Plasmodium infection; however, exacerbated host immune responses and inflammation induced by the parasite, contribute to the pathology accompanying infection (4, 5). CD4+ T-cell responses have been associated with control of erythrocytic stage parasites, but a small number of studies indicate a helper role also in pre-erythrocytic immunity (6–11). Parasite biology, host cell and tissue tropism, and kinetics of parasite growth differ between pre-erythrocytic and erythrocytic stages within the vertebrate host and, accordingly, the particular CD4+ T-cell responses elicited also differ. Herein, we discuss activation of different CD4+ T-cell subsets during malaria, their role in the control of the infection and the interplay between different subsets, with a particular emphasis on the concept of CD4+ T-cell plasticity.
CD4+ T-Cell Subsets Activated by Pre-Erythrocytic Stages
Very little is known about the CD4+ T-cell response to Plasmodium pre-erythrocytic stages, or its regulation in natural infection either in humans or in experimental models. Clearly, since IgG antibodies and memory B-cells are generated to a wide range of pre-erythrocytic antigens, including those with expression restricted primarily to these stages, such as circumsporozoite protein (CSP), liver-stage antigen 1 (LSA1), and sporozoite threonine–asparagine-rich protein (STARP) (12–14), CD4+ T-cells must be induced by these stages of the infection. Indeed, CD4+ T-cells specific for pre-erythrocytic antigens have been documented, and in some cases, have been shown to correlate with protection in humans following natural infection (15) and immunization (11). However, we have few details of their functional heterogeneity.
CD4+ T-cells of undefined Th1/Th2 phenotype have been shown to confer protection against the pre-erythrocytic stages of P. yoelii even in the absence of CD8+ T-cells (9), and CD4+ T-cell clones recognizing peptides of CSP protected against a P. yoelii sporozoite challenge in mice, irrespective of their Th1 or Th2 phenotype (6, 7). The location of priming of CD4+ T-cell specific for pre-erythrocytic stages is still a matter of debate, and there is little evidence as yet on priming of CD4+ T-cell in the liver. As protective CD8+ T-cells specific for a peptide of CSP from P. yoelii can be primed by dendritic cells (DCs) in lymph nodes after infection with sporozoites (16), it is likely that DCs in lymph nodes might also be critical for priming CD4+ T-cell responses to Plasmodium pre-erythrocytic stages.
As well as possible direct killing of infected hepatocytes, subsets of CD4+ T-cells provide crucial help for both B-cell and CD8+ T-cell responses (Figure 1B). In a very few studies, CD4+ T-cells were shown to be necessary to ensure survival of protective effector and memory CD8+ T-cells induced by irradiation-attenuated sporozoites (8, 10). This mechanism is dependent on STAT-6 and IL-4, suggesting that Th2 CD4+ T-cells may be in charge of providing help to CD8+ T-cells. More recently, CSP-specific CD4+ T-cells expressing CD107a (LAMP-1), a marker for cytotoxic degranulation, were shown to be induced and associated with protection against the pre-erythrocytic stages after immunizations of healthy volunteers by bites from P. falciparum-infected mosquitoes during chloroquine chemoprophylaxis (11). Nothing is yet known about induction of Tfh cells, the subset that provides help for T-cell dependent B-cell responses (17), which are presumably activated by, and would be important for, generation of high affinity IgG antibodies.
Altogether, these data suggest a role of CD4+ T-cells in protective immunity to pre-erythrocytic-stage infection, the mechanisms of which are not yet completely understood. With the current emphasis on pre-erythrocytic vaccines, it is important that we understand more about the potential contribution of diverse CD4+ T-cell populations on direct killing of infected cells, in the B-cell and CD8+ T-cell responses, as well as their regulatory roles in immunity to the pre-erythrocytic stages of Plasmodium.
CD4+ T-Cell Subsets Activated by Erythrocytic Stages
The Th1/Th2 paradigm proposed by Mosmann and Coffman postulated stable lineages of activated CD4+ T-cells with distinctive cytokine production patterns and functional capacity; IFN-γ-producing Th1 cells being crucial mediators of host immunity against intracellular pathogens, while IL-4-producing Th2 cells mediating immunity toward extracellular pathogens and collaboration with B-cells for antibody production (18). As Plasmodium invades red blood cells (Figure 1A), which do not express MHC class I or II, it was difficult to envisage parasites at this stage as direct targets of Th1 or Th2 cells. Nonetheless, it was possible to draw parallels with the original Mosmann and Coffman model; malaria researchers observed that the erythrocytic stages triggered a strong IFN-γ response during acute infections in P. berghei, P. yoelii, and P. chabaudi infections in mice, as well as in P. falciparum infection in humans (19–24).
Since that time, the Th1/Th2 paradigm has gained in complexity with the identification of novel CD4+ T-cell subsets with distinctive characteristics and transcriptional programs in charge of driving the different cell fates (25). These differentiation programs are governed predominantly by signals derived from antigen-presenting cells (APC) and the microenvironment at the time of CD4+ T-cell activation. DCs are necessary for effective priming of the T-cell response in erythrocytic-stage malaria (26), and two subsets of splenic DCs, CD8− and CD4+ classical DCs, have been shown to present antigen for the activation of CD4+ T-cells during an erythrocytic-stage infection with P. chabaudi and P. berghei, respectively (26–29). Although it is known that IL-12 is an important cytokine in the induction of a protective response in experimental malaria infections (30), understanding of the regulation of this cytokine or other factors in DCs necessary for effective Plasmodium-antigen presentation to different subsets of CD4+ T-cells is still lacking.
IFN-γ, a defining cytokine of Th1 cells expressing the transcription factor T-bet, has proven to be important for controlling the acute erythrocytic stage of Plasmodium infection in rodent models (31–34). IFN-γ-producing CD4+ effector (E) and effector memory (EM) CD4+ T-cells both confer partial protection from P. chabaudi infection (35). In general agreement with this, IFN-γ from CD4+ T-cells has been shown to be important in maintaining strain-transcending blood-stage immunity (36). However, IFN-γ is not only produced by T-bet+ Th1 cells but also by NK cells, NKT cells, and γδ T-cells (37, 38) as well as CD8+ T-cells, and it is not always clear whether Th1 cells, IFN-γ, or IFN-γ from Th1 cells per se are the main players in early protection or pathology in experimental malaria. Studies so far to address these questions have given conflicting results. One study using P. berghei ANKA has shown that the enhanced IFNγ+ T-bet+ CD4+ T-cell responses observed in mice lacking Type I IFN signaling are associated with better control of P. berghei ANKA infections, resulting in lower morbidity and mortality (39, 40). In contrast, others have shown that in the absence of T-bet, essential for Th1 commitment, cerebral pathology of P. berghei ANKA infections is ameliorated, and the number of IFNγ+ CD4+ T-cells is reduced. However, control of parasite replication is lost and mice succumb to hyper-parasitemia and anemia (41). In a different rodent model of erythrocytic-stage malaria, P. yoelii 17X(NL), although activation of T-bet was detected on CD4+ T-cells early in infection (42), the infection can still be controlled in T-bet-deficient mice (43).
The erythrocytic stages of Plasmodium are also able to activate CD4+ T-cells that are very effective helpers for Plasmodium-specific antibody production, but produce little or no IFN-γ (21). This response was shown to coincide with the appearance of IL-4-producing CD4+ T-cells (21). The association between IL-4-producing CD4+ T-cells and antibody responses toward the parasite was also observed in P. falciparum immune subjects (44). An erythrocytic-stage P. chabaudi infection, the only mouse model that generates a chronic phase of infection (3), presents a biphasic CD4+ T-cell activation, with a large IFN-γ-producing CD4+ T-cell response during the acute phase, followed by an antibody-helper/IL-4-producing CD4+ T-cell response during the chronic phase (Figure 1C) (21, 24, 45, 46). These data were interpreted as an early activation of Th1 cells able to control parasitemia through the activation of effector mechanisms such as macrophages, followed by a Th2 response in charge of activating B-cell responses to complete the clearance of the parasite (47, 48). However, the frequency of CD4+ T-cells able to help B-cells to produce Plasmodium-specific antibodies was much higher than the frequency of IL-4-producing CD4+ T-cells (21). Furthermore, control of a P. chabaudi infection and specific IgG responses, including IgG1 antibodies, was possible even in the complete absence of IL-4 (45). Therefore, it was clear, despite its attractiveness as a model, that the simple Th1/Th2 paradigm was not sufficient to explain the full complexity of CD4+ T-cell activation in the erythrocytic stages of Plasmodium. More recently, a subset of CD4+ T-cells, Tfh cells, has been described that produce IL-21, as well as other cytokines originally associated with other Th subsets, such as IFN-γ and IL-4 (17). We believe that the Tfh program, and not a Th2 response, is the critical one for B-cell help and activation of protective B-cell responses against the erythrocytic stages of Plasmodium infection (Figure 1D). However, there are very few data on Tfh or its crucial signature cytokine, IL-21, in malaria. Lymphocytes closely resembling Tfh have been observed in peripheral blood of humans (49), although not yet in people exposed to malaria. However, IL-21-producing CD4+ T-cells have been demonstrated in blood of immune adults living in endemic areas of P. falciparum transmission (50–52). Given the importance of the humoral response in protective immunity to the erythrocytic stages of Plasmodium, understanding the activation and maintenance of Tfh cells during malaria is of outstanding interest for vaccine design.
Recently, the Th17 subset of CD4+ T-cells, defined by the expression of the transcription factor RORγt, has gained attention among malaria researchers because of its role in autoimmune diseases and chronic inflammation and in responses to extracellular pathogens such as bacteria and fungi (53). CD4+ IL-17A+ RORγt+ Th17 cells are activated during acute P. berghei ANKA and P. yoelii infection, but the function of these cells during infection was not explored (54). Ishida and colleagues demonstrated no association of Th17 cells and cerebral malaria in P. berghei ANKA-infected IL-17-deficient mice (55). We have also observed the presence of IL-17A and IL-17F-producing CD4+ T-cells mainly in the liver during acute erythrocytic-stage P. chabaudi infection; however, IL-17A-deficient mice showed no significant alterations in the course of P. chabaudi infection (56). Therefore, despite activation, Th17 cells have so far not been shown to have a defined role during Plasmodium infections (Figure 1D).
Additional CD4+ T-cell subsets, such as that producing IL-22 (Th22), continue to be identified (57, 58). IL-22 has been implicated in both host defense against bacterial infections and tissue repair (59). Interestingly, IL-22 single-nucleotide polymorphisms associated with resistance and susceptibility to severe malaria have been identified (60). We have observed that IL-22-producing CD4+ T-cells are activated, albeit in low frequency, in both spleen and liver during acute erythrocytic-stage P. chabaudi infections, and that IL-22-deficient mice infected with P. chabaudi show exacerbated pathology (56) (Figure 1D). Research is ongoing to explore in greater detail the origins of these cells, their location, and the mechanisms underlying the observed pathology.
With the discovery of this wide array of possible CD4+ T-cell subsets and their different activation requirements and functional capacities, it is becoming clear that CD4+ T-cells may not be simply defined as individual subsets of Th cells producing a single cytokine, but rather they represent components of a dynamic and interactive response, in which these cells can be multifunctional, flexible, and plastic depending on the disease/infection and activation environment (61). The multifunctional capacity of T-cells, or the ability to perform more than one function (e.g., production of different cytokines) at the single-cell level, and its association with the capacity to control infections was first recognized in HIV-infected subjects (62) and in a mouse model of vaccination against Leishmania major (63). This association between multifunctional capacity of T-cells and control of infections is not limited to HIV and Leishmania, and was soon observed in several other chronic infections, including viral, parasitic, and mycobacterial infections (64). Immunization of subjects with P. falciparum apical membrane antigen 1 (AMA1) (65), and immunizations of mice with full-length P. falciparum CSP protein (66) also activates multifunctional CD4+ T-cells responses. Moreover, multi-parameter flow cytometric analyses of human PBMC from children and adults exposed to malaria infection reveal the existence of CD4+ T-cells co-expressing several cytokines characteristic of many CD4+ T-cell subsets (52, 67), demonstrating the complexity of the CD4+ T-cell response activated by the erythrocytic stages of Plasmodium.
There is an important body of evidence suggesting that, far from being terminally differentiated stable lineages, the different Th subsets have an extensive capacity to interconvert further between different phenotypes, a concept known as plasticity (68). The most recent studies suggest that CD4+ T-cell activation with overlapping characteristics of different Th subsets is the norm rather than the exception (61), and this is likely to be reflected in complex diseases such as malaria. Although this has not been explored in any detail in the context of malaria, the concept of Th plasticity opens new possibilities for studying the function and regulation of CD4+ T-cells in the control of Plasmodium infection and related immunopathology. One subset known for its remarkable plasticity is the Tfh subset. It has been shown that Th1, Th2, and Th17 cells can migrate into the B-cell areas of secondary lymphoid organs and acquire the functional capacity and biomarkers of Tfh cells and, conversely, the Tfh subset can become Th1, Th2, and Th17 (69). In the context of Plasmodium infection, this would imply that not only could Plasmodium parasites activate Tfh responses directly but also the Tfh subset could potentially arise from any of the CD4+ T-cell subsets already activated by the erythrocytic stages.
In light of the multifunctional and plastic capacities of the CD4+ T-cells, a scenario can be envisaged in which Th subsets required for the control of parasite burden, such as Th1 cells, have the capacity to acquire a regulatory phenotype depending on the context, contributing to control of the inflammation and thus to protection of tissues and organs and preventing any potentially harmful effects of the response. This would allow a fine-tuning of the CD4+ T-cell response to guarantee the control of Plasmodium infection without causing deleterious side effects. One mechanism of self-regulation by CD4+ Th1 cells in malaria is the induction of IL-10. IFN-γ+IL-10+T-bet+ Th1 CD4+ T-cells can prevent pathology during P. chabaudi infection (70) (Figure 1D). In addition, IL-10 from CD4+ T-cells distinct from regulatory T (Treg) cells is able to control pathology in a P. yoelii infection in mice, but in this case, these cells do not co-express IFN-γ (71). IL-10 is produced in these cells in response to IL-27 (70), although the signals responsible for the induction of IL-27 remain unknown. The occurrence of IFN-γ+IL-10+T-bet+ CD4+ T-cells during Plasmodium infections is not restricted to mouse models; they have been reported to be present in PBMC of children living in highly malaria-endemic regions (72–74) and their proportion is higher in children with uncomplicated malaria compared to children with severe malaria (72). IL-10 can also be induced in IL-17-producing CD4+ T-cells, as yet by unknown pathways (75–77), and thus, IL-10 may be a more general mechanism for regulating any subset of CD4+ T-cells in malaria. CD4+ T-cells, particularly Th1 cells, can also be controlled by Type I IFNs. In the P. berghei ANKA model, Type I IFN signaling suppresses Th1 responses by directly acting on classical DCs (40). Given that type I IFN signaling can also promote the expression of IL-10 on CD4+ T-cells (78–81), we hypothesize that these two regulatory mechanisms might share some common activation signals during Plasmodium infection.
CD4+ T-cell responses may also be controlled by the expression of surface molecules associated with exhaustion. Elevated frequencies of PD-1+ LAG-3+ CD4+ T-cells have been reported in P. falciparum-infected subjects (82, 83), and combined blockade of PD-1 and LAG-3 accelerated clearance of erythrocytic-stage Plasmodium infection in a mouse model (83). In agreement with these observations, PD-1-deficient mice show better control of an erythrocytic-stage P. chabaudi infection with higher frequencies of IFN-γ+ and T-bet+ CD4+ T-cells during the chronic phase (84). The kinetics of PD-1+ CD4+ T-cells during the acute erythrocytic-stage P. yoelii 17X(NL) infection are similar to those observed during P. chabaudi infection (42, 84). However, some caution should be exercised in assuming that expression of PD-1 automatically means exhaustion, as in some subsets of activated CD4+ T-cells, in particular Tfh cells, PD1 is expressed without affecting their functional capacity. It may be that the triggering of PD-1 by its ligand PDL-1 (85) is the key to whether the cell is programed for cell death.
Many of the CD4+ T-cells activated in a Plasmodium infection may undergo interconversion between defined cell subsets depending on antigen dose, APC, location, and cytokine/chemokine environment, such as that described for Treg and Th17 subsets (86, 87). Thus, it is possible that the Th17 cells found in the spleen of malaria-infected mice gain a regulatory phenotype in other organs or tissues such as brain and liver. The capacity to identify and manipulate these possible mechanisms of CD4+ T-cell plasticity during Plasmodium infections would be of great value for the design of novel therapeutic strategies.
Concluding Remarks
The identification of two CD4+ T-cell subsets with different well-defined functions represented an attractive organizational system with which to rationalize CD4+ T-cell responses to Plasmodium infections. However, such a model has not been sufficient to reflect fully the complexity of CD4+ T-cell biology observed in human or experimental malaria. In particular, control of Plasmodium infection requires strictly regulated immune responses that are able to prevent parasite replication without causing detrimental side effects of uncontrolled inflammation. Plasmodium species have different stages with different tissue tropisms and this complex life cycle challenges the idea that a single static group of terminally differentiated CD4+ T-cells would be able to perform all the tasks required to control this infection. In order to cope with these tasks, the CD4+ T-cell response has to adapt to the changing scenarios presented as the infection evolves. The newer concept of CD4+ T-cell plasticity would add substantially to our understanding of induction and regulation of CD4+ T-cell responses in malaria, and it is highly probable that some of the CD4+ Th programs not yet explored in depth, such as the Tfh response, might play critical roles in the outcome of the infection. The combination of potent tools such as multi-parameter flow cytometry, in vivo imaging, systems analyses of transcriptome, proteome, and metabolome, together with T-cell receptor transgenic mice and peptide-MHC II tetramers will give us the chance to explore the complexity of the CD4+ T-cell responses to malaria in in vivo models (74, 88–97). In addition, confocal microscopy and intravital imaging techniques make now possible to follow sporozoites injected via the mosquito bite into the skin, or by injection of attenuated sporozoites through to their arrival in the lymphoid organs and liver (98), and to study the consequent activation of CD4+ T-cells and their subsequent effector functions. Field studies of natural human Plasmodium infections and mouse models should complement each other to get a deeper understanding of the complex CD4+ T-cell response activated by these infections. A detailed delineation on how CD4+ T-cells modulate the activation of effector cells such as CD8+ T-cells, macrophages, and B-cells in response to Plasmodium infection is critical to achieve the goal of generating protective treatments to control malaria.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Deirdre Cunningham, Jan Sodenkamp, Wiebke Nahrendorf, and Jingwen Lin for their helpful comments and critical reading of the manuscript. The excellent support of Biological Services, NIMR Flow Facility, and PhotoGraphics is much appreciated. Recent work cited from the authors’ laboratory was funded by the Medical Research Council, UK (U117584248), the Wellcome Trust (048684), and the European Union (FP7/2007–2013) under grant agreement 242095-EVIMalaR.
References
2. Landau I. Life cycles and morphology. In: Killick-Kendrick R, Peters W, editors. Rodent Malaria. London: Academic Press (1978). p. 53–84.
3. Stephens R, Culleton RL, Lamb TJ. The contribution of Plasmodium chabaudi to our understanding of malaria. Trends Parasitol (2012) 28(2):73–82. doi: 10.1016/j.pt.2011.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Lamb TJ, Brown DE, Potocnik AJ, Langhorne J. Insights into the immunopathogenesis of malaria using mouse models. Expert Rev Mol Med (2006) 8(06):1–22. doi:10.1017/S1462399406010581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Schofield L. Intravascular infiltrates and organ-specific inflammation in malaria pathogenesis. Immunol Cell Biol (2007) 85(2):130–7. doi:10.1038/sj.icb.7100040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Rénia L, Grillot D, Marussig M, Corradin G, Miltgen F, Lambert PH, et al. Effector functions of circumsporozoite peptide-primed CD4+ T cell clones against Plasmodium yoelii liver stage. J Immunol (1993) 150(4):1471–8.
7. Takita-Sonoda Y, Tsuji M, Kamboj K, Nussenzweig RS, Clavijo P, Zavala F. Plasmodium yoelii: peptide immunization induces protective CD4+ T cells against a previously unrecognized cryptic epitope of the circumsporozoite protein. Exp Parasitol (1996) 84(2):223–30. doi:10.1006/expr.1996.0108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Carvalho LH, Sano G-I, Hafalla JCR, Morrot A, Curotto de Lafaille MA, Zavala F. IL-4-secreting CD4+ T cells are crucial to the development of CD8+ T-cell responses against malaria liver stages. Nat Med (2002) 8(2):166–70. doi:10.1038/nm0202-166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Oliveira GA, Kumar KA, Calvo-Calle JM, Othoro C, Altszuler D, Nussenzweig V, et al. Class II-restricted protective immunity induced by malaria sporozoites. Infect Immun (2008) 76(3):1200–6. doi:10.1128/IAI.00566-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Overstreet MG, Chen Y-C, Cockburn IA, Tse S-W, Zavala F. CD4+ T cells modulate expansion and survival but not functional properties of effector and memory CD8+ T cells induced by malaria sporozoites. PLoS One (2011) 6(1):e15948. doi:10.1371/journal.pone.0015948
11. Bijker EM, Teirlinck AC, Schats R, van Gemert G-J, van de Vegte-Bolmer M, van Lieshout L, et al. Cytotoxic markers associate with protection against malaria in human volunteers immunized with Plasmodium falciparum sporozoites. J Infect Dis (2014) 9(11):e112910. doi:10.1093/infdis/jiu293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Crompton PD, Kayala MA, Traore B, Kayentao K, Ongoiba A, Weiss GE, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci U S A (2010) 107(15):6958–63. doi:10.1073/pnas.1001323107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Felgner PL, Roestenberg M, Liang L, Hung C, Jain A, Pablo J, et al. Pre-erythrocytic antibody profiles induced by controlled human malaria infections in healthy volunteers under chloroquine prophylaxis. Sci Rep (2013) 3:3549. doi:10.1038/srep03549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Nahrendorf W, Scholzen A, Bijker EM, Teirlinck AC, Bastiaens GJH, Schats R, et al. Memory B-cell and antibody responses induced by Plasmodium falciparum sporozoite immunization. J Infect Dis (2014) 210(12):1981–90. doi:10.1093/infdis/jiu354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Reece WHH, Pinder M, Gothard PK, Milligan P, Bojang K, Doherty T, et al. A CD4(+) T-cell immune response to a conserved epitope in the circumsporozoite protein correlates with protection from natural Plasmodium falciparum infection and disease. Nat Med (2004) 10(4):406–10. doi:10.1038/nm1009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Sacci JB, Zavala F. CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat Med (2007) 13(9):1035–41. doi:10.1038/nm1628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
18. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol (1989) 7:145–73. doi:10.1146/annurev.iy.07.040189.001045
19. Huang KY, Schultz WW, Gordon FB. Interferon induced by Plasmodium berghei. Science (1968) 162(3849):123–4. doi:10.1126/science.162.3849.123
20. Troye-Blomberg M, Andersson G, Stoczkowska M, Shabo R, Romero P, Patarroyo ME, et al. Production of IL 2 and IFN-gamma by T cells from malaria patients in response to Plasmodium falciparum or erythrocyte antigens in vitro. J Immunol (1985) 135(5):3498–504.
21. Langhorne J, Gillard S, Simon B, Slade S, Eichmann K. Frequencies of CD4+ T cells reactive with Plasmodium chabaudi chabaudi: distinct response kinetics for cells with Th1 and Th2 characteristics during infection. Int Immunol (1989) 1(4):416–24. doi:10.1093/intimm/1.4.416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Shear HL, Srinivasan R, Nolan T, Ng C. Role of IFN-gamma in lethal and nonlethal malaria in susceptible and resistant murine hosts. J Immunol (1989) 143(6):2038–44.
23. Troye-Blomberg M, Riley EM, Perlmann H, Andersson G, Larsson A, Snow RW, et al. T and B cell responses of Plasmodium falciparum malaria-immune individuals to synthetic peptides corresponding to sequences in different regions of the P. falciparum antigen Pf155/RESA. J Immunol (1989) 143(9):3043–8.
24. Stevenson MM, Tam MF. Differential induction of helper T cell subsets during blood-stage Plasmodium chabaudi AS infection in resistant and susceptible mice. Clin Exp Immunol (1993) 92(1):77–83. doi:10.1111/j.1365-2249.1993.tb05951.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Coomes SM, Pelly VS, Wilson MS. Plasticity within the αβ+CD4+ T-cell lineage: when, how and what for? Open Biol (2013) 3(1):120157. doi:10.1098/rsob.120157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, et al. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc Natl Acad Sci U S A (2008) 105(38):14509–14. doi:10.1073/pnas.0806727105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Sponaas AM, Cadman ET, Voisine C, Harrison V, Boonstra A, O’Garra A, et al. Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J Exp Med (2006) 203(6):1427–33. doi:10.1084/jem.20052450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, et al. Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J Immunol (2007) 178(10):6033–7. doi:10.4049/jimmunol.178.10.6033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Voisine C, Mastelic B, Sponaas A-M, Langhorne J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int J Parasitol (2010) 40(6):711–9. doi:10.1016/j.ijpara.2009.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Su Z, Stevenson MM. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J Immunol (2002) 168(3):1348–55. doi:10.4049/jimmunol.168.3.1348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Sauvaget F, Fauconnier B. The protective effect of endogenous interferon in mouse malaria, as demonstrated by the use of anti-interferon globulins. Biomedicine (1978) 29(6):184–7.
32. Meding SJ, Cheng SC, Simon-Haarhaus B, Langhorne J. Role of gamma interferon during infection with Plasmodium chabaudi chabaudi. Infect Immun (1990) 58(11):3671–8.
33. Simon-Haarhaus B, Langhorne J, Meding S. CD4+T cell-dependent effector mechanisms important in the immune response to the erythrocytic stages of Plasmodium chabaudi chabaudi (AS). Behring Inst Mitt (1991) 88:94–8.
34. Su Z, Stevenson MM. Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect Immun (2000) 68(8):4399–406. doi:10.1128/IAI.68.8.4399-4406.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Stephens R, Langhorne J. Effector memory Th1 CD4 T cells are maintained in a mouse model of chronic malaria. PLoS Pathog (2010) 6(11):e1001208. doi:10.1371/journal.ppat.1001208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. da Silva HB, de Salles EM, Panatieri RH, Boscardin SB, Rodriguez-Málaga SM, Álvarez JM, et al. IFN-γ-induced priming maintains long-term strain-transcending immunity against blood-stage Plasmodium chabaudi malaria. J Immunol (2013) 191(10):5160–9. doi:10.4049/jimmunol.1300462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. McCall MBB, Sauerwein RW. Interferon-γ–central mediator of protective immune responses against the pre-erythrocytic and blood stage of malaria. J Leukoc Biol (2010) 88(6):1131–43. doi:10.1189/jlb.0310137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Villegas-Mendez A, Greig R, Shaw TN, De SOUZA JB, Gwyer Findlay E, Stumhofer JS, et al. IFN-γ-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J Immunol (2012) 189(2):968–79. doi:10.4049/jimmunol.1200688
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Haque A, Best SE, Ammerdorffer A, Desbarrieres L, de Oca MM, Amante FH, et al. Type I interferons suppress CD4+ T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol (2011) 41(9):2688–98. doi:10.1002/eji.201141539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Haque A, Best SE, Montes de Oca M, James KR, Ammerdorffer A, Edwards CL, et al. Type I IFN signaling in CD8- DCs impairs Th1-dependent malaria immunity. J Clin Invest (2014) 124(6):2483–96. doi:10.1172/JCI70698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Oakley MS, Sahu BR, Lotspeich-Cole L, Solanki NR, Majam V, Pham PT, et al. The transcription factor T-bet regulates parasitemia and promotes pathogenesis during Plasmodium berghei ANKA murine malaria. J Immunol (2013) 191(9):4699–708. doi:10.4049/jimmunol.1300396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Chandele A, Mukerjee P, Das G, Ahmed R, Chauhan VS. Phenotypic and functional profiling of malaria-induced CD8 and CD4 T cells during blood-stage infection with Plasmodium yoelii. Immunology (2011) 132(2):273–86. doi:10.1111/j.1365-2567.2010.03363.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Oakley MS, Sahu BR, Lotspeich-Cole L, Majam V, Thao Pham P, Sengupta Banerjee A, et al. T-bet modulates the antibody response and immune protection during murine malaria. Eur J Immunol (2014) 44(9):2680–91. doi:10.1002/eji.201344437
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Troye-Blomberg M, Riley EM, Kabilan L, Holmberg M, Perlmann H, Andersson U, et al. Production by activated human T cells of interleukin 4 but not interferon-gamma is associated with elevated levels of serum antibodies to activating malaria antigens. Proc Natl Acad Sci U S A (1990) 87(14):5484–8. doi:10.1073/pnas.87.14.5484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. von der Weid T, Kopf M, Köhler G, Langhorne J. The immune response to Plasmodium chabaudi malaria in interleukin-4-deficient mice. Eur J Immunol (1994) 24(10):2285–93. doi:10.1002/eji.1830241004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Linke A, Kühn R, Müller W, Honarvar N, Li C, Langhorne J. Plasmodium chabaudi chabaudi: differential susceptibility of gene-targeted mice deficient in IL-10 to an erythrocytic-stage infection. Exp Parasitol (1996) 84(2):253–63. doi:10.1006/expr.1996.0111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Langhorne J, Meding SJ, Eichmann K, Gillard SS. The response of CD4+ T cells to Plasmodium chabaudi chabaudi. Immunol Rev (1989) 112:71–94. doi:10.1111/j.1600-065X.1989.tb00553.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. von der Weid T, Langhorne J. The roles of cytokines produced in the immune response to the erythrocytic stages of mouse malarias. Immunobiology (1993) 189(3–4):397–418. doi:10.1016/S0171-2985(11)80367-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39(4):758–69. doi:10.1016/j.immuni.2013.08.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Mewono L, Matondo Maya DW, Matsiegui P-B, Agnandji ST, Kendjo E, Barondi F, et al. Interleukin-21 is associated with IgG1 and IgG3 antibodies to erythrocyte-binding antigen-175 peptide 4 of Plasmodium falciparum in Gabonese children with acute falciparum malaria. Eur Cytokine Netw (2008) 19(1):30–6. doi:10.1684/ecn.2008.0114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Mewono L, Agnandji ST, Matondo Maya DW, Mouima A-MN, Iroungou BA, Issifou S, et al. Malaria antigen-mediated enhancement of interleukin-21 responses of peripheral blood mononuclear cells in African adults. Exp Parasitol (2009) 122(1):37–40. doi:10.1016/j.exppara.2009.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Roetynck S, Olotu A, Simam J, Marsh K, Stockinger B, Urban B, et al. Phenotypic and functional profiling of CD4 T cell compartment in distinct populations of healthy adults with different antigenic exposure. PLoS One (2013) 8(1):e55195. doi:10.1371/journal.pone.0055195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Chen Z, Laurence A, O’Shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin Immunol (2007) 19(6):400–8. doi:10.1016/j.smim.2007.10.015
54. Keswani T, Bhattacharyya A. Differential role of T regulatory and Th17 in Swiss mice infected with Plasmodium berghei ANKA and Plasmodium yoelii. Exp Parasitol (2014) 141:82–92. doi:10.1016/j.exppara.2014.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Ishida H, Matsuzaki-Moriya C, Imai T, Yanagisawa K, Nojima Y, Suzue K, et al. Development of experimental cerebral malaria is independent of IL-23 and IL-17. Biochem Biophys Res Commun (2010) 402(4):790–5. doi:10.1016/j.bbrc.2010.10.114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Mastelic B, do Rosario APF, Veldhoen M, Renauld JC, Jarra W, Sponaas A-M, et al. IL-22 protects against liver pathology and lethality of an experimental blood-stage malaria infection. Front Immunol (2012) 3:85. doi:10.3389/fimmu.2012.00085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol (2009) 10(8):857–63. doi:10.1038/ni.1767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol (2009) 10(8):864–71. doi:10.1038/ni.1770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol (2011) 12(5):383–90. doi:10.1038/ni.2025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Koch O, Rockett K, Jallow M, Pinder M, Sisay-Joof F, Kwiatkowski D. Investigation of malaria susceptibility determinants in the IFNG/IL26/IL22 genomic region. Genes Immun (2005) 6(4):312–8. doi:10.1038/sj.gene.6364214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol (2012) 24(3):297–302. doi:10.1016/j.coi.2012.01.014
62. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood (2006) 107(12):4781–9. doi:10.1182/blood-2005-12-4818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Darrah PA, Patel DT, De Luca PM, Lindsay RWB, Davey DF, Flynn BJ, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med (2007) 13(7):843–50. doi:10.1038/nm1592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol (2008) 8(4):247–58. doi:10.1038/nri2274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Huaman MC, Mullen GED, Long CA, Mahanty S. Plasmodium falciparum apical membrane antigen 1 vaccine elicits multifunctional CD4 cytokine-producing and memory T cells. Vaccine (2009) 27(38):5239–46. doi:10.1016/j.vaccine.2009.06.066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Kastenmuller K, Espinosa DA, Trager L, Stoyanov C, Salazar AM, Pokalwar S, et al. Full-Length Plasmodium falciparum circumsporozoite protein administered with long-chain poly(I∙C) or the Toll-like receptor 4 agonist glucopyranosyl lipid adjuvant-stable emulsion elicits potent antibody and cd4+ t cell immunity and protection in mice. Infect Immun (2013) 81(3):789–800. doi:10.1128/IAI.01108-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Metenou S, Dembele B, Konate S, Dolo H, Coulibaly YI, Diallo AA, et al. Filarial infection suppresses malaria-specific multifunctional Th1 and Th17 responses in malaria and filarial coinfections. J Immunol (2011) 186(8):4725–33. doi:10.4049/jimmunol.1003778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol (2010) 11(8):674–80. doi:10.1038/ni.1899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Lu KT, Kanno Y, Cannons JL, Handon R, Bible P, Elkahloun AG, et al. Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro-generated and in vivo-derived follicular T helper cells. Immunity (2011) 35(4):622–32. doi:10.1016/j.immuni.2011.07.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Freitas do Rosário AP, Lamb T, Spence P, Stephens R, Lang A, Roers A, et al. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: a critical mechanism for protection from severe immunopathology during malaria infection. J Immunol (2012) 188(3):1178–90. doi:10.4049/jimmunol.1102755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Couper KN, Blount DG, Wilson MS, Hafalla JC, Belkaid Y, Kamanaka M, et al. IL-10 from CD4+CD25-Foxp3-CD127- adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog (2008) 4(2):e1000004. doi:10.1371/journal.ppat.1000004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Walther M, Jeffries D, Finney OC, Njie M, Ebonyi A, Deininger S, et al. Distinct roles for FOXP3+ and FOXP3- CD4+ T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog (2009) 5(4):e1000364. doi:10.1371/journal.ppat.1000364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Jagannathan P, Eccles-James I, Bowen K, Nankya F, Auma A, Wamala S, et al. IFNγ/IL-10 co-producing cells dominate the CD4 response to malaria in highly exposed children. PLoS Pathog (2014) 10(1):e1003864. doi:10.1371/journal.ppat.1003864
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Portugal S, Moebius J, Skinner J, Doumbo S, Doumtabe D, Kone Y, et al. Exposure-dependent control of malaria-induced inflammation in children. PLoS Pathog (2014) 10(4):e1004079. doi:10.1371/journal.ppat.1004079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi:10.1038/ni1539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. O’Garra A, Vieira PT. (H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol (2007) 7(6):425–8. doi:10.1038/nri2097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature (2013) 484(7395):514–8. doi:10.1038/nature10957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Levings MK, Sangregorio R, Galbiati F, Squadrone S, de Waal Malefyt R, Roncarolo MG. IFN-α and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol (2001) 166(9):5530–9. doi:10.4049/jimmunol.166.9.5530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Dikopoulos N, Bertoletti A, Kroger A, Hauser H, Schirmbeck R, Reimann J. Type I IFN negatively regulates CD8+ T cell responses through IL-10-producing CD4+ T regulatory 1 cells. J Immunol (2004) 174(1):99–109. doi:10.4049/jimmunol.174.1.99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Zhang L, Yuan S, Cheng G, Guo B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One (2011) 6(12):e28432. doi:10.1371/journal.pone.0028432
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Corre B, Perrier J, El Khouri M, Cerboni S, Pellegrini S, Michel F. Type I interferon potentiates T-cell receptor mediated induction of IL-10-producing CD4+ T cells. Eur J Immunol (2013) 43(10):2730–40. doi:10.1002/eji.201242977
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Illingworth J, Butler NS, Roetynck S, Mwacharo J, Pierce SK, Bejon P, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T Cell exhaustion. J Immunol (2013) 190(3):1038–47. doi:10.4049/jimmunol.1202438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol (2012) 13(2):188–95. doi:10.1038/ni.2180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Horne-Debets JM, Faleiro R, Karunarathne DS, Liu XQ, Lineburg KE, Poh CM, et al. PD-1 dependent exhaustion of CD8+ T cells drives chronic malaria. Cell Rep (2013) 5(5):1204–13. doi:10.1016/j.celrep.2013.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Cubas RA, Mudd JC, Savoye A-L, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med (2013) 19(4):494–9. doi:10.1038/nm.3109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol (2010) 40(7):1830–5. doi:10.1002/eji.201040391
87. Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev (2013) 252(1):52–77. doi:10.1111/imr.12029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Stephens R, Albano FR, Quin S, Pascal BJ, Harrison V, Stockinger B, et al. Malaria-specific transgenic CD4(+) T cells protect immunodeficient mice from lethal infection and demonstrate requirement for a protective threshold of antibody production for parasite clearance. Blood (2005) 106(5):1676–84. doi:10.1182/blood-2004-10-4047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Olszewski KL, Morrisey JM, Wilinski D, Burns JM, Vaidya AB, Rabinowitz JD, et al. Host-parasite interactions revealed by Plasmodium falciparum metabolomics. Cell Host Microbe (2009) 5(2):191–9. doi:10.1016/j.chom.2009.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kafsack BFC, Llinás M. Eating at the table of another: metabolomics of host-parasite interactions. Cell Host Microbe (2010) 7(2):90–9. doi:10.1016/j.chom.2010.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Cockburn IA, Amino R, Kelemen RK, Kuo SC, Tse S-W, Radtke A, et al. In vivo imaging of CD8+ T cell-mediated elimination of malaria liver stages. Proc Natl Acad Sci U S A (2013) 110(22):9090–5. doi:10.1073/pnas.1303858110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Painter HJ, Altenhofen LM, Kafsack BFC, Llinás M. Whole-genome analysis of Plasmodium spp. Utilizing a new Agilent technologies DNA microarray platform. Methods Mol Biol (2013) 923:213–9. doi:10.1007/978-1-62703-026-7_14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Tse S-W, Cockburn IA, Zhang H, Scott AL, Zavala F. Unique transcriptional profile of liver-resident memory CD8+ T cells induced by immunization with malaria sporozoites. Genes Immun (2013) 14(5):302–9. doi:10.1038/gene.2013.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Salinas JL, Kissinger JC, Jones DP, Galinski MR. Metabolomics in the fight against malaria. Mem Inst Oswaldo Cruz (2014) 109(5):589–97. doi:10.1590/0074-0276140043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Franke-Fayard B, Waters AP, Janse CJ. Real-time in vivo imaging of transgenic bioluminescent blood stages of rodent malaria parasites in mice. Nat Protoc (2006) 1(1):476–85. doi:10.1038/nprot.2006.69
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Tran TM, Samal B, Kirkness E, Crompton PD. Systems immunology of human malaria. Trends Parasitol (2012) 28(6):248–57. doi:10.1016/j.pt.2012.03.006
98. Ménard R, Tavares J, Cockburn I, Markus M, Zavala F, Amino R. Looking under the skin: the first steps in malarial infection and immunity. Nat Rev Microbiol (2013) 11(10):701–12. doi:10.1038/nrmicro3111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: malaria, Plasmodium, multifunctional CD4 T-cells, CD4 T-cell subsets, Tfh, Th1, Th2, Th22
Citation: Perez-Mazliah D and Langhorne J (2015) CD4 T-cell subsets in malaria: TH1/TH2 revisited. Front. Immunol. 5:671. doi: 10.3389/fimmu.2014.00671
Received: 03 September 2014; Accepted: 15 December 2014;
Published online: 12 January 2015.
Edited by:
Dragana Jankovic, National Institutes of Health, USAReviewed by:
Urszula Krzych, Walter Reed Army Institute of Research, USAMauricio Martins Rodrigues, Federal University of São Paulo, Brazil
David K. Cole, Cardiff University, UK
Copyright: © 2015 Perez-Mazliah and Langhorne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean Langhorne, Division of Parasitology, MRC National Institute for Medical Research, The Ridgeway, London NW7 1AA, UK e-mail:amxhbmdob0BuaW1yLm1yYy5hYy51aw==