 Anna von Rossum1
Anna von Rossum1 Jonathan C. Choy
Jonathan C. Choy- 1Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC, Canada
- 2Department of Anaesthesiology, Pharmacology and Therapeutics, University of British Columbia, Vancouver, BC, Canada
Solid organ transplantation is the only treatment for end-stage organ failure but this life-saving procedure is limited by immune-mediated rejection of most grafts. Blood vessels within transplanted organs are targeted by the immune system and the resultant vascular damage is a main contributor to acute and chronic graft failure. The vasculature is a unique tissue with specific immunological properties. This review discusses the interactions of the immune system with blood vessels in transplanted organs and how these interactions lead to the development of transplant arteriosclerosis, a leading cause of heart transplant failure.
Introduction
The success of organ transplantation as a curative therapy is hindered by the eventual failure of almost all grafts due largely to immune-mediated rejection (1). Also, transplant recipients need to take non-specific immunosuppressive drugs that are associated with many morbid side effects. A better understanding of how immune responses that are directed toward foreign organ grafts cause transplant failure is needed to develop strategies that specifically prolong survival and increase quality of life of graft recipients.
By its nature, organ transplantation results in the exposure of the immune system to an abundance of foreign antigens associated with inflammation, the former being a result of genetic differences between organ donors and recipients and the latter being caused by ischemic and mechanical damage during the transplantation procedure. The result is that T and B cells of the adaptive immune system specifically recognize graft-derived antigens (an alloimmune response) and become activated to elicit effector responses that reject transplanted organs. T cells recognize graft-derived peptides bound to major histocompatibility complexes [pMHCs; human leukocyte antigens (HLAs) in humans] expressed by graft cells and recipient antigen-presenting cells (e.g., dendritic cells and macrophages). This leads to the induction of cell- and antibody-mediated alloimmune responses (2, 3). Alloimmune-mediated arterial injury and dysfunction causes the development of transplant arteriosclerosis (TA), a vascular occlusive condition that causes ischemic graft failure. TA is prevalent in all solid organ transplants and is the main challenge in heart transplants because its incidence has not been diminished by advancements in current immunosuppressive drug therapies despite their ability to prevent acute rejection (1, 4). This may be because the immune response in arteries has unique features that necessitate distinct approaches for intervention. We review the current knowledge on the mechanisms by which alloimmune responses lead to vascular cell injury and dysfunction, the alarmin molecules released in response to alloimmune-mediated cell injury, and how these processes drive the development of TA.
Immune Targeting of Vascular Cells
In response to T cell recognition of pMHC molecules, T cells undergo rapid activation, proliferation, and differentiation into effector cells. Effector CD8 T cells are equipped to specifically induce cell death of target cells by expressing cytotoxic molecules, such as granzymes and perforin, which are contained within cytotoxic granules. Death ligands, such as FasL and TRAIL, are also expressed on CD8 T cell membranes (5–8). In addition to cytotoxic effector mechanisms, CD8 T cells also secrete interferon-γ (IFNγ), which induces cellular changes that lead to tissue remodeling. Effector properties of CD4 T cells mainly involve the production of cytokines to induce inflammation and that alter cell function in tissues. Three main types of CD4 effector T cells have been described in peripheral tissues: Th1 (that produce IFNγ), Th2 (that produce IL-4, IL-5, and IL-13), and Th17 (that produce IL-17, IL17F, IL-21, and IL-22) (9). Follicular helper T cells (Tfh; that produce IL-21 and express ICOS and CXCR5) reside within lymphoid tissues and control antibody production and class switching (10). Finally, antibodies that are secreted by B cells bind antigens within tissues and damage cells through complement-mediated cell lysis, activation of inflammation, and antibody-dependent cell cytotoxicity that is mediated by natural killer (NK) cells (11).
In addition to providing a conduit for tissue oxygenation and delivery of nutrients, blood vessels also interact intimately with the immune system to control the outcome of immune responses. Endothelial changes in the microvasculature are essential for leukocyte migration into sites of inflammation. Human endothelial cells (ECs) also basally express MHC class I and II molecules and up-regulate both molecules in response to inflammatory cytokines such as IFNγ and TNF, thereby enabling them to directly present alloantigens to T cells and be targeted by alloreactive effector T cells (12–16). Further, this vascular cell type expresses a variety of co-stimulatory molecules and, as such, human ECs are able to directly activate alloreactive memory T cells within the vessel wall (17). In addition to ECs, most arteries also contain resident dendritic cells that elicit immune activation after activation by inflammatory stimuli (18–20). Importantly, animal models have established that antigen presentation by both human ECs and arterial dendritic cells activate immunopathological T cell responses within arteries that lead to arteriosclerotic thickening (21–23). In contrast to activating T cells, the endothelium also expresses some immunoregulatory cell surface and soluble factors that inhibit effector T cell responses and some types of dendritic cells induce tolerance to arterial antigens (19, 24–29). Vascular smooth muscle cells within arteries also interact with T cells. This vascular cell type basally expresses MHC class I molecules and can be induced to express MHC class II molecules in response to inflammatory cytokines (30). T cell recognition of alloantigens presented by vascular smooth muscle cells attenuates T cell activation through the production of indoleamine 2,3-dioxygenase (IDO) and a lack of co-stimulatory molecule expression, implying that vascular smooth muscle cells possess properties that may define immunoprivilege-like status in arteries (31–33). All together, the distinct combination of immune-stimulatory and immune-regulatory features of vascular cells and artery-associated dendritic cells may define the uniqueness of immune responses in blood vessels.
Once activated by alloantigens, immune targeting of the graft vasculature occurs through processes mediated by cytotoxic CD8 T cells, effector CD4 T cells, and B cell-derived antibodies. Cytotoxic T cells induce EC death through a granzyme/perforin mechanism that is inhibited by Bcl-2. Moreover, granzyme B and perforin are sufficient to induce rapid cell death of human ECs in vitro and granzyme B alone is capable of inducing EC death in a delayed fashion by proteolyzing extracellular proteins required for adhesion-mediated cell survival (34–38). With regard to death receptors, vascular ECs express low levels of Fas and are relatively resistant to FasL-mediated apoptosis due to their expression of c-FLIP, which is an endogenous inhibitor of caspase-8 (39, 40). However, IFNγ and oxidized low-density lipoproteins (which are present in human TA lesions) sensitize ECs to Fas-mediated cell death by down-regulating expression of c-FLIP (41–43). The death ligand TRAIL, which is expressed by some types of T and NK cells, induces EC death in vitro so may also induce EC death in certain inflammatory settings (44). In addition to cytotoxic T cells, ECs also activate alloreactive CD4 T cells, which lead to the production of mainly IFNγ and IL-2, although a small subset of T cells produce IL-17 (22, 45).
B cell responses contribute to allograft injury through the production of graft-reactive antibodies (46). The presence of anti-donor antibodies is associated with a high rate of rejection and poor long-term outcome (47, 48). The histological description of antibody-mediated rejection (AMR) is vascular in nature including morphological changes to the microvascular endothelium, such as EC swelling, and the intravascular accumulation of monocytes. The observation of complement deposition in the vascular compartment of biopsies adds additional prognostic value (49). Foreign HLA molecules are the predominant antigens recognized by pathologic antibodies in the setting of transplantation but some non-HLA molecules are also targeted (50–53).
There are several cellular mechanisms by which antibodies can cause pathological changes in ECs. One of the main effector processes triggered by antibodies is complement activation. The presence of complement-binding anti-HLA antibodies is associated with extremely poor kidney graft survival as compared with the presence of non-complement-binding antibodies or the absence of donor anti-HLA antibodies (54). Also, grafts and/or recipients that are unable to activate complement fail to reject grafts in preclinical models, and therapeutic inhibition of complement with blocking antibodies prevents acute AMR in preclinical studies and clinical trials (55–59). Although vascular deposition of complement is used as a diagnostic feature of AMR, complement-mediated lysis of ECs is rarely observed (60, 61). Instead, membrane deposition of the membrane attack complex of the complement cascade augments immune responses by increasing inflammation and supporting the activation of T cells by the endothelium (62). The complement fragments C3a and C5a also have pro-inflammatory effects that increase the ability of antigen-presenting cells to activate alloreactive T cells, which oppose the induction of regulatory T cells, and that directly amplify the activation of effector T cells (63, 64).
Binding of antibodies to HLA antigens on ECs also initiates complement-independent processes that cause phenotypic changes in vascular cells. Cross-linking of HLA I molecules by antibodies triggers the downstream activation of Rho kinase and ERK1/2 signaling pathways (65). This leads to phenotypic changes that include cell proliferation, survival, and migration (66–68). HLA cross-linking also induces the rapid cell surface presentation of P-selectin and secretion of von Willebrand factor, which increases transendothelial migration of leukocytes (69, 70). Other effects of HLA cross-linking include up-regulation of cell adhesion molecules such as ICAM, chemokines such as IL-8 and RANTES, and cytokines such as IL-6 (71). This could result in prolonged activation of the endothelium that supports leukocyte recruitment and chronic inflammation.
In the discussion above, we have introduced the mechanisms by which alloimmune responses damage the graft vasculature. The effect on transplantation of these processes depends on whether the microvasculature or macrovasculature is affected. Microvascular injury results in hemorrhage and thrombosis, thereby causing ischemic graft damage that leads to acute graft failure or chronic fibrosis (72, 73). EC death also results in the release of fibrotic factors that can directly drive tissue fibrosis (74, 75). Macrovascular damage of arteries and arterioles triggers the development of TA, as discussed below (76).
Immunopathological Mechanisms in TA
Transplant arteriosclerosis is characterized by intimal hyperplasia and vasomotor dysfunction that develops as a result of immunological targeting of vascular endothelial and smooth muscle cells. The intima in TA is formed by the accumulation of smooth muscle cells, CD4 and CD8 T cells, B cells, macrophages, dendritic cells, and occasional NK cells. Structurally, there is concentric intimal expansion, alteration of extracellular matrix composition, aberrant lipid deposition, and intraplaque hemorrhage (77–79). In addition to intimal thickening, vasodilatory function is compromised in allograft arteries (80). The combination of intimal thickening and vasodilatory dysfunction occludes the arterial lumen, resulting in reduced blood flow and ischemic damage of downstream tissues (81).
It is clear that the development of TA is driven by alloimmune targeting of the graft vasculature because intimal thickening is confined to the graft and does not develop in experimental models in which grafts are placed in genetically identical animals or in recipients that lack adaptive immune responses (82, 83). Targeting of graft arteries by T cells and antibodies causes the development of intimal thickening through the induction of vascular cell injury, and cytokine- and antibody-mediated alteration of graft vascular cell phenotypes. T cells also cause vasoregulatory dysfunction of allograft arteries. These processes are discussed below and summarized in the Table 1.
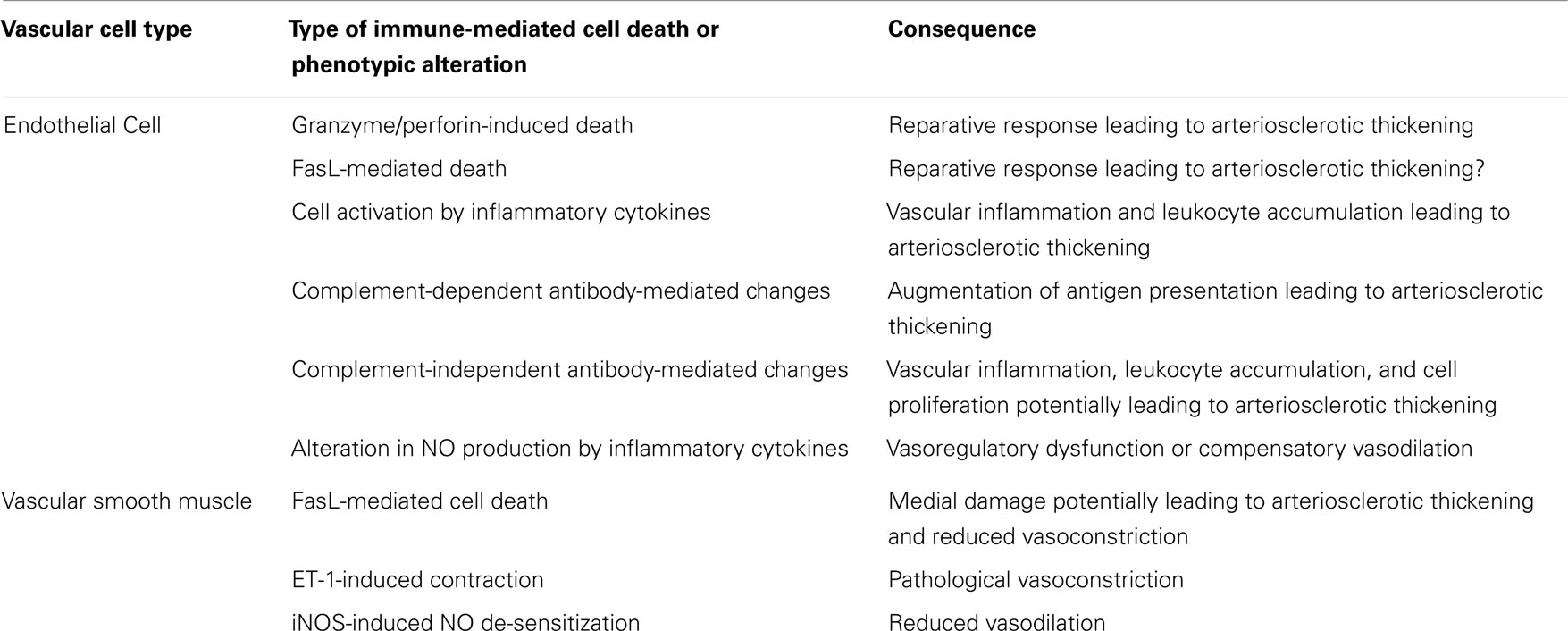
Table 1. Immunological effects on vascular cells and their consequences to arterial structure and function in TA.
Vascular Cell Injury in TA
Elegant studies in the 1970s and 1980s examining the response of arteries to mechanical injury demonstrated that vascular damage can initiate a “response-to-injury” process that culminates in the development of arteriosclerotic thickening (84). In these models, intimal thickening is caused by vascular damage that triggers the migration of leukocytes and platelets to regions of injury. The resultant production of cytokines and growth factors, such as PDGF and bFGF, by infiltrating leukocytes and injured vascular cells in turn stimulates smooth muscle cells to migrate into the intima and proliferate, thereby forming the nexus of hyperplastic intimal thickening (85–91). In addition to triggering the production of growth factors from infiltrating leukocytes and neighboring vascular cells, EC death also increases smooth muscle cell accumulation through the caspase-mediated generation of a bioactive fragment of the cell matrix protein perlecan, which inhibits smooth muscle cell death (52, 92). The concept that arteriosclerotic changes are driven by a reparative response in the arterial wall was proposed to be generalizable to several forms of vascular occlusive diseases, including TA (93).
Clinical and experimental findings show that immunologic injury of endothelial and/or smooth muscle cells is a main trigger for the development of TA. Detailed histopathological analysis of clinical specimens of TA initially identified the presence of apoptotic luminal ECs in these arteries and cytotoxic T cells expressing perforin were present in the subendothelial space immediately underlying dying ECs (94–96). Granzyme B and FasL were also abundant in the intima of allograft arteries with TA and their presence was correlated with increased vascular cell death (94, 97).
Examination of clinical specimens of TA provides valuable insight into potential pathological mechanisms driving disease development. Pairing such observations with experimental investigations is needed to establish causative processes. Consistent with a role for EC death in the initiation of TA, early endothelial disruption characterized by missing cells, intracellular gaps, and exposed extracellular matrix is observed in arteries from heterotopic rat heart transplants very early after transplantation (98). Experiments studying the transplantation of grafts across a complete MHC barrier established a role for CD4 T cells and B cells in TA but CD8 T cells do not appear to be needed in these models (82, 83). However, it is clear in humans that many grafts reject through antibody-independent mechanisms that are mediated by T cell effector processes. The transplantation of grafts across minor histocompatibility antigen mismatch barriers leads to immunological rejection of grafts that depends on T cells but not antibodies (99). In these models, the development of TA is triggered by EC death induced by CD8 T cells, perforin, and granzyme B suggesting that this type of cytotoxic T cell response is primarily responsible for the induction of EC death in TA (Figure 1A) (37, 100–102).
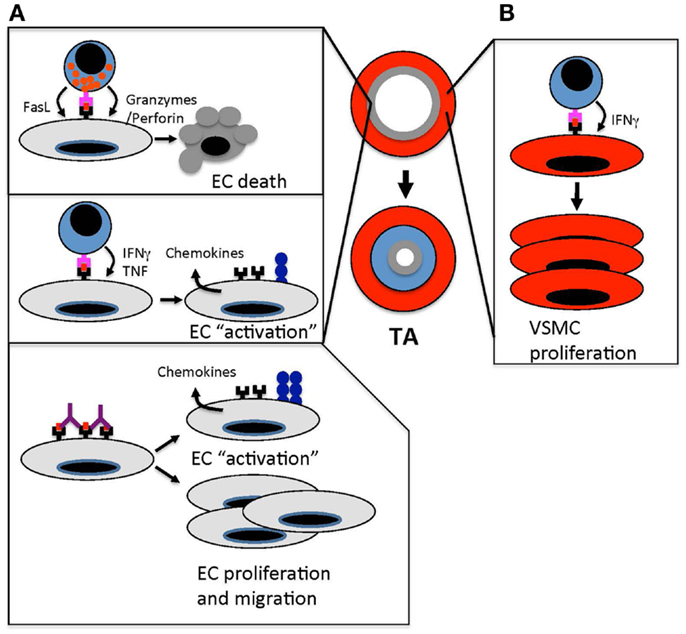
Figure 1. Immune-mediated vascular changes that cause TA. (A). Effector T cells target ECs in arteries of transplanted organs. Cytotoxic T cells kill arterial ECs through granzyme/perforin- and FasL-mediated pathways. This endothelial damage triggers a “response-to-injury” process that involves leukocyte and smooth muscle cell migration into the arterial intima that drives intimal thickening and occludes the arterial lumen. In addition to cytotoxic T cell responses, T cell recognition of allogeneic ECs results in the secretion of effector cytokines, such as IFNγ and TNF, which “activates” the endothelium to up-regulate MHC class I and II, cell adhesion molecules, and chemokines. These changes amplify the recruitment of leukocytes into allograft arteries. Anti-MHC antibodies amplify immune responses in allograft arteries by cross-linking MHC molecules, which induces cell signaling pathways that up-regulate adhesion molecules and von Willebrand factor. This enhances leukocyte transendothelial migration. Anti-MHC antibodies also induce proliferation and migration of ECs, which could trigger the remodeling of allograft arteries. (B) The secretion of IFNγ from effector T cells stimulates the proliferation of vascular smooth muscle cells in the intima of allograft arteries. This increases the accumulation of vascular smooth muscle cells and intimal thickening.
Models of arteriosclerosis also suggest that medial smooth muscle cell injury can trigger or promote the development of intimal thickening (103–105). Medial smooth muscle cell death is observed in some models of arterial vascular rejection and TA, and depletion of CD8 T cells or blockade of the Fas/FasL pathway prevents medial smooth muscle cell death and intimal thickening (106, 107). Inflammatory cytokines, such as IFNγ, sensitize smooth muscle cells to FasL by relocating the Fas receptor to the cell surface (108, 109). Finally, a mechanism by which vascular smooth muscle cell death in allograft arteries triggers intimal thickening may be through the induction of stromal cell-derived factor-1 (CXCL12) production by dying and neighboring cells, which initiates the migration and proliferation of mesenchymal stem cells into the intima (110).
In addition to triggering a pathological reparative response that initiates intimal thickening, cell injury results in the release of alarmins that stimulate inflammation and immune activation. This serves to initiate or propagate immunological responses and, as such, could contribute to immunopathology. Alarmins are intracellular molecules that bind pattern recognition receptors, such as toll-like receptors (TLRs), to activate antigen-presenting cells and vascular cells (111). They also stimulate the recruitment of antigen-presenting cells to sites of injury or infection. Several alarmins have been implicated in the regulation of allogeneic responses and these include HMGB1, IL-1α, endogenous RNA and DNA, and IL-33.
HMGB1 is a chromatin binding protein that is universally expressed in cells. It is released into the extracellular space after necrotic cell death but can also be secreted in a regulated manner by macrophages. It binds to TLR4 and receptor of advanced glycation end-products (RAGE) to activate the immune stimulating properties of antigen-presenting cells (112). HMGB1 is released by dying ECs and this promotes allogeneic T cell responses by inducing the release of IL-1β from monocytes and IL-1α from neighboring ECs (113). Systemic blockade of extracellular HMGB1 prevents the development of chronic heart transplant failure and TA in a mouse model (114). IL-1α is also released from dying ECs in allograft arteries in response to ischemic and immune-mediated damage whereupon it promotes the development of intimal thickening (22). Nuclei acids are another type of endogenous molecule that is released after cellular damage and that act as alarmins. DNA and RNA from pathogens can be recognized by various TLRs as well as the STING pathway in the cytosol for DNA and the MDA5/RIG-I cytosolic RNA receptors (115). Although TLR7/8/9 may also be able to recognize self-RNA and -DNA Tellides and colleagues (116) showed that self-RNA is detected by vascular smooth muscle cells primarily through the MDA5/RIG-I pathway and that this augments inflammation within human coronary arteries.
The above examples highlight the role of alarmins in the activation of allogeneic immune responses and development of TA. Recent evidence has established a novel role for the alarmin IL-33 in preventing allogeneic immune responses, cardiac transplant failure, and TA. IL-33 is expressed in non-hematopoeitic cells including ECs, is released after cellular damage, and has been established to promote the activation of protective immune responses following virus infection (117). In contrast to the role of IL-33 in promoting immune activation in response to pathogen infection, several groups have made the unexpected observation that it prevents cardiac transplant rejection and TA (118–120). Although it promotes the development of Th2 responses, the mechanism by which IL-33 is protective in transplantation is likely through the generation of suppressive myeloid cells and T regs (120, 121).
Cytokine-Mediated Alteration of Graft Vascular Cell Phenotypes
During immune responses, the endothelium is “activated” by cytokines to express cell adhesion molecules and chemokines that facilitate the recruitment of leukocytes from the blood into tissues. During this process, the endothelium also undergoes morphological changes that increase vascular permeability to plasma proteins (122). These vascular changes are essential for the development and localization of immune responses. Organ grafts are characterized by heightened inflammation and immunity resulting from ischemic and surgical damage as well as from immunological targeting of the graft. The resultant production of cytokines alters the function and phenotype of vascular cells and, in this way, remodels blood vessels (123).
Profiling of the immune response in clinical samples of TA has revealed a predominance of Th1 cytokines and associated chemokines (124). These findings suggest a pathological role of Th1 cytokines, of which IFNγ is the prototypical member, in vascular changes associated with TA. Indeed, experimental studies confirm that IFNγ has an unequivocal role in the development of TA (125, 126). IFNγ has broad-ranging effects on both ECs and vascular smooth muscle cells (Figures 1A,B). Stimulation of ECs with IFNγ up-regulates the cell surface expression of MHC class I and II molecules. This enhances the activation of T cells by graft ECs as well as the recognition and targeting of blood vessels by effector T cells (12, 127). The induction of chemokines, such as IP-10, by IFNγ supports the migration of T cells into allograft arteries (128). IFNγ also increases EC susceptibility to FasL-mediated cell death (39, 129, 130). IFNγ signaling in smooth muscle cells is also important in the development of intimal thickening, as supported by findings that IFNγ is sufficient to cause intimal thickening by promoting vascular smooth muscle cell mitogenesis in a humanized mouse model of arteriosclerosis (131). Recent studies indicate that IFNγ-stimulated smooth muscle cell proliferation is mediated by PI3K activation of mammalian target of rapamcyin (mTOR) and attenuated by ASK1-interacting protein 1, which is a Ras GTPase-activating protein family member that antagonizes JAK-STAT signaling (132, 133).
The inflammatory cytokines IL-1β, IL-1α, and TNF are also abundant in TA lesions and all are induced early after transplantation (134–136). These cytokines activate similar signaling pathways, have overlapping effects on vascular cells (137, 138), and contribute to the development of TA in animal models (22, 139). Distinctions in the effects of these cytokines may arise from differences in their cell source within arteries. While IL-1β and TNF are mainly expressed by infiltrating or tissue resident macrophages, TNF is secreted by some T cells and IL-1α is released mainly from injured parenchymal and vascular cells (140). Both TNF and IL-1 induce the rapid up-regulation of MHC molecules on ECs and support the transmigration of leukocytes into allograft arteries (137, 138). TNF can be cytotoxic to vascular cells in some situations (141). IL-17 is another inflammatory cytokine that has been implicated in the development of TA (142, 143). Both TNF and IL-17 activate similar cell signaling pathways. However, IL-17 by itself has little effect on inflammatory “activation” of ECs but it synergizes with TNF to up-regulate cell adhesion molecules and facilitate leukocyte transendothelial migration (144). Graft infiltrating Th17 cells have been associated with increased chronic allograft failure and lymphoid neogenesis in kidney transplantation, suggesting that Th17 responses that produce IL-17 could augment alloimmune responses locally within the graft (145). Although TA is a component of chronic kidney graft failure, other pathogenic mechanisms are also involved so future studies need to be performed to determine the potential effect of Th17 cells on lymphoid neogenesis as it relates specifically to the development of TA.
Antibody-Mediated Alteration of Graft Vascular Cell Phenotypes
Due to their polymorphic nature and cell surface abundance, donor HLA molecules are a major class of antigens recognized by antibodies in graft recipients (146, 147). The presence of HLA reactive antibodies predicts the development of TA and studies in mouse models show that anti-MHC class I antibodies are sufficient to induce the development of this vascular condition (148–151). These antibodies may drive the development of TA by triggering phenotypic changes, such as increased proliferation and migration, in endothelial and smooth muscle cells (66, 152). These phenotypic changes could lead to structural alterations in arteries that remodel the vessel wall (153). In addition, anti-HLA antibodies amplify the immunogenic properties of the endothelium by up-regulating cell adhesion molecules and von Willebrand factor, which facilitates immune cell transmigration into the arterial wall. Anti-HLA antibodies also amplify T cell responses toward allograft arteries by increasing the antigen-presenting capabilities of the endothelium through a complement-dependent mechanism (Figure 1A) (62, 69, 154). NK cells have also been implicated in the development of TA through an antibody-mediated mechanism (151).
Some graft recipients also develop graft-reactive antibodies toward non-HLA molecules. This likely arises from an aversion of tolerance stemming from broad-scale inflammation that triggers activation of autoreactive lymphocytes and/or that creates neo-antigens through the proteolytic cleavage of self-proteins. Non-HLA antibodies that target vascular cells bind to vimentin, a novel fragment of perlecan termed LG3, and angiotensin II type 1 receptor (51–53). Increased levels of antibodies reactive to all mentioned antigens correlate with poor graft outcome in humans and these antibodies induce or accelerate TA and/or vascular dysfunction in animal models (52, 155–157).
Vasoregulatory Dysfunction in Allograft Arteries
Besides intimal thickening, another change in allograft arteries that ultimately drives ischemic graft failure is vasomotor dysfunction. The vascular endothelium is essential for regulating arterial vasomotor function, acting on vascular smooth muscle cells to control the dilation and constriction of blood vessels (158–160). The balance between vasodilatory and vasoconstrictive factors, as well as the inherent myogenic properties of the smooth muscle, determines blood flow through arteries. This balance is disturbed in allograft arteries (Figure 2) (161–163).
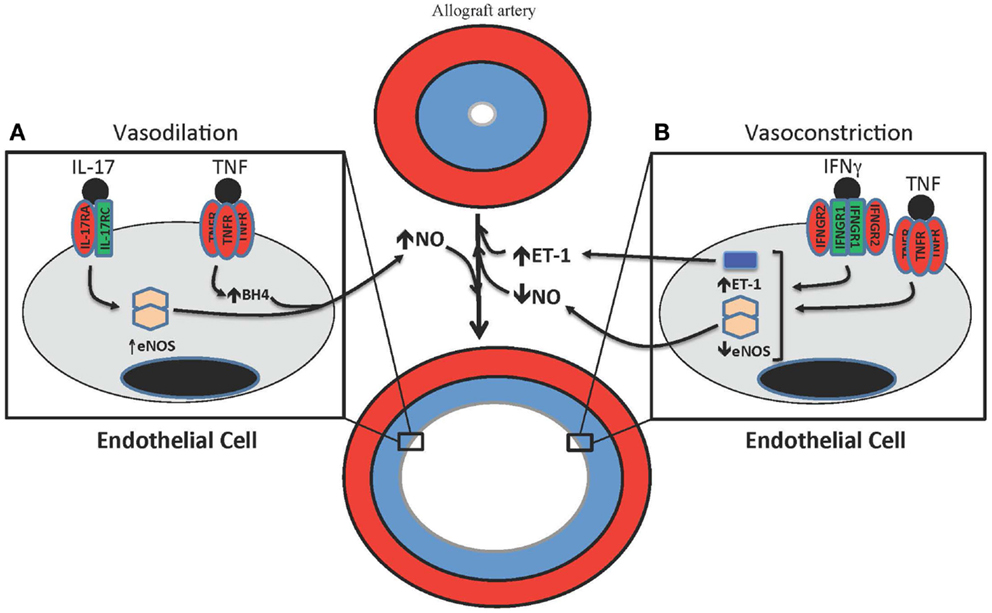
Figure 2. T cell-mediated effects on vasodilation and vasoconstriction in allograft arteries. (A) TNF and IL-17 are produced by T cells in allograft arteries. TNF acutely increases NO production from the endothelium by increasing eNOS activity through the up-regulation of tetrahydrobiopterin (BH4) synthesis. IL-17 increases NO production by increasing expression of eNOS. (B) IFNγ and TNF contribute to the vasoconstriction of allograft arteries by inhibiting the expression of eNOS, which reduces the levels of bioactive NO, as well as by increasing the production of the vasoconstrictive peptide ET-1.
Immunological effects on vasodilation
Nitric oxide is an important endothelial-derived factor that induces arterial dilation (164). In blood vessels, this bioactive gas is produced by endothelial nitric oxide synthase (eNOS) expressed in the endothelium (165–168). Human Th1 effector CD4 T cells inhibit nitric oxide (NO) production from ECs by attenuating eNOS expression through the effects of both IFNγ and TNF (169, 170). This attenuation of eNOS expression by T cells compromises vasodilation, thereby providing an explanation for the early endothelial dysfunction observed after transplantation (171). Arterial dilation is also affected in allograft arteries by the production of NO by T cells that express inducible NOS (iNOS). NO production through this mechanism desensitizes smooth muscle cells to NO-mediated relaxation (170, 172, 173).
Th17 cells have been implicated in vascular changes that occur in TA (143), prompting us to investigate the effect of IL-17 on eNOS expression in ECs. IL-17 increased eNOS expression and NO production by human ECs through the coordinated activation of NF-κB, MEK1, and JNK signaling pathways. Further, eNOS expression was significantly correlated with increased levels of IL-17 in clinical specimens of TA and the abundance of this cytokine correlated with increased lumen size, but not intimal thickening, and increased eNOS expression (174). These data suggest that IL-17 could support outward arterial expansion in TA although a study examining the effects of IL-17 neutralization in a humanized model of allograft artery rejection did not observe any effects on total vessel area diameter (22). Also, IL-17 has been reported to reduce eNOS activity in mouse ECs and to contribute to hypertension in mouse models (175, 176). Additional studies are needed to determine the exact role of IL-17 in vascular changes and how this relates to TA.
Endothelial regulation of vasodilation is dependent on NOS co-factor availability. The “uncoupling” of eNOS that occurs when co-factors are limiting results in reduced NO bioactivity and a concomitant increase in reactive oxygen species (177). Both effects exacerbate pathological constriction of arteries in the presence of immunological responses. Interestingly, stimulation of human ECs with TNF, IL-1, or IFNγ augments eNOS activity by increasing tetrahydrobiopterin (BH4) levels through the induction of GTP cyclohydrolase I, the rate limiting enzyme in the BH4 synthesis pathway (178, 179). T cells also express GTP cyclohydrolase I and produce BH4 after activation through the T cell receptor (180, 181). It seems paradoxical that TNF and IFNγ both inhibit eNOS expression and increase its enzymatic activity, but it is possible that the induction of BH4 biosynthesis by these cytokines may support the recovery of endothelial-dependent vasodilation during inflammation (182).
Immunologic effects on vasoconstriction
The production of endothelium-derived constrictor factors (EDCFs) is also a key regulatory component of arterial vasomotor function. A discussion of EDCFs tends to be focused on endothelin (ET-1) since it is one of the most potent endogenous vasoconstrictors produced in humans and there are clear clinical indications for pathophysiological roles of ET-1, including in TA (183–186). TNF, IL-1, IFNγ, and IL-6 induce ET-1 production by ECs (187, 188).
Small arteries in humans and other mammals exist in a state of partial constriction. This allows a basal level of endothelial-independent arterial tone against vasodilators and vasoconstrictors that can act to influence arterial diameter and resultant blood flow. Allogeneic immune responses induce smooth muscle cell death in the media of allograft arteries and this likely reduces the functional properties of arteries (107). In a rat model of TA, myogenic constriction and dilation were both compromised at late time-points indicating that immune-mediated medial damage and dysfunction are prevalent (189). The loss of pressure-induced myogenic constriction in cardiac allografts could increase intravascular hydrostatic pressure and concomitant fluid leakage into the interstitial space, leading to loss of ventricular compliance and organ failure. Immunosuppression with cyclosporine reduces medial smooth muscle cell death and preserves myogenic activity (190).
Cytoprotective and Immunoregulatory Features of the Graft Vasculature
The expression of cytoprotective and immunoregulatory proteins by tissue cells is essential for preventing pathological tissue damage and resultant immunopathology that can occur during immune responses toward pathogens. Similar processes may also be operational in a transplant setting and, as such, are pertinent to any discussion of alloimmune-mediated vascular injury. Profiling gene expression in non-rejecting or tolerized grafts has identified the increased expression of “cytoprotective” genes A20, hemeoxygenase-1 (HO-1), Bcl-xL, and Bcl-2 in non-rejecting grafts, suggesting a role for them in accommodating graft survival (191, 192).
A20 is a TNF-inducible zinc finger protein expressed by ECs and vascular smooth muscle cells (193). It inhibits EC death induced through both death receptor- and mitochondria-mediated mechanisms by preventing caspase activation and cytochrome c release from the mitochondria (194). As such, A20 may be able to prevent most forms of immune-mediated EC death that are operational in allogeneic responses. In addition to being cytoprotective in ECs, A20 is also able to reduce inflammatory responses by inhibiting NF-κB activation (195). This prevents the up-regulation of cell adhesion molecules and chemokines in ECs that facilitate leukocyte transendothelial migration into allografts. In vascular smooth muscle cells, A20 is anti-inflammatory through the inhibition of NF-κB activation but it also promotes cell death of intimal vascular smooth muscle cells, which is in contrast to its cytoprotective effects in ECs (196). All together, the overlapping but distinct functions of A20 in ECs and vascular smooth muscle cells may inhibit intimal thickening by preventing EC damage and ameliorating vascular smooth muscle cell accumulation in the intima. Indeed, A20 expression in vessel wall cells correlates with protection against TA and overexpression of A20 in donor artery segments in a mouse model of TA reduces the development of this vascular occlusive condition (197, 198).
HO-1 is an enzyme that catalyzes the conversion of heme to free iron, biliverdin, and carbon monoxide (199). It is expressed in several cells, including ECs, and has cytoprotective and immune-inhibitory effects. In transplantation, some studies have shown an association of HO-1 gene promoter polymorphisms with better kidney graft function and survival, although others have failed to observe this association (200–202). There is no apparent association between HO-1 gene promoter polymorphisms and TA in cardiac transplants (203). In preclinical studies, HO-1 in allografts has been shown to mediate graft survival (204). Importantly, the expression of HO-1 in vascular cells prevents the development of TA (205, 206). Experimental evidence further indicates that HO-1 prevents EC death, inhibits inflammatory responses, and attenuates adaptive immune responses (207). These effects are mediated through the actions of CO and biliverdin, which can prevent NF-κB and NFAT activation and may induce activation-induced cell death of T cells (208, 209).
In addition to the cytoprotective genes described above, graft vascular cells can also be induced to express immunoregulatory genes that are known to play a role in tolerance induction. These include IDO, programed cell death ligand-1 (PD-L1), and immunoglobulin-like transcript-3 and -4 (ILT3/4). IDO is an enzyme that degrades tryptophan, resulting in the release of kynurenines. The depletion of local levels of tryptophan can inhibit the proliferation of immune cells and kynurenines can actively inhibit immune cell activation (210). The induction of IDO by IFNγ in both ECs and vascular smooth muscle cells inhibits the activation of allogeneic T cells (211). IDO expression in ECs is also responsible for the development of cardiac allograft tolerance induced by the deoxyspergualine analog LF15-0195 in a rat model and its expression in vascular smooth muscle cells prevents alloimmune-mediated medial damage (32, 212). PD-L1 is also induced by IFNγ in ECs and vascular smooth muscle cells, and it inhibits the activation of effector T cell responses by binding to PD-1 on T cells (24, 213). The PD-L1/PD-1 system is an essential component of peripheral tolerance as indicated by the development or exacerbation of autoimmune-like manifestations in mice that lack PD-1 (214). In allografts, the expression of PD-L1 on arterial ECs reduces the development of TA (26, 215). Finally, Sucia-Foca and colleagues have identified the up-regulation of ILT3 and ILT4 on human ECs by CD8 T suppressor cells and IL-10, and established that these molecules inhibit allogeneic T cell activation (28, 216). The mechanism by which ILT3/4 on ECs is immunoregulatory remains to be fully elucidated but may involve the inhibition of T cell co-stimulatory signals (29).
Therapeutic Opportunities in TA
Therapeutic prevention of TA requires a combination of strategies to specifically inhibit immune responses and to directly prevent hyperplastic responses of vessel wall cells. Non-specific immunosupression with cyclosporine is a mainstay in heart transplantation but the eventual failure of most grafts and the association of this drug with side effects necessitate the need for improved approaches. The “holy grail” of transplantation therapy is the induction of tolerance to specifically prevent immune activation toward transplanted grafts while maintaining protective immunity. Many preclinical studies demonstrate the induction of tolerance and prevention of TA using co-stimulatory blockade (217–219). In clinical studies, co-stimulatory blockade with CTLA4-Ig is effective for preventing kidney graft rejection although functional tolerance does not appear to develop (220). Similar studies have not been performed in heart transplant recipients to evaluate TA specifically. Clinical trials also suggest that it may be possible to induce tolerance toward kidney grafts by concurrently performing bone marrow transplantation that induces transient chimerism, although studies in non-human primates show that this approach may not lead to tolerance of heart grafts (221–223). A promising approach for the specific prevention of TA by immunomodulation has been established in a humanized mouse model of arterial rejection in which the delivery of ex vivo-expanded regulatory T cells, which suppress effector T cell responses, can prevent arterial remodeling reflective of TA (224, 225).
In addition to preventing immune activation, anti-proliferative drugs such as mTOR inhibitors inhibit smooth muscle proliferation and resultant intimal thickening in TA. These inhibitors first found use in the prevention of intimal hyperplasia after restenosis (226, 227). Studies in heart transplantation subsequently established that mTOR inhibition with everolimus reduced immune activation as well as intimal thickenining in TA through inhibiting smooth muscle cell proliferation in allograft arteries (228). The inclusion of everolimus in the immunosuppression regimen can reduce the dose of cyclosporine needed and, as a consequence, reduce cyclosporine-associated renal damage (229). Other studies have examined the ex vivo modification of allograft artery cells using viral transduction for the prevention of TA since it is likely that inhibition of vascular cell death by forced expression of protective genes, such as A20, can reduce the development of TA in preclinical models (198). Although such strategies seem ideal for therapies involving the ex vivo modification of graft cells prior to transplantation, such therapeutic approaches yet to be translated into a clinical setting.
Conclusion
Blood vessels possess unique immunological features that define the outcome of immune responses. Cytotoxic damage of vessel wall cells and the alteration of vascular cell phenotypes by different components of the allogeneic immune response drives the remodeling of arteries in transplanted organs and this culminates in the development of TA. Further understanding these pathogenic mechanisms will be essential for future advancements that are able to specifically prevent immune activation toward allograft blood vessels and the hyperplastic responses of vessel wall cells. Also, similar immunopathological mechanisms that contribute to the development of TA are also involved in other immune-mediated arteriosclerotic conditions, such as giant cell arteritis and atherosclerosis, so insights obtained from studies on TA could also be informative for these diseases (76, 230).
Author Contributions
Anna Von Rossum, Ismail Laher, and Jonathan C. Choy wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors’ work was funded by the Canadian Institutes of Health Research (CIHR), Heart and Stroke Foundation of British Columbia and Yukon, and the Natural Sciences and Engineering Research Council of Canada. Jonathan C. Choy is a recipient of a Michael Smith Foundation for Health Research Scholar award.
Abbreviations
AMR, antibody-mediated rejection; APC, antigen-presenting cell; EC, endothelial cell; EDCF, endothelial-derived constrictor factors; EDRF, endothelial-derived relaxation factor; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; HLA, human leukocyte antigen; IDO, indoleamine 2,3-dioxygenase; IFNγ, intergeron-γ; ILT, immunoglobulin-like transcript; MHC, major histocompatibility complex; NO, nitric oxide; PD-1, programed cell death-1; PD-L1, programed cell death ligand-1; TA, transplant arteriosclerosis; TNF, tumor necrosis factor.
References
1. Taylor DO, Edwards LB, Boucek MM, Trulock EP, Waltz DA, Keck BM, et al. Registry of the international society for heart and lung transplantation: twenty-third official adult heart transplantation report – 2006. J Heart Lung Transplant (2006) 25:869–79. doi: 10.1016/j.healun.2006.05.002
2. Choy JC. Granzymes and perforin in solid organ transplant rejection. Cell Death Differ (2010) 17:567–76. doi:10.1038/cdd.2009.161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Valenzuela NM, McNamara JT, Reed EF. Antibody-mediated graft injury: complement-dependent and complement-independent mechanisms. Curr Opin Organ Transplant (2014) 19:33–40. doi:10.1097/MOT.0000000000000040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Radio S, Wood S, Wilson J, Lin H, Winters G, McManus B. Allograft vascular disease: comparison of heart and other grafted organs. Transplant Proc (1996) 28:496–9.
5. Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol (2003) 3:361–70. doi:10.1038/nri1083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Lopez JA, Brennan AJ, Whisstock JC, Voskoboinik I, Trapani JA. Protecting a serial killer: pathways for perforin trafficking and self-defence ensure sequential target cell death. Trends Immunol (2012) 33:406–12. doi:10.1016/j.it.2012.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Susanto O, Trapani JA, Brasacchio D. Controversies in granzyme biology. Tissue Antigens (2012) 80:477–87. doi:10.1111/tan.12014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J (2009) 23:1625–37. doi:10.1096/fj.08-111005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Kanno Y, Vahedi G, Hirahara K, Singleton K, O’Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol (2012) 30:707–31. doi:10.1146/annurev-immunol-020711-075058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41:529–42. doi:10.1016/j.immuni.2014.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Wasowska BA. Mechanisms involved in antibody- and complement-mediated allograft rejection. Immunol Res (2010) 47:25–44. doi:10.1007/s12026-009-8136-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Pober JS, Collins T, Gimbrone MA Jr, Cotran RS, Gitlin JD, Fiers W, et al. Lymphocytes recognize human vascular endothelial and dermal fibroblast Ia antigens induced by recombinant immune interferon. Nature (1983) 305:726–9. doi:10.1038/305726a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Page CS, Holloway N, Smith H, Yacoub M, Rose ML. Alloproliferative responses of purified CD4+ and CD8+ T cells to endothelial cells in the absence of contaminating accessory cells. Transplantation (1994) 57:1628–37. doi:10.1097/00007890-199457110-00016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Epperson DE, Pober JS. Antigen-presenting function of human endothelial cells. Direct activation of resting CD8 T cells. J Immunol (1994) 153:5402–12.
15. Hasegawa S, Becker G, Nagano H, Libby P, Mitchell RN. Pattern of graft- and host-specific MHC class II expression in long-term murine cardiac allografts: origin of inflammatory and vascular wall cells. Am J Pathol (1998) 153:69–79. doi:10.1016/S0002-9440(10)65547-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Jutte NH, Heijse P, van Batenburg MH, Vaessen LM, Mochtar B, Balk AH, et al. Donor heart endothelial cells as targets for graft infiltrating lymphocytes after clinical cardiac transplantation. Transpl Immunol (1993) 1:39–44. doi:10.1016/0966-3274(93)90057-F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Shiao SL, McNiff JM, Pober JS. Memory T cells and their costimulators in human allograft injury. J Immunol (2005) 175:4886–96. doi:10.4049/jimmunol.175.8.4886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation (2008) 118:1276–84. doi:10.1161/CIRCULATIONAHA.108.789172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Choi JH, Cheong C, Dandamudi DB, Park CG, Rodriguez A, Mehandru S, et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity (2011) 35:819–31. doi:10.1016/j.immuni.2011.09.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Cheong C, Choi JH. Dendritic cells and regulatory T cells in atherosclerosis. Mol Cells (2012) 34:341–7. doi:10.1007/s10059-012-0128-9
21. Lorber MI, Wilson JH, Robert ME, Schechner JS, Kirkiles N, Qian HY, et al. Human allogeneic vascular rejection after arterial transplantation and peripheral lymphoid reconstitution in severe combined immunodeficient mice. Transplantation (1999) 67:897–903. doi:10.1097/00007890-199903270-00018
22. Rao DA, Eid RE, Qin L, Yi T, Kirkiles-Smith NC, Tellides G, et al. Interleukin (IL)-1 promotes allogeneic T cell intimal infiltration and IL-17 production in a model of human artery rejection. J Exp Med (2008) 205:3145–58. doi:10.1084/jem.20081661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med (2004) 199:173–83. doi:10.1084/jem.20030850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Mazanet MM, Hughes CC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol (2002) 169:3581–8. doi:10.4049/jimmunol.169.7.3581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation (2007) 116:2062–71. doi:10.1161/CIRCULATIONAHA.107.709360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Bolinger B, Engeler D, Krebs P, Miller S, Firner S, Hoffmann M, et al. IFN-gamma-receptor signaling ameliorates transplant vasculopathy through attenuation of CD8(+) T-cell-mediated injury of vascular endothelial cells. Eur J Immunol (2010) 40(3):733–43. doi:10.1002/eji.200939706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Escalante NK, von Rossum A, Lee M, Choy JC. CD155 on human vascular endothelial cells attenuates the acquisition of effector functions in CD8 T cells. Arterioscler Thromb Vasc Biol (2011) 31:1177–84. doi:10.1161/ATVBAHA.111.224162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Manavalan JS, Kim-Schulze S, Scotto L, Naiyer AJ, Vlad G, Colombo PC, et al. Alloantigen specific CD8+CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int Immunol (2004) 16:1055–68. doi:10.1093/intimm/dxh107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Gleissner CA, Zastrow A, Klingenberg R, Kluger MS, Konstandin M, Celik S, et al. IL-10 inhibits endothelium-dependent T cell costimulation by up-regulation of ILT3/4 in human vascular endothelial cells. Eur J Immunol (2007) 37:177–92. doi:10.1002/eji.200636498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Warner SJ, Friedman GB, Libby P. Regulation of major histocompatibility gene expression in human vascular smooth muscle cells. Arteriosclerosis (1989) 9:279–88. doi:10.1161/01.ATV.9.3.279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Murray AG, Libby P, Pober JS. Human vascular smooth muscle cells poorly co-stimulate and actively inhibit allogeneic CD4+ T cell proliferation in vitro. J Immunol (1995) 154:151–61.
32. Cuffy MC, Silverio AM, Qin L, Wang Y, Eid R, Brandacher G, et al. Induction of indoleamine 2,3-dioxygenase in vascular smooth muscle cells by interferon-gamma contributes to medial immunoprivilege. J Immunol (2007) 179:5246–54. doi:10.4049/jimmunol.179.8.5246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Zhang P, Manes TD, Pober JS, Tellides G. Human vascular smooth muscle cells lack essential costimulatory molecules to activate allogeneic memory T cells. Arterioscler Thromb Vasc Biol (2010) 10:10. doi:10.1161/ATVBAHA.109.200758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Zheng L, Ben LH, Pober JS, Bothwell AL. Porcine endothelial cells, unlike human endothelial cells, can be killed by human CTL via Fas ligand and cannot be protected by Bcl-2. J Immunol (2002) 169:6850–5. doi:10.4049/jimmunol.169.12.6850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Krupnick AS, Kreisel D, Popma SH, Balsara KR, Szeto WY, Krasinskas AM, et al. Mechanism of T cell-mediated endothelial apoptosis. Transplantation (2002) 74:871–6. doi:10.1097/00007890-200209270-00022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Zheng L, Gibson TF, Schechner JS, Pober JS, Bothwell AL. Bcl-2 transduction protects human endothelial cell synthetic microvessel grafts from allogeneic T cells in vivo. J Immunol (2004) 173:3020–6. doi:10.4049/jimmunol.173.5.3020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Choy JC, Cruz RP, Kerjner A, Geisbrecht J, Sawchuk T, Fraser SA, et al. Granzyme B induces endothelial cell apoptosis and contributes to the development of transplant vascular disease. Am J Transplant (2005) 5:494–9. doi:10.1111/j.1600-6143.2004.00710.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, et al. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem (2005) 280:23549–58. doi:10.1074/jbc.M412001200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Sata M, Suhara T, Walsh K. Vascular endothelial cells and smooth muscle cells differ in expression of Fas and Fas ligand and in sensitivity to Fas ligand-induced cell death: implications for vascular disease and therapy. Arterioscler Thromb Vasc Biol (2000) 20:309–16. doi:10.1161/01.ATV.20.2.309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Bannerman DD, Tupper JC, Ricketts WA, Bennett CF, Winn RK, Harlan JM. A constitutive cytoprotective pathway protects endothelial cells from lipopolysaccharide-induced apoptosis. J Biol Chem (2001) 276:14924–32. doi:10.1074/jbc.M100819200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Stefanescu R, Bassett D, Modarresi R, Santiago F, Fakruddin M, Laurence J. Synergistic interactions between interferon-gamma and TRAIL modulate c-FLIP in endothelial cells, mediating their lineage-specific sensitivity to thrombotic thrombocytopenic purpura plasma-associated apoptosis. Blood (2008) 112:340–9. doi:10.1182/blood-2007-10-119552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Sata M, Walsh K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J Clin Invest (1998) 102:1682–9. doi:10.1172/JCI3531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Sata M, Walsh K. Endothelial cell apoptosis induced by oxidized LDL is associated with the down-regulation of the cellular caspase inhibitor FLIP. J Biol Chem (1998) 273:33103–6. doi:10.1074/jbc.273.50.33103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Li JH, Kirkiles-Smith NC, McNiff JM, Pober JS. TRAIL induces apoptosis and inflammatory gene expression in human endothelial cells. J Immunol (2003) 171:1526–33. doi:10.4049/jimmunol.171.3.1526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Ma W, Pober JS. Human endothelial cells effectively costimulate cytokine production by, but not differentiation of, naive CD4+ T cells. J Immunol (1998) 161:2158–67.
46. Terasaki PI, Cai J. Humoral theory of transplantation: further evidence. Curr Opin Immunol (2005) 17:541–5. doi:10.1016/j.coi.2005.07.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lawrence C, Willicombe M, Brookes PA, Santos-Nunez E, Bajaj R, Cook T, et al. Preformed complement-activating low-level donor-specific antibody predicts early antibody-mediated rejection in renal allografts. Transplantation (2013) 95:341–6. doi:10.1097/TP.0b013e3182743cfa
48. Uber WE, Self SE, Van Bakel AB, Pereira NL. Acute antibody-mediated rejection following heart transplantation. Am J Transplant (2007) 7:2064–74. doi:10.1111/j.1600-6143.2007.01900.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Stewart S, Winters GL, Fishbein MC, Tazelaar HD, Kobashigawa J, Abrams J, et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J Heart Lung Transplant (2005) 24:1710–20. doi:10.1016/j.healun.2005.03.019
50. Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, et al. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant (2012) 12:1157–67. doi:10.1111/j.1600-6143.2012.04013.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Jurcevic S, Ainsworth ME, Pomerance A, Smith JD, Robinson DR, Dunn MJ, et al. Antivimentin antibodies are an independent predictor of transplant-associated coronary artery disease after cardiac transplantation. Transplantation (2001) 71:886–92. doi:10.1097/00007890-200104150-00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Soulez M, Pilon EA, Dieude M, Cardinal H, Brassard N, Qi S, et al. The perlecan fragment LG3 is a novel regulator of obliterative remodeling associated with allograft vascular rejection. Circ Res (2012) 110:94–104. doi:10.1161/CIRCRESAHA.111.250431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med (2005) 352:558–69. doi:10.1056/NEJMoa035717
54. Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med (2013) 369:1215–26. doi:10.1056/NEJMoa1302506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med (2002) 8:582–7. doi:10.1038/nm0602-582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Qian Z, Jakobs FM, Pfaff-Amesse T, Sanfilippo F, Baldwin WM III. Complement contributes to the rejection of complete and class I major histocompatibility complex – incompatible cardiac allografts. J Heart Lung Transplant (1998) 17:470–8.
57. Wang H, Arp J, Liu W, Faas SJ, Jiang J, Gies DR, et al. Inhibition of terminal complement components in presensitized transplant recipients prevents antibody-mediated rejection leading to long-term graft survival and accommodation. J Immunol (2007) 179:4451–63. doi:10.4049/jimmunol.179.7.4451
58. Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant (2011) 11:2405–13. doi:10.1111/j.1600-6143.2011.03757.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, et al. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant (2009) 9:231–5. doi:10.1111/j.1600-6143.2008.02451.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Fukami N, Ramachandran S, Narayanan K, Liu W, Nath DS, Jendrisak M, et al. Mechanism of accommodation in a sensitized human leukocyte antigen transgenic murine cardiac transplant model. Transplantation (2012) 93:364–72. doi:10.1097/TP.0b013e3182406a6b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Jin YP, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, et al. Anti-HLA class I antibody-mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum Immunol (2004) 65:291–302. doi:10.1016/j.humimm.2004.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-kappaB signaling in endothelial cells. Circulation (2013) 128:2504–16. doi:10.1161/CIRCULATIONAHA.113.002972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med (2013) 210:257–68. doi:10.1084/jem.20121525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant (2013) 13:2530–9. doi:10.1111/ajt.12405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Ziegler ME, Jin YP, Young SH, Rozengurt E, Reed EF. HLA class I-mediated stress fiber formation requires ERK1/2 activation in the absence of an increase in intracellular Ca2+ in human aortic endothelial cells. Am J Physiol Cell Physiol (2012) 303:C872–82. doi:10.1152/ajpcell.00199.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Jindra PT, Jin YP, Rozengurt E, Reed EF. HLA class I antibody-mediated endothelial cell proliferation via the mTOR pathway. J Immunol (2008) 180:2357–66. doi:10.4049/jimmunol.180.4.2357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Galvani S, Trayssac M, Auge N, Thiers JC, Calise D, Krell HW, et al. A key role for matrix metalloproteinases and neutral sphingomyelinase-2 in transplant vasculopathy triggered by anti-HLA antibody. Circulation (2011) 124:2725–34. doi:10.1161/CIRCULATIONAHA.111.021790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Galvani S, Auge N, Calise D, Thiers JC, Canivet C, Kamar N, et al. HLA class I antibodies provoke graft arteriosclerosis in human arteries transplanted into SCID/beige mice. Am J Transplant (2009) 9:2607–14. doi:10.1111/j.1600-6143.2009.02804.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, et al. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A (2007) 104:1301–6. doi:10.1073/pnas.0602035104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Valenzuela NM, Hong L, Shen XD, Gao F, Young SH, Rozengurt E, et al. Blockade of p-selectin is sufficient to reduce MHC I antibody-elicited monocyte recruitment in vitro and in vivo. Am J Transplant (2013) 13:299–311. doi:10.1111/ajt.12016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Naemi FM, Carter V, Kirby JA, Ali S. Anti-donor HLA class I antibodies: pathways to endothelial cell activation and cell-mediated allograft rejection. Transplantation (2013) 96:258–66. doi:10.1097/TP.0b013e3182985504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Bhan AK, Mihm MC Jr, Dvorak HF. T cell subsets in allograft rejection. In situ characterization of T cell subsets in human skin allografts by the use of monoclonal antibodies. J Immunol (1982) 129:1578–83.
73. Forbes RD, Lowry RP, Darden AG, Gomersall M, Marghesco DM. Morphologic studies of acute rat cardiac allograft rejection across an isolated major histocompatibility complex class I (RT1A) disparity. Transplantation (1988) 45:943–8. doi:10.1097/00007890-198805000-00021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Laplante P, Raymond MA, Gagnon G, Vigneault N, Sasseville AM, Langelier Y, et al. Novel fibrogenic pathways are activated in response to endothelial apoptosis: implications in the pathophysiology of systemic sclerosis. J Immunol (2005) 174:5740–9. doi:10.4049/jimmunol.174.9.5740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Laplante P, Sirois I, Raymond MA, Kokta V, Beliveau A, Prat A, et al. Caspase-3-mediated secretion of connective tissue growth factor by apoptotic endothelial cells promotes fibrosis. Cell Death Differ (2010) 17:291–303. doi:10.1038/cdd.2009.124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Rahmani M, Cruz RP, Granville DJ, McManus BM. Allograft vasculopathy versus atherosclerosis. Circ Res (2006) 99:801–15. doi:10.1161/01.RES.0000246086.93555.f3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Lin H, Wilson JE, Roberts CR, Horley KJ, Winters GL, Costanzo MR, et al. Biglycan, decorin, and versican protein expression patterns in coronary arteriopathy of human cardiac allograft: distinctness as compared to native atherosclerosis. J Heart Lung Transplant (1996) 15:1233–47.
78. McManus BM, Horley KJ, Wilson JE, Malcom GT, Kendall TJ, Miles RR, et al. Prominence of coronary arterial wall lipids in human heart allografts. Implications for pathogenesis of allograft arteriopathy. Am J Pathol (1995) 147:293–308.
79. Castellani C, Angelini A, de Boer OJ, van der Loos CM, Fedrigo M, Frigo AC, et al. Intraplaque hemorrhage in cardiac allograft vasculopathy. Am J Transplant (2014) 14:184–92. doi:10.1111/ajt.12517
80. Anderson TJ, Meredith IT, Uehata A, Mudge GH, Selwyn AP, Ganz P, et al. Functional significance of intimal thickening as detected by intravascular ultrasound early and late after cardiac transplantation. Circulation (1993) 88:1093–100. doi:10.1161/01.CIR.88.3.1093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Tsutsui H, Ziada KM, Schoenhagen P, Iyisoy A, Magyar WA, Crowe TD, et al. Lumen loss in transplant coronary artery disease is a biphasic process involving early intimal thickening and late constrictive remodeling: results from a 5-year serial intravascular ultrasound study. Circulation (2001) 104:653–7. doi:10.1161/hc3101.093867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Shi C, Lee WS, He Q, Zhang D, Fletcher DL Jr, Newell JB, et al. Immunologic basis of transplant-associated arteriosclerosis. Proc Natl Acad Sci U S A (1996) 93:4051–6. doi:10.1073/pnas.93.9.4051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Russell PS, Chase CM, Winn HJ, Colvin RB. Coronary atherosclerosis in transplanted mouse hearts. II. Importance of humoral immunity. J Immunol (1994) 152:5135–41.
84. Ross R, Glomset J, Harker L. Response to injury and atherogenesis. Am J Pathol (1977) 86:675–84.
85. Reidy MA, Schwartz SM. Endothelial regeneration. III. Time course of intimal changes after small defined injury to rat aortic endothelium. Lab Invest (1981) 44:301–8.
86. Reidy MA. A reassessment of endothelial injury and arterial lesion formation. Lab Invest (1985) 53:513–20.
87. Fingerle J, Johnson R, Clowes AW, Majesky MW, Reidy MA. Role of platelets in smooth muscle cell proliferation and migration after vascular injury in rat carotid artery. Proc Natl Acad Sci U S A (1989) 86:8412–6. doi:10.1073/pnas.86.21.8412
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Lindner V, Majack RA, Reidy MA. Basic fibroblast growth factor stimulates endothelial regrowth and proliferation in denuded arteries. J Clin Invest (1990) 85:2004–8. doi:10.1172/JCI114665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Shimokado K, Raines EW, Madtes DK, Barrett TB, Benditt EP, Ross R. A significant part of macrophage-derived growth factor consists of at least two forms of PDGF. Cell (1985) 43:277–86. doi:10.1016/0092-8674(85)90033-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Bowen-Pope DF, Ross R, Seifert RA. Locally acting growth factors for vascular smooth muscle cells: endogenous synthesis and release from platelets. Circulation (1985) 72:735–40. doi:10.1161/01.CIR.72.4.735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science (1991) 253:1129–32. doi:10.1126/science.1653454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Raymond MA, Desormeaux A, Laplante P, Vigneault N, Filep JG, Landry K, et al. Apoptosis of endothelial cells triggers a caspase-dependent anti-apoptotic paracrine loop active on VSMC. FASEB J (2004) 18:705–7.
93. Choy JC, Podor TJ, Yanagawa B, Lai JC, Granville DJ, Walker DC, et al. The regulation and consequences of immune-mediated cell death in atheromatous diseases. Cardiovasc Toxicol (2003) 3:269–82. doi:10.1385/CT:3:3:269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Dong C, Wilson JE, Winters GL, McManus BM. Human transplant coronary artery disease: pathological evidence for Fas-mediated apoptotic cytotoxicity in allograft arteriopathy. Lab Invest (1996) 74:921–31.
95. Hruban RH, Beschorner WE, Baumgartner WA, Augustine SM, Ren H, Reitz BA, et al. Accelerated arteriosclerosis in heart transplant recipients is associated with a T-lymphocyte-mediated endothelialitis. Am J Pathol (1990) 137:871–82.
96. Hameed A, Olsen KJ, Cheng L, Fox WM III, Hruban RH, Podack ER. Immunohistochemical identification of cytotoxic lymphocytes using human perforin monoclonal antibody. Am J Pathol (1992) 140:1025–30.
97. Choy JC, McDonald PC, Suarez AC, Hung VH, Wilson JE, McManus BM, et al. Granzyme B in atherosclerosis and transplant vascular disease: association with cell death and atherosclerotic disease severity. Mod Pathol (2003) 16:460–70. doi:10.1097/01.MP.0000067424.12280.BC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Lai JC, Tranfield EM, Walker DC, Dyck J, Kerjner A, Loo S, et al. Ultrastructural evidence of early endothelial damage in coronary arteries of rat cardiac allografts. J Heart Lung Transplant (2003) 22:993–1004. doi:10.1016/S1053-2498(02)01163-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Miller C, DeWitt CW. Cellular and humoral responses to major and minor histocompatibility antigens. Transplant Proc (1973) 5:303–5.
100. Choy JC, Hung VH, Hunter AL, Cheung PK, Motyka B, Goping IS, et al. Granzyme B induces smooth muscle cell apoptosis in the absence of perforin: involvement of extracellular matrix degradation. Arterioscler Thromb Vasc Biol (2004) 24:2245–50. doi:10.1161/01.ATV.0000147162.51930.b7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Choy JC, Kerjner A, Wong BW, McManus BM, Granville DJ. Perforin mediates endothelial cell death and resultant transplant vascular disease in cardiac allografts. Am J Pathol (2004) 165:127–33. doi:10.1016/S0002-9440(10)63281-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Schnickel GT, Whiting D, Hsieh GR, Yun JJ, Fischbein MP, Fishbein MC, et al. CD8 lymphocytes are sufficient for the development of chronic rejection. Transplantation (2004) 78:1634–9. doi:10.1097/01.TP.0000141362.33931.40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Fingerle J, Au YP, Clowes AW, Reidy MA. Intimal lesion formation in rat carotid arteries after endothelial denudation in absence of medial injury. Arteriosclerosis (1990) 10:1082–7. doi:10.1161/01.ATV.10.6.1082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med (2006) 12:1075–80. doi:10.1038/nm1459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Yu H, Clarke MC, Figg N, Littlewood TD, Bennett MR. Smooth muscle cell apoptosis promotes vessel remodeling and repair via activation of cell migration, proliferation, and collagen synthesis. Arterioscler Thromb Vasc Biol (2011) 31:2402–9. doi:10.1161/ATVBAHA.111.235622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Legare JF, Issekutz T, Lee TD, Hirsch G. CD8+ T lymphocytes mediate destruction of the vascular media in a model of chronic rejection. Am J Pathol (2000) 157:859–65. doi:10.1016/S0002-9440(10)64599-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Hart-Matyas M, Nejat S, Jordan JL, Hirsch GM, Lee TD. IFN-gamma and Fas/FasL pathways cooperate to induce medial cell loss and neointimal lesion formation in allograft vasculopathy. Transpl Immunol (2010) 22:157–64. doi:10.1016/j.trim.2009.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Chan SW, Hegyi L, Scott S, Cary NR, Weissberg PL, Bennett MR. Sensitivity to Fas-mediated apoptosis is determined below receptor level in human vascular smooth muscle cells. Circ Res (2000) 86:1038–46. doi:10.1161/01.RES.86.10.1038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Rosner D, Stoneman V, Littlewood T, McCarthy N, Figg N, Wang Y, et al. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am J Pathol (2006) 168:2054–63. doi:10.2353/ajpath.2006.050473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Li J, Liu S, Li W, Hu S, Xiong J, Shu X, et al. Vascular smooth muscle cell apoptosis promotes transplant arteriosclerosis through inducing the production of SDF-1alpha. Am J Transplant (2012) 12:2029–43. doi:10.1111/j.1600-6143.2012.04082.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81:1–5. doi:10.1189/jlb.0306164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol (2010) 28:367–88. doi:10.1146/annurev.immunol.021908.132603
113. Rao DA, Tracey KJ, Pober JS. IL-1alpha and IL-1beta are endogenous mediators linking cell injury to the adaptive alloimmune response. J Immunol (2007) 179:6536–46. doi:10.4049/jimmunol.179.10.6536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Zou H, Yang Y, Gao M, Zhang B, Ming B, Sun Y, et al. HMGB1 is involved in chronic rejection of cardiac allograft via promoting inflammatory-like mDCs. Am J Transplant (2014) 14:1765–77. doi:10.1111/ajt.12781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol (2009) 21:317–37. doi:10.1093/intimm/dxp017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Ahmad U, Ali R, Lebastchi AH, Qin L, Lo SF, Yakimov AO, et al. IFN-gamma primes intact human coronary arteries and cultured coronary smooth muscle cells to double-stranded RNA- and self-RNA-induced inflammatory responses by upregulating TLR3 and melanoma differentiation-associated gene 5. J Immunol (2010) 185:1283–94. doi:10.4049/jimmunol.0902283
117. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science (2012) 335:984–9. doi:10.1126/science.1215418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Yin H, Li XY, Jin XB, Zhang BB, Gong Q, Yang H, et al. IL-33 prolongs murine cardiac allograft survival through induction of TH2-type immune deviation. Transplantation (2010) 89:1189–97. doi:10.1097/TP.0b013e3181d720af
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, Fichtner-Feigl S. Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int (2011) 24:1027–39. doi:10.1111/j.1432-2277.2011.01306.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol (2011) 187:4598–610. doi:10.4049/jimmunol.1100519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 Is an unconventional alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol (2014) 193:4010–20. doi:10.4049/jimmunol.1400481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7:803–15. doi:10.1038/nri2171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Mitchell RN, Libby P. Vascular remodeling in transplant vasculopathy. Circ Res (2007) 100:967–78. doi:10.1161/01.RES.0000261982.76892.09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. van Loosdregt J, van Oosterhout MF, Bruggink AH, van Wichen DF, van Kuik J, de Koning E, et al. The chemokine and chemokine receptor profile of infiltrating cells in the wall of arteries with cardiac allograft vasculopathy is indicative of a memory T-helper 1 response. Circulation (2006) 114:1599–607. doi:10.1161/CIRCULATIONAHA.105.597526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Russell PS, Chase CM, Winn HJ, Colvin RB. Coronary atherosclerosis in transplanted mouse hearts. III. Effects of recipient treatment with a monoclonal antibody to interferon-gamma. Transplantation (1994) 57:1367–71. doi:10.1097/00007890-199405150-00014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Stadlbauer TH, Wagner AH, Holschermann H, Fiedel S, Fingerhuth H, Tillmanns H, et al. AP-1 and STAT-1 decoy oligodeoxynucleotides attenuate transplant vasculopathy in rat cardiac allografts. Cardiovasc Res (2008) 79:698–705. doi:10.1093/cvr/cvn135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Pober JS, Gimbrone MA Jr, Cotran RS, Reiss CS, Burakoff SJ, Fiers W, et al. Ia expression by vascular endothelium is inducible by activated T cells and by human gamma interferon. J Exp Med (1983) 157:1339–53. doi:10.1084/jem.157.4.1339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Burns WR, Wang Y, Tang PC, Ranjbaran H, Iakimov A, Kim J, et al. Recruitment of CXCR3+ and CCR5+ T cells and production of interferon-gamma-inducible chemokines in rejecting human arteries. Am J Transplant (2005) 5:1226–36. doi:10.1111/j.1600-6143.2005.00892.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Li JH, Kluger MS, Madge LA, Zheng L, Bothwell AL, Pober JS. Interferon-gamma augments CD95(APO-1/Fas) and pro-caspase-8 expression and sensitizes human vascular endothelial cells to CD95-mediated apoptosis. Am J Pathol (2002) 161:1485–95. doi:10.1016/S0002-9440(10)64424-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Li JH, Pober JS. The cathepsin B death pathway contributes to TNF plus IFN-gamma-mediated human endothelial injury. J Immunol (2005) 175:1858–66. doi:10.4049/jimmunol.175.3.1858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, et al. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature (2000) 403:207–11. doi:10.1038/35003221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Wang Y, Bai Y, Qin L, Zhang P, Yi T, Teesdale SA, et al. Interferon-gamma induces human vascular smooth muscle cell proliferation and intimal expansion by phosphatidylinositol 3-kinase dependent mammalian target of rapamycin raptor complex 1 activation. Circ Res (2007) 101:560–9. doi:10.1161/CIRCRESAHA.107.151068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Yu L, Qin L, Zhang H, He Y, Chen H, Pober JS, et al. AIP1 prevents graft arteriosclerosis by inhibiting interferon-gamma-dependent smooth muscle cell proliferation and intimal expansion. Circ Res (2011) 109:418–27. doi:10.1161/CIRCRESAHA.111.248245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Dallman MJ, Larsen CP, Morris PJ. Cytokine gene transcription in vascularised organ grafts: analysis using semiquantitative polymerase chain reaction. J Exp Med (1991) 174:493–6. doi:10.1084/jem.174.2.493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Arbustini E, Grasso M, Diegoli M, Bramerio M, Foglieni AS, Albertario M, et al. Expression of tumor necrosis factor in human acute cardiac rejection. An immunohistochemical and immunoblotting study. Am J Pathol (1991) 139:709–15.
136. Wu CJ, Lovett M, Wong-Lee J, Moeller F, Kitamura M, Goralski TJ, et al. Cytokine gene expression in rejecting cardiac allografts. Transplantation (1992) 54:326–32. doi:10.1097/00007890-199208000-00024
137. Pober JS, Bevilacqua MP, Mendrick DL, Lapierre LA, Fiers W, Gimbrone MA Jr. Two distinct monokines, interleukin 1 and tumor necrosis factor, each independently induce biosynthesis and transient expression of the same antigen on the surface of cultured human vascular endothelial cells. J Immunol (1986) 136:1680–7.
138. Pober JS, Gimbrone MA Jr, Lapierre LA, Mendrick DL, Fiers W, Rothlein R, et al. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor, and immune interferon. J Immunol (1986) 137:1893–6.
139. Clausell N, Molossi S, Sett S, Rabinovitch M. In vivo blockade of tumor necrosis factor-alpha in cholesterol-fed rabbits after cardiac transplant inhibits acute coronary artery neointimal formation. Circulation (1994) 89:2768–79. doi:10.1161/01.CIR.89.6.2768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Rao DA, Pober JS. Endothelial injury, alarmins, and allograft rejection. Crit Rev Immunol (2008) 28:229–48. doi:10.1615/CritRevImmunol.v28.i3.40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Lee HY, Youn SW, Kim JY, Park KW, Hwang CI, Park WY, et al. FOXO3a turns the tumor necrosis factor receptor signaling towards apoptosis through reciprocal regulation of c-Jun N-terminal kinase and NF-kappaB. Arterioscler Thromb Vasc Biol (2008) 28:112–20. doi:10.1161/ATVBAHA.107.153304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Tang JL, Subbotin VM, Antonysamy MA, Troutt AB, Rao AS, Thomson AW. Interleukin-17 antagonism inhibits acute but not chronic vascular rejection. Transplantation (2001) 72:348–50. doi:10.1097/00007890-200107270-00035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M, et al. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med (2008) 205:3133–44. doi:10.1084/jem.20081937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol (2012) 188:6287–99. doi:10.4049/jimmunol.1200385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol (2010) 184:5344–51. doi:10.4049/jimmunol.0902999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Djamali A, Kaufman DB, Ellis TM, Zhong W, Matas A, Samaniego M. Diagnosis and management of antibody-mediated rejection: current status and novel approaches. Am J Transplant (2014) 14:255–71. doi:10.1111/ajt.12589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Zhang X, Valenzuela NM, Reed EF. HLA class I antibody-mediated endothelial and smooth muscle cell activation. Curr Opin Organ Transplant (2012) 17:446–51. doi:10.1097/MOT.0b013e328355f1c2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Petrossian GA, Nichols AB, Marboe CC, Sciacca R, Rose EA, Smith CR, et al. Relation between survival and development of coronary artery disease and anti-HLA antibodies after cardiac transplantation. Circulation (1989) 80:III122–5.
149. Cherry R, Nielsen H, Reed E, Reemtsma K, Suciu-Foca N, Marboe CC. Vascular (humoral) rejection in human cardiac allograft biopsies: relation to circulating anti-HLA antibodies. J Heart Lung Transplant (1992) 11:24–9.
150. Lee CY, Lotfi-Emran S, Erdinc M, Murata K, Velidedeoglu E, Fox-Talbot K, et al. The involvement of FcR mechanisms in antibody-mediated rejection. Transplantation (2007) 84:1324–34. doi:10.1097/01.tp.0000287457.54761.53
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Hirohashi T, Chase CM, Della Pelle P, Sebastian D, Alessandrini A, Madsen JC, et al. A novel pathway of chronic allograft rejection mediated by NK cells and alloantibody. Am J Transplant (2012) 12:313–21. doi:10.1111/j.1600-6143.2011.03836.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Jin YP, Korin Y, Zhang X, Jindra PT, Rozengurt E, Reed EF. RNA interference elucidates the role of focal adhesion kinase in HLA class I-mediated focal adhesion complex formation and proliferation in human endothelial cells. J Immunol (2007) 178:7911–22. doi:10.4049/jimmunol.178.12.7911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Choy JC, Granville DJ, Hunt DW, McManus BM. Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol (2001) 33:1673–90. doi:10.1006/jmcc.2001.1419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Lucchiari N, Panajotopoulos N, Xu C, Rodrigues H, Ianhez LE, Kalil J, et al. Antibodies eluted from acutely rejected renal allografts bind to and activate human endothelial cells. Hum Immunol (2000) 61:518–27. doi:10.1016/S0198-8859(00)00109-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Mahesh B, Leong HS, McCormack A, Sarathchandra P, Holder A, Rose ML. Autoantibodies to vimentin cause accelerated rejection of cardiac allografts. Am J Pathol (2007) 170:1415–27. doi:10.2353/ajpath.2007.060728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Mahesh B, Leong HS, Nair KS, McCormack A, Sarathchandra P, Rose ML. Autoimmunity to vimentin potentiates graft vasculopathy in murine cardiac allografts. Transplantation (2010) 90:4–13. doi:10.1097/TP.0b013e3181dfa694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Lukitsch I, Kehr J, Chaykovska L, Wallukat G, Nieminen-Kelha M, Batuman V, et al. Renal ischemia and transplantation predispose to vascular constriction mediated by angiotensin II type 1 receptor-activating antibodies. Transplantation (2012) 94:8–13. doi:10.1097/TP.0b013e3182529bb7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature (1980) 288:373–6. doi:10.1038/288373a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Griffith TM, Edwards DH, Lewis MJ, Newby AC, Henderson AH. The nature of endothelium-derived vascular relaxant factor. Nature (1984) 308:645–7. doi:10.1038/308645a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. De Mey JG, Vanhoutte PM. Heterogeneous behavior of the canine arterial and venous wall. Importance of the endothelium. Circ Res (1982) 51:439–47. doi:10.1161/01.RES.51.4.439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Pethig K, Heublein B, Wahlers T, Haverich A. Mechanism of luminal narrowing in cardiac allograft vasculopathy: inadequate vascular remodeling rather than intimal hyperplasia is the major predictor of coronary artery stenosis. Working group on cardiac allograft vasculopathy. Am Heart J (1998) 135:628–33. doi:10.1016/S0002-8703(98)70278-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Hollenberg SM, Tamburro P, Klein LW, Burns D, Easington C, Costanzo MR, et al. Discordant epicardial and microvascular endothelial responses in heart transplant recipients early after transplantation. J Heart Lung Transplant (1998) 17:487–94.
163. Hollenberg SM, Klein LW, Parrillo JE, Scherer M, Burns D, Tamburro P, et al. Coronary endothelial dysfunction after heart transplantation predicts allograft vasculopathy and cardiac death. Circulation (2001) 104:3091–6. doi:10.1161/hc5001.100796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature (1987) 327:524–6. doi:10.1038/327524a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Pollock JS, Forstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, et al. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci U S A (1991) 88:10480–4. doi:10.1073/pnas.88.23.10480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Zembowicz A, Hecker M, Macarthur H, Sessa WC, Vane JR. Nitric oxide and another potent vasodilator are formed from NG-hydroxy-L-arginine by cultured endothelial cells. Proc Natl Acad Sci U S A (1991) 88:11172–6. doi:10.1073/pnas.88.24.11172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Janssens SP, Shimouchi A, Quertermous T, Bloch DB, Bloch KD. Cloning and expression of a cDNA encoding human endothelium-derived relaxing factor/nitric oxide synthase. J Biol Chem (1992) 267:14519–22.
168. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature (1995) 377:239–42. doi:10.1038/377239a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Anderson HD, Rahmutula D, Gardner DG. Tumor necrosis factor-alpha inhibits endothelial nitric-oxide synthase gene promoter activity in bovine aortic endothelial cells. J Biol Chem (2004) 279:963–9. doi:10.1074/jbc.M309552200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Koh KP, Wang Y, Yi T, Shiao SL, Lorber MI, Sessa WC, et al. T cell-mediated vascular dysfunction of human allografts results from IFN-gamma dysregulation of NO synthase. J Clin Invest (2004) 114:846–56. doi:10.1172/JCI21767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Mitchell RN, Lichtman AH. The link between IFN-gamma and allograft arteriopathy: is the answer NO? J Clin Invest (2004) 114:762–4. doi:10.1172/JCI200422927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Choy JC, Yi T, Rao DA, Tellides G, Fox-Talbot K, Baldwin WM III, et al. CXCL12 induction of inducible nitric oxide synthase in human CD8 T cells. J Heart Lung Transplant (2008) 27:1333–9. doi:10.1016/j.healun.2008.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Choy JC, Wang Y, Tellides G, Pober JS. Induction of inducible NO synthase in bystander human T cells increases allogeneic responses in the vasculature. Proc Natl Acad Sci U S A (2007) 104:1313–8. doi:10.1073/pnas.0607731104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Liu AC, Lee M, McManus BM, Choy JC. Induction of endothelial nitric oxide synthase expression by IL-17 in human vascular endothelial cells: implications for vascular remodeling in transplant vasculopathy. J Immunol (2012) 188:1544–50. doi:10.4049/jimmunol.1102527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res (2013) 97:696–704. doi:10.1093/cvr/cvs422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension (2010) 55:500–7. doi:10.1161/HYPERTENSIONAHA.109.145094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J (2012) 33:837a–837a. doi:10.1093/eurheartj/ehr304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Rosenkranz-Weiss P, Sessa WC, Milstien S, Kaufman S, Watson CA, Pober JS. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J Clin Invest (1994) 93:2236–43. doi:10.1172/JCI117221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Katusic ZS, Stelter A, Milstien S. Cytokines stimulate GTP cyclohydrolase I gene expression in cultured human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol (1998) 18:27–32. doi:10.1161/01.ATV.18.1.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Chen W, Li L, Brod T, Saeed O, Thabet S, Jansen T, et al. Role of increased guanosine triphosphate cyclohydrolase-1 expression and tetrahydrobiopterin levels upon T cell activation. J Biol Chem (2011) 286:13846–51. doi:10.1074/jbc.M110.191023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Ziegler I, Schott K, Lubbert M, Herrmann F, Schwulera U, Bacher A. Control of tetrahydrobiopterin synthesis in T lymphocytes by synergistic action of interferon-gamma and interleukin-2. J Biol Chem (1990) 265:17026–30.
182. Antoniades C, Cunnington C, Antonopoulos A, Neville M, Margaritis M, Demosthenous M, et al. Induction of vascular GTP-cyclohydrolase I and endogenous tetrahydrobiopterin synthesis protect against inflammation- induced endothelial dysfunction in human atherosclerosis. Circulation (2011) 124:1860–70. doi:10.1161/CIRCULATIONAHA.111.029272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Virdis A, Ghiadoni L, Taddei S. Human endothelial dysfunction: EDCFs. Pflugers Arch (2010) 459:1015–23. doi:10.1007/s00424-009-0783-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Ravalli S, Szabolcs M, Albala A, Michler RE, Cannon PJ. Increased immunoreactive endothelin-1 in human transplant coronary artery disease. Circulation (1996) 94:2096–102. doi:10.1161/01.CIR.94.9.2096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Weis M, Wildhirt SM, Schulze C, Rieder G, Wilbert-Lampen U, Wolf WP, et al. Endothelin in coronary endothelial dysfunction early after human heart transplantation. J Heart Lung Transplant (1999) 18:1071–9. doi:10.1016/S1053-2498(99)00081-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Larose E, Behrendt D, Kinlay S, Selwyn AP, Ganz P, Fang JC. Endothelin-1 is a key mediator of coronary vasoconstriction in patients with transplant coronary arteriosclerosis. Circ Heart Fail (2009) 2:409–16. doi:10.1161/CIRCHEARTFAILURE.108.836759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Lamas S, Michel T, Collins T, Brenner BM, Marsden PA. Effects of interferon-gamma on nitric oxide synthase activity and endothelin-1 production by vascular endothelial cells. J Clin Invest (1992) 90:879–87. doi:10.1172/JCI115963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Kahaleh MB, Fan PS. Effect of cytokines on the production of endothelin by endothelial cells. Clin Exp Rheumatol (1997) 15:163–7.
189. Skarsgard PL, Wang X, McDonald P, Lui AH, Lam EK, McManus BM, et al. Profound inhibition of myogenic tone in rat cardiac allografts is due to eNOS- and iNOS-based nitric oxide and an intrinsic defect in vascular smooth muscle contraction. Circulation (2000) 101:1303–10. doi:10.1161/01.CIR.101.11.1303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Moien-Afshari F, Choy JC, McManus BM, Laher I. Cyclosporine treatment preserves coronary resistance artery function in rat cardiac allografts. J Heart Lung Transplant (2004) 23:193–203. doi:10.1016/S1053-2498(03)00113-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Avihingsanon Y, Ma N, Csizmadia E, Wang C, Pavlakis M, Giraldo M, et al. Expression of protective genes in human renal allografts: a regulatory response to injury associated with graft rejection. Transplantation (2002) 73:1079–85. doi:10.1097/00007890-200204150-00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Bach FH, Ferran C, Hechenleitner P, Mark W, Koyamada N, Miyatake T, et al. Accommodation of vascularized xenografts: expression of “protective genes” by donor endothelial cells in a host Th2 cytokine environment. Nat Med (1997) 3:196–204. doi:10.1038/nm0297-196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Opipari AW Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem (1990) 265:14705–8.
194. Daniel S, Arvelo MB, Patel VI, Longo CR, Shrikhande G, Shukri T, et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood (2004) 104:2376–84. doi:10.1182/blood-2003-02-0635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem (1996) 271:18068–73. doi:10.1074/jbc.271.30.18068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Patel VI, Daniel S, Longo CR, Shrikhande GV, Scali ST, Czismadia E, et al. A20, a modulator of smooth muscle cell proliferation and apoptosis, prevents and induces regression of neointimal hyperplasia. FASEB J (2006) 20:1418–30. doi:10.1096/fj.05-4981com
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Kunter U, Floege J, von Jurgensonn AS, Stojanovic T, Merkel S, Grone HJ, et al. Expression of A20 in the vessel wall of rat-kidney allografts correlates with protection from transplant arteriosclerosis. Transplantation (2003) 75:3–9. doi:10.1097/00007890-200301150-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Siracuse JJ, Fisher MD, da Silva CG, Peterson CR, Csizmadia E, Moll HP, et al. A20-mediated modulation of inflammatory and immune responses in aortic allografts and development of transplant arteriosclerosis. Transplantation (2012) 93:373–82. doi:10.1097/TP.0b013e3182419829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Katori M, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 system in organ transplantation. Transplantation (2002) 74:905–12. doi:10.1097/00007890-200210150-00001
200. Exner M, Bohmig GA, Schillinger M, Regele H, Watschinger B, Horl WH, et al. Donor heme oxygenase-1 genotype is associated with renal allograft function. Transplantation (2004) 77:538–42. doi:10.1097/01.TP.0000113467.36269.F8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Ozaki KS, Marques GM, Nogueira E, Feitoza RQ, Cenedeze MA, Franco MF, et al. Improved renal function after kidney transplantation is associated with heme oxygenase-1 polymorphism. Clin Transplant (2008) 22:609–16. doi:10.1111/j.1399-0012.2008.00832.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Courtney AE, McNamee PT, Middleton D, Heggarty S, Patterson CC, Maxwell AP. Association of functional heme oxygenase-1 gene promoter polymorphism with renal transplantation outcomes. Am J Transplant (2007) 7:908–13. doi:10.1111/j.1600-6143.2006.01726.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Ullrich R, Exner M, Schillinger M, Zuckermann A, Raith M, Dunkler D, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter and cardiac allograft vasculopathy. J Heart Lung Transplant (2005) 24:1600–5. doi:10.1016/j.healun.2004.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Soares MP, Bach FH. Heme oxygenase-1 in organ transplantation. Front BioSci (2007) 12:4932–45. doi:10.2741/2439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Clarke HM, Shrivastava S, Motterlini R, Sawle P, Chen D, Dorling A. Donor HO-1 expression inhibits intimal hyperplasia in unmanipulated graft recipients: a potential role for CD8+ T-cell modulation by carbon monoxide. Transplantation (2009) 88:653–61. doi:10.1097/TP.0b013e3181b2fd83
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Hancock WW, Buelow R, Sayegh MH, Turka LA. Antibody-induced transplant arteriosclerosis is prevented by graft expression of anti-oxidant and anti-apoptotic genes. Nat Med (1998) 4:1392–6. doi:10.1038/3982
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med (2000) 192:1015–26. doi:10.1084/jem.192.7.1015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Pae HO, Oh GS, Choi BM, Chae SC, Kim YM, Chung KR, et al. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J Immunol (2004) 172:4744–51. doi:10.4049/jimmunol.172.8.4744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. McDaid J, Yamashita K, Chora A, Ollinger R, Strom TB, Li XC, et al. Heme oxygenase-1 modulates the allo-immune response by promoting activation-induced cell death of T cells. FASEB J (2005) 19:458–60.
210. Durr S, Kindler V. Implication of indolamine 2,3 dioxygenase in the tolerance toward fetuses, tumors, and allografts. J Leukoc Biol (2013) 93:681–7. doi:10.1189/jlb.0712347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Beutelspacher SC, Tan PH, McClure MO, Larkin DF, Lechler RI, George AJ. Expression of indoleamine 2,3-dioxygenase (IDO) by endothelial cells: implications for the control of alloresponses. Am J Transplant (2006) 6:1320–30. doi:10.1111/j.1600-6143.2006.01324.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Thebault P, Condamine T, Heslan M, Hill M, Bernard I, Saoudi A, et al. Role of IFNgamma in allograft tolerance mediated by CD4+CD25+ regulatory T cells by induction of IDO in endothelial cells. Am J Transplant (2007) 7:2472–82. doi:10.1111/j.1600-6143.2007.01960.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. LaGier AJ, Pober JS. Immune accessory functions of human endothelial cells are modulated by overexpression of B7-H1 (PDL1). Hum Immunol (2006) 67:568–78. doi:10.1016/j.humimm.2006.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Koga N, Suzuki J, Kosuge H, Haraguchi G, Onai Y, Futamatsu H, et al. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler Thromb Vasc Biol (2004) 24:2057–62. doi:10.1161/01.ATV.0000145015.23656.e4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Kim-Schulze S, Seki T, Vlad G, Scotto L, Fan J, Colombo PC, et al. Regulation of ILT3 gene expression by processing of precursor transcripts in human endothelial cells. Am J Transplant (2006) 6:76–82. doi:10.1111/j.1600-6143.2005.01162.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Russell ME, Hancock WW, Akalin E, Wallace AF, Glysing-Jensen T, Willett TA, et al. Chronic cardiac rejection in the LEW to F344 rat model. Blockade of CD28-B7 costimulation by CTLA4Ig modulates T cell and macrophage activation and attenuates arteriosclerosis. J Clin Invest (1996) 97:833–8. doi:10.1172/JCI118483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Glysing-Jensen T, Raisanen-Sokolowski A, Sayegh MH, Russell ME. Chronic blockade of CD28-B7-mediated T-cell costimulation by CTLA4Ig reduces intimal thickening in MHC class I and II incompatible mouse heart allografts. Transplantation (1997) 64:1641–5. doi:10.1097/00007890-199712270-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
219. Sun H, Subbotin V, Chen C, Aitouche A, Valdivia LA, Sayegh MH, et al. Prevention of chronic rejection in mouse aortic allografts by combined treatment with CTLA4-Ig and anti-CD40 ligand monoclonal antibody. Transplantation (1997) 64:1838–43. doi:10.1097/00007890-199712270-00035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Kirk AD, Guasch A, Xu H, Cheeseman J, Mead SI, Ghali A, et al. Renal transplantation using belatacept without maintenance steroids or calcineurin inhibitors. Am J Transplant (2014) 14:1142–51. doi:10.1111/ajt.12712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med (2008) 358:353–61. doi:10.1056/NEJMoa071074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Kawai T, Sachs DH, Sprangers B, Spitzer TR, Saidman SL, Zorn E, et al. Long-term results in recipients of combined HLA-mismatched kidney and bone marrow transplantation without maintenance immunosuppression. Am J Transplant (2014) 14:1599–611. doi:10.1111/ajt.12731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
223. Kawai T, Cosimi AB, Wee SL, Houser S, Andrews D, Sogawa H, et al. Effect of mixed hematopoietic chimerism on cardiac allograft survival in cynomolgus monkeys. Transplantation (2002) 73:1757–64. doi:10.1097/00007890-200206150-00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Nadig SN, Wieckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med (2010) 16:809–13. doi:10.1038/nm.2154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Hester J, Schiopu A, Nadig SN, Wood KJ. Low-dose rapamycin treatment increases the ability of human regulatory T cells to inhibit transplant arteriosclerosis in vivo. Am J Transplant (2012) 12:2008–16. doi:10.1111/j.1600-6143.2012.04065.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Farb A, John M, Acampado E, Kolodgie FD, Prescott MF, Virmani R. Oral everolimus inhibits in-stent neointimal growth. Circulation (2002) 106:2379–84. doi:10.1161/01.CIR.0000033973.06059.04
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Grube E, Sonoda S, Ikeno F, Honda Y, Kar S, Chan C, et al. Six- and twelve-month results from first human experience using everolimus-eluting stents with bioabsorbable polymer. Circulation (2004) 109:2168–71. doi:10.1161/01.CIR.0000128850.84227.FD
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N Engl J Med (2003) 349:847–58. doi:10.1056/NEJMoa022171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
229. Costanzo MR, Dipchand A, Starling R, Anderson A, Chan M, Desai S, et al. The international society of heart and lung transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant (2010) 29:914–56. doi:10.1016/j.healun.2010.05.034
230. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol (2013) 9:731–40. doi:10.1038/nrrheum.2013.161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: organ transplantation, transplant arteriosclerosis, blood vessels, endothelium, T cell, antibodies
Citation: von Rossum A, Laher I and Choy JC (2015) Immune-mediated vascular injury and dysfunction in transplant arteriosclerosis. Front. Immunol. 5:684. doi: 10.3389/fimmu.2014.00684
Received: 23 October 2014; Accepted: 18 December 2014;
Published online: 12 January 2015.
Edited by:
Philippe Saas, Etablissement Français du Sang, FranceReviewed by:
Nuala Mooney, Centre National de la Recherche Scientifique, FrancePhilippe Saas, Etablissement Français du Sang, France
Marcelo Hill, Institut Pasteur de Montevideo, Uruguay
Copyright: © 2015 von Rossum, Laher and Choy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan C. Choy, Department of Molecular Biology and Biochemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC V5A 1S6, Canada e-mail:am9uYXRoYW4uY2hveUBzZnUuY2E=