 Nicole M. Chapman
Nicole M. Chapman Hongbo Chi
Hongbo Chi- Department of Immunology, St. Jude Children’s Research Hospital, Memphis, TN, USA
T cell fate decisions play an integral role in maintaining the health of organisms under homeostatic and inflammatory conditions. The localized microenvironment in which developing and mature T cells reside provides signals that serve essential functions in shaping these fate decisions. These signals are derived from the immune compartment, including antigens, co-stimulation, and cytokines, and other factors, including growth factors and nutrients. The mechanistic target of rapamycin (mTOR), a vital sensor of signals within the immune microenvironment, is a central regulator of T cell biology. In this review, we discuss how various environmental cues tune mTOR activity in T cells, and summarize how mTOR integrates these signals to influence multiple aspects of T cell biology.
Introduction
T lymphocytes are comprised of heterogeneous populations that include conventional αβ T cells, γδ T cells, invariant natural killer T (iNKT) cells, and Foxp3+ regulatory T (Treg) cells. These functionally and phenotypically distinct T cell populations are involved in immune homeostasis and tolerance, pathogen clearance, and elimination of cancerous cells. T cell fate decisions are shaped by environmental signals received from nutrients, growth factors, cytokines, and cell–cell interactions. The serine/threonine kinase, mechanistic target of rapamycin (mTOR; formerly known as the mammalian target of rapamycin), integrates these environmental cues. The mTOR kinase exists in two, multi-protein complexes: mTOR complex 1 (mTORC1) where mTOR associates with Raptor, or mTOR complex 2 (mTORC2) where Rictor and mSin1 bind mTOR (1, 2). mTORC1 activity is sensitive to, while mTORC2 activity is largely insensitive to, rapamycin treatment. Additionally, the upstream activating stimuli and downstream effector functions differ between these complexes (1, 2).
While the signaling pathways inducing mTORC2 activation in T cells are poorly understood, in other cell lineages, mTORC2 associated with ribosomes is strongly activated, while ER stress or GSK3-β-mediated phosphorylation of Rictor inhibits its activation (3, 4). Upstream positive regulators of mTORC1 activation include the PI3K–PDK1–Akt pathway, the RasGRP–Ras–MAPKK (also known as MEK)-ERK1/2 kinase cascade, and the small GTPase, RHEB. By contrast, the phosphatase, PTEN, TSC1/TSC2, and the LKB1–AMPK pathway antagonize mTORC1 function (1, 2).
When activated, mTORC1 signaling promotes S6K function and suppresses 4E-BP1 activation, while mTORC2 regulates Akt, SGK1, and PKC catalytic activity (1, 2, 5–8). mTOR signaling also activates transcription factors, such as c-MYC, hypoxia inducible factor 1-α (HIF1-α), and sterol regulatory element-binding proteins (SREBPs) (1, 2). Ultimately, the activation of mTOR-induced pathways impacts gene expression, protein translation, cell metabolism, growth, proliferation, survival, or migration in multiple cell lineages, including T lymphocytes (1, 2). Because of these critical biological effects, dysfunctional mTOR signaling is also linked to autoimmunity, obesity, and cancer, among other conditions (2, 9, 10).
Here, we review the multifactorial roles of mTOR in T cell biology. We first discuss how different environmental stimuli activate mTOR within T cells. Second, we describe the role of mTOR in thymocyte development. We then reveal how mTOR function is coupled to peripheral T cell quiescence, functional activation, and differentiation. The ability of mTOR to dampen the immune response by modulating Treg cell function is also discussed. We then review the known functions mTOR serves in regulating T cell trafficking under homeostasis and upon infection. Finally, we highlight how future studies will further advance our understanding of mTOR functions in T cells, and how these findings may be applied therapeutically.
Multiple Signals within the Immune Microenvironment Tune mTOR Activity in T Cells
Specialized signals derived from immune microenvironments shape T cell biology. To develop into mature T cells or gain effector functions, T cells require stimulation by immune receptors, including the TCR and co-stimulatory receptors. Soluble factors, such as cytokines, adipokines, growth factors, and nutrients, also affect T cell development and functional activation (1). mTOR integrates these immunological and environmental cues to ultimately shape T cell development, activation, and differentiation into effector or long-lived, antigen-experienced memory T cells. Below, we discuss how various factors within the immune microenvironment tune mTOR activity, and a select summary of these pathways is shown in Figure 1.
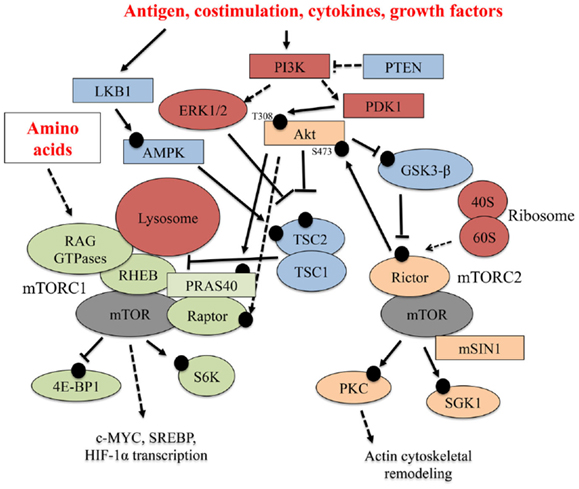
Figure 1. Select upstream regulators and downstream effectors of mTOR signaling. Multiple signaling pathways emanating from the TCR, co-stimulatory receptors, cytokines, and nutrients (amino acids) tune mTOR activation in T cells. In this figure, black circles represent phosphorylation events. Solid arrows indicate a direct, activating phosphorylation event mediated by an upstream kinase, while dashed arrows indicate an indirect, positive regulatory role for a protein in a particular pathway. Inhibitory phosphorylation events or control of pathway activation are indicated by solid or dashed flat-ended arrows, respectively.
TCR and Co-Stimulatory Receptors
When occurring in the presence of co-stimulation, TCR recognition of self and non-self peptides expressed in the context of MHC molecules is critical for T cell development and functional activation, respectively (11–15). TCR and co-stimulatory receptor triggering activate mTOR in multiple thymocyte populations, peripheral CD4+ and CD8+ T cells, and Foxp3+ Treg cells. Many studies have aimed to elucidate the mechanisms underlying this activation. PI3K catalyzes the conversion of membrane-bound phosphatidylinositol (PtdIns)-(4,5)-bisphosphate (PIP2) into PtdIns-(3,4,5)-triphosphate (PIP3), which acts as a second messenger to recruit the enzymes, PDK1 and Akt, to the plasma membrane (13). As we discuss in greater detail below, the PI3K–PDK1–Akt signaling axis promotes mTORC1 activation by inactivating the TSC1/TSC2 complex, thereby driving RHEB activation (2). However, downstream of the TCR, RHEB is only required for early (e.g., during the first 4 h of stimulation) mTORC1 activation (16), suggesting further mechanisms by which PI3K–Akt regulates mTORC1 activation. In this regard, PRAS40 is a steric inhibitor of mTOR, and its direct phosphorylation by Akt releases its suppressive activity and promotes mTOR activation (17). Akt also indirectly promotes mTOR phosphorylation by inducing IκB kinase α (IKKα) activity, resulting in the formation of stable Raptor–mTOR interactions that support mTORC1 function (18). The requirement for Akt in regulating mTOR activation may differ between different T cell populations, as TCR-induced mTORC1 activity is controlled by a PI3K–PDK1-dependent, Akt-independent pathway in effector CD8+ T cells (19). This Akt-independent pathway is linked to IL-2 induced metabolic reprograming and T cell proliferation (20). PI3K–Akt signaling is antagonized by PTEN, and loss of PTEN enhances mTOR activation (1, 2). Thus, the PI3K signaling axis is a critical regulator of mTORC1 activation at multiple levels.
In addition to PI3K, the modification of membrane-associated lipids is also controlled by PLC-γ1. Early after TCR stimulation, PLC-γ1 is activated, resulting in the cleavage of PIP2 into inositol triphosphate (IP3) and diacylglycerol (DAG). DAG supports the functional activation of the RasGRP–Ras–MAPKK–ERK1/2 pathway (13), and may cooperate with mTORC2 to induce PKC-θ activity (5). The Ras–MAPKK–ERK1/2 pathway promotes mTORC1 activation via the ERK1/2-dependent phosphorylation of TSC2 (21). T cells that lack DAG kinase (DGK)-α and DGK-ζ, which terminate DAG signaling, have elevated mTORC1 and mTORC2 activation (22). However, whether the DAG–RasGRP–Ras–MAPKK–ERK1/2 pathway acts independently or in concert with PI3K signaling is unknown, as the catalytic function of PI3K positively regulates TCR-induced ERK1/2 activation in mouse and human T cells (23, 24). Inducible Tec kinase (Itk), which directly phosphorylates and activates PLC-γ1, also promotes TCR-induced mTOR activation by inducing microRNAs that suppress PTEN expression (25). These studies indicate that many signaling pathways regulate mTOR activity downstream of the TCR.
Although TCR stimulation is necessary for effective T cell development and activation, co-stimulatory receptors must also be ligated to fully promote these processes and overcome a state of TCR-induced hypo-responsiveness called anergy (13). The classical co-stimulatory receptor for naïve T cells is CD28, which binds CD80–CD86 on antigen presenting cells (APC). However, other co-stimulatory receptors are expressed on activated T cells and Treg cells, including OX40 and ICOS (26). OX40 has been demonstrated to augment TCR-induced PI3K activation to potentiate and sustain mTORC1 activity (27), further demonstrating the critical importance of the PI3K pathway in tuning mTOR activation.
Non-enzymatic proteins also regulate mTOR activation in response to antigen and co-stimulation. The CARD-containing membrane-associated protein 1 (CARMA1)-mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) scaffolding complex is a recently identified, positive regulator of mTORC1 activation (28, 29). Because IKKα is known to associate with these proteins (14), this scaffolding complex may regulate the IKKα-dependent phosphorylation of mTORC1 in T cells. Additionally, the Hsp90 chaperone protein prevents Raptor protein degradation, thus promoting mTORC1 activation downstream of the TCR (30). However, the detailed mechanism by which Hsp90 prevents Raptor degradation remains unexplored.
Cytokines
The cytokine milieu is another crucial environmental component regulating T cell fate decisions. Within the thymus and in the periphery, IL-7 signaling via IL-7R drives T cell development and homeostasis, respectively (31). In a STAT5-dependent manner, IL-7 promotes low, transient mTORC1 activation that is critical to support IL-7 function in conventional T cells (32, 33). IL-12 activates mTOR via a STAT4-dependent mechanism in activated CD8+ T cells (34), while IL-4 and IL-1 promote mTOR activation in TH2 and TH17 cells, respectively, to induce cell cycling (35, 36). The cytokine IL-15 regulates memory T cell formation (31, 37); however, although it activates mTOR via the PI3K pathway, IL-15-induced mTOR activation driving naïve, CD8+ homeostatic proliferation is not necessary for memory T cell formation (38). Finally, IL-2 is a crucial cytokine that induces clonal expansion in activated T cells and supports Treg cell development and function (31). After cells express high levels of the high affinity IL-2 receptor (e.g., CD25 coupled with CD127), IL-2 signaling strongly activates transcriptional and metabolic reprograming via the Jak3–STAT5 and PI3K–Akt–mTORC1 pathways (1, 31). Itk is also required for efficient mTOR activation following IL-2 stimulation via mechanisms that are not fully elucidated (25). Like co-stimulatory receptor signaling in conventional T cells, IL-2 signaling also synergizes with TCR-dependent signals to enhance mTOR activation in Treg cells (1, 39).
Amino Acids
As we will discuss throughout this review, amino acids also regulate T cell activation. Relatively little is known about how amino acids control mTOR activation in T cells, but RHEB is an essential regulator of amino acid-induced mTORC1 activation in other cell lineages (40, 41). Mechanistically, amino acids drive mTORC1 activation by recruiting the heterodimeric complex of GTP-bound RAG GEFs (RAGA, RAGB, RAGC, and RAGD) to the lysosomes via the Ragulator complex (40, 41). This process is antagonized by the GAP activity of TSC2, which, when associated with lysosomes in the absence of PI3K–Akt signaling, inactivates RHEB (42). Indeed, TSC1-deficient T cells have hyper-elevated mTORC1 signaling (43), but it should be noted that amino acids can activate mTORC1 in a TSC1-independent fashion in other cell lineages (44).
Precisely how amino acids regulate T cell responses remains uncertain. In the absence of TCR and CD28 stimulation, amino acids promote mTORC1 activation in effector CD8+ T cells (45). Moreover, amino acids enhance TCR and CD28-induced mTORC1 activation (29), and IL-7 or TCR and IL-2 stimulation also increases amino acids transport to promote efficient CD8+ T cell responses (45). However, TCR and CD28-induced mTORC1 activation is controlled by RHEB-dependent and RHEB-independent mechanisms (16). One potential explanation for these data is that amino acids localize mTORC1 to the lysosome to potentiate the early activation of mTORC1 via RHEB. After prolonged antigen exposure, however, other TCR and CD28-induced signaling pathways are sufficient to sustain mTOR activation independently of RHEB (16). Future work will continue to dissect the mechanisms by which amino acids activate mTORC1 in T cells and other cell linages, but they may regulate CARMA1–MALT1–Bcl10 complex composition and function (28, 29).
NOTCH
NOTCH signaling promotes thymocyte proliferation and survival, and aids in their differentiation into terminally differentiated T cells (15). We discuss the process of thymocyte development in greater detail in the next section. Ligation of NOTCH activates mTOR activation through PI3K–Akt (46). Interestingly, aberrant NOTCH signaling is observed in both human and murine T cell acute lymphoblastic leukemia (T-ALL), and NOTCH inhibition in T-ALL lines suppresses mTOR activation by inhibiting c-MYC expression (47). However, the precise mechanisms by which this occurs remain undefined.
Leptin and Sphingosine 1-Phosphate (S1P)
Leptin is an adipocyte-derived cytokine, or adipokine, and serves multiple roles in T cells as discussed throughout this review. Recently, it was demonstrated that leptin receptor signaling contributes to the high levels of mTORC1 signaling that inhibits their IL-2-induced proliferation in vitro (39, 48). We describe how mTOR controls Treg cell development, differentiation, and function in a later section. The lipid chemokine, S1P, signals via S1PR1 and drives mTORC1 activation in a PI3K–Akt-dependent manner (49–51). These studies indicate that multiple, immune-mediated signals regulate mTOR activation within T cell populations. Below, we discuss how the integration of these signals via mTOR regulates T cell development, functional activation, suppressive function, and migration.
Role of mTOR Signaling in Thymocyte Development
Overview of Thymocyte Development
T cell development occurs within the thymus and results in the generation of mature, conventional αβ CD8+ or CD4+ T cells or non-conventional T cell populations, including CD4+ Foxp3+ thymic-derived Treg (tTreg) cells, γδ T cells, and iNKT cells. Thymocytes destined to become any T cell lineage begin as CD4−CD8− double negative (DN) thymocytes, which can be further divided into substages: DN1, DN2a, DN2b, DN3a, DN3b, and DN4. NOTCH signals drive early proliferation and T cell lineage commitment by inducing expression of the pre-TCR (e.g., a rearranged TCRβ chain with a surrogate α chain) or the γδTCR in DN thymocytes. DN2 cells that upregulate the expression of the γδTCR in the presence of high levels of IL-7R signaling will become mature γδ T cells. By contrast, to develop into conventional αβ T cells, the DN3a cells must receive signals through the pre-TCR and NOTCH to undergo β-selection. DN cells next progress into the CD4+CD8+ double positive (DP) stage. Then, these cells receive positive and negative selection signals from the TCR to become CD4+ or CD8+ single positive (SP) cells. These SP will migrate to peripheral tissues as quiescent, mature CD4+ or CD8+ T cells. Foxp3+ tTreg cells differentiate from DP cells upon receiving intermediate affinity TCR signals in the presence of IL-2 and/or IL-15. The coordination of receptor-mediated signals and transcription factor networks driving T cell development are discussed in other reviews (14, 15).
iNKT cells are a specialized, non-conventional subset of αβ T cells, and are harmful or protective in a variety of diseases (12). In both humans and mice, the TCR repertoire is restricted to Vα18–Jα18 chain paired with a limited number of Vβ chains (12). This TCR recognizes lipid antigens expressed in the context of the non-classical MHC molecule, CD1d. iNKT cell development also occurs in the thymus, diverging from the conventional αβ T cells at the DP stage in response to strong, CD1d-presented TCR signals in combination with SLAM ligation (12). In mice, the development of these cells is tracked by the expression of CD24, CD44, and NK1.1: immature stage 0 (CD24+CD44−NK1.1−), transitional stages 1 (CD24−CD44−NK1.1−) and 2 (CD24−CD44+NK1.1−), and mature stage 3 (CD24−CD44+NK1.1+). The transcription factors PLZF, GATA3, T-bet, and ROR-γt are expressed at different levels in these stages, determining their IL-4-producing NKT-2, IFN-γ-producing NKT-1, and IL-17-producing NKT-17 cell fate commitments (12, 52). NKT-2, NKT-17, and NKT-1 cells are enriched in stages 1/2, stage 2, and stage 3, respectively (52).
mTOR Controls Conventional αβ T Cell Development
To date, many studies have determined the impacts of mTOR inhibition at different stages of thymopoiesis. The conditional deletion of Raptor early during thymocyte development results in less cell cycling and proliferation, more apoptosis, and severe thymic atrophy (53). By contrast, abrogation of mTORC1 function does not appear to affect later stages of thymocytes development, as no major developmental defects are observed when mTOR is deleted in the DP stage (54) or when Raptor is deleted in the DN3 or DP stage by Lck-Cre and CD4-Cre, respectively (16, 53). Thus, mTORC1 activation serves different functions throughout thymocyte development (Figure 2).
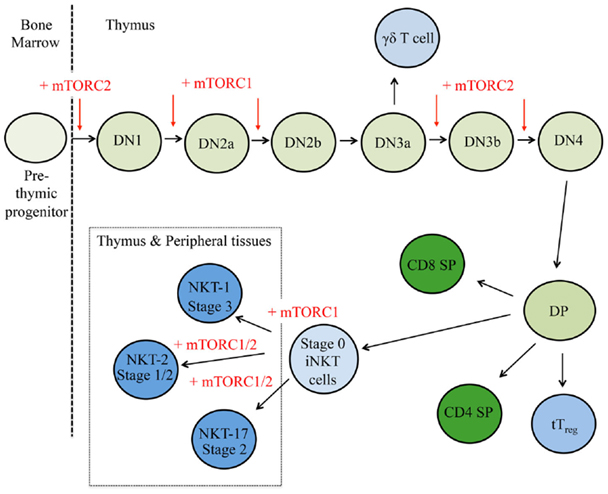
Figure 2. mTOR is a critical regulator of thymocyte development. T cell progenitors first develop within the bone marrow and migrate to the thymus. Here, cells respond to multiple environmental stimuli and progress through CD4−CD8− double negative (DN) stages 1–4 to the double positive (DP) stage. These DP thymocytes will then adopt different cellular fates in response to additional cues. Red arrows indicate where mTORC1 and/or mTORC2 control thymocyte fate decisions, where plus signs (+) represent positive regulation and minus signs (−) depict negative regulation.
mTORC2 is also critical for thymocyte development, but it appears that the mechanisms by which mTORC2 supports thymocyte development differ from mTORC1 (Figure 2). Three different genetic models (e.g., whole animal, hematopoietic-specific deletion, and T cell precursor-specific deletion) have shown loss of Rictor at different stages compromises thymocyte development and leads to thymic atrophy (53, 55, 56). Mechanistically, mTORC2 activity is connected to the stability, de novo synthesis, and/or posttranscriptional modifications of proteins involved in thymic selection, including CD4, CD8, pre-TCR, TCR, NOTCH, and CD147, a receptor expressed on highly proliferative DN4 cells (56). Collectively, these studies reveal discrete functions of mTORC1 and mTORC2 in regulating thymocyte development.
Negative regulators of mTOR also influence T cell development. Pten−/− T cells undergo malignant transformation regulated, in part, by elevated Akt and mTOR activation (57, 58). PTEN deficiency does not affect conventional T cell development, although only CD4 SP thymocyte frequencies were reported (59). However, another study demonstrated that loss of PTEN leads to the accumulation of DN, DP, and CD4 SP thymocytes, and a reduction in negative selection at the DP stage (60). These data are in subtle contrast to the positive roles Akt and mTOR play in thymocyte development (16, 53–55, 61, 62). Work from our lab and others have shown that T cell-specific deletion of TSC1 does not inhibit thymocyte development (43, 63, 64). By contrast, Lkb1−/− thymocytes have a severe developmental block linked to defects in proliferation and survival (65, 66), but these effects appear to be independent of the known substrates of LKB1, AMPK1α or the related protein, MAP/microtubule affinity-regulating kinase 2 (MARK2) (65, 67, 68). Whether LKB1 controls thymocyte development via AMPK-independent pathways or AMPK family members are functionally redundant in thymocyte development is currently unresolved.
mTOR Supports Non-Conventional iNKT Cell and Treg Cell Development
γδ T cell
Treating human peripheral blood mononuclear cells with rapamycin increases the TCR-driven expansion and effector functions of γδ T cell (69), while rapamycin treatment in vivo suppresses the functional activation of skin-resident, murine γδ T cells (70). However, the functional role mTOR signaling serves in γδ T cell development is currently unknown.
iNKT cells
mTORC1 and mTORC2 are critical regulators of iNKT cell development. Rptor−/− iNKT cells accumulate in stages 0 and 1, leading to a severe reduction of mature iNKT cells in the periphery (71, 72), whereas Rictor−/− iNKT cells are developmentally blocked at stage 2 (73, 74). The lineage commitment of iNKT cells is compromised by loss of Raptor, as the frequency of IFN-γ-producing, T-bet+ NKT-1 cells is reduced (72). By contrast, Rictor deficiency does not diminish NKT-1 cell differentiation. Loss of Rictor, however, does suppress NKT-17 cell and/or NKT-2 cell development (73, 74). Mechanistically, mTORC1 regulates iNKT cell proliferation (72), whereas mTORC2 drives TCR-induced proliferation at stage 1 and protects from TCR-induced apoptosis (73, 74). These data indicate that mTORC1 and mTORC2 serve important, yet distinct, functions in iNKT cell development.
Elevated mTOR signaling also alters iNKT cell development. Compared to conventional T cells, iNKT cells express higher levels of Tsc1 and Tsc2 mRNA (75). Importantly, this high level of TSC1/TSC2 expression regulates the terminal maturation of iNKT cells, as Tsc1−/− thymocytes have severe limitations in developing past stage 2 and into functional NKT-1 cells (75). Recent work has also demonstrated that folliculin-interacting protein 1 (Fnip1) is required for iNKT cell progression beyond stage 2 (76). Mechanistically, Fnip1−/− iNKT cells are more sensitive to apoptosis, which may be attributed to excessive mTOR signaling and mitochondrial disruption (76). Finally, PTEN also regulates iNKT cell development and function. Suzuki and co-workers demonstrated that PTEN deficiency blocks progression from stage 2 to stage 3 and also abrogates TCR-induced IFN-γ production in these cells (77). Moreover, we have recently demonstrated that NKT-17 cell development is enhanced in the absence of PTEN, in part because mTORC2 signaling is elevated in these cells (74). These studies demonstrate a pivotal role for mTOR signaling in controlling iNKT cell development.
Foxp3+ tTreg cells
In addition to iNKT cells, Foxp3+ tTreg represent a non-conventional T cell population that develops within the thymus (14). It has been reported that mTOR conditional knockout mice have normal frequencies of Treg cells (54). Conditionally deleting PTEN within T cells does not dramatically alter Treg cell development, although PTEN does suppress the IL-2-induced expansion of these cells (59). Moreover, TSC1 deficiency within the total T cell or Treg cell compartments does not alter thymic or peripheral Treg cell ratios (43, 78), but does impair their function as we discuss below. It is noteworthy that these studies did not distinguish between tTreg and peripherally induced Treg cells (pTreg), which differentiate from naïve CD4+ T cells following antigen stimulation in the presence of select cytokines. We discuss the pharmacological and genetic evidence linking mTOR signaling to pTreg differentiation later in this review. Additional studies should explore the effects of LKB1–AMPK signaling on tTreg cell development.
mTOR Controls Peripheral T Cell Homeostasis, Activation, and Differentiation
In the periphery, naïve T cells undergoing IL-7–IL-7R-driven homeostatic proliferation are maintained in a quiescent state (11). Upon receiving the appropriate antigen, co-stimulatory, cytokine, and nutrient signals, these T cells rapidly proliferate, generating multiple, antigen-specific T cell clones capable of inducing effective adaptive immune responses (13, 79, 80). These signals also induce the expression of transcription factors, including T-bet, GATA3, ROR-γt, Bcl-6, and Foxp3, which promote CD4+ T helper (TH)1, TH2, TH17, T follicular helper (TFH), and pTreg cell differentiation, respectively (79). Similarly, these signals drive CD8+ T cell differentiation into short-lived effector T cells [SLECs; T-bethiEomesodermin (EOMES)+Blimp-1hiKLRG1+IL-7Rαlo] or memory precursor cells (MPECs; T-betloEOMESloBlimp-1hiKLRG1loIL-7Rαhi) (80, 81). The switch from naïve to activated to memory T cells is coordinated by an intricate network of epigenetic, transcriptional, and metabolic programs, many of which are directly influenced by mTOR activation (1, 82, 83). Below, we discuss how alterations in mTOR signaling affect mature T cell quiescence, functional activation, and differentiation. A summary is shown in Figure 3.
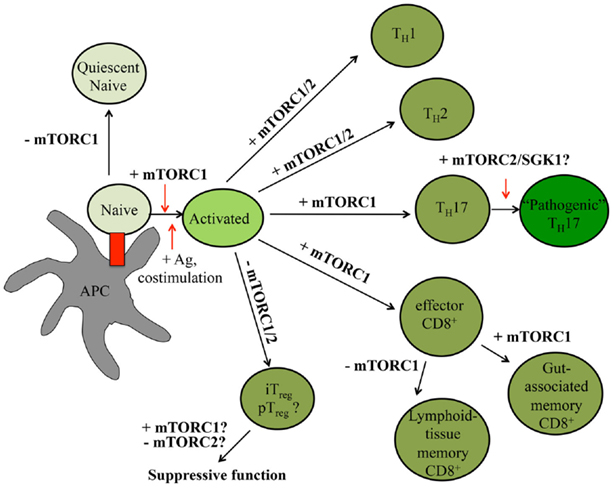
Figure 3. mTOR signaling controls peripheral T cell fate decisions. In the peripheral tissues, T cell quiescence is controlled by low levels of mTORC1 signaling. Upon receiving antigen and co-stimulatory signals, T cells rapidly expand. In the presence of select cytokines, CD4+ T cells further differentiate into different effector CD4+ T cell lineages. CD8+ T cells will become effector T cells before becoming memory T cells. The roles mTORC1 and mTORC2 serve in various T cell states are indicated within the figure, with positive roles shown with plus (+) signs and negative roles indicated by minus (−) signs. Question marks (?) indicate pathways requiring further investigation.
T Cell Homeostasis Requires Low Levels of mTORC1 Signaling
Tonic TCR signaling induced by host-derived antigens in combination with IL-7R signaling maintains T cell homeostasis (11). Moreover, recent work has linked PI3K–Akt–mTOR signaling to the homeostatic proliferation of NKT-17 cells, which preferentially require IL-7 for their homeostasis (84). While mTOR, Raptor, or Rictor-deficient T cells have no alterations in steady-state peripheral T cell homeostasis (16, 54), low levels of mTOR signaling appear to maintain CD4+ and CD8+ T cell quiescence. In support of this idea, Tsc1−/− T cells have excessive mTORC1 signaling, which promotes aberrant cell cycling (43, 63, 64, 85). Tsc1−/− T cells have reduced homeostatic proliferation in response to IL-7 signaling and are hyper-responsive to TCR-induced apoptotic signals (43, 63, 64, 85). Bcl2 overexpression rescues this defect in apoptosis, but does not restore quiescence (43).
PTEN and LKB1 are also regulators of peripheral T cell homeostasis. Mature PTEN-deficient T cells are hyper-proliferative, resistant to apoptosis, and drive autoimmunity (86). Similar to Tsc1−/− T cells, peripheral Lkb1−/− T cells are hyper-activated and are more sensitive to TCR-induced apoptosis (87). Moreover, anti-CD3 and anti-CD28 antibody, but not IL-7, induced proliferation is impaired in the absence of LKB1 (66). Although TSC1 and LKB1 have similar defects, multiple metabolic pathways, including mitochondrial functions, are dysregulated in Tsc1−/− T cells (43, 63), while glycolysis is enhanced in the absence of LKB1 (87). Thus, TSC1 and LKB1 are both critical to maintain quiescence, but they control naïve T cell homeostasis by different mechanisms.
mTOR Signaling is Coupled to T Cell Clonal Expansion
It has been demonstrated that mTOR, RHEB, and Raptor-deficient T cells have defects in antigen-driven proliferation (16, 54). This effect is largely dependent upon mTORC1-mediated signaling driving cell cycle entry from quiescence, as loss of Raptor or rapamycin treatment in naïve, but not proliferating, T cells blocks clonal expansion and instead promotes T cell anergy (16, 88). Rapamycin-treated, human T cells also have reduced proliferation (89), further supporting the idea that mTORC1 is a critical regulator of T cell proliferation. Raptor-deficient T cells have reduced c-MYC and SREBP expression and activation, respectively, leading to decreased glycolysis, oxidative phosphorylation, and/or lipogenesis (16, 90).
In addition to TCR and co-stimulatory signals, amino acids also regulate mTORC1 activation to promote T cell proliferation. Depletion of select amino acids, including arginine, leucine, or tryptophan, impairs T cell proliferation (91–93). Similarly, glutamine uptake is required for efficient T cell responses, and deletion of leucine transporters, including CD98, Sla7a5, and ASCT2, reduces mTOR activation and T cell clonal expansion (29, 94–98). Interestingly, leucine and glucose import appear to be linked, as ASCT2-deficient T cells have reduced expression of the glucose receptor, Glut1 (29). This observation may functionally link amino acid sensing to proliferation, as glucose uptake and glycolysis are intimately linked to this process (99). Collectively, these studies reveal that mTOR signaling is a crucial determinant of T cell activation.
mTOR Regulates Transcriptional and Metabolic Programs to Control T Cell Differentiation
CD4+ T cells
In addition to driving T cell proliferation, mTORC1 and mTORC2 also serve different roles in priming effector CD4+ T cell differentiation in response to antigen, co-stimulatory, and cytokine signals. In the absence of mTOR function, TH1, TH2, and TH17 polarization are all impaired (16, 54, 89, 100–102). mTORC1 activity controls TH1 and TH17 differentiation (100, 101). However, whether mTORC2 is also required for TH1 generation remains controversial (100, 101). TH2 polarization and function are severely impaired in the absence of Raptor (16), but are retained in RHEB-deficient T cells that exhibit a partial loss of mTORC1 activity (16, 101). Interestingly, although other studies link Rictor–mTORC2 to TH2 differentiation, rapamycin treatment of Rictor−/− T cells diminishes TH2 polarization more profoundly than Rictor deficiency alone (16, 100, 101). These data highlight the central role of mTORC1 in shaping TH2 differentiation.
Additional work has aimed to determine the mechanisms by which mTOR links immunological signaling to effector CD4+ T cell differentiation. Rapamycin treatment impairs TCR and CD28-induced T-bet and GATA3 upregulation, and also abrogates permissive de-methylation of the Ifng and Il4 gene loci (103). These results may explain why deleting various mTOR-related proteins inhibits TH1 and TH2 differentiation. We have demonstrated that Rptor−/− CD4+ T cells have profound defects in metabolic reprograming driven by the transcription factors, c-MYC and SREBP (16), which impairs the functional activation and differentiation of these cells. TH17 differentiation is reduced in the absence of HIF-1α, a transcription factor functionally regulated by mTORC1 activity (104, 105). Interestingly, although Rictor−/− CD4+ T cells do not exhibit defective TH17 differentiation, recent studies link the mTORC2 substrate, SGK1, to the IL-23-driven generation of highly inflammatory, “pathogenic” TH17 cells that can promote autoimmune disease development in mice (106, 107). Future work will investigate if mTORC2 regulates IL-23R signaling to facilitate this process.
Environmental cues also signal to mTOR, supporting the differentiation of CD4+ T cells. It has been demonstrated that Asct2−/− T cells have reduced TH1 and TH17 differentiation and function as a result of reduced leucine import (29, 98). This defect is linked to attenuated TCR and CD28-induced mTORC1 activation (29). Slc7a5-deficient T cells, which have impaired amino acid transport, also have reductions in TH1 and TH17 differentiation (45). Moreover, S1PR1 signaling promotes TH1 differentiation (51), while leptin receptor signaling drives TH1 and TH17 differentiation (108, 109). Future work will explore the detailed mechanisms by which these and other environmental signals, including additional amino acids, influence effector CD4+ T cell differentiation. We describe studies implicating how mTOR signaling shapes pTreg differentiation in a later section of this review.
CD8+ T cells
In CD8+ T cells, mTORC1 inhibition or deletion increases memory CD8+ T cell formation or maintenance by regulating the expression of various transcription factors, including FoxO1, T-bet, and Blimp-1 (38, 110–113). Memory CD8+ T cells may arise due to asymmetric cell division or impaired differentiation from effector CD8+ T cells (81, 114). However, knocking down Raptor in activated CD8+ T cells also potentiates memory functional CD8+ T cell differentiation (113), and deleting TSC1 from activated CD8+ T cells impairs memory differentiation and function (115). Thus, mTORC1-mediated control of memory CD8+ T cell differentiation appears to be linked to defective effector to memory differentiation. mTORC1 signaling regulates CD8+ T cell differentiation, in part, by controlling glycolytic and oxidative phosphorylation metabolism following IL-15 stimulation (115). However, it should be noted that IL-15-independent functions for mTOR in controlling CD8+ T cell memory formation have been described (38). For instance, mTORC1 imparts control over effector versus memory T cell fate decisions by regulating the expression of NOTCH on naïve CD8+ T cells (116). Thus, mTORC1 utilizes multiple mechanisms to influence effector versus memory CD8+ T cell differentiation and function.
Recent data revealed a site-specific role for mTOR signaling in the generation of CD8+ T cell memory. Marzo and colleagues found that rapamycin treatment enhances memory CD8+ T cell differentiation in the blood and spleen, but the number of memory CD8+ T cells in the lungs and peripheral lymph nodes are not affected (117). In fact, mucosal CD8+ T cells isolated from the small intestine lamina propria are reduced in numbers upon rapamycin treatment, in part due to defects in T cell trafficking as discussed below. Collectively, these data indicate a critical role for mTOR in modulating tissue-specific, effector versus memory fate decisions in CD8+ T cells.
In response to chronic infections, CD8+ T cells become functionally impaired or exhausted (118). Kaech and colleagues recently demonstrated that Akt and mTOR signaling are impaired in CD8+ effector T cells following a chronic viral infection as compared to an acute infection (119). This event leads to the FoxO1-dependent upregulation of PD-1 and promotes the survival of terminally differentiated, exhausted CD8+ T cells. Signaling downstream of PD-1 antagonizes mTOR activation (120), which drives CD8+ T cell exhaustion (119). Consistent with this idea, PD-1 blockade restores function in exhausted, CD8+ T cells in an mTOR-dependent manner (119). Therefore, in addition to supporting CD8+ T cell effector versus memory formation, the mTOR–FoxO1 axis also regulates CD8+ T cell exhaustion.
mTOR Maintains Immune Tolerance by Controlling Treg Cell Function and Stability
Foxp3+ Treg cells maintain T cell homeostasis in the periphery, and their loss of function causes severe, multi-organ autoimmunity in humans and mice (121). Interestingly, mTOR signaling serves discrete functions in Treg cell differentiation and function. Several groups demonstrated that Treg cell differentiation is potentiated in vitro (called iTreg cells) in the presence of rapamycin (54, 122–128). An inhibitory role for mTOR in the generation of iTreg cells was further supported using Mtor−/− T cells (54), with mTORC1 and mTORC2 serving functionally redundant roles in suppressing iTreg differentiation (54, 101). Likewise, HIF-1α deficiency enhances Treg cell differentiation (104, 105). However, the functional capacities of Mtor−/− or rapamycin-expanded Treg cells require further investigation, as the source of the Treg cells used in the in vitro suppression assays were not a highly purified population of Foxp3+ Treg cells. The in vivo suppressive activity of these cells also remains largely unexplored, although rapamycin-expanded, human Treg cells are functional in a xenograft transfer model (129).
Regulatory T cells have high, basal levels of mTOR signaling compared to their naïve T cell counterparts (39, 48). However, the proper threshold of mTOR signaling is critical to support their suppressive function in vitro and in vivo. We recently demonstrated that Raptor-deficient Treg cells lose suppressive activity in vitro and in vivo, the latter of which contributes to rampant autoimmunity and lethality in mice (39). Mechanistically, Raptor-mTORC1 signaling is linked to cholesterol biosynthesis and lipid metabolism, processes that are important to support the expression of the Treg cell effector molecules, ICOS and cytotoxic T lymphocyte antigen (CTLA)-4. These effects are not observed in Treg cells lacking Rictor, and combined loss of Raptor and Rictor partially restores the suppressive function of Treg cells in vitro and in vivo (39). Thus, loss of mTORC1, but not mTORC2, activity is linked to Treg cell dysfunction.
While these studies show that loss of mTORC1 activity is deleterious to Treg cell function, excessive mTOR signaling within Treg cells also compromises their function and affects their stability. TSC1-deficient Treg cells are impaired in their ability to suppress inflammatory responses, as they lose Foxp3 expression and acquire TH17 cell effector-like functions in vitro and in vivo (78). Consistent with this study, recent work demonstrated that patients with autoimmune diseases have elevated mTOR activation within their Treg cells (10). Although they proliferate more robustly following IL-2 stimulation, Pten−/− Treg cells appear to retain their suppressive capacity in vitro and can suppress colitis development in vivo (59). However, the role of PTEN in Treg cells has not been specifically addressed using a conditional deletion model. Thus, distinct negative regulators of mTOR activity appear to serve different functions in Treg cells.
Several pathways have mechanistically been shown to modulate mTOR activity within Treg cells to regulate their proliferation, differentiation, and function. Leptin receptor signaling restrains TCR and/or IL-2 stimulation-induced Treg proliferation in vitro (48, 130), suggesting that leptin levels may be a critical factor influencing Treg cell proliferation in vivo (131). Maintenance of Foxp3 expression is required for Treg suppressive function (132). Transient TCR stimulation drives PI3K–Akt–mTOR signaling that antagonizes Foxp3 expression (133), and rapamycin treatment enhances Foxp3 expression by modulating DNA methylation within the Foxp3 locus (103). Through multiple mechanisms, Treg cells can modulate amino acid availability within a microenvironment (92, 121, 134). Interestingly, mTOR inhibition and amino acid deprivation synergize with TGF-β signaling to augment Foxp3 expression in vitro (91, 92). Finally, S1PR1 signaling to mTORC1 restrains Treg differentiation in the thymus and periphery, and limits their suppressive function in vitro and in vivo during homeostasis and inflammation (50, 51).
mTOR Regulates T Cell Trafficking
After an infection occurs, chemokine and adhesion receptors localize T cells to the proper anatomical location. The adhesion receptor CD62L and chemokine receptors, CCR7 and S1PR1, allow T cells to enter and be retained in peripheral lymph nodes such that T cell activation may occur (49, 135). As with T cell development and activation, mTOR signaling is also a critical regulator of T cell trafficking. PI3K or mTORC1 inhibition in activated CD8+ T cells reduces IL-2-induced downregulation of CCR7, CD62L, and S1PR1 expression (136), which causes these cells to traffic to lymph nodes (34). By contrast, the downregulation of these molecules occurs more efficiently in the absence of PTEN or TSC1 (43, 115, 136, 137). These trafficking defects may partially account for why rapamycin treatment enhances and TSC1 deficiency suppresses memory CD8+ T cell differentiation (34, 115, 136). Although the precise mechanisms by which mTOR signaling regulates trafficking are not known, mTOR modulates the expression of Kruppel-like factor 2 (KLF2) and HIF-1α, two transcription factors that modulate the expression of lymph node homing receptors (19, 136). Further, mTORC2 may inhibit FoxO1 function by enhancing Akt activity, and FoxO1 transcriptional activity modulates the expression of lymph node homing receptors (137). Finally, mTORC1 activity induces T-bet expression (34), which drives CXCR3 upregulation and subsequently localizes T cells to sites of infection (138, 139). Thus, mTOR activity regulates T cell trafficking via multiple mechanisms (Figure 4).
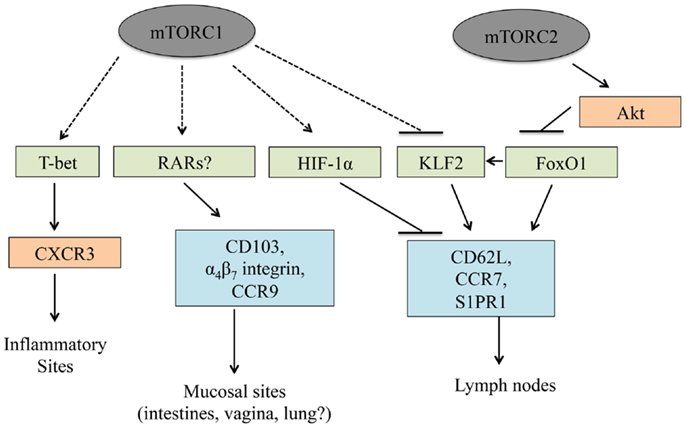
Figure 4. T cell trafficking is linked to mTOR. mTORC1 and mTORC2 control T cell trafficking by regulating the expression and/or functional activation of multiple transcription factors. In this manner, mTOR signaling regulates trafficking into inflammatory sites, lymph nodes, and mucosal sites.
Recent work also demonstrates a role for mTOR in T cell trafficking to non-lymphoid tissues. Trafficking into the gut-associated mucosa is regulated by CCR9, the α4β7 integrin, and CD103 (140). In CD8+ T cells, rapamycin treatment suppresses the expression and/or function of these molecules, leading to a severe reduction in these cells within mucosal sites (117). Similarly, knocking down mTOR within activated CD8+ T cells also reduces trafficking to the small intestine. Although it was not mechanistically determined how mTOR controls mucosal site homing, the retinoic acid receptors (RARs) induce CCR9 and α4β7 integrin expression in activated T cells (141, 142). As Treg cells, TH17 cells, and iNKT cells play pivotal roles in gut-associated lymphoid reactions (143, 144), future work will need to explore how mTOR inhibition or hyper-activation influences trafficking to mucosal sites within these cell lineages.
Concluding Remarks
Current work has highlighted the critical role the environmental sensor mTOR plays in T cell biology. mTORC1 and mTORC2 both support thymocyte development, but integrate distinct and overlapping signals and impart discrete functions to facilitate this process. In contrast to thymocytes, mTORC1 is the dominant regulator of the functional activation and differentiation of conventional T cells in the periphery. mTORC1 activation is critical for clonal expansion, effector CD4+ T cell differentiation, and Treg cell function, while mTORC2 also contributes to these processes but with limited effects. However, further work is needed to determine the role mTORC1 and mTORC2 serve in the induction of site-specific immune responses, including the generation of TFH cells and tissue-specific Treg cell populations, the latter of which play critical functions in dampening immune responses in mucosal sites, adipose tissues, and tumors (121, 145).
From a clinical perspective, it will be critical to determine the impacts of mTOR inhibition on the specific immunity to pathogens, tumors, and auto-antigens. Hyper- or hypo-activation of mTOR has a profound impact on T cell development and activation, so these investigations will provide insight into how rapamycin, its rapalogs, and other next generation mTOR inhibitors will influence localized and systemic immune responses in different disease settings. Given the intricate link between mTOR function and T cell fate decisions, it is feasible that one could modulate mTOR activation within specific inflammatory sites and/or immune cell types to modulate the immune response in states where both mTOR and T cells are dysfunctional. These studies will be key toward determining if mTOR suppression in T cells is a viable target for treating autoimmunity, cancers, and infectious diseases, or for boosting memory CD8+ T cell responses to enhance vaccine efficacy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by US NIH (AI101407, AI105887, CA176624 and NS064599), National Multiple Sclerosis Society, Crohn’s & Colitis Foundation of America, the American Lebanese Syrian Associated Charities (Hongbo Chi) and by The Hartwell Foundation Biomedical Research Fellowship (Nicole M. Chapman).
References
1. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol (2012) 12:325–38. doi: 10.1038/nri3198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi:10.1016/j.cell.2012.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, et al. ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Sci Signal (2011) 4:ra10. doi:10.1126/scisignal.2001731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell (2011) 144:757–68. doi:10.1016/j.cell.2011.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol (2004) 6:1122–8. doi:10.1038/ncb1183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol (2004) 14:1296–302. doi:10.1016/j.cub.2004.06.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307:1098–101. doi:10.1126/science.1106148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J (2008) 416:375–85. doi:10.1042/BJ20081668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Daley SR, Coakley KM, Hu DY, Randall KL, Jenne CN, Limnander A, et al. Rasgrp1 mutation increases naive T-cell CD44 expression and drives mTOR-dependent accumulation of Helios+ T cells and autoantibodies. Elife (2013) 2:e01020. doi:10.7554/eLife.01020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Carbone F, De Rosa V, Carrieri PB, Montella S, Bruzzese D, Porcellini A, et al. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med (2014) 20:69–74. doi:10.1038/nm.3411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol (2011) 12:478–84. doi:10.1038/ni.2018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13:101–17. doi:10.1038/nri3369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol (2013) 13:257–69. doi:10.1038/nri3403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Gerondakis S, Fulford TS, Messina NL, Grumont RJ. NF-kappaB control of T cell development. Nat Immunol (2014) 15:15–25. doi:10.1038/ni.2785
15. Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol (2014) 14:529–45. doi:10.1038/nri3702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity (2013) 39:1043–56. doi:10.1016/j.immuni.2013.09.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol (2007) 9:316–23. doi:10.1038/ncb1547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, Baldwin AS. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IkappaB kinase alpha (IKKalpha). J Biol Chem (2014) 289:25227–40. doi:10.1074/jbc.M114.554881
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med (2012) 209:2441–53. doi:10.1084/jem.20112607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity (2011) 34:224–36. doi:10.1016/j.immuni.2011.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell (2005) 121:179–93. doi:10.1016/j.cell.2005.02.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood (2011) 117:4022–31. doi:10.1182/blood-2010-08-300731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Robertson LK, Mireau LR, Ostergaard HL. A role for phosphatidylinositol 3-kinase in TCR-stimulated ERK activation leading to paxillin phosphorylation and CTL degranulation. J Immunol (2005) 175:8138–45. doi:10.4049/jimmunol.175.12.8138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Cruz-Orcutt N, Houtman JC. PI3 kinase function is vital for the function but not formation of LAT-mediated signaling complexes. Mol Immunol (2009) 46:2274–83. doi:10.1016/j.molimm.2009.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med (2014) 211:529–43. doi:10.1084/jem.20131459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Ford ML, Adams AB, Pearson TC. Targeting co-stimulatory pathways: transplantation and autoimmunity. Nat Rev Nephrol (2014) 10:14–24. doi:10.1038/nrneph.2013.183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. So T, Choi H, Croft M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J Immunol (2011) 186:3547–55. doi:10.4049/jimmunol.1003156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signal (2014) 7:ra55. doi:10.1126/scisignal.2005169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi:10.1016/j.immuni.2014.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Delgoffe GM, Kole TP, Cotter RJ, Powell JD. Enhanced interaction between Hsp90 and raptor regulates mTOR signaling upon T cell activation. Mol Immunol (2009) 46:2694–8. doi:10.1016/j.molimm.2009.05.185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol (2009) 9:480–90. doi:10.1038/nri2580
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol (2001) 167:6869–76. doi:10.4049/jimmunol.167.12.6869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood (2008) 111:2101–11. doi:10.1182/blood-2007-06-096297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity (2010) 32:67–78. doi:10.1016/j.immuni.2009.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Stephenson LM, Park DS, Mora AL, Goenka S, Boothby M. Sequence motifs in IL-4R alpha mediating cell-cycle progression of primary lymphocytes. J Immunol (2005) 175:5178–85. doi:10.4049/jimmunol.175.8.5178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity (2010) 32:54–66. doi:10.1016/j.immuni.2009.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med (2000) 191:771–80. doi:10.1084/jem.191.5.771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Li Q, Rao RR, Araki K, Pollizzi K, Odunsi K, Powell JD, et al. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity (2011) 34:541–53. doi:10.1016/j.immuni.2011.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499:485–90. doi:10.1038/nature12297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med (2012) 18:524–33. doi:10.1016/j.molmed.2012.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol (2013) 14:133–9. doi:10.1038/nrm3522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell (2014) 156:771–85. doi:10.1016/j.cell.2013.11.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol (2011) 12:888–97. doi:10.1038/ni.2068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Peng M, Yin N, Li MO. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell (2014) 159:122–33. doi:10.1016/j.cell.2014.08.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14:500–8. doi:10.1038/ni.2556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res (2006) 66:4715–24. doi:10.1158/0008-5472.CAN-05-3830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood (2007) 110:278–86. doi:10.1182/blood-2006-08-039883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity (2010) 33:929–41. doi:10.1016/j.immuni.2010.11.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Chi H, Flavell RA. Cutting edge: regulation of T cell trafficking and primary immune responses by sphingosine 1-phosphate receptor 1. J Immunol (2005) 174:2485–8. doi:10.4049/jimmunol.174.5.2485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, et al. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol (2009) 10:769–77. doi:10.1038/ni.1743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol (2010) 11:1047–56. doi:10.1038/ni.1939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol (2013) 14:1146–54. doi:10.1038/ni.2731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Hoshii T, Kasada A, Hatakeyama T, Ohtani M, Tadokoro Y, Naka K, et al. Loss of mTOR complex 1 induces developmental blockage in early T-lymphopoiesis and eradicates T-cell acute lymphoblastic leukemia cells. Proc Natl Acad Sci U S A (2014) 111:3805–10. doi:10.1073/pnas.1320265111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi:10.1016/j.immuni.2009.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Lee K, Nam KT, Cho SH, Gudapati P, Hwang Y, Park DS, et al. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J Exp Med (2012) 209:713–28. doi:10.1084/jem.20111470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Chou PC, Oh WJ, Wu CC, Moloughney J, Ruegg MA, Hall MN, et al. Mammalian target of rapamycin complex 2 modulates alphabetaTCR processing and surface expression during thymocyte development. J Immunol (2014) 193:1162–70. doi:10.4049/jimmunol.1303162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Finlay DK, Sinclair LV, Feijoo C, Waugh CM, Hagenbeek TJ, Spits H, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med (2009) 206:2441–54. doi:10.1084/jem.20090219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Guo W, Schubbert S, Chen JY, Valamehr B, Mosessian S, Shi H, et al. Suppression of leukemia development caused by PTEN loss. Proc Natl Acad Sci U S A (2011) 108:1409–14. doi:10.1073/pnas.1006937108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Walsh PT, Buckler JL, Zhang J, Gelman AE, Dalton NM, Taylor DK, et al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Invest (2006) 116:2521–31. doi:10.1172/JCI28057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity (2001) 14:523–34. doi:10.1016/S1074-7613(01)00134-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Juntilla MM, Wofford JA, Birnbaum MJ, Rathmell JC, Koretzky GA. Akt1 and Akt2 are required for alphabeta thymocyte survival and differentiation. Proc Natl Acad Sci U S A (2007) 104:12105–10. doi:10.1073/pnas.0705285104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Mao C, Tili EG, Dose M, Haks MC, Bear SE, Maroulakou I, et al. Unequal contribution of Akt isoforms in the double-negative to double-positive thymocyte transition. J Immunol (2007) 178:5443–53. doi:10.4049/jimmunol.178.9.5443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. O’Brien TF, Gorentla BK, Xie D, Srivatsan S, Mcleod IX, He YW, et al. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur J Immunol (2011) 41:3361–70. doi:10.1002/eji.201141411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Wu Q, Liu Y, Chen C, Ikenoue T, Qiao Y, Li CS, et al. The tuberous sclerosis complex-mammalian target of rapamycin pathway maintains the quiescence and survival of naive T cells. J Immunol (2011) 187:1106–12. doi:10.4049/jimmunol.1003968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Cao Y, Li H, Liu H, Zheng C, Ji H, Liu X. The serine/threonine kinase LKB1 controls thymocyte survival through regulation of AMPK activation and Bcl-XL expression. Cell Res (2010) 20:99–108. doi:10.1038/cr.2009.141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Tamas P, Macintyre A, Finlay D, Clarke R, Feijoo-Carnero C, Ashworth A, et al. LKB1 is essential for the proliferation of T-cell progenitors and mature peripheral T cells. Eur J Immunol (2010) 40:242–53. doi:10.1002/eji.200939677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Hurov JB, Stappenbeck TS, Zmasek CM, White LS, Ranganath SH, Russell JH, et al. Immune system dysfunction and autoimmune disease in mice lacking Emk (Par-1) protein kinase. Mol Cell Biol (2001) 21:3206–19. doi:10.1128/MCB.21.9.3206-3219.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol (2008) 38:948–56. doi:10.1002/eji.200738045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Li H, Pauza CD. Rapamycin increases the yield and effector function of human gammadelta T cells stimulated in vitro. Cancer Immunol Immunother (2011) 60:361–70. doi:10.1007/s00262-010-0945-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Mills RE, Taylor KR, Podshivalova K, Mckay DB, Jameson JM. Defects in skin gamma delta T cell function contribute to delayed wound repair in rapamycin-treated mice. J Immunol (2008) 181:3974–83. doi:10.4049/jimmunol.181.6.3974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Shin J, Wang S, Deng W, Wu J, Gao J, Zhong XP. Mechanistic target of rapamycin complex 1 is critical for invariant natural killer T-cell development and effector function. Proc Natl Acad Sci U S A (2014) 111:E776–83. doi:10.1073/pnas.1315435111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Zhang L, Tschumi BO, Corgnac S, Ruegg MA, Hall MN, Mach JP, et al. Mammalian target of rapamycin complex 1 orchestrates invariant NKT cell differentiation and effector function. J Immunol (2014) 193:1759–65. doi:10.4049/jimmunol.1400769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Prevot N, Pyaram K, Bischoff E, Sen JM, Powell JD, Chang CH. Mammalian target of rapamycin complex 2 regulates invariant NK T cell development and function independent of promyelocytic leukemia zinc-finger. J Immunol (2014) 194(1):223–30. doi:10.4049/jimmunol.1401985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Wei J, Yang K, Chi H. Cutting edge: discrete functions of mTOR signaling in invariant NKT cell development and NKT17 fate decision. J Immunol (2014) 193:4297–301. doi:10.4049/jimmunol.1402042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Wu J, Shin J, Xie D, Wang H, Gao J, Zhong XP. Tuberous sclerosis 1 promotes invariant NKT cell anergy and inhibits invariant NKT cell-mediated antitumor immunity. J Immunol (2014) 192:2643–50. doi:10.4049/jimmunol.1302076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Park H, Tsang M, Iritani BM, Bevan MJ. Metabolic regulator Fnip1 is crucial for iNKT lymphocyte development. Proc Natl Acad Sci U S A (2014) 111:7066–71. doi:10.1073/pnas.1406473111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Kishimoto H, Ohteki T, Yajima N, Kawahara K, Natsui M, Kawarasaki S, et al. The Pten/PI3K pathway governs the homeostasis of Valpha14iNKT cells. Blood (2007) 109:3316–24. doi:10.1182/blood-2006-07-038059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Park Y, Jin HS, Lopez J, Elly C, Kim G, Murai M, et al. TSC1 regulates the balance between effector and regulatory T cells. J Clin Invest (2013) 123:5165–78. doi:10.1172/JCI69751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol (2013) 13:777–89. doi:10.1038/nri3536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12:749–61. doi:10.1038/nri3307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Yang K, Chi H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin Immunol (2012) 24:421–8. doi:10.1016/j.smim.2012.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Zeng H, Chi H. mTOR and lymphocyte metabolism. Curr Opin Immunol (2013) 25:347–55. doi:10.1016/j.coi.2013.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Webster KE, Kim HO, Kyparissoudis K, Corpuz TM, Pinget GV, Uldrich AP, et al. IL-17-producing NKT cells depend exclusively on IL-7 for homeostasis and survival. Mucosal Immunol (2014) 7:1058–67. doi:10.1038/mi.2013.122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Zhang L, Zhang H, Li L, Xiao Y, Rao E, Miao Z, et al. TSC1/2 signaling complex is essential for peripheral naive CD8+ T cell survival and homeostasis in mice. PLoS One (2012) 7:e30592. doi:10.1371/journal.pone.0030592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Liu X, Karnell JL, Yin B, Zhang R, Zhang J, Li P, et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J Clin Invest (2010) 120:2497–507. doi:10.1172/JCI42382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol (2011) 187:4187–98. doi:10.4049/jimmunol.1100367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, et al. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol (2007) 178:2163–70. doi:10.4049/jimmunol.178.4.2163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Kopf H, De La Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol (2007) 7:1819–24. doi:10.1016/j.intimp.2007.08.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Fallarino F, Grohmann U, You S, Mcgrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176:6752–61. doi:10.4049/jimmunol.176.11.6752
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A (2009) 106:12055–60. doi:10.1073/pnas.0903919106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183:6095–101. doi:10.4049/jimmunol.0803510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Utsunomiya-Tate N, Endou H, Kanai Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J Biol Chem (1996) 271:14883–90. doi:10.1074/jbc.271.25.14883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185:1037–44. doi:10.4049/jimmunol.0903586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Cantor J, Slepak M, Ege N, Chang JT, Ginsberg MH. Loss of T cell CD98 H chain specifically ablates T cell clonal expansion and protects from autoimmunity. J Immunol (2011) 187:851–60. doi:10.4049/jimmunol.1100002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Fotiadis D, Kanai Y, Palacin M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med (2013) 34:139–58. doi:10.1016/j.mam.2012.10.007
98. Poffenberger MC, Jones RG. Amino acids fuel T cell-mediated inflammation. Immunity (2014) 40:635–7. doi:10.1016/j.immuni.2014.04.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13:907–15. doi:10.1038/ni.2386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity (2010) 32:743–53. doi:10.1016/j.immuni.2010.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12:295–303. doi:10.1038/ni.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep (2012) 1:360–73. doi:10.1016/j.celrep.2012.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Tomasoni R, Basso V, Pilipow K, Sitia G, Saccani S, Agresti A, et al. Rapamycin-sensitive signals control TCR/CD28-driven Ifng, Il4 and Foxp3 transcription and promoter region methylation. Eur J Immunol (2011) 41:2086–96. doi:10.1002/eji.201041130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature (2013) 496:518–22. doi:10.1038/nature11868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature (2013) 496:513–7. doi:10.1038/nature11984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature (1998) 394:897–901. doi:10.1038/29795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Chuang HC, Sheu WH, Lin YT, Tsai CY, Yang CY, Cheng YJ, et al. HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat Commun (2014) 5:4602. doi:10.1038/ncomms5602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. doi:10.1038/nature08155
111. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi:10.1038/nature08097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Turner AP, Shaffer VO, Araki K, Martens C, Turner PL, Gangappa S, et al. Sirolimus enhances the magnitude and quality of viral-specific CD8+ T-cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant (2011) 11:613–8. doi:10.1111/j.1600-6143.2010.03407.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest (2014) 124:188–97. doi:10.1172/JCI69856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science (2007) 315:1687–91. doi:10.1126/science.1139393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Shrestha S, Yang K, Wei J, Karmaus PW, Neale G, Chi H. Tsc1 promotes the differentiation of memory CD8+ T cells via orchestrating the transcriptional and metabolic programs. Proc Natl Acad Sci U S A (2014) 111:14858–63. doi:10.1073/pnas.1404264111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Backer RA, Helbig C, Gentek R, Kent A, Laidlaw BJ, Dominguez CX, et al. A central role for Notch in effector CD8 T cell differentiation. Nat Immunol (2014) 15(12):1143–51. doi:10.1038/ni.3027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Sowell RT, Rogozinska M, Nelson CE, Vezys V, Marzo AL. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J Immunol (2014) 193:2067–71. doi:10.4049/jimmunol.1400074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8+ T cells during chronic infection. Immunity (2014) 41:802–14. doi:10.1016/j.immuni.2014.10.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi:10.1084/jem.20090847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105:4743–8. doi:10.1182/blood-2004-10-3932
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol (2006) 177:8338–47. doi:10.4049/jimmunol.177.12.8338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Valmori D, Tosello V, Souleimanian NE, Godefroy E, Scotto L, Wang Y, et al. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T cells. J Immunol (2006) 177:944–9. doi:10.4049/jimmunol.177.2.944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, Strom TB. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant (2007) 7:1722–32. doi:10.1111/j.1600-6143.2007.01842.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Kang J, Huddleston SJ, Fraser JM, Khoruts A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. J Leukoc Biol (2008) 83:1230–9. doi:10.1189/jlb.1207851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Bocian K, Borysowski J, Wierzbicki P, Wyzgal J, Klosowska D, Bialoszewska A, et al. Rapamycin, unlike cyclosporine A, enhances suppressive functions of in vitro-induced CD4+CD25+ Tregs. Nephrol Dial Transplant (2010) 25:710–7. doi:10.1093/ndt/gfp586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS One (2011) 6:e15868. doi:10.1371/journal.pone.0015868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Tresoldi E, Dell’Albani I, Stabilini A, Jofra T, Valle A, Gagliani N, et al. Stability of human rapamycin-expanded CD4+CD25+ T regulatory cells. Haematologica (2011) 96:1357–65. doi:10.3324/haematol.2011.041483
130. De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity (2007) 26:241–55. doi:10.1016/j.immuni.2007.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med (2003) 198:249–58. doi:10.1084/jem.20030315
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10:490–500. doi:10.1038/nri2785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A (2008) 105:7797–802. doi:10.1073/pnas.0800928105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4:1206–12. doi:10.1038/ni1003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol (2011) 11:109–17. doi:10.1038/nri2888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol (2008) 9:513–21. doi:10.1038/ni.1603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, Depinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol (2009) 10:176–84. doi:10.1038/ni.1689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Taqueti VR, Grabie N, Colvin R, Pang H, Jarolim P, Luster AD, et al. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. J Immunol (2006) 177:5890–901. doi:10.4049/jimmunol.177.9.5890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10:595–602. doi:10.1038/ni.1731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Islam SA, Luster AD. T cell homing to epithelial barriers in allergic disease. Nat Med (2012) 18:705–15. doi:10.1038/nm.2760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity (2004) 21:527–38. doi:10.1016/j.immuni.2004.08.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Svensson M, Johansson-Lindbom B, Zapata F, Jaensson E, Austenaa LM, Blomhoff R, et al. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunol (2008) 1:38–48. doi:10.1038/mi.2007.4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Harrison OJ, Powrie FM. Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb Perspect Biol (2013) 5:7. doi:10.1101/cshperspect.a018341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Ussher JE, Klenerman P, Willberg CB. Mucosal-associated invariant T-cells: new players in anti-bacterial immunity. Front Immunol (2014) 5:450. doi:10.3389/fimmu.2014.00450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Lehtimaki S, Lahesmaa R. Regulatory T cells control immune responses through their non-redundant tissue specific features. Front Immunol (2013) 4:294. doi:10.3389/fimmu.2013.00294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mTOR, T cells, iNKT cell, Treg cells
Citation: Chapman NM and Chi H (2015) mTOR links environmental signals to T cell fate decisions. Front. Immunol. 5:686. doi: 10.3389/fimmu.2014.00686
Received: 02 December 2014; Accepted: 20 December 2014;
Published online: 20 January 2015.
Edited by:
Anne L. Astier, The University of Edinburgh, UKReviewed by:
Jonathan Powell, Johns Hopkins University, USAGiuseppe Matarese, Università di Salerno, Italy
Copyright: © 2015 Chapman and Chi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongbo Chi, Department of Immunology, St. Jude Children’s Research Hospital, MS 351, Memphis, TN 38105, USA e-mail:aG9uZ2JvLmNoaUBzdGp1ZGUub3Jn