 Graziela Gorete Romagnoli1
Graziela Gorete Romagnoli1 Bruna Barbosa Zelante1
Bruna Barbosa Zelante1 Patrícia Argenta Toniolo1
Patrícia Argenta Toniolo1 Isabella Katz Migliori1
Isabella Katz Migliori1 José Alexandre M. Barbuto1,2*
José Alexandre M. Barbuto1,2*- 1Laboratory of Tumor Immunology, Department of Immunology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil
- 2Center for Cellular and Molecular Studies and Therapy (NETCEM), University of São Paulo, São Paulo, Brazil
Dendritic cells (DCs) have been attracting attention in cancer immunotherapy because of their role in inducing and modulating effective immune responses. Besides the direct contact with other cell types and the secretion of cytokines, it is becoming clear that nanovesicles, such as exosomes (Exo), secreted by DCs also have a role in their function. Conversely, tumor-derived Exo carry antigens and have been used as a source of specific stimulus for the immune response against tumors. At the same time, several works have shown that different cells types incorporate DC-derived Exo (DC-Exo), resulting in modifications of their phenotype and function. Since DC-Exo carry many of the immune function-associated molecules of DCs, their incorporation by tumor cells could turn tumor cells into immunogenic targets. We have, therefore, treated human breast adenocarcinoma cells (SK-BR-3) with DCs-Exo and used these to stimulate previously SK-BR-3-primed CD3+ T-cells. Sensitized T-cells cultured with DC-Exo-treated tumor cells showed a significantly higher percentage of IFN-γ-secreting cells (as measured by ELISPOT), when compared to the frequency of cells responding to non-DC-Exo-treated cells. These data show that the incorporation of DC-Exo by the tumor cells increased their ability to activate T-cells for a possibly more effective response, thus showing that DC-Exo may become another tool in cancer immunotherapy.
Introduction
Dendritic cells (DCs) are key players in the immune response; they are able to capture antigens with their pattern-recognition receptors, process and present them to naïve T-cells, inducing their activation (1, 2), thus, building an essential bridge between innate and adaptive responses. The central role that DCs play in the immune response, and the possibility of their in vitro generation has pathways for immunotherapy, in particular, for the treatment of cancer (3–8). However, the use of DCs outside clinical studies is hampered by the difficulties inherent to cell therapy strategies and, furthermore, in the case of DCs specifically against cancer, also by the compromised function of these cells in cancer patients (9–12). Not surprisingly, therefore, the general appraisal of DC-based strategies against cancer has been negative (10, 13). On the other hand, tumor cells do present potentially immunogenic antigens (14), which, when recognized by T-cells in immunotherapeutic approaches, seem to be associated with lasting tumor remissions (15). Therefore, strategies aimed at exposing tumor antigens to the immune system, bypassing the need for very active DCs, but in such a way that it leads to the establishment of T-cell responses, would be a potentially effective approach to harness the immune system to fight cancer.
In this context, therefore, it is relevant to note that, as most other cell types, DCs secrete nanovesicles, among which are the exosomes (Exo) (16–19). Exo are secreted vesicles that originate in the late endosomal compartment and result from the fusion of multivesicular bodies with the plasma membrane (20) and which can be acquired by other cells, at least in ex vivo cell cultures (21–24). These nanovesicles contain membrane proteins and genetic material, which, upon capture by other cells, contribute to the intercellular communication in the body (25–27). In fact, membrane traffic between DCs via Exo has been shown to occur (22), and Exo-carried antigens can be reprocessed for presentation or simply transferred directly to the membrane, in a process called cross-dressing (28). Furthermore, Exo transfer has been reported also to happen between cells of different types (25, 29, 30). Indeed, we demonstrated previously that Exo originated from DCs may be incorporated by tumor cells in vitro and that these tumor cells, after treatment with DC-derived Exo (DC-Exo), expressed molecules involved with antigen presentation, such as HLA-DR and CD86 (21).
Therefore, in this paper, we investigated if DC-Exo have the capacity to turn tumor cells into better targets for the immune system. We show that, indeed, DC-Exo treated tumor cells are able to induce tumor-sensitized T-cells to secrete higher levels of IFN-γ than non-DC-Exo-treated tumor cells. This observation supports our hypothesis and indicates that, as a minimum, DC-Exo used in cancer immunotherapy may act as a means to sensitize tumor cells to other immune effectors, thus enhancing the effectiveness of different immunotherapeutic approaches.
Exosomes from Dendritic Cells and Their Role in Anti-Tumor Response
Raposo et al. (20) were the first to describe that Exo (originating from EBV-transformed B cells) contained functional MHC-II molecules, which carried peptides to which the cells were exposed and were able to stimulate peptide-specific CD4+ T-cells. From this initial observation, many others indicated a role for Exo in immune response to various stimuli, including tumors. Actually, in a mouse model, DC-Exo, containing class I major histocompatibility antigens (MHC-I) complexed with tumor-derived peptides were shown to induce a cytotoxic T lymphocyte (CTL) response, which inhibited tumor growth and rejected established tumors (16, 19, 22), probably due to the incorporation of the Exo by host DCs in vivo (31). Also, in a clinical trial, ascites-derived Exo administered with GM-CSF induced CEA-specific-T lymphocytes (17), confirming the potential of Exo to carry and deliver effectively tumor antigens, as observed in various other settings (16, 18).
Exosomes from DCs Could Turn Tolerogenic Tumor Cells into Immunogenic Targets?
Though the generation of effective T lymphocytes responses is one of the main goals of cancer immunotherapy and the use of DC-Exo has been shown to be a means to achieve this goal, directly (19, 32, 33) or indirectly, by their incorporation by the recipients DCs (31, 33), the effectiveness of these immune effector cells depends on other factors. Actually, this is well illustrated by the fact that tumor-specific immune responses are frequently detected in patients who, nonetheless, have a progressive disease. This observation, essentially, has challenged for a long time, the potential of immunotherapy to control cancer. However, more recent experimental and clinical data have led to the realization that the immune system can, indeed, control and, probably, eliminate tumors, if “properly” engaged (34). In various clinical settings, this “proper” engagement of the immune system has been achieved, so far, either by the elimination of immune checkpoints, with monoclonal antibodies directed to immunoregulatory surface molecules (35), or by “arming” T lymphocytes with tumor-specific chimeric receptors, a strategy that, likewise, bypasses physiological controls of the immune response (36). However, these strategies depend, on one hand, on the existence of tumor cell targets already recognized by the immune effectors and, on the other hand, on the establishment of strong enough interactions between the immune effectors and the tumor targets.
In this perspective, our observation that tumor cells incorporate DC-Exo and, thereafter, express various surface molecules involved in the interaction between antigen-presenting cells and T lymphocytes (21) gains a new relevance. Not only could DC-Exo-treated tumor cells gain antigen-presenting capabilities, which might induce de novo anti-tumor responses, but also such cells could become better targets to any immune effector cell, either naturally induced by the presence of the tumor in the patient or induced by immunotherapeutic approaches (Figure 1).
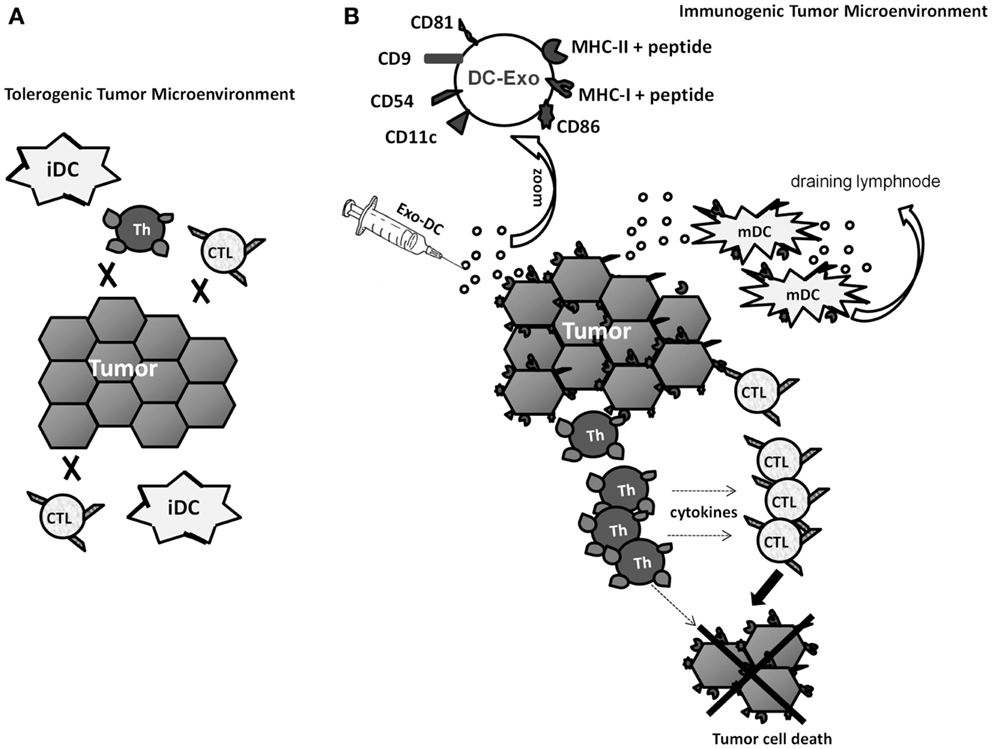
Figure 1. Hypothetical scheme of the tumor microenvironment changed by treatment with Exo from immunogenic DCs (DC-Exo). (A) Low immunogenic tumor cells and immature DCs (iDCs) cannot induce specific-T lymphocyte response and, if such effectors do exist, they are not sufficiently stimulated by the tumor cells. (B) Enrichment of the tumor microenvironment with Exo-DC modifies tumor cells, enhances their immunogenic potential and turns them into better targets for any immune effector cell. Furthermore, immature DCs present in the tumor could also capture DC-Exo, acquire a mature phenotype and migrate to draining lymph nodes, where they could set up and amplify the immune response.
To test this hypothesis, we sensitized human CD3+ T-cells against the breast carcinoma cell line SK-BR-3. Monocyte-derived DCs were pulsed with tumor cell Exo for 24 h, in the presence of IL-1, IL-6, TNF-α, and PGE2. Tumor-Exo were used as antigen source, since it was shown that they induce anti-tumor responses more efficiently than irradiated tumor cells, apoptotic bodies, or tumor cells lysate (37). Next, these DCs were co-cultured with autologous T-cells, in the presence of IL-2 and IL-7 for 14 days. After that, tumor-Exo sensitized T-cells were exposed to SK-BR-3 cells that were treated or not with DCs-Exo (Exo Control in the figure) or Exo obtained from DCs exposed to tumor Exo (Exo Tex in the figure). After 2 days, the IFN-γ-producing T-cells were quantified by ELISPOT (Figure 2). It is interesting to note that SK-BR-3 cells, alone, seemed to induce some cytokine production by sensitized T-cells, but this production was not statistically different from that of T-cells cultured alone. However, the treatment of the tumor cells with Exo (both Exo control and Exo Tex) increased this response to a significant level. It is noteworthy that these responses were modest, but nevertheless, significant, thus in agreement with our hypothesis: tumor cells treated with Exo derived from DCs became better targets for an already existing immune effector cell population – those T-cells that had been sensitized in vitro against the tumor cells, but whose response was not strong enough to raise above the background. Furthermore, when the Exo were obtained from DCs that were treated with tumor cell exosomes (Tex), the enhancement of IFN-γ production seemed to be even higher. Though speculative, it is possible to recognize, here, an antigenic enrichment of the targets, since cells may load their Exo not only with their co-stimulatory molecules but also with antigen-loaded MHC complexes (20), which would be captured by the tumor cells, hence turning these into even better targets for the antigen-specific-T-cells.
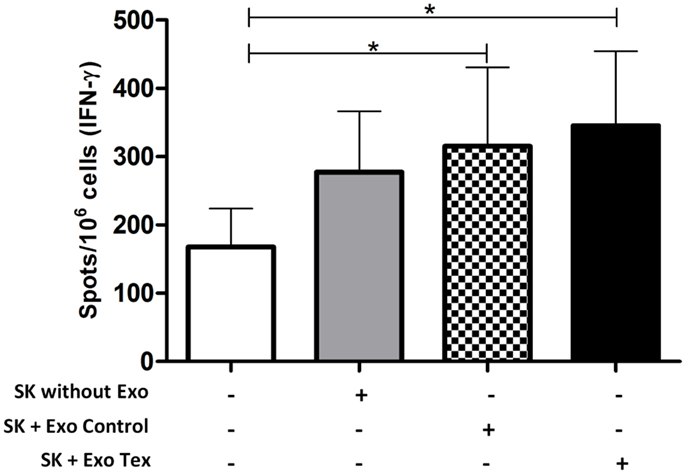
Figure 2. IFN-γ-production by tumor-sensitized T-cells is enhanced when tumor target cells are treated with Exo obtained from DCs. Cells of the human breast adenocarcinoma cell line SK-BR-3 were pre-treated or not with Exo (130 μg/106 cells), obtained from control DCs (Exo Control) of from DCs exposed to tumor cell Exo (Exo Tex) and co-cultured for 2 days with previously sensitized CD3+ T-cells (autologous to the DC source of the Exo). After that, the cells were harvested and the number of IFN-γ-producing cells was quantified by ELISPOT. T-cells pre-sensitization was obtained by co-culture with autologous tumor cell exosome-treated DCs, for 14 days, in presence of IL-2 and IL-7. Average and SD of the number of cytokine-producing cells are represented (n = 6; ANOVA *p < 0.0028, flowing Dunnett’s Multiple Comparison Test).
Discussion
Tumor cells are poorly immunogenic and this has hampered the development of effective cancer immunotherapy. Yet, new insights on the role of different components of the immune response have broadened the field, so that, today, effective immune strategies to treat cancer are not longer seen as a remote possibility, but as a concrete breakthrough. However, though such approaches, as the use of checkpoint inhibitors or chimeric antigen receptor-transfected T-cells have reached astounding successes in some situations, the need for improvement is still pressing.
As depicted in Figure 1, even the presence (naturally or by transfer) of tumor-specific immune effectors within the tumor microenvironment may not be enough to hamper tumor growth and development. An immunosuppressive environment and targets that are poor in both antigens and co-stimulatory molecules may decrease significantly the effectiveness of tumor-specific responses. Therefore, any strategy that achieves the enhancement of tumor cell antigenicity or, even better, immunogenicity, should make it easier to control the disease in cancer patients. Such strategies could do so either by directly inducing tumor-specific responses, if the increase in tumor immunogenicity is strong enough, and, if not, by sensitizing tumors to borderline responses that, by themselves are not able to control the disease, but in face of more readily recognizable targets would do it.
Here, we tested the hypothesis that DC-Exo would perform such a role, increasing tumor cell antigenicity/immunogenicity. The transfer of information between cells via Exo is being recognized as a significant phenomenon for the body intercellular communication (38, 39), since Exo can interact with several cells types (26) and transfer information by means of fusion with the plasma membrane (40). Furthermore, this transfer seems not to be random, but controlled by molecules expressed by Exo and the “target cells,” as we have shown for the incorporation of DC-Exo by tumor cells, where the incorporation of DC-Exo by tumor cells was directly proportional to the expression CD9 molecules by the latter (21). In other setting, Exo capture, by the monocytes lineage, THP-1, has been shown to depend on toll like receptors, TLR-2 and TLR-4, leading to activation of NF-κB and STAT3, and to cytokine secretion (41).
Since Exo carry several molecules of their cells of origin (27), they would be a possible way to load tumor cells with antigen presentation-associated molecules from DCs. Indeed, we did observe the transfer of HLA-DR and CD86 molecules from DC-Exo to tumor cells (21). Furthermore, when we analyzed tumor cells treated with DC-Exo, we noticed that these had an increased expression of ICAM-molecules, which could facilitate their interaction with T lymphocytes, since the interaction between ICAM-I and LFA on lymphocytes accentuates TCR/MHC/peptide interaction (32).
Thus, we tested if SK-BR-3 tumor cells treated with DC-Exo would be able to induce a T-cell response more effectively. Accordingly, we observed that treatment of the tumor cell line with DC-Exo enhanced their ability to activate tumor-sensitized T-cells to secrete IFN-γ. We chose this cytokine because of its role in anti-tumor immune responses (42, 43) being associated with both CD8 and CD4 T-cell responses (44, 45), and essential for priming long-lived memory CD8+ T-cells (46, 47). It must be recognized, however, that the response we obtained though significant, when compared to the non-stimulated T-cell response, was modest, showing just a tendency to statistical significance, when compared to the response induced by the non-Exo-treated cells. This is, actually, in agreement with our hypothesis, since the expected action of the nanovesicles would be to enhance just enough the sensitivity of the targets to already existing immune effector mechanisms – and not, necessarily, the induction of new responses. Another issue that should be noted is that the clinical application of such a strategy would be restricted to situations where tumors might be directly accessed, since the systemic inoculation of Exo would hardly deliver them to the tumor cells, but, rather, to other cells along their distribution through the body.
On the positive side, however, it is worth noting that this strategy, of treating tumor cells with DC-Exo, eliminates the need of specific tumor antigens identification, since it transfers to the patients’ immune system this task. Through their association with DC-derived molecules, tumor antigens that would be poorly presented otherwise, would have their “visibility” increased, and, therefore, would be more likely to become effective targets of the patient’s immune response. Furthermore, the local injection of these DC-Exo could allow them to be captured also by the patient’s DCs in the tumor microenvironment. This would have a positive effect in the tumor immune response since in cancer patients, tumor-infiltrating DCs have functional deviations, maintaining an immature phenotype (9, 48–51). Thus, if these DCs incorporate Exo derived from immunogenic DCs, a phenomenon shown to occur (22), they might even become able to induce the response of tumor-specific naïve T lymphocytes.
Finally, even if the modifications of tumor cells immunogenicity caused by their incorporation of DC-Exo are not enough to give rise to de novo tumor-specific immune responses in the patients, they would, as we show here, turn the tumor cells into more effective targets to immune effectors induced by any other means. Thus, it is possible to suggest that treatment of tumors with DC-Exo could contribute to the effectiveness of any immunotherapeutic strategy, be it active, like vaccination protocols or passive, like the transfer of tumor-specific effectors. In face of this hypothesis and the data we obtained, we believe that this approach should receive consideration and be further investigated, in order to evaluate better its possible effectiveness, and to determine the most effective Exo doses, the length of its effect on tumor cells, and, mainly, the precise activation status of the DC, used as Exo source.
Author Contributions
Graziela Gorete Romagnoli: project development, participation in all experiments, analyses, and discussions. Bruna Barbosa Zelante: participation in the experiments, discussions, and drafting the article. Patrícia Argenta Toniolo: participation in the experiments, discussions, and drafting the article. Isabella Katz Migliori: participation in the experiments and discussions. José Alexandre M. Barbuto: project development, discussions, drafting, and revising the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants (#04/09956-0; #07/58597-1; #09/54599-5) from the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq (#303731/2007-9).
References
1. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi: 10.1038/32588
2. Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol (1991) 9:271–96. doi:10.1146/annurev.iy.09.040191.001415
3. Barbuto JA, Ensina LF, Neves AR, Bergami-Santos P, Leite KR, Marques R, et al. Dendritic cell-tumor cell hybrid vaccination for metastatic cancer. Cancer Immunol Immunother (2004) 53:1111–8. doi:10.1007/s00262-004-0551-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Berzofsky JA, Terabe M, Oh S, Belyakov IM, Ahlers JD, Janik JE, et al. Progress on new vaccine strategies for the immunotherapy and prevention of cancer. J Clin Invest (2004) 113:1515–25. doi:10.1172/JCI21926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Murphy G, Tjoa B, Ragde H, Kenny G, Boynton A. Phase I clinical trial: T-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate (1996) 29:371–80. doi:10.1002/(SICI)1097-0045(199612)29:6<371::AID-PROS5>3.3.CO;2-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med (1998) 4:328–32. doi:10.1038/nm0398-328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer (2012) 12:265–77. doi:10.1038/nrc3258
8. Wang K, Zhou Q, Guo AL, Xu CR, An SJ, Wu YL. An autologous therapeutic dendritic cell vaccine transfected with total lung carcinoma RNA stimulates cytotoxic T lymphocyte responses against non-small cell lung cancer. Immunol Invest (2009) 38:665–80. doi:10.1080/08820130903070528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Baleeiro RB, Anselmo LB, Soares FA, Pinto CA, Ramos O, Gross JL, et al. High frequency of immature dendritic cells and altered in situ production of interleukin-4 and tumor necrosis factor-alpha in lung cancer. Cancer Immunol Immunother (2008) 57:1335–45. doi:10.1007/s00262-008-0468-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Barbuto JA. Are dysfunctional monocyte-derived dendritic cells in cancer an explanation for cancer vaccine failures? Immunotherapy (2013) 5:105–7. doi:10.2217/imt.12.153
11. Neves AR, Ensina LF, Anselmo LB, Leite KR, Buzaid AC, Camara-Lopes LH, et al. Dendritic cells derived from metastatic cancer patients vaccinated with allogeneic dendritic cell-autologous tumor cell hybrids express more CD86 and induce higher levels of interferon-gamma in mixed lymphocyte reactions. Cancer Immunol Immunother (2005) 54:61–6. doi:10.1007/s00262-004-0550-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Ramos RN, Chin LS, Dos Santos AP, Bergami-Santos PC, Laginha F, Barbuto JA. Monocyte-derived dendritic cells from breast cancer patients are biased to induce CD4+CD25+Foxp3+ regulatory T cells. J Leukoc Biol (2012) 92:673–82. doi:10.1189/jlb.0112048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Yi DH, Appel S. Current status and future perspectives of dendritic cell-based cancer immunotherapy. Scand J Immunol (2013) 78:167–71. doi:10.1111/sji.12060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Urban JL, Schreiber H. Tumor antigens. Annu Rev Immunol (1992) 10:617–44. doi:10.1146/annurev.iy.10.040192.003153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res (2014) 20:3401–10. doi:10.1158/1078-0432.CCR-14-0433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Chaput N, Schartz NE, Andre F, Taieb J, Novault S, Bonnaventure P, et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol (2004) 172:2137–46. doi:10.4049/jimmunol.172.4.2137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther (2008) 16:782–90. doi:10.1038/mt.2008.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Viaud S, Thery C, Ploix S, Tursz T, Lapierre V, Lantz O, et al. Dendritic cell-derived exosomes for cancer immunotherapy: what’s next? Cancer Res (2010) 70:1281–5. doi:10.1158/0008-5472.CAN-09-3276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med (1998) 4:594–600. doi:10.1038/nm0598-594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med (1996) 183:1161–72. doi:10.1084/jem.183.3.1161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Romagnoli GG, Toniolo PA, Migliori IK, Caldini ÉG, Ferreira MA, Pizzo CR, et al. Tumour cells incorporate exosomes derived from dendritic cells through a mechanism involving the tetraspanin CD9. Exosomes Microvesicles (2013) 1:4. doi:10.5772/52069
22. Hao S, Bai O, Li F, Yuan J, Laferte S, Xiang J. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology (2007) 120:90–102. doi:10.1111/j.1365-2567.2006.02483.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Skogberg G, Gudmundsdottir J, van der Post S, Sandstrom K, Bruhn S, Benson M, et al. Characterization of human thymic exosomes. PLoS One (2013) 8:e67554. doi:10.1371/journal.pone.0067554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Suntres ZE, Smith MG, Momen-Heravi F, Hu J, Zhang X, Wu Y, et al. Therapeutic uses of exosomes. Exosomes Microvesicles (2013) 1:5. doi:10.5772/56522
25. Denzer K, van Eijk M, Kleijmeer MJ, Jakobson E, de Groot C, Geuze HJ. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol (2000) 165:1259–65. doi:10.4049/jimmunol.165.3.1259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Ludwig AK, Giebel B. Exosomes: small vesicles participating in intercellular communication. Int J Biochem Cell Biol (2012) 44:11–5. doi:10.1016/j.biocel.2011.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics (2010) 73:1907–20. doi:10.1016/j.jprot.2010.06.006
28. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol (2014) 14:195–208. doi:10.1038/nri3622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Hannafon BN, Ding WQ. Intercellular communication by exosome-derived microRNAs in cancer. Int J Mol Sci (2013) 14:14240–69. doi:10.3390/ijms140714240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Nolte-‘t Hoen EN, Buschow SI, Anderton SM, Stoorvogel W, Wauben MH. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood (2009) 113:1977–81. doi:10.1182/blood-2008-08-174094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Hao S, Bai O, Yuan J, Qureshi M, Xiang J. Dendritic cell-derived exosomes stimulate stronger CD8+ CTL responses and antitumor immunity than tumor cell-derived exosomes. Cell Mol Immunol (2006) 3:205–11.
32. Sprent J. Direct stimulation of naive T cells by antigen-presenting cell vesicles. Blood Cells Mol Dis (2005) 35:17–20. doi:10.1016/j.bcmd.2005.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Viaud S, Ploix S, Lapierre V, Thery C, Commere PH, Tramalloni D, et al. Updated technology to produce highly immunogenic dendritic cell-derived exosomes of clinical grade: a critical role of interferon-gamma. J Immunother (2011) 34:65–75. doi:10.1097/CJI.0b013e3181fe535b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Blank CU. The perspective of immunotherapy: new molecules and new mechanisms of action in immune modulation. Curr Opin Oncol (2014) 26:204–14. doi:10.1097/CCO.0000000000000054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood (2014) 123:2625–35. doi:10.1182/blood-2013-11-492231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med (2001) 7:297–303. doi:10.1038/85438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kourembanas S. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol (2014). doi:10.1146/annurev-physiol-021014-071641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol (2014) 30:255–89. doi:10.1146/annurev-cellbio-101512-122326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kooijmans SA, Vader P, van Dommelen SM, van Solinge WW, Schiffelers RM. Exosome mimetics: a novel class of drug delivery systems. Int J Nanomedicine (2012) 7:1525–41. doi:10.2147/IJN.S29661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Bretz NP, Ridinger J, Rupp AK, Rimbach K, Keller S, Rupp C, et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via toll-like receptor signaling. J Biol Chem (2013) 288:36691–702. doi:10.1074/jbc.M113.512806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Barth RJ Jr, Mule JJ, Spiess PJ, Rosenberg SA. Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med (1991) 173:647–58. doi:10.1084/jem.173.3.647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21:137–48. doi:10.1016/j.immuni.2004.07.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Teixeira LK, Fonseca BP, Vieira-de-Abreu A, Barboza BA, Robbs BK, Bozza PT, et al. IFN-gamma production by CD8+ T cells depends on NFAT1 transcription factor and regulates Th differentiation. J Immunol (2005) 175:5931–9. doi:10.4049/jimmunol.175.9.5931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science (2002) 295:338–42. doi:10.1126/science.1065543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature (2003) 421:852–6. doi:10.1038/nature01441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science (2003) 300:337–9. doi:10.1126/science.1082305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Della Bella S, Gennaro M, Vaccari M, Ferraris C, Nicola S, Riva A, et al. Altered maturation of peripheral blood dendritic cells in patients with breast cancer. Br J Cancer (2003) 89:1463–72. doi:10.1038/sj.bjc.6601243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol (2004) 4:941–52. doi:10.1038/nri1498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Gottfried E, Kreutz M, Mackensen A. Tumor-induced modulation of dendritic cell function. Cytokine Growth Factor Rev (2008) 19:65–77. doi:10.1016/j.cytogfr.2007.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: dendritic cells, exosomes, cancer immunotherapy, immunomodulation, tumor immune response, tumor cell immunogenicity
Citation: Romagnoli GG, Zelante BB, Toniolo PA, Migliori IK and Barbuto JAM (2015) Dendritic cell-derived exosomes may be a tool for cancer immunotherapy by converting tumor cells into immunogenic targets. Front. Immunol. 5:692. doi: 10.3389/fimmu.2014.00692
Received: 26 November 2014; Accepted: 31 December 2014;
Published online: 19 January 2015.
Edited by:
Matías Sáenz-Cuesta, Biodonostia Health Research Institute, SpainReviewed by:
Lesley Ann Smyth, King’s College London, UKElke Pogge Von Strandmann, University Clinic Cologne, Germany
Copyright: © 2015 Romagnoli, Zelante, Toniolo, Migliori and Barbuto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José Alexandre M. Barbuto, Laboratory of Tumor Immunology, Department of Immunology, Institute of Biomedical Sciences of University of São Paulo, Av. Prof. Lineu Prestes 1730, São Paulo, São Paulo 05508-000, Brazil e-mail:amJhcmJ1dG9AaWNiLnVzcC5icg==