 Erika Héninger
Erika Héninger Timothy E. G. Krueger
Timothy E. G. Krueger Joshua M. Lang
Joshua M. Lang- 1University of Wisconsin Carbone Cancer Center, Madison, WI, USA
- 2Department of Medicine, University of Wisconsin, Madison, WI, USA
Epigenetic silencing of immune-related genes is a striking feature of the cancer genome that occurs in the process of tumorigenesis. This phenomena impacts antigen processing and antigen presentation by tumor cells and facilitates evasion of immunosurveillance. Further modulation of the tumor microenvironment by altered expression of immunosuppressive cytokines impairs antigen-presenting cells and cytolytic T-cell function. The potential reversal of immunosuppression by epigenetic modulation is therefore a promising and versatile therapeutic approach to reinstate endogenous immune recognition and tumor lysis. Pre-clinical studies have identified multiple elements of the immune system that can be modulated by epigenetic mechanisms and result in improved antigen presentation, effector T-cell function, and breakdown of suppressor mechanisms. Recent clinical studies are utilizing epigenetic therapies prior to, or in combination with, immune therapies to improve clinical outcomes.
Introduction
Immune evasion is a complex phenomenon that entails alterations in cancer cells and the microenvironment to inhibit recognition of tumor cells by immune infiltrating cells. This process includes altered expression and presentation of tumor-associated antigens (TAAs) and secretion of cytokines that promote a regulatory/inhibitory milieu of antigen-presenting cells (APC) and cytolytic T cells (CTL). This complex process is driven by a multitude of factors including altered epigenetic marks in tumor cells that control gene expression. A growing body of literature also suggests that epigenetic alterations can alter immune cell phenotype and function, for both regulatory and cytolytic function. These epigenetic modifications include alterations in DNA methylation and histone modifications, such as acetylation and methylation. The accumulation of epigenetic alterations during tumorigenesis contributes to profound changes in genome-wide transcriptional regulation and genetic stability that promotes immune evasion. The availability of state-of-the-art technologies to screen epigenetic alterations across a variety of malignancies has advanced our understanding of defects in tumor regulation, and new therapeutic approaches have been devised and studied to reverse epigenetic silencing. Since site-specific epigenomic patterns may associate with disease progression, epigenetics has also come into focus for biomarker research (1). These reciprocal fields identify potential therapeutic strategies with integrated biomarkers focused on epigenetic mechanisms to improve antitumor immune responses (Figure 1).
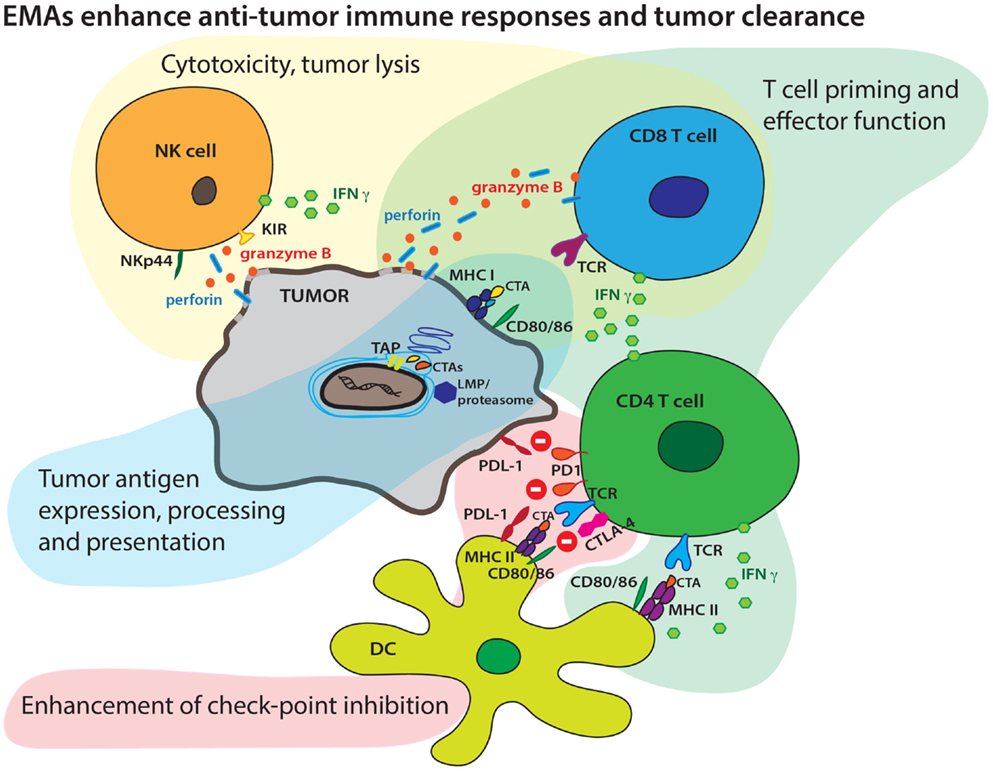
Figure 1. Epigenetic modifying agents can enhance multiple aspects of an antitumor immune response. EMAs may boost tumor antigen expression, endogenous antigen processing, increase surface CTA display in context of MHC molecules, and boost presentation to T cells by increasing expression of co-stimulatory molecules. EMAs may also enhance both cellular and cytokine-mediated effector T-cell mechanisms and tumor lysis. EMAs may alter checkpoint inhibition targeting the PD1/PD-L1 and CTLA-4/CD28 axis resulting in more efficient effector T-cell mechanisms.
Introduction to Epigenetics
Methylation and Hypomethylating Agents
DNA methylation is one of the most studied epigenetic phenomenon and involves the enzymatic conversion of cytosine residues to 5-methylcytosine. This reaction is catalyzed by the five known mammalian DNA methyltransferases (DNMTs) DNMT1, 3A, 3A2, 3B, and 3L (2). The conversion is typically restricted to cytosine residues of cytosine-guanine dinucleotides called CpG sites. CpG islands contain a high density of CpG sites, which are mostly unmethylated in normal tissues (3). However, most cancers are characterized by localized aberrant DNA hypermethylation of CpG islands within the promoters of various genes (4), including tumor suppressors involved in cell cycle control, cell growth, apoptosis, cell adhesion, DNA repair, angiogenesis, and cell adhesion (5). Furthermore, DNA hypermethylation inhibits gene expression, as evidenced by numerous studies correlating promoter methylation with transcriptional repression during both normal processes as well as tumorigenesis (6–8).
Hypomethylating agents can be used to counteract hypermethylation and restore gene expression. Hypomethylating agents can generally be grouped into (a) nucleosidic or (b) non-nucleosidic DNA methylation inhibitors (Table 1).
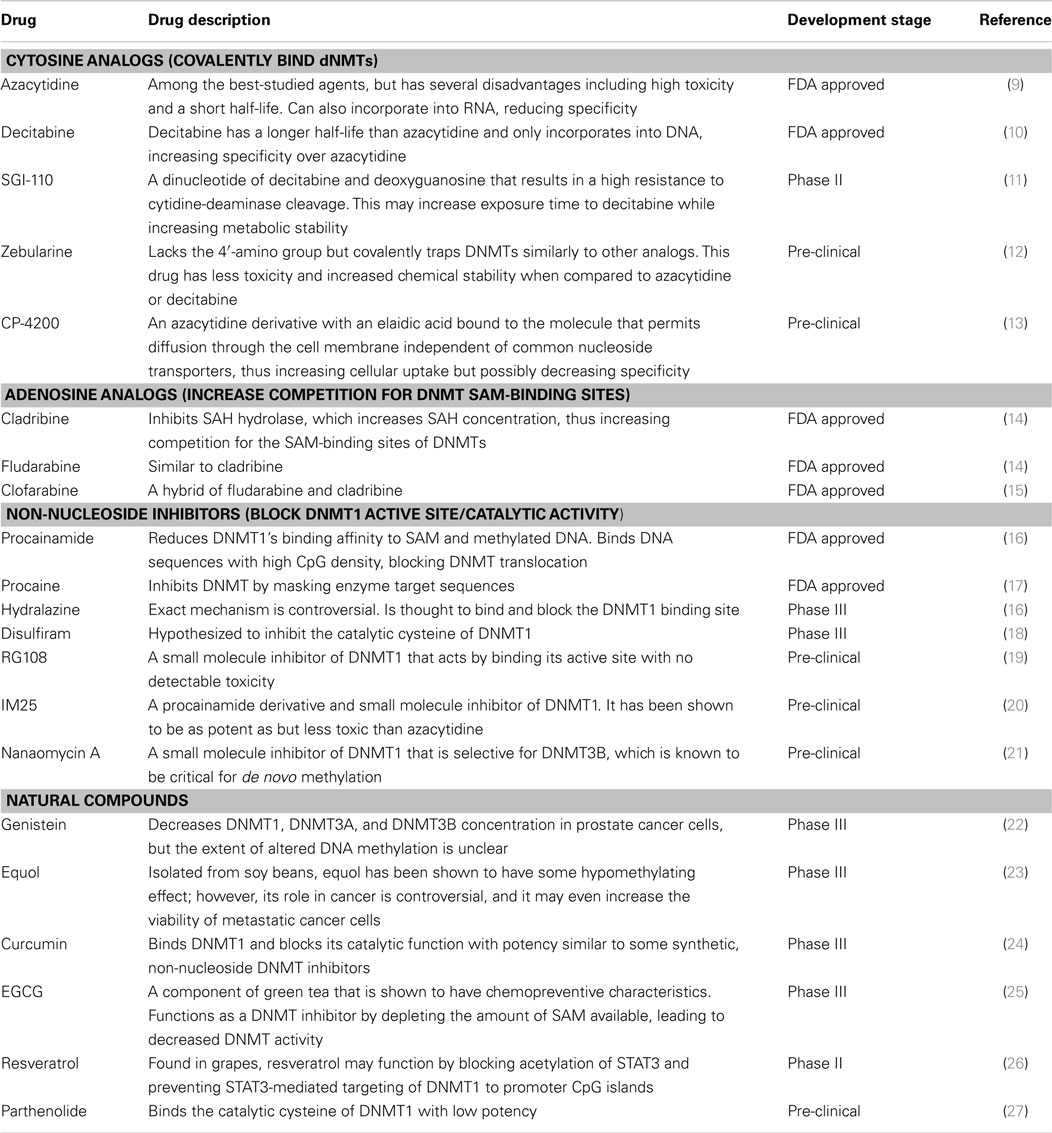
Table 1. Hypomethylating agents currently in development.
Nucleosidic DNA methylation inhibitors are incorporated into the genome during DNA replication. Thus, this class of agents acts only in tumor cells actively undergoing cell division. Agents such as Azacitidine (AZA) and 5-aza-2′-deoxycitidine (5AZA2) were originally synthetized in the 1960s to use as cytotoxic drugs with potential anti-leukemic activity (9, 28, 29). However, their effect on DNA methylation was not identified until later in the process of drug development. 5AZA2 incorporates into DNA in place of cytidine during S-phase and covalently binds DNMTs during the process of DNA replication to ultimately prevent DNA methylation. 5AZA2 has a dual, dose-dependent antineoplastic action. At high doses, it covalently traps DNMT into DNA leading to cytotoxicity. At lower doses, it suppresses tumor growth primarily via hypomethylation of promoter CpG islands of tumor-suppressor specific loci (9, 30). AZA is similar to 5AZA2 but can also incorporate into RNA in the form of azacytidine-triphosphate and directly inhibit protein synthesis.
The restoration of gene expression mediated by hypomethylating agents can impact tumor growth in a wide variety of mechanisms. In prostate cancer (PC), 5AZA2 targets multiple genes including the tumor-suppressor miR-146a microRNA and the androgen receptor (AR). 5AZA2-induced miR-146a induction correlated with both delayed tumor growth and disease progression of castrate-resistant PC (CRPC) in an LNCap xenograft model. The miR46a promoter methylation pattern was also suggested as a biomarker for progression from androgen-dependent to androgen-independent phases of PC (1). Hypermethylation of the AR promoter was shown to associate with PC tumorigenicity and the therapeutic potential of epigenetic agents in addition to anti-androgen therapy has been suggested in several pre-clinical studies both in vitro and in vivo. 5AZA2 reduced tumorigenicity and cell proliferation of PC cell lines and PC stem/progenitor cells via AR promoter demethylation and AR induction (31). 5AZA2 also restored the antiproliferative and pro-apoptotic effects of the AR-antagonist bicalutamide (BCLT) in both in vitro and in vivo xenograft models (32). A second-generation derivative, 5AZA2-p-deoxyguanosine (SGI-110) was formulated to protect 5AZA2 from cytidine-deaminase inactivation and prolong half-life. SG-110 efficiently retarded tumor growth in an EJ6 bladder cancer xenograft model with less toxicity compared to 5AZA2 in vivo (33). Zebularine is a cytidine analog displaying both cytidine-deaminase and DNMT inhibitor properties (34). An in vitro study treated breast cancer cell lines with zebularine, potentiating the antitumor effects of other epigenetic drugs including 5AZA2 and SAHA by inhibiting tumor proliferation and clonogenic potential. Other pre-clinical studies in AML and solid tumors found growth inhibition by zebularine via cell cycle arrest and apoptosis induction via various pathways including p53-dependent endoplasmic reticulum (ER) stress (35, 36).
Non-nucleosidic DNA methylation inhibitors directly inhibit DNMT activity without incorporating into nucleic acids. The best-studied agents in this class include hydralazine, procaine, and procainamide. Hydralazine has been studied alone or in combination with valproate acid/magnesium valproate in refractory solid tumors, and it was shown to restore chemosensitivity in gemcitabine-resistant CaLo cervical cancer cell lines via histone methyltransferase inhibition (37, 38). Hydralazine treatment resulted in significant dose- and time-dependent growth inhibition, increased apoptosis, DNA damage, cell cycle arrest, and decreased invasiveness of DU145 PC cells via blockage of the EGF-receptor pathway (39).
Procaine and procainamide are both derivatives of 4-aminobenzoic acid, ester- and amide-, respectively. Procainamide is a competitive inhibitor of DNMT1 hemimethylase activity (40). In an MDA-231 xenograft model, both procainamide and hydralazine demonstrated potent tumor-suppressor reactivation including demethylation and re-expression of the estrogen receptor (41). Procaine suppressed growth of MCF-1 breast cancer cells simultaneously with demethylation events (17). New DNMT inhibitors developed by conjugation of procainamide to L-RG08 or phthalimide showing strong cytotoxicity against DU145 and HCT116 cell lines (42).
Natural plant-derived compounds have been also identified as non-nucleosidic DNA methylation inhibitors and have been extensively studied for global DNA methylation and tumor inhibitory effects. Curcumin was shown to reactivate expression of the Neurog1 gene via promoter CpG site demethylation in LNCap cells. The promoter methylation status of Neurog1 was proposed as a potential biomarker to detect early PC (43). Genistein inhibited the AKT signaling pathway in PC cells via demethylation and deacetylation of histone-H3-lysine 9 at the PTEN, CYLD, p53, and FOXO3 tumor-suppressor gene promoters (44). Resveratrol was also shown to demethylate several tumor-suppressor promoters and restore estrogen sensitivity in triple-negative breast cancer cells by increasing estrogen receptor expression via the inhibition of STAT3 acetylation (26). A clinical trial has been initiated to analyze tissue gene expression patterns in PC patients after genistein treatment (NCT01126879).
Histone Acetylation and Histone Deacetylase Inhibitors
Histone modifications can increase spatial separation of DNA from protein in the nucleosome to permit transcription factor binding to promoter regions, leading to enhanced global gene expression. Chromatin acetylation is regulated by the balanced action between histone acetyltransferases (HAT) and deacetylases (HDAC). Histone deacetylase inhibitors (HDIs) block the catalytic domain of HDACs preventing chromatin condensation and transcriptional repression. HDIs have been intensively studied as potential anticancer compounds both alone and in combination with other therapies in a wide variety of solid tumors and hematologic malignancies with strong pre-clinical evidence for antitumor activity (Table 2). In addition to increasing gene expression of various tumor suppressors, HDIs have also been shown to exert tumor-selective apoptosis induction and proliferation arrest via multiple mechanisms including induction of p21 and the TRAIL-pathway (45). One HDI, suberoylanilide hydroxamic acid (SAHA), suppressed tumor growth, invasion, and migration of highly aggressive ovarian carcinomas by modulating a variety of phenotype-related molecules including members of the caspase pathway, the cell cycle regulator Cyclin B1, tumor-suppressor genes (p21, p53), and tissue remodeling MMP-9 enzyme, among others (46). SAHA promoted autophagy both alone and in combination with 5AZA2 (47, 48) and enhanced T-cell and NK cell-mediated tumor cell targeting by re-sensitizing tumor cells for the TRAIL/Apo2L death receptor pathway in various cancer types (49–54). Panobinostat (or LBH589) displays robust growth inhibition of a wide range of melanoma phenotypes via direct cytotoxicity and cell cycle arrest and is known to induce cell death independently from the apoptotic machinery or death receptor pathways, likely via mitochondrial damage (55, 56). Entinostat has been tested in combination with AZA and SG-110 in lung cancer models, and these combination therapies highlighted robust gene expression changes in key pathways of antitumor mechanisms including cell cycle, apoptosis, tissue remodeling, and DNA damage (57, 58).
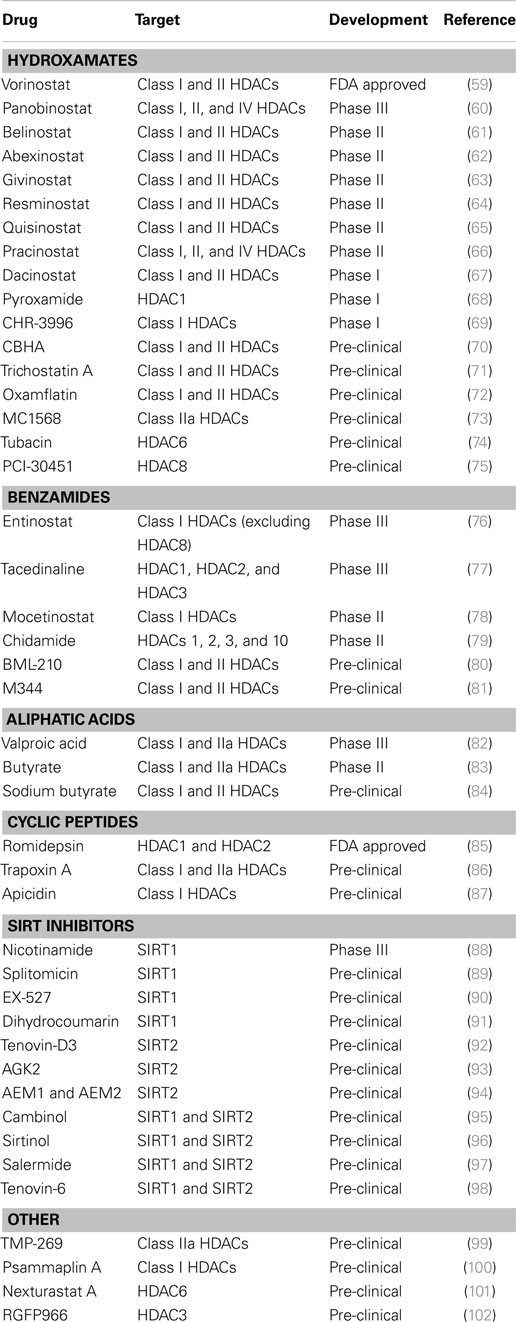
Table 2. Histone deacetylase inhibitors.
Romidepsin is a unique natural compound originally isolated in the early 1990s from Gram-negative Chromobacterium violaceum from a Japanese soil sample and was FDA approved in 2009 for the treatment of cutaneous T-cell lymphoma. Romidepsin was originally described as an anti-Ras molecule and later identified as an HDAC inhibitor. Romidepsin induces G1 phase cell cycle arrest (103) primarily via p21 induction (103, 104).
Sirtuins are potent regulators of cell division that promote survival. SIRT inhibitors have been tested as potential candidates for novel anticancer agents in pre-clinical studies. Sirtinol, a SIRT1/2 inhibitor, suppressed growth of MCF-7 breast cancer cells via various pathways including G1 cell cycle arrest and induction of apoptotic machinery by PARP cleavage, cytochrome c release, Bax up-regulation, and BCL-2 down-regulation. Furthermore, sirtinol elevated autophagy-related markers and increased tumor-suppressor p53 acetylation (105). Inhibition of p53 deacetylation was also shown as a key pathway of tumor suppression and cytotoxicity exerted by two novel SIRT2-specific inhibitors, AEM1 and AEM2 (94).
Epigenetic Modifying Agents in Cancer Therapy
Clinical utility of epigenetic modifying agents (EMAs) has been demonstrated in a growing body of clinical research studies. Current FDA-approved EMAs include DNMT inhibitors Vidaza (AZA), Dacogen (5AZA2) for AML and MDS, and HDAC inhibitors Istodax (romidepsin) and Zolinza (vorinostat) for treatment of cutaneous T-cell lymphoma. AZA and 5AZA2 have provided a significant advancement in treatment of high-risk hematologic malignancies; however, their clinical efficacy and therapeutic value in solid tumors is limited. This is due to many factors including both the relative molecular instability and significant toxicity of these agents. These dose-limiting toxicities include myelosuppression, fatigue, and infection (106). Furthermore, the lack of clinical benefit at the maximally tolerated doses (MTD) of these agents in patients with solid tumors significantly hindered interest in further clinical development. However, recent studies utilizing EMAs with doses lower than the MTD have found significant antitumor effects renewing interest in these agents. Further pre-clinical work has led to the development of alternative formulations to decrease toxicity and improve pharmacokinetics of these agents (107, 108). For example, the pharmacokinetic profile of the second-generation derivative, 5-aza-2′-deoxycitidine-p-deoxyguanosine (SGI-110) is promising, and this agent is being tested in AML, CMML, and MDS in a Phase I/II dose escalation study (NCT 01261312).
Epigenetic aberrations have been shown to associate with resistance to chemotherapy. Several studies addressed the potential of EMAs to reinstate chemosensitivity. When 5AZA2 was administered prior to standard chemotherapy for AML patients, the overall complete remission was 83% (NCT00538876) (109). A recently concluded Phase I/II study on metastatic, docetaxel-resistant CRPC studied the effect of AZA in combination with docetaxel and prednisone and found significant PSA response, favorable clinical outcome, and demethylation of tumor-derived DNA (110). The efficacy of hydralazine and magnesium valproate treatment prior to re-challenge with chemotherapy was addressed in a Phase II clinical trial with patients with refractory, chemoresistant solid tumors (NCT00404508). This study reported a reduction in global DNA methylation, histone deacetylase activity, and 80% of the patients showed clinical benefits with partial response or stable disease (111). A Phase I study in chemoresistant metastatic melanoma (NCT00925132) assessed the potential of sequential epigenetic therapy including 5AZA2 and panobinostat combined with temozolomide chemotherapy. This regimen was generally well tolerated by the cohort with no patient reaching dose-limiting toxicity (112) and has advanced to Phase II testing to further evaluate if EMAs may modify chemosensitivity and apoptosis. A Phase II clinical study of hydralazine and valproic acid in combination with neoadjuvant cytotoxic chemotherapy in locally advanced Stage IIB and IIIA breast carcinoma (NCT00395655) reported an overall response of 81% with complete clinical response in 31% of the patients (113), as well as increased efficacy of conventional cytotoxic agents. A significant decrease in global DNA methylation and in HDAC enzymatic activity was also observed. This study is being continued in a randomized ongoing Phase III study to analyze the efficacy of epigenetic cancer therapy. A Phase II evaluation of efficacy of AZA and/or lenalidomide in relapsed/refractory follicular and marginal zone lymphoma (NCT 01121757) is also ongoing.
Other novel EMAs are in development, such as chromosome-remodeling bromodomain inhibitors (114). These agents have promising pre-clinical data with robust, targeted antitumor effects in a broad range of malignancies. Further clinical development, including evaluation of optimal dosing strategies and toxicity evaluations, will be required to evaluate potential combinatorial strategies with other agents. However, great potential exists for these novel agents to be deployed in a range of clinical contexts.
Epigenetics and Antitumor Immune Responses
During tumorigenesis, epigenetic alterations play a key role in the suppression of immune recognition and immune surveillance to promote immune evasion via alterations in both the tumor and microenvironment. Immunosuppression is a striking feature of the global methylation pattern of the cancer genome, and it is also a common feature that extends across heterogenous cancer phenotypes. The potential for EMAs to reverse these phenomena is an exciting therapeutic approach to reinstate immune recognition and enhance endogenous tumor clearance. Multiple studies have indicated the potential of epigenetic therapies prior to or in combination with immune therapies, which can act through a variety of mechanisms to enhance antitumor immune responses. These include improving immune recognition via expression, processing, and presentation of TAAs in tumor cells and efficient recognition, T-cell activation, and lysis of tumor targets by immune cells. It is within this complexity of an effective antitumor immune response that epigenetic therapies could play multiple roles.
Novel Tumor-Associated Antigens
Tumor immune evasion is due, in part, to tolerance for self-antigens and reduced expression of neoantigens. In 1943, Gross published a study demonstrating that foreign antigens expressed by tumor cells may induce immune-mediated rejection of syngeneic tumor grafts (115). The identification of these TAAs and their potential to enhance immunogenicity has been well studied in the field of tumor immunology. These novel TAAs include cancer testis antigens (CTAs) or germline antigens as potential candidates for novel vaccine therapies. CTAs are ideal targets as their expression occurs during tumorigenesis with expression confined to the tumor. The otherwise limited expression in normal tissue beyond the blood-testis barrier suggests these TAAs may be highly immunogenic. However, expression of CTAs in medullary thymic epithelial cells was previously reported (116), and further evidence supported the existence of some level of central tolerance against these germline antigens (117). CTA expression has been detected in a wide variety of hematologic and solid tumors types, although expression levels often vary between disease models, and significant heterogeneity can be observed at different tumor loci within the same host or even the same lesion (118, 119). Lung, ovarian, head and neck, bladder, PC, melanoma, and multiple myeloma have been shown to express a considerable amount of CTAs, and clinical trials have been testing CTA-based therapies with promising results in these groups. Genome-wide analysis has described CTAs as a heterogenous group of antigens characterized by three distinct expression profiles: testis-restricted, testis/brain-restricted, and testis-selective (120). Almost half of the discovered CTAs are linked to the X chromosome (CG-X), which are predominantly testis-restricted. Other CTAs located on various autosomes are typically involved in later stages of germ-cell differentiation with lower antigenic potential (121, 122).
Although their function is not completely understood, prior studies have demonstrated that CTAs play a role in orchestrating cell differentiation processes during germline development. CTAs interact with transcriptional factors driving various signaling pathways involved in gametogenesis, maintenance of genomic integrity, mRNA regulation, metabolic activity, meiosis, and sperm motility. Interestingly, most of the known CTAs show redundancy in germline development as shown in small-animal knock-out models. While the role of CTAs in tumor pathogenesis is even less characterized, data suggest that besides regulating transcriptional activity, CTAs also promote tumor development by supporting cell cycle processes and the mitotic machinery including centrosome formation, mitotic spindle assembly, chromosomal alignment, and nuclear envelope breakdown. Additionally, CTAs promote tumor growth by suppressing apoptosis signaling cascades, inducing aberrant gene expression patterns and impairing response to cancer treatment drugs.
Cancer testis antigens have been proposed for use as biomarkers of disease progression in multiple disease models. Microarray screening of both primary and CRPC revealed remarkable differences in CTA expression. The MAGEA/CSAG family was up-regulated in CRPC but not in primary cancer. Interestingly, PAGEA4 was shown as a strong marker of primary tumors and was silenced in CRPC (123). A study on tumor biopsies demonstrated that SSX expression was an exclusive marker of metastatic lesions and was not detected in primary tumor tissue (124). The expression pattern of 30 CTA antigens was evaluated in glioblastoma samples compared to normal brain tissue. Glioblastoma lines co-expressing three to four CTAs were found to be associated with significantly better overall survival (125). A study in patients with PC (126) detected CTA-specific IgG in sera including NY-ESO-1, LAGE-1, NFX-2, and SSX-2. SSX-2 mRNA levels were also significantly elevated in metastatic PC tissue compared to primary tumors or to benign prostate tissue (126). SSX proteins were also up-regulated in MCH class I-deficient germline cells and in various types of advanced cancers with poor prognosis. SSX-2 was most frequently expressed across prostate cell lines, but SSX-1 and SSX-5 were also inducible after 5AZA2 treatment. SSX expression detected by immunohistochemical tissue arrays in patient tumor samples was restricted to metastatic lesions with no expression detected in primary prostate tumors. Cross-reactive immune responses to a dominant HLA-A2 SSX epitope (p103-111) were observed after immunization of A2/DR1 transgenic mice with SSX vaccines (124).
This tumor-specific expression suggests CTAs may be ideal antigens for tumor-targeting vaccines. In fact, the first CTA was discovered by studying a patient with unusually favorable cytolytic immune responses against melanoma. The CD8-restricted antigen was identified as MAGE-A1 (127). The MAGE family was subsequently recognized as a potent target to enhance tumor-specific CTL cell responses (128) and have been extensively studied as a promising candidate for therapeutic approaches. In 1994, Weber et al. found that 40–50% of tissue samples obtained from patients with advanced melanoma were positive for MAGE-1 from both early and metastatic stages while a wide range of normal tissue samples including tumor-infiltrating lymphocytes, peripheral blood from patients with metastatic melanoma, melanocytes, and benign nevus showed no expression of MAGE-1. Re-induction of MAGE-1 expression in non-expressing cell lines led to HLA-A1 restricted antigen presentation and epitope-specific lysis by CTL (129). HLA-A2-restricted T-cell receptors cloned from SSX-2-seropositive melanoma patients showed epitope-specific reactivity, tumor cell recognition, and tetramer binding when engineered into peripheral blood leukocytes (130). In a DNA vaccine study, improved SSX-2 immunogenicity was reported by introducing peptide ligand modifications to increase binding affinity to HLA-A2 molecules. This enhanced both the magnitude and efficiency of the Th1-type antitumor CD8 T-cell responses and also resulted in a more diverse T-cell-derived cytokine profile (124). Such improved overall efficiency and quality of the epitope-specific responses makes this a promising strategy to enhance tumor-targeting CTA vaccination strategies.
A key regulatory mechanism of CTA expression in both normal and tumor tissue is epigenetic modification (131). Epigenetic silencing is also one of the key changes associated with tumorigenesis (132), and hypermethylation in CTA promoter regions has been observed in various cancer types (133–136). Knockdown models of DNMT1 and DNMT3b demonstrated the role of these enzymes in mediating CG-X antigen gene repression and promoter methylation (135). A considerable body of literature on multiple tumor types has demonstrated that EMAs have the potential to increase immunogenicity via the re-expression of numerous CTAs (126, 129, 135, 137–139). In 1994, Weber et al. reported that 5AZA2 treatment induced MAGE-1 antigen expression in non-expressing cell lines, but not in normal blood cells or melanocytes, and led to HLA-A1-restricted, epitope-specific lysis by CTL (129). Similarly, inducibility of SSX-2 gene in PC LNCap and DU145 cell lines was found following 5AZA2 treatment but not in normal prostate epithelium cell line RWPE1 (126). A subsequent study of the same CTA family demonstrated that While SSX-2 was expressed most frequently in PC cell lines, SSX1 and SSX5 expression was also induced after 5AZA2 treatment (124). In a global screening study of human epithelial cell lines, low-dose AZA treatment up-regulated a wide selection of CTAs, including several members of the MAGE, SSX, SPANX, PAGE families, which were induced in all three tumor types analyzed (breast, colorectal, and ovarian) (140). The inhibition of histone lysine methylation enhanced expression of NY-ESO1, MAGE-A1, and MAGE-A3 expression in H841 lung cancer cells and enhanced tumor cell targeting and lysis by MAGE-A3 and NY-ESO1 epitope-specific T cells (136). HDIs Trichostatin A and depsipeptide FR901228 were both shown to synergize with 5AZA2 to activate CG-X antigen expression in various thoracic and colorectal cancer cell lines (135, 141, 142).
Increased expression of CTAs following treatment with EMAs has been identified in several clinical trials. In a Phase II study for patients with multiple myeloma, sequential AZA and a cytotoxicity-enhancer led to a significant increase of MAGEA4, MAGEA6, SPA17, and AKAP4 in bone marrow compared to pre-therapy samples. The treatment also resulted in enhanced CTAGB1 epitope-specific IFNγ response by patient PBMCs tested ex vivo in response to monocyte-derived, CTAGB1-pulsed dendritic cells (DCs) (143). NCT01483274 was proposed to test 5AZA2 efficiency in up-regulating CTA expression followed by a donor lymphocyte infusion and an autologous transfer of MAGE-A1, MAGE-A3, and NY-ESO1 peptide-pulsed DCs in patients with AML who had relapsed after an allogeneic stem cell transplant. Study outcomes are specified as tolerance of study treatment, clinical disease response, and assessment of immune responses to vaccine peptides. The same center proposed a Phase I study with the same treatment to treat relapsed high-risk neuroblastoma, Ewing’s sarcoma, osteogenic sarcoma, rhabdomyosarcoma, or synovial sarcoma. Both of these studies are registered but not yet recruiting.
CTAs have been identified as promising candidates for highly selective tumor-targeting by enhancement of endogenous antitumor responses. Conversely, a vaccine clinical trial with antigenic targets from the same MAGE family, namely MAGE-A3/A9/A12 was just recently terminated due to adverse side effects including substantial neurotoxicity with two vaccination induced fatalities with necrotizing white matter tissue damage and CD8+CD3+ T-cell neuro-infiltration. Follow-up studies revealed a previously unrecognized expression of MAGE A12 in normal brain tissue, which could be the potential target for the vaccine-mediated neuroinflammatory response detected in this trial. This case has alerted for the need of refining CTA characterization to help identify suitable and safe targets for systemic immunization against cancer (144).
Antigen Processing and Antigen Presentation
Beyond sufficient expression of TAAs, effector antitumor T-cell responses also require the processing and loading of TAAs onto major histocompatibility complex (MHC) I complexes in the context of co-stimulatory molecules. The MHC I antigen-presenting machinery samples designated ubiquitin-tagged endogenous proteins and delivers them to the proteasome complex low molecular mass polypeptide (LMP) 2, LMP7, LMP10 for preprocessing into up to 25-meric peptides. This peptide-pool is then further cleaved by cytosolic aminopeptidases and delivered to the ER via the transporter associated with antigen processing (TAP) complex (TAP1, TAP2) for subsequential trimming by ER aminopeptidase associated with antigen processing (ERAAP) followed by chaperone (calnexin, calreticulin, ERp57, and tapasin)-mediated assembly and loading onto the antigen-MHC complex. After peptide loading, the chaperones are released from the peptide-MHC complex, which is then trafficked to the Golgi via vesicle transport and delivered for surface membrane display (145). Defects of the MHC I antigen presentation system lead to impairment of immune surveillance, which has been linked to both tumorigenesis and poor clinical outcomes (146–150). A tissue microarray analysis on 71 PC patient samples revealed a significant decrease in beta 2 microglobulin (B2M) expression compared to normal surrounding tissue (145). A study on diffuse large B cell lymphoma revealed aberrant B2M protein expression in 75% of the examined biopsies, which was associated with the loss of HLA-I surface expression (151–153).
Epigenetic alterations have been identified as one of the mechanisms underlying deficient antigen presentation in pre-clinical tumor models. Histone acetylation of the TAP1 promoter was proposed as a potential repressor mechanism accounting for TAP1 deficiency in various carcinoma cell lines. The level of acetyl-histone H3 strongly correlated with the level of TAP1 expression and with metastatic features in malignant carcinomas (154). The reversal of tumor antigen presentation impairment and MHC-complex deficiencies may promote tumor killing. EMA has been shown as a potential approach to reverse such defect and enhance tumor immune responses. Li et al. analyzed the global response to low-dose AZA in 63 human epithelial cancer cell lines and found that B2M, HLA-B, HLA-C, CTSS, NSF2, TAP1, and proteasome proteins PMSB8 and PMSB9 were up-regulated in at least three cell lines each (140). Using a high-throughput bioinformatics approach, Kortenhorst et al. identified a comprehensive list of genes and pathways affected by HDI treatment in PC cell lines and showed a significant up-regulation of MHC genes including HLA-Class I molecules and B2M (145). Antigen processing and presentation is also enhanced by AZA treatment via up-regulation of the interferon type I and type II families including interferon-gamma receptor 1 and STAT1 as was shown in an in vivo mouse model for HPV-16-associated tumors and in NSCLC tumor cell lines (153, 155).
Dendritic cells play a pivotal role in TAA sampling, processing, and presentation to T cells. Selective DC targeting makes antigen delivery to the draining lymph nodes more efficient. Fusion proteins with high-affinity DC-specific binding components facilitate DC loading and shield antigens from biodegradation allowing for lower vaccine doses. DC targeting has been shown to be a promising novel vaccination strategy that results in enhanced, durable, and overall higher quality immune responses (156). A phase I trial study has been registered (NCT01834248) and is currently recruiting to test immune response to DEC-205/NY-ESO1 fusion protein (CDX-1401) and 5AZA2 in patients with MDS or AML. CDX-1401 is a full length NY-ESO1 protein sequence fused to a monoclonal antibody against DEC-205, a surface marker present on professional APCs to enhance targeted delivery of peptide antigen to the antigen processing machinery and to enhance the efficacy of DC-mediated T-cell induction. NY-ESO1-specific primed T cells are expected to target tumor more efficiently due to an increase in 5AZA2-induced in situ NY-ESO1 expression by the cancer cells. In addition to addressing safety, efficacy, tolerability, and vaccine immune responses, the study also aims to assess the molecular epigenetic response to 5AZA2.
Epigenetics and Adaptive Immune Responses
Epigenetic silencing of immune-related genes is a striking feature of the global methylation pattern of the cancer genome. The impact of epigenetic alterations in tumorigenesis can foster an immunosuppressive tumor microenvironment. These alterations are potentially modifiable with EMAs. A large-scale study on epithelial cancer cell lines treated with low-dose AZA have identified 80 up-regulated gene sets similar across three cancer types analyzed including ovarian, colorectal, and breast cancer and among those there were 15 commonly up-regulated immune gene sets including elements of the interferon signaling, antigen presentation, influenza, and the chemokine and cytokine families. A similar analysis of lung cancer cell lines then also showed a 95% overlap of the gene sets up-regulated by AZA as well as a relative dominance among those by immune-related pathways. The same study also analyzed primary tumor data in public gene expression data bases and found that the above identified AZA-responder immunomodulatory genes showed clustering into a “low” and “high” expression subgroup across all three epithelial cancer types independent of clinical stage or of most tumor subtypes. Similar findings were concluded when looking at NSCLC and melanoma databases. Strikingly, 11 of the 15 AZA-responder immunomodulatory gene clusters were up-regulated in patient biopsies following an 8-week AZA and entinostat combination therapy. These findings suggest that gene expression profiling may help identify patients with immune evasion phenotype as candidates potentially benefiting from epigenetic therapy (140).
Effective T-cell priming requires the antigen presented in the MHC complex in the context of co-stimulatory molecules, which define the phenotype of T-cell responses. Modifying co-stimulatory patterns in the immune synapsis can effectively alter the magnitude of both effector and regulatory T-cell responses (157). Therapeutic targeting of negative or positive co-stimulatory molecules to enhance antitumor immune responses has been tested in both pre-clinical and clinical studies with promising outcomes. Low-dose in vitro 5AZA2 treatment of EL4 cells increased co-stimulatory CD80 expression by the tumor cells, which led to increased immunogenicity detected after engraftment into C57BL/6 mice (158). The addition of AZA and entinostat to treatment with checkpoint-inhibitor anti-PD-1/anti-CTLA-4 antibodies led to remarkable tumor regression in a syngeneic mouse model with checkpoint-inhibitor-resistant metastatic cancer (159). A study in human leukemia cell lines demonstrated that AZA treatment increased the expression of immune-checkpoint molecules PD-1, PD-L1, PD-L2, and CTLA-4 (160). AZA increased both transcript and surface expression levels of PD-L1 on a NSCLC cell line (153).
Targeting regulatory immune responses to enhance antitumor immune responses has been addressed in several recent clinical trials. Clinical response and prolonged stabilization were reported in a substantial proportion of patients with diverse tumor types even with treatment-refractory, metastatic types otherwise considered as non-responsive to immunotherapy. Notably, patients with PD-L1 negative tumors showed no response to therapy (161). A follow-up study of three patients for more than 3 years after cessation of anti-PD-1 therapy showed durable, stabile, or re-inducible complete remission (162). Patients showing no up-regulation of checkpoint elements after treatment with combination therapy of AZA and vorinostat had increased survival (160). The analysis of PD-L1 and PD-1 expression patterns within the tumor microenvironment revealed a strong correlation between tumor cell PD-L1 expression and both the magnitude of intratumoral immune cell infiltration and their PD-1 expression. These findings suggested a strong correlation between tumor PD-L1 expression and clinical response (163). Since EMA treatment has a potential to restore PD-L1 expression on tumor cells, combinatorial EMA treatment may expand the group of candidates for PD-L1 checkpoint-inhibitor therapy. A Phase II study in NSCLC (NCT01928576) is currently recruiting to analyze the efficacy of entinostat and/or azacitidine prior to PD-1 blocker nivolumab treatment.
Enhancement of effector immune mechanisms can benefit antitumor interventions and therapeutic approaches to improve clinical outcomes. Combining EMAs provides a novel alternative to improve antitumor immune responses. Importantly, data from immunodeficient animal models have demonstrated that the tumor inhibitory effect of EMAs requires an intact immune system. The antitumor effect of HDI vorinostat and panobinostat treatment was diminished in RAG2γC-/- and IFNγR-/- immunocompromised animals (164, 165). Pre-clinical studies have demonstrated that hypomethylating agents enhance the effector function of both T cells and NK cells. 5AZA2-treated tumor cells induced a higher yield of tumor-infiltrating CD4, CD8 T cells, and NK cells and IFNγ production was also elevated in both T-cell compartments. Mechanistic experiments within the same study identified a CD8-dependent tumor rejection mechanism induced by 5AZA2 (158). 5AZA2 treatment had a biphasic effect on both the phenotype and function of ex vivo expanded normal human peripheral NK cells. After treatment, KIR and NKp44 expression were increased while NKG2D decreased. The 5AZA2-induced hypomethylation followed a U-shaped dose-response curve similar to the effects on cytotoxicity (166). Human peripheral NK cells showed increased cytotoxicity when exposed to exosomes isolated from MS-275-treated HEPG2 cells compared to untreated cancer cells. The TAA chaperone HSP70 and the MHC-related MICA, MICB content of the exosomes was elevated by MS-275 treatment suggesting a non-antigen-specific mechanism selective for NK cell activation (167). Combinatorial panobinostat treatment significantly reduced tumor burden and enhanced TH1 cytokine profile and effector function of adoptively transferred tumor-specific T cells in a melanoma model. This treatment also resulted in a dramatic increase in the tumor-infiltrating effector cell to regulatory T-cell ratio (168).
Previous studies have suggested that EMAs can impact T-cell proliferation, differentiation, and function (169–171). Animal studies support the concept that HDIs can promote a cytotoxic antitumor immune response (172) but numerous studies have shown that patients with PRCA have a profound impairment in the function of circulating and tumor-infiltrating T cells (173, 174). Whether EMAs can promote antitumor responses in this immunosuppressive environment in patients with PRCA is unknown. Another challenge to this hypothesis lies in the clinical findings of myelosuppression due to EMA treatment when given at high doses or intervals (175–177). However, low doses of EMAs may promote T-cell-mediated cell lysis (169).
As with any systemic therapy, there are potential toxicities from EMAs that could inhibit antitumor immune responses or complicate patient outcomes. For example, dose-dependent toxicities with EMAs include leukopenia, granulocytopenia, and thrombocytopenia. Altering dose and schedule of these agents can ameliorate some of these toxicities but must be closely monitored. In addition, reactivation of chronic viral infections, including HIV type 1, was reported following treatment with SAHA in an in vitro model for latent infection (178). Virus-related illnesses were also observed in a multi-institutional phase II clinical trial of romidepsin, including EBV, HBV, and VZV reactivation (179). Finally, epigenetic modification has been also suggested to regulate immune mechanisms involved in chronic inflammation and autoimmunity. The extent to which these toxicities may impact patient outcomes is unclear and is the subject of ongoing clinical studies.
Biomarkers for Epigenetic Therapies
Studies have identified a wide variety of impacts that EMAs can have on both the tumor and immune compartments. However, the toxicities associated with these agents when dosed at traditional MTDs will likely mitigate effective immune responses. Thus, optimal biologic dosing strategies may be key to the success of these therapeutic strategies. Integration of biomarkers for the purpose of assessing methylation and histone modification status in tumor and immune compartments as well as expression of relevant immune-related genes, could identify patient-specific dosing strategies targeting immune activation. State-of-the-art tools are available to dissect and measure immune responses quickly and efficiently using multi-parameter analysis platforms and high-throughput technologies. Improving tumor analytics for these purposes, whether through traditional tumor biopsies or alternate assays for tumor cells in circulation, may further identify optimal biologic dosing strategies to modify gene expression. Given the potential of these agents to alter expression of a wide range of genetic targets, incorporation of discovery biomarkers to identify novel targets would have further clinical and experimental utility. For example, large clusters of genes involved in effector immune mechanisms have been identified as loci accumulating genetic aberrations in the cancer genome. Only a limited number of individual genes from these clusters have been tested so far in either pre-clinical or clinical studies in cancer research. Thus, careful integration of biomarkers into these therapeutic strategies will be critical to advance these clinical hypotheses.
Conclusion
In recent years, the development of combination immunotherapies has branched out as a robust new approach to novel cancer therapeutics. The interplay between tumor and host during tumor pathogenesis is a complex process and epigenetic modifications mark many components of the pathologic changes leading to tumor evasion. Epigenetic therapies have the potential to reverse this process at multiple levels. Immune therapies provide a significant benefit compared to standard cancer therapies by allowing for better accommodation of tumor heterogeneity and the patient’s individual immune repertoire. Epigenetic agents can complement immunotherapies by enhancing many underlying mechanisms of the antitumor immune response (Figure 1). However, the potential benefits of these agents must be balanced by the potential toxicities of these therapies. Biologic dosing strategies are likely the best approach to maximize benefit while limiting toxicity. Integrated biomarkers across tumor and immune compartments may allow determination of biologic doses, though thoughtful approaches to these assays must be considered.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wang X, Gao H, Ren L, Gu J, Zhang Y, Zhang Y. Demethylation of the miR-146a promoter by 5-Aza-2′-deoxycytidine correlates with delayed progression of castration-resistant prostate cancer. BMC Cancer (2014) 14:308. doi:10.1186/1471-2407-14-308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Cheng X, Blumenthal RM. Mammalian DNA methyltransferases: a structural perspective. Structure (2008) 16:341–50. doi:10.1016/j.str.2008.01.004
3. Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev (1995) 5:309–14. doi:10.1016/0959-437X(95)80044-1
4. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet (2000) 16:168–74. doi:10.1016/S0168-9525(99)01971-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer (2004) 4:143–53. doi:10.1038/nrc1279
6. Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature (1993) 366:362–5. doi:10.1038/366362a0
7. Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet (2001) 10:687–92. doi:10.1093/hmg/10.7.687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet (2008) 9:465–76. doi:10.1038/nrg2341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell (1980) 20:85–93. doi:10.1016/0092-8674(80)90237-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene (2002) 21:5483–95. doi:10.1038/sj.onc.1205699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res (2007) 67:6400–8. doi:10.1158/0008-5472.CAN-07-0251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol (2002) 321:591–9. doi:10.1016/S0022-2836(02)00676-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Brueckner B, Rius M, Markelova MR, Fichtner I, Hals PA, Sandvold ML, et al. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol Cancer Ther (2010) 9:1256–64. doi:10.1158/1535-7163.MCT-09-1202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Pettitt AR. Mechanism of action of purine analogues in chronic lymphocytic leukaemia. Br J Haematol (2003) 121:692–702. doi:10.1046/j.1365-2141.2003.04336.x
15. Jeha S, Gandhi V, Chan KW, Mcdonald L, Ramirez I, Madden R, et al. Clofarabine, a novel nucleoside analog, is active in pediatric patients with advanced leukemia. Blood (2004) 103:784–9. doi:10.1182/blood-2003-06-2122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol (1988) 140:2197–200.
17. Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res (2003) 63:4984–9.
18. Iljin K, Ketola K, Vainio P, Halonen P, Kohonen P, Fey V, et al. High-throughput cell-based screening of 4910 known drugs and drug-like small molecules identifies disulfiram as an inhibitor of prostate cancer cell growth. Clin Cancer Res (2009) 15:6070–8. doi:10.1158/1078-0432.CCR-09-1035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res (2005) 65:6305–11. doi:10.1158/0008-5472.CAN-04-2957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Lin YS, Shaw AY, Wang SG, Hsu CC, Teng IW, Tseng MJ, et al. Identification of novel DNA methylation inhibitors via a two-component reporter gene system. J Biomed Sci (2011) 18:3. doi:10.1186/1423-0127-18-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Kuck D, Caulfield T, Lyko F, Medina-Franco JL. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol Cancer Ther (2010) 9:3015–23. doi:10.1158/1535-7163.MCT-10-0609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Majid S, Dar AA, Shahryari V, Hirata H, Ahmad A, Saini S, et al. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-cell translocation gene 3 in prostate cancer. Cancer (2010) 116:66–76. doi:10.1002/cncr.24662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Bosviel R, Durif J, Dechelotte P, Bignon YJ, Bernard-Gallon D. Epigenetic modulation of BRCA1 and BRCA2 gene expression by equol in breast cancer cell lines. Br J Nutr (2012) 108:1187–93. doi:10.1017/S000711451100657X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, et al. Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett (2009) 19:706–9. doi:10.1016/j.bmcl.2008.12.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol (2005) 68:1018–30. doi:10.1124/mol.104.008367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci U S A (2012) 109:7765–9. doi:10.1073/pnas.1205132109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Liu Z, Liu S, Xie Z, Pavlovicz RE, Wu J, Chen P, et al. Modulation of DNA methylation by a sesquiterpene lactone parthenolide. J Pharmacol Exp Ther (2009) 329:505–14. doi:10.1124/jpet.108.147934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Sorm F, Piskala A, Cihak A, Vesely J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia (1964) 20:202–3. doi:10.1007/BF02135399
29. Sorm F, Vesely J. Effect of 5-aza-2′-deoxycytidine against leukemic and hemopoietic tissues in AKR mice. Neoplasma (1968) 15:339–43.
30. Momparler RL, Onetto-Pothier N, Momparler LF. Comparison of antineoplastic activity of cytosine arabinoside and 5-aza-2′-deoxycytidine against human leukemic cells of different phenotype. Leuk Res (1990) 14:755–60. doi:10.1016/0145-2126(90)90068-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Fialova B, Smesny Trtkova K, Paskova L, Langova K, Kolar Z. Effect of histone deacetylase and DNA methyltransferase inhibitors on the expression of the androgen receptor gene in androgen-independent prostate cancer cell lines. Oncol Rep (2013) 29:2039–45. doi:10.3892/or.2013.2344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Gravina GL, Marampon F, Di Staso M, Bonfili P, Vitturini A, Jannini EA, et al. 5-Azacitidine restores and amplifies the bicalutamide response on preclinical models of androgen receptor expressing or deficient prostate tumors. Prostate (2010) 70:1166–78. doi:10.1002/pros.21151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Chuang JC, Warner SL, Vollmer D, Vankayalapati H, Redkar S, Bearss DJ, et al. S110, a 5-Aza-2′-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther (2010) 9:1443–50. doi:10.1158/1535-7163.MCT-09-1048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Marquez VE, Barchi JJ Jr, Kelley JA, Rao KV, Agbaria R, Ben-Kasus T, et al. Zebularine: a unique molecule for an epigenetically based strategy in cancer chemotherapy. The magic of its chemistry and biology. Nucleosides Nucleotides Nucleic Acids (2005) 24:305–18. doi:10.1081/NCN-200059765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Billam M, Sobolewski MD, Davidson NE. Effects of a novel DNA methyltransferase inhibitor zebularine on human breast cancer cells. Breast Cancer Res Treat (2010) 120:581–92. doi:10.1007/s10549-009-0420-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Yang PM, Lin YT, Shun CT, Lin SH, Wei TT, Chuang SH, et al. Zebularine inhibits tumorigenesis and stemness of colorectal cancer via p53-dependent endoplasmic reticulum stress. Sci Rep (2013) 3:3219. doi:10.1038/srep03219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Candelaria M, De La Cruz-Hernandez E, Taja-Chayeb L, Perez-Cardenas E, Trejo-Becerril C, Gonzalez-Fierro A, et al. DNA methylation-independent reversion of gemcitabine resistance by hydralazine in cervical cancer cells. PLoS One (2012) 7:e29181. doi:10.1371/journal.pone.0029181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Yamanegi K, Yamane J, Kobayashi K, Kato-Kogoe N, Ohyama H, Nakasho K, et al. Valproic acid cooperates with hydralazine to augment the susceptibility of human osteosarcoma cells to Fas- and NK cell-mediated cell death. Int J Oncol (2012) 41:83–91. doi:10.3892/ijo.2012.1438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Graca I, Sousa EJ, Costa-Pinheiro P, Vieira FQ, Torres-Ferreira J, Martins MG, et al. Anti-neoplastic properties of hydralazine in prostate cancer. Oncotarget (2014) 5:5950–64.
40. Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem (2005) 280:40749–56. doi:10.1074/jbc.M505593200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res (2003) 9:1596–603.
42. Halby L, Champion C, Senamaud-Beaufort C, Ajjan S, Drujon T, Rajavelu A, et al. Rapid synthesis of new DNMT inhibitors derivatives of procainamide. Chembiochem (2012) 13:157–65. doi:10.1002/cbic.201100522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Herbst A, Rahmig K, Stieber P, Philipp A, Jung A, Ofner A, et al. Methylation of NEUROG1 in serum is a sensitive marker for the detection of early colorectal cancer. Am J Gastroenterol (2011) 106:1110–8. doi:10.1038/ajg.2011.6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, et al. Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer (2008) 123:552–60. doi:10.1002/ijc.23590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med (2005) 11:77–84. doi:10.1038/nm1161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Chen S, Zhao Y, Gou WF, Zhao S, Takano Y, Zheng HC. The anti-tumor effects and molecular mechanisms of suberoylanilide hydroxamic acid (SAHA) on the aggressive phenotypes of ovarian carcinoma cells. PLoS One (2013) 8:e79781. doi:10.1371/journal.pone.0079781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Chen MY, Liao WS, Lu Z, Bornmann WG, Hennessey V, Washington MN, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer (2011) 117:4424–38. doi:10.1002/cncr.26073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Chiao MT, Cheng WY, Yang YC, Shen CC, Ko JL. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy (2013) 9:1509–26. doi:10.4161/auto.25664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene (2004) 23:6261–71. doi:10.1038/sj.onc.1207830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Lakshmikanthan V, Kaddour-Djebbar I, Lewis RW, Kumar MV. SAHA-sensitized prostate cancer cells to TNFalpha-related apoptosis-inducing ligand (TRAIL): mechanisms leading to synergistic apoptosis. Int J Cancer (2006) 119:221–8. doi:10.1002/ijc.21824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lillehammer T, Engesaeter BO, Prasmickaite L, Maelandsmo GM, Fodstad O, Engebraaten O. Combined treatment with Ad-hTRAIL and DTIC or SAHA is associated with increased mitochondrial-mediated apoptosis in human melanoma cell lines. J Gene Med (2007) 9:440–51. doi:10.1002/jgm.1036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Carlisi D, Lauricella M, D’anneo A, Emanuele S, Angileri L, Di Fazio P, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. Eur J Cancer (2009) 45:2425–38. doi:10.1016/j.ejca.2009.06.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Lauricella M, Ciraolo A, Carlisi D, Vento R, Tesoriere G. SAHA/TRAIL combination induces detachment and anoikis of MDA-MB231 and MCF-7 breast cancer cells. Biochimie (2012) 94:287–99. doi:10.1016/j.biochi.2011.06.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Jazirehi AR, Arle D. Epigenetic regulation of the TRAIL/Apo2L apoptotic pathway by histone deacetylase inhibitors: an attractive approach to bypass melanoma immunotherapy resistance. Am J Clin Exp Immunol (2013) 2:55–74.
55. Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood (2009) 114:380–93. doi:10.1182/blood-2008-10-182758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Woods DM, Woan K, Cheng F, Wang H, Perez-Villarroel P, Lee C, et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res (2013) 23(5):341–8. doi:10.1097/CMR.0b013e328364c0ed
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Belinsky SA, Grimes MJ, Picchi MA, Mitchell HD, Stidley CA, Tesfaigzi Y, et al. Combination therapy with vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Res (2011) 71:454–62. doi:10.1158/0008-5472.CAN-10-3184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Tellez CS, Grimes MJ, Picchi MA, Liu Y, March TH, Reed MD, et al. SGI-110 and entinostat therapy reduces lung tumor burden and reprograms the epigenome. Int J Cancer (2014) 135:2223–31. doi:10.1002/ijc.28865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin-6 analysis: a study by the department of defense prostate cancer clinical trial consortium and University of Chicago phase 2 consortium. Cancer (2009) 115:5541–9. doi:10.1002/cncr.24597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett (2009) 280:233–41. doi:10.1016/j.canlet.2009.02.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, et al. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther (2003) 2:721–8.
62. Buggy JJ, Cao ZA, Bass KE, Verner E, Balasubramanian S, Liu L, et al. CRA-024781: a novel synthetic inhibitor of histone deacetylase enzymes with antitumor activity in vitro and in vivo. Mol Cancer Ther (2006) 5:1309–17. doi:10.1158/1535-7163.MCT-05-0442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med (2005) 11:1–15. doi:10.2119/2006-00005.Dinarello
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Mandl-Weber S, Meinel FG, Jankowsky R, Oduncu F, Schmidmaier R, Baumann P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br J Haematol (2010) 149:518–28. doi:10.1111/j.1365-2141.2010.08124.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Arts J, King P, Marien A, Floren W, Belien A, Janssen L, et al. JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res (2009) 15:6841–51. doi:10.1158/1078-0432.CCR-09-0547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Novotny-Diermayr V, Sangthongpitag K, Hu CY, Wu X, Sausgruber N, Yeo P, et al. SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol Cancer Ther (2010) 9:642–52. doi:10.1158/1535-7163.MCT-09-0689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood (2003) 102:2615–22. doi:10.1182/blood-2003-01-0233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Butler LM, Webb Y, Agus DB, Higgins B, Tolentino TR, Kutko MC, et al. Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase. Clin Cancer Res (2001) 7:962–70.
69. Moffat D, Patel S, Day F, Belfield A, Donald A, Rowlands M, et al. Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl]amino}bicyclo[3.1.0]hex-3-yl)-N-hydroxypyrim idine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J Med Chem (2010) 53:8663–78. doi:10.1021/jm101177s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5:769–84. doi:10.1038/nrd2133
71. Yoshida M, Horinouchi S, Beppu T. Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays (1995) 17:423–30. doi:10.1002/bies.950170510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Kim YB, Lee KH, Sugita K, Yoshida M, Horinouchi S. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene (1999) 18:2461–70. doi:10.1038/sj.onc.1202564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Duong V, Bret C, Altucci L, Mai A, Duraffourd C, Loubersac J, et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol Cancer Res (2008) 6:1908–19. doi:10.1158/1541-7786.MCR-08-0299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A (2010) 107:20003–8. doi:10.1073/pnas.1013754107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia (2008) 22:1026–34. doi:10.1038/leu.2008.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Takai N, Ueda T, Nishida M, Nasu K, Narahara H. Anticancer activity of MS-275, a novel histone deacetylase inhibitor, against human endometrial cancer cells. Anticancer Res (2006) 26:939–45.
77. Kraker AJ, Mizzen CA, Hartl BG, Miin J, Allis CD, Merriman RL. Modulation of histone acetylation by [4-(acetylamino)-N-(2-amino-phenyl) benzamide] in HCT-8 colon carcinoma. Mol Cancer Ther (2003) 2:401–8.
78. Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther (2008) 7:759–68. doi:10.1158/1535-7163.MCT-07-2026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, et al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol (2012) 69:901–9. doi:10.1007/s00280-011-1766-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Savickiene J, Borutinskaite VV, Treigyte G, Magnusson KE, Navakauskiene R. The novel histone deacetylase inhibitor BML-210 exerts growth inhibitory, proapoptotic and differentiation stimulating effects on the human leukemia cell lines. Eur J Pharmacol (2006) 549:9–18. doi:10.1016/j.ejphar.2006.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Yeung A, Bhargava RK, Ahn R, Bahna S, Kang NH, Lacoul A, et al. HDAC inhibitor M344 suppresses MCF-7 breast cancer cell proliferation. Biomed Pharmacother (2012) 66:232–6. doi:10.1016/j.biopha.2011.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J (2001) 20:6969–78. doi:10.1093/emboj/20.24.6969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell (1978) 14:105–13. doi:10.1016/0092-8674(78)90305-7
85. Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J Antibiot (Tokyo) (1994) 47:301–10. doi:10.7164/antibiotics.47.301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem (1993) 268:22429–35.
87. Han JW, Ahn SH, Park SH, Wang SY, Bae GU, Seo DW, et al. Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res (2000) 60:6068–74.
88. Peled T, Shoham H, Aschengrau D, Yackoubov D, Frei G, Rosenheimer GN, et al. Nicotinamide, a SIRT1 inhibitor, inhibits differentiation and facilitates expansion of hematopoietic progenitor cells with enhanced bone marrow homing and engraftment. Exp Hematol (2012) 40(342–355):e341. doi:10.1016/j.exphem.2011.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci U S A (2001) 98:15113–8. doi:10.1073/pnas.261574398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Napper AD, Hixon J, Mcdonagh T, Keavey K, Pons JF, Barker J, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem (2005) 48:8045–54. doi:10.1021/jm050522v
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Olaharski AJ, Rine J, Marshall BL, Babiarz J, Zhang L, Verdin E, et al. The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases. PLoS Genet (2005) 1:e77. doi:10.1371/journal.pgen.0010077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. McCarthy AR, Sachweh MC, Higgins M, Campbell J, Drummond CJ, Van Leeuwen IM, et al. Tenovin-D3, a novel small-molecule inhibitor of sirtuin SirT2, increases p21 (CDKN1A) expression in a p53-independent manner. Mol Cancer Ther (2013) 12:352–60. doi:10.1158/1535-7163.MCT-12-0900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Rotili D, Tarantino D, Nebbioso A, Paolini C, Huidobro C, Lara E, et al. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J Med Chem (2012) 55:10937–47. doi:10.1021/jm3011614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Hoffmann G, Breitenbucher F, Schuler M, Ehrenhofer-Murray AE. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J Biol Chem (2014) 289:5208–16. doi:10.1074/jbc.M113.487736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res (2006) 66:4368–77. doi:10.1158/0008-5472.CAN-05-3617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem (2001) 276:38837–43. doi:10.1074/jbc.M106779200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, et al. Salermide, a sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene (2009) 28:781–91. doi:10.1038/onc.2008.436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell (2008) 13:454–63. doi:10.1016/j.ccr.2008.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol (2013) 9:319–25. doi:10.1038/nchembio.1223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Kim DH, Shin J, Kwon HJ. Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Exp Mol Med (2007) 39:47–55. doi:10.1038/emm.2007.6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, Kozikowski AP. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J Med Chem (2012) 55:9891–9. doi:10.1021/jm301098e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, Chagot B, et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One (2013) 8:e68915. doi:10.1371/journal.pone.0068915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res (2003) 63:3637–45.
104. Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, et al. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer (2000) 83:817–25. doi:10.1054/bjoc.2000.1327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Wang J, Kim TH, Ahn MY, Lee J, Jung JH, Choi WS, et al. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int J Oncol (2012) 41:1101–9. doi:10.3892/ijo.2012.1534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Quagliana JM, O’bryan RM, Baker L, Gottlieb J, Morrison FS, Eyre HJ, et al. Phase II study of 5-azacytidine in solid tumors. Cancer Treat Rep (1977) 61:51–4.
107. Reed MD, Tellez CS, Grimes MJ, Picchi MA, Tessema M, Cheng YS, et al. Aerosolised 5-azacytidine suppresses tumour growth and reprogrammes the epigenome in an orthotopic lung cancer model. Br J Cancer (2013) 109:1775–81. doi:10.1038/bjc.2013.575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Naldi I, Taranta M, Gherardini L, Pelosi G, Viglione F, Grimaldi S, et al. Novel epigenetic target therapy for prostate cancer: a preclinical study. PLoS One (2014) 9:e98101. doi:10.1371/journal.pone.0098101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Scandura JM, Roboz GJ, Moh M, Morawa E, Brenet F, Bose JR, et al. Phase 1 study of epigenetic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood (2011) 118:1472–80. doi:10.1182/blood-2010-11-320093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin Genitourin Cancer (2015) 13(1):22–31. doi:10.1016/j.clgc.2014.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Candelaria M, Gallardo-Rincon D, Arce C, Cetina L, Aguilar-Ponce JL, Arrieta O, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol (2007) 18:1529–38. doi:10.1093/annonc/mdm204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Xia C, Leon-Ferre R, Laux D, Deutsch J, Smith BJ, Frees M, et al. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother Pharmacol (2014) 74:691–7. doi:10.1007/s00280-014-2501-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Arce C, Perez-Plasencia C, Gonzalez-Fierro A, De La Cruz-Hernandez E, Revilla-Vazquez A, Chavez-Blanco A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One (2006) 1:e98. doi:10.1371/journal.pone.0000098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer (2012) 12:465–77. doi:10.1038/nrc3256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Gross L. The importance of dosage in the intradermal immunization against transplantable neoplasms. Cancer Res (1943) 3:770–8.
116. Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J Exp Med (2004) 199:155–66. doi:10.1084/jem.20031677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Huijbers IJ, Soudja SM, Uyttenhove C, Buferne M, Inderberg-Suso EM, Colau D, et al. Minimal tolerance to a tumor antigen encoded by a cancer-germline gene. J Immunol (2012) 188:111–21. doi:10.4049/jimmunol.1002612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A (2008) 105:20422–7. doi:10.1073/pnas.0810777105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Woloszynska-Read A, Mhawech-Fauceglia P, Yu J, Odunsi K, Karpf AR. Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clin Cancer Res (2008) 14:3283–90. doi:10.1158/1078-0432.CCR-07-5279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Kalejs M, Erenpreisa J. Cancer/testis antigens and gametogenesis: a review and “brain-storming” session. Cancer Cell Int (2005) 5:4. doi:10.1186/1475-2867-5-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer (2005) 5:615–25. doi:10.1038/nrc1669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Whitehurst AW. Cause and consequence of cancer/testis antigen activation in cancer. Annu Rev Pharmacol Toxicol (2014) 54:251–72. doi:10.1146/annurev-pharmtox-011112-140326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Suyama T, Shiraishi T, Zeng Y, Yu W, Parekh N, Vessella RL, et al. Expression of cancer/testis antigens in prostate cancer is associated with disease progression. Prostate (2010) 70:1778–87. doi:10.1002/pros.21214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Smith HA, Cronk RJ, Lang JM, Mcneel DG. Expression and immunotherapeutic targeting of the SSX family of cancer-testis antigens in prostate cancer. Cancer Res (2011) 71:6785–95. doi:10.1158/0008-5472.CAN-11-2127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Freitas M, Malheiros S, Stavale JN, Biassi TP, Zamuner FT, De Souza Begnami M, et al. Expression of cancer/testis antigens is correlated with improved survival in glioblastoma. Oncotarget (2013) 4:636–46.
126. Dubovsky JA, McNeel DG. Inducible expression of a prostate cancer-testis antigen, SSX-2, following treatment with a DNA methylation inhibitor. Prostate (2007) 67:1781–90. doi:10.1002/pros.20665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van Den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (1991) 254:1643–7. doi:10.1126/science.1840703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Boon T, Cerottini JC, Van Den Eynde B, Van Der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol (1994) 12:337–65. doi:10.1146/annurev.iy.12.040194.002005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2′-deoxycytidine. Cancer Res (1994) 54:1766–71.
130. Abate-Daga D, Speiser DE, Chinnasamy N, Zheng Z, Xu H, Feldman SA, et al. Development of a T cell receptor targeting an HLA-A*0201 restricted epitope from the cancer-testis antigen SSX2 for adoptive immunotherapy of cancer. PLoS One (2014) 9:e93321. doi:10.1371/journal.pone.0093321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene (2002) 21:5496–503. doi:10.1038/sj.onc.1205602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. De Smet C, De Backer O, Faraoni I, Lurquin C, Brasseur F, Boon T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci U S A (1996) 93:7149–53. doi:10.1073/pnas.93.14.7149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Yu J, Ni M, Xu J, Zhang H, Gao B, Gu J, et al. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer (2002) 2:29. doi:10.1186/1471-2407-2-29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Yu J, Zhang H, Gu J, Lin S, Li J, Lu W, et al. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer (2004) 4:65. doi:10.1186/1471-2407-4-65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. James SR, Link PA, Karpf AR. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene (2006) 25:6975–85. doi:10.1038/sj.onc.1209678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Rao M, Chinnasamy N, Hong JA, Zhang Y, Zhang M, Xi S, et al. Inhibition of histone lysine methylation enhances cancer-testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res (2011) 71:4192–204. doi:10.1158/0008-5472.CAN-10-2442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Loriot A, De Plaen E, Boon T, De Smet C. Transient down-regulation of DNMT1 methyltransferase leads to activation and stable hypomethylation of MAGE-A1 in melanoma cells. J Biol Chem (2006) 281:10118–26. doi:10.1074/jbc.M510469200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Natsume A, Wakabayashi T, Tsujimura K, Shimato S, Ito M, Kuzushima K, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine activates NY-ESO-1 antigenicity in orthotopic human glioma. Int J Cancer (2008) 122:2542–53. doi:10.1002/ijc.23407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, Karbach J, Pfeifer D, Jager E, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk Res (2010) 34:899–905. doi:10.1016/j.leukres.2010.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget (2014) 5:587–98.
141. Weiser TS, Guo ZS, Ohnmacht GA, Parkhurst ML, Tong-On P, Marincola FM, et al. Sequential 5-Aza-2 deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1. J Immunother (2001) 24:151–61. doi:10.1097/00002371-200103000-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Weiser TS, Ohnmacht GA, Guo ZS, Fischette MR, Chen GA, Hong JA, et al. Induction of MAGE-3 expression in lung and esophageal cancer cells. Ann Thorac Surg (2001) 71:295–301. doi:10.1016/S0003-4975(00)02421-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Toor AA, Payne KK, Chung HM, Sabo RT, Hazlett AF, Kmieciak M, et al. Epigenetic induction of adaptive immune response in multiple myeloma: sequential azacitidine and lenalidomide generate cancer testis antigen-specific cellular immunity. Br J Haematol (2012) 158:700–11. doi:10.1111/j.1365-2141.2012.09225.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi:10.1097/CJI.0b013e3182829903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Kortenhorst MS, Wissing MD, Rodriguez R, Kachhap SK, Jans JJ, Van Der Groep P, et al. Analysis of the genomic response of human prostate cancer cells to histone deacetylase inhibitors. Epigenetics (2013) 8:907–20. doi:10.4161/epi.25574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Watson NF, Ramage JM, Madjd Z, Spendlove I, Ellis IO, Scholefield JH, et al. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int J Cancer (2006) 118:6–10. doi:10.1002/ijc.21303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Cathro HP, Smolkin ME, Theodorescu D, Jo VY, Ferrone S, Frierson HF Jr. Relationship between HLA class I antigen processing machinery component expression and the clinicopathologic characteristics of bladder carcinomas. Cancer Immunol Immunother (2010) 59:465–72. doi:10.1007/s00262-009-0765-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Seliger B, Stoehr R, Handke D, Mueller A, Ferrone S, Wullich B, et al. Association of HLA class I antigen abnormalities with disease progression and early recurrence in prostate cancer. Cancer Immunol Immunother (2010) 59:529–40. doi:10.1007/s00262-009-0769-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Ayshamgul H, Ma H, Ilyar S, Zhang LW, Abulizi A. Association of defective HLA-I expression with antigen processing machinery and their association with clinicopathological characteristics in Kazak patients with esophageal cancer. Chin Med J (Engl) (2011) 124:341–6.
150. Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst (2013) 105:1172–87. doi:10.1093/jnci/djt184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Procko E, Gaudet R. Antigen processing and presentation: TAPping into ABC transporters. Curr Opin Immunol (2009) 21:84–91. doi:10.1016/j.coi.2009.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, et al. Combined genetic inactivation of beta2-microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell (2011) 20:728–40. doi:10.1016/j.ccr.2011.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget (2013) 4:2067–79.
154. Setiadi AF, David MD, Seipp RP, Hartikainen JA, Gopaul R, Jefferies WA. Epigenetic control of the immune escape mechanisms in malignant carcinomas. Mol Cell Biol (2007) 27:7886–94. doi:10.1128/MCB.01547-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Simova J, Pollakova V, Indrova M, Mikyskova R, Bieblova J, Stepanek I, et al. Immunotherapy augments the effect of 5-azacytidine on HPV16-associated tumours with different MHC class I-expression status. Br J Cancer (2011) 105:1533–41. doi:10.1038/bjc.2011.428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Trumpfheller C, Longhi MP, Caskey M, Idoyaga J, Bozzacco L, Keler T, et al. Dendritic cell-targeted protein vaccines: a novel approach to induce T-cell immunity. J Intern Med (2012) 271:183–92. doi:10.1111/j.1365-2796.2011.02496.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Dilek N, Poirier N, Hulin P, Coulon F, Mary C, Ville S, et al. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS One (2013) 8:e83139. doi:10.1371/journal.pone.0083139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, Xu YH, et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One (2013) 8:e62924. doi:10.1371/journal.pone.0062924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A (2014) 111:11774–9. doi:10.1073/pnas.1410626111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Yang H, Bueso-Ramos C, Dinardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia (2014) 28:1280–8. doi:10.1038/leu.2013.355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, Mcdermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res (2013) 19:462–8. doi:10.1158/1078-0432.CCR-12-2625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res (2014) 20:5064–74. doi:10.1158/1078-0432.CCR-13-3271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, et al. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res (2013) 73:7265–76. doi:10.1158/0008-5472.CAN-13-0890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology (2014) 3:e27414. doi:10.4161/onci.27414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Kopp LM, Ray A, Denman CJ, Senyukov VS, Somanchi SS, Zhu S, et al. Decitabine has a biphasic effect on natural killer cell viability, phenotype, and function under proliferative conditions. Mol Immunol (2013) 54:296–301. doi:10.1016/j.molimm.2012.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Xiao W, Dong W, Zhang C, Saren G, Geng P, Zhao H, et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur J Med Res (2013) 18:61. doi:10.1186/2047-783X-18-61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Lisiero DN, Soto H, Everson RG, Liau LM, Prins RM. The histone deacetylase inhibitor, LBH589, promotes the systemic cytokine and effector responses of adoptively transferred CD8+ T cells. J Immunother Cancer (2014) 2:8. doi:10.1186/2051-1426-2-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J Immunol (2008) 180:8102–8. doi:10.4049/jimmunol.180.12.8102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol (2009) 183:1695–704. doi:10.4049/jimmunol.0900592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Lal G, Zhang N, Van Der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol (2009) 182:259–73. doi:10.4049/jimmunol.182.1.259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res (2009) 69:8693–9. doi:10.1158/0008-5472.CAN-09-1456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res (2007) 13:6947–58. doi:10.1158/1078-0432.CCR-07-0842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Yokokawa J, Cereda V, Remondo C, Gulley JL, Arlen PM, Schlom J, et al. Enhanced functionality of CD4+CD25(high)FoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clin Cancer Res (2008) 14:1032–40. doi:10.1158/1078-0432.CCR-07-2056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Sonpavde G, Aparicio AM, Zhan F, North B, Delaune R, Garbo LE, et al. Azacitidine favorably modulates PSA kinetics correlating with plasma DNA LINE-1 hypomethylation in men with chemonaïve castration-resistant prostate cancer. Urol Oncol (2011) 29(6):682–9. doi:10.1016/j.urolonc.2009.09.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Thibault A, Figg WD, Bergan RC, Lush RM, Myers CE, Tompkins A, et al. A phase II study of 5-aza-2′-deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori (1998) 84:87–9.
177. Deborah B, Dana R, Rodney D, Walter MS, Glenn L, David CS, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862). Cancer (2009) 115:5541–9. doi:10.1002/cncr.24597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Edelstein LC, Micheva-Viteva S, Phelan BD, Dougherty JP. Short communication: activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res Hum Retroviruses (2009) 25:883–7. doi:10.1089/aid.2008.0294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Ritchie D, Piekarz RL, Blombery P, Karai LJ, Pittaluga S, Jaffe ES, et al. Reactivation of DNA viruses in association with histone deacetylase inhibitor therapy a case series report. Haematologica (2009) 94:1618–22. doi:10.3324/haematol.2009.008607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: epigenetics, tumor immunotherapy, histone acetylation, antigen presentation, methylation
Citation: Héninger E, Krueger TEG and Lang JM (2015) Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol. 6:29. doi: 10.3389/fimmu.2015.00029
Received: 02 December 2014; Accepted: 14 January 2015;
Published online: 04 February 2015.
Edited by:
William L. Redmond, Earle A. Chiles Research Institute, USACopyright: © 2015 Héninger, Krueger and Lang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua M. Lang, Wisconsin Institutes for Medical Research, Room 7151, 1111 Highland Avenue, Madison, WI 53705, USA e-mail:am1sYW5nQG1lZGljaW5lLndpc2MuZWR1