 Mariana Aris
Mariana Aris María Marcela Barrio
María Marcela Barrio- Centro de Investigaciones Oncológicas-Fundación Cáncer, Buenos Aires, Argentina
Cutaneous melanoma arises from the malignant transformation of skin melanocytes; its incidence and mortality have been increasing steadily over the last 50 years, now representing 3% of total tumors. Once melanoma metastasizes, prognosis is somber and therapeutic options are limited. However, the discovery of prevalent BRAF mutations in at least 50% of melanoma tumors led to development of BRAF-inhibitors, and other drugs targeting the MAPK pathway including MEK-inhibitors, are changing this reality. These recently approved treatments for metastatic melanoma have made a significant impact on patient survival; though the results are shadowed by the appearance of drug-resistance. Combination therapies provide a rational strategy to potentiate efficacy and potentially overcome resistance. Undoubtedly, the last decade has also born a renaissance of immunotherapy, and encouraging advances in metastatic melanoma treatment are illuminating the road. Immune checkpoint blockades, such as CTLA-4 antagonist-antibodies, and multiple cancer vaccines are now invaluable arms of anti-tumor therapy. Recent work has brought to light the delicate relationship between tumor biology and the immune system. Host immunity contributes to the anti-tumor activity of oncogene-targeted inhibitors within a complex network of cytokines and chemokines. Therefore, combining immunotherapy with oncogene-targeted drugs may be the key to melanoma control. Here, we review ongoing clinical studies of combination therapies using both oncogene inhibitors and immunotherapeutic strategies in melanoma patients. We will revisit the preclinical evidence that tested sequential and concurrent schemes in suitable animal models and formed the basis for the current trials. Finally, we will discuss potential future directions of the field.
Introduction
The American Cancer Society projected 76,100 new cases of melanoma in the United States in 2014 (1). These cases represent <2% of total skin cancer diagnoses, but account for an overwhelming proportion of skin cancer deaths. Melanoma incidence has been increasing for at least 30 years, and between 2006 and 2010 the incidence rate among Caucasians increased by 2.7% per year. Though incidence is rising, mortality has been declining rapidly in Caucasians under 50: from 2006 to 2010, mortality rates decreased by 2.6% per year in men and by 2.0% per year in women. In contrast, among Caucasians 50 and older, mortality increased by 1.1% per year in men and by 0.2% per year in women during this same time period. Even with this decline in mortality, the American Cancer Society predicted 9,710 deaths from melanoma in 2014 (1).
Standard cutaneous melanoma (CM) treatment consists of surgical removal of the primary tumor and surrounding normal tissue; a sentinel lymph node is routinely biopsied to determine stage (2). More extensive surgery may be needed if the lymph nodes are compromised. Melanomas with deep local invasion, or that have spread to the lymph nodes, may be treated with surgery, immunotherapy, chemotherapy, and/or radiation therapy. Advanced cases may be treated with palliative surgery, chemotherapy and/or radiation therapy, and newer targeted or immunotherapy drugs.
Melanoma is highly curable if detected in its earliest stages and treated properly (1, 2). However, melanoma is likely to spread to other parts of the body. The 5- and 10-year relative survival rates for melanoma patients are 91 and 89%, respectively. For localized melanoma (84% of cases), the 5-year survival rate is 98%; survival declines to 62 and 16% for regional and distant stage disease, respectively. Patients with tumors that have invaded the deep dermis or spread to the draining lymph nodes have a high risk of recurrence after surgery, and safe and effective therapeutic options are still limited as adjuvant treatments. The approval of oncogene-targeted drugs such as vemurafenib and dabrafenib for BRAFV600 mutated melanoma (3, 4), and of monoclonal antibodies (mAbs) targeting immunomodulatory molecules such as ipilimumab for CTLA-4 (5) and pembrolizumab for PD-1 (6), has dramatically changed the treatment of advanced melanoma in recent years. All of these agents have demonstrated a positive impact on overall survival (OS) and impressive clinical responses in advanced melanoma patients. However, related toxicity and emerging resistance are ongoing challenges. There is increasing evidence that a combination therapy using oncogene-targeted drugs and immunotherapy could produce long-lasting responses in a broader spectrum of patients.
In the present work, we will revisit the latest, promising oncogene-targeted drugs and several immunotherapeutic approaches that are under intense research, as well as their clinical effects on melanoma patients. We will discuss the preclinical and clinical evidence supporting the rationale for the combination of oncogene-targeted therapies and immunotherapy to improve patient outcome.
Advances in Melanoma Treatment
Oncogene-Targeted Drugs
Cutaneous melanoma has the highest mutational frequency of any tumor pathology. Tumor transformation and progression is believed to be sustained by the emergent combination of genetic changes (7). Whole-genome sequencing studies reveal that the point mutation load is proportional to UV exposure rays (C > T/G > A), with the highest rates in patients with a documented history of chronic sun exposure; however, there are also non-related sun exposure mutations (8). A recent report describing a panel of cancer mutations in clinical melanoma samples of different subtypes, including cutaneous, acral, mucosal, and unknown primary melanomas, revealed that the number of mutations per tumor was associated with melanoma subtype (9). Here, we will describe the most prevalent mutations in melanoma that resulted in development of targeted therapies, some with impressive clinical results.
The canonical MAPK signaling pathway – receptor tyrosine kinase (RTK)–RAS–RAF–MEK–ERK – promotes survival, growth, migration, and resistance to apoptosis. The MAPK pathway initiates in the cell membrane, either by ligand interaction with a RTK, or by the adhesion of integrins to the extracellular matrix. Mutations in the serine/threonine–protein kinase BRAF were identified a decade ago as the most frequent in CM, with a prevalence of ~40–60% clinical cases (10). Updated data reveal that the T1799A mutation, resulting in the BRAFV600E constitutively active isoform, accounts for ~75% of BRAFV600 gene mutations (11). Other less frequent variants include BRAFV600K and BRAFV600R, which are present in ~20 and ~4% of BRAFV600 cases. In melanoma, NRAS mutations are rare in BRAFV600 tumors (1.6%). Among 20% of melanoma tumors are mutated in NRAS, especially in exon 2 (NRASQ61R/Q61K/Q61L). Additionally, 16% of melanoma tumors present TP53 mutations, and concurrent TP53 mutations are the most frequent events in tumors with BRAFV600 or NRAS mutations. BRAFV600 and TP53 mutations are significantly associated with cutaneous primary tumor location, while NRAS and c-KIT alterations were associated with acral and mucosal melanoma (9). Other sequencing studies indicate that 8% of MEK1 and MEK2 mutations may occur simultaneously with BRAF (60%) or NRAS (10%) mutations (12). The most frequent MEK mutants, MEK1P124S and MEK1E203K, are related to ERK phosphorylation. All of these driver mutations lead to the constitutive activation of the MAPK pathway; thus, they arose as appropriate druggable targets with specific inhibitors.
Vemurafenib (PLX-4032, Zelboraf, Roche) is a small, orally bioavailable molecule that selectively binds the ATP-binding site of BRAFV600E kinase and inhibits its activity (13). Vemurafenib efficacy was assessed in a randomized clinical trial against Dacarbazine in metastatic CM patients carrying the BRAFV600E mutation. Vemurafenib produced a higher response rate (48 vs. 5%), and an increase in OS (84 vs. 64%) and disease-free survival (DFS) (5.3 vs. 1.6 months). Impressive rapid tumor remissions were observed, with a median time-to-response of 1.45 months (3). Skin complications were frequently associated with treatment: 24% of patients from the vemurafenib arm developed low-grade cutaneous squamous-cell carcinomas or keratoacanthomas, through paradoxical ERK activation. These tumors required excision and continuous dermatologic evaluation during treatment. Fortunately, patients with BRAFV600 mutations other than BRAFV600E will respond to vemurafenib, including BRAFV600K and BRAFV600R (14, 15). Vemurafenib became one of the cornerstones of metastatic or unresectable CM treatment with its approval in 2011 by the Federal Drug Administration (FDA) and in 2012 by the European Medicines Agency (EMEA). Despite the impressive initial tumor remissions observed with vemurafenib, drug-resistance has limited the duration of remissions; therefore, great efforts are being directed toward revealing and overcoming the mechanisms of resistance to BRAF inhibition (16).
Dabrafenib (Tafinlar, GlaxoSmithKline) is another orally bioavailable BRAFV600E small-molecule inhibitor, which was approved by the FDA and EMEA in 2013 for treatment of unresectable or metastatic CM with BRAFV600E mutation. In a randomized trial of advanced CM patients with BRAFV600E determined tumors, dabrafenib significantly improved progression-free survival (PFS) compared to Dacarbazine (5.1 vs. 2.7 months) (4). Although 6% of patients from the dabrafenib arm developed keratoacanthoma or squamous-cell skin carcinoma, they did not require dose modification or interruption. Trametinib (Mekinist, GlaxoSmithKline) is an oral MEK1 and MEK2 inhibitor targeting the MAPK pathway downstream. This inhibitor provided longer PFS than Dacarbazine and Paclitaxel (4.8 vs. 1.5 months) for CM patients with unresectable metastatic BRAFV600E tumors, and was therefore approved for CM treatment by the FDA in 2013, and by the EMEA in 2014 (17). The combination of dabrafenib and trametinib proved superior to monotherapy and produced fewer side effects, which led to FDA approval in January 2014.
Upstream in the MAPK pathway, there are altered RTKs in melanoma, including c-KIT, EGFR, and PDGFR. c-KIT is involved in the development and maintenance of melanocytes, activating the MAPK, PI3K–AKT, and Janus kinases (JAK)–signal transducers and activators of transcription (STAT) proliferation and survival pathways. Amplifications or mutations in c-KIT account for 4% of melanomas, and are most frequently found in acral, mucosal, and chronically sun-damaged skin (18). Although less prevalent in Caucasian populations, these subtypes constitute approximately 65% of the melanomas observed in Asians and African American populations. A large mutational analysis of RTKs performed in metastatic CM samples revealed that growth factor receptor ERBB4 was mutated in 19% of the samples. This receptor is involved in AKT signaling, and can be down-regulated by either ERBB4 knockdown or inhibition with Lapatinib (19).
Imatinib (Gleevec, Novartis) is an orally available, chemical ATP-competitive RTK inhibitor, which prevents phosphorylation and the subsequent activation of growth receptors and their downstream signal transduction pathways. Its efficacy was initially demonstrated in the BCR-ABL oncogene in hematological malignancies, and in RTKs such as c-KIT and PDGFR, which are frequently altered in gastrointestinal tumors. A phase II study in metastatic melanoma patients with mutated or amplified c-KIT showed a 23% overall response rate (ORR) with imatinib therapy (20). Dasatinib (Sprycel, Bristol-Myers Squibb) is another synthetic broad-spectrum multi-kinase inhibitor, with potent effect on BCR-ABL, SRC, c-KIT, PDGFR, and ephrin TK. Dasatinib has demonstrated only modest clinical efficacy in melanoma patients as a single agent (21). Tumor regression was noted in approximately 14% of total patients without c-kit mutations, and in one of two patients with tumor c-kit mutations. This level of activity suggests that biomarker-based patient pre-selection, in this case by c-KIT alteration, may identify a subset of patients that can potentially derive benefit from dasatinib.
Monoclonal Antibodies
Tumor evolution is the result of continuous feedback between tumor cells and their environment. The immune system is a crucial player, since it can both repress and enable tumor growth and is capable of shifting from an anti-tumor immune environment to a tumor-permissive one, as described by the theory of cancer immunoediting (22). There is significant evidence of molecules and immune populations involved in tumor immunoediting in CM (23). In particular, immune checkpoints refer to the circuit of inhibitory pathways that the immune system uses to modulate the duration and amplitude of immune responses. Many of these signals are initiated by ligand–receptor interactions and many tumors express these ligands as a mechanism of immune escape, particularly against tumor specific T cells. However, these ligand–receptor interactions can be blocked by specific antibodies, which then halt the immune checkpoint blockade and favor an effector anti-tumor immune response. The binding of co-stimulatory B7 molecules in Antigen-Presenting Cells (APC) to the CD28 receptor on naïve T cells induces the expression of CTLA-4 (cytotoxic T-lymphocyte-associated antigen-4). The CTLA-4 molecule has a higher affinity for B7 than CD28 and has an inhibitory function, thereby helping to extinguish the signal. Another inhibitory molecule is PD-1 (Programed death-1), which is a co-receptor expressed in activated and exhausted T and B cells. Activated PD-1 negatively regulates T cell activation through the suppression of the PI3K/Akt pathway. PD-1 interacts with two ligands: B7-H1 (PD-L1), the main mediator of the immunosuppressive response, and B7-H2 (PD-L2). In tumor pathology, PD-L1 expression is increased on tumor cells and APCs, while the expression of CTLA-4 is increased on APCs and effector T cells (24, 25).
Ipilimumab (Yervoy, Bristol-Myers Squibb) is a monoclonal antibody (IgG1) directed against CTLA-4 that was developed for systemic anti-tumor immunotherapy. The effects of ipilimumab in unresectable melanoma patients were assessed in a randomized trial against a gp100-peptide vaccine. Patients receiving ipilimumab plus gp100-peptide vaccine showed improved OS in comparison to those receiving the vaccine alone (10.1 vs. 6.4 months), with a median time-to-response of 3.3 months. ORR after ipilimumab administration ranged from 10 to 20%. Furthermore, complete regression continued throughout the 2 years of follow-up. No differences in OS were observed between ipilimumab treatment alone and ipilimumab plus the gp100-peptide vaccine (5). Because ipilimumab stimulates T cells, there is substantial risk of immune-related adverse events (irAEs). In this trial, 10–15% of patients presented grade 3/4 irAEs, with seven related deaths. Management of irAEs included initiation of high-dose corticosteroids and discontinuation of ipilimumab. In 2011, the FDA and EMEA approved the use of ipilimumab for the treatment of unresectable or metastatic melanoma, contributing to the renaissance of immunotherapy in cancer treatment. Given the potential for toxicity, ipilimumab approval was predicated on a risk evaluation and mitigation strategy. Tremelimumab (CP-675206, Pfizer/MedImmune) is another recently developed CTLA-4-blocking monoclonal antibody (IgG2). Although tremelimumab generated durable responses in metastatic melanoma patients in phase I and II trials, it failed to demonstrate a survival advantage in a randomized trial against standard-of-care chemotherapy (26).
Pembrolizumab (formerly Lambrolizumab, Keytruda, Merck), an IgG4 monoclonal antibody against PD-1, was recently approved in September 2014 by the FDA for treatment of patients with unresectable or metastatic melanoma. Approval was based on the results obtained in a randomized trial comparing two doses of pembrolizumab in metastatic melanoma patients whose cancer had progressed following treatment with ipilimumab or targeted therapy in BRAFV600 tumors (NCT01295827) (6). Key exclusionary criteria included any autoimmune disease, induced immunosuppression, and/or a history of severe irAEs from treatment with ipilimumab. The ORR was 26% at both doses, with a median time-to-response of 12 months. Treatment was well tolerated; the only drug-related grade 3 adverse event was fatigue, which was reported by 3% of patients in the 2 mg/kg pembrolizumab group. Currently, there are two ongoing, randomized, confirmatory trials for pembrolizumab in advanced melanoma patients (NCT01866319, NCT02083484). Nivolumab (Bristol-Myers Squibb) is another antagonist monoclonal antibody (IgG4) specific to PD-1. A phase I trial in several solid tumors showed a cumulative response in 28% of melanoma patients (27). Interestingly, only patients with PD-L1-positive tumors achieved an objective response, suggesting a relationship between PD-L1 expression on tumor cells and clinical response. Only one patient experienced a serious AE, inflammatory colitis. Controlled phase III trials of nivolumab vs. standard-of-care chemotherapy with prospective survival end points are currently underway (NCT01721772, NCT01721746) in melanoma patients. In a phase I trial in metastatic melanoma patients, co-administration of nivolumab and ipilimumab, followed by nivolumab, resulted in rapid ORR in 40% of patients, with an acceptable level of AEs (28). Currently, a phase III study comparing nivolumab or nivolumab plus ipilimumab vs. ipilimumab alone in advanced melanoma patients is underway (NCT01844505). Furthermore, specific biomarkers are being studied, including baseline variations in activated and memory T cells, interferon inducible factors, and tumor T cell infiltration (CD4+ and CD8+ cells) (NCT01621490).
Another strategy for interference with the PD-1 ↔ PD-L1 axis is BMS-936559 (Bristol-Myers Squibb), a monoclonal antibody (IgG4) targeting PD-L1. In the initial trial, BMS-936559 induced tumor regression (ORR of 6–17%) and disease stabilization (rates of 12–41% at 24 weeks) in patients with advanced cancers, including non-small-cell lung cancer, melanoma, and renal cancer (29). Serious AEs were detected in 9% patients. Other antagonist mAbs against PD-L1 are currently being assessed in clinical trials for metastatic treatment, such as MEDI4736 (Astrazeneca, NCT01693562) and RG7446 (Roche, NCT01375842).
Therapeutic Vaccines
Cutaneous melanoma is an immunogenic tumor; several tumor antigens (Ags) have been identified and evidence of tumor immunoediting has been observed in patients (23). Immunotherapy has emerged as an option of interest for CM treatment. Immunotherapy seeks to stimulate, restore, manage, and even complement the patient’s own immune system to control tumor growth and dissemination. Among other immunotherapeutic approaches, vaccines could be administered as an adjuvant therapy after tumor excision, with the purpose of eliciting long-lasting immunity and controlling micro-metastatic foci. The rational basis for such vaccines is that tumor Ags must be captured by APCs, which migrate to the lymph nodes where they further activate CD4+ and CD8+ cells. This triggering of the adaptive immune response would also result in the development of immunological memory. The main objectives of active immunotherapy against melanoma are to overcome the immunosuppression produced by the tumor and its microenvironment, to stimulate specific immune effectors that can destroy tumor cells, and to increase immunogenicity to tumor Ag. Pre-existing anti-tumor T cells may be ineffective at rejecting the tumor either because their frequency is too low, because tumor cells were selected to escape recognition, or because such lymphocytes are functionally deficient. Vaccination can induce cytokine cascades both locally and systemically, resulting in the activation and proliferation of anti-melanoma Ag precursors, and infiltration of effector immune populations into tumors. In general, vaccines require two critical components, the Ag source and the adjuvant (30). Therapeutic vaccines include the use of different Ag sources, such as peptides, proteins, nucleic acids, tumor lysates, recombinant virus, or whole irradiated cells. Alternatively, dendritic cell (DC) vaccines are comprised of patients’ DC in vitro loaded with an Ag source, ex vivo matured and then given back to the patient.
Peptide vaccines are directed at one or several representative CM Ag. To induce T cell responses, it is necessary to immunize with adjuvants or Toll-like receptor (TLR) ligands. Numerous clinical trials were conducted with this strategy, and although some encouraging results were observed, peptide vaccines promote the selection of tumor Ag-negative resistant populations. Peptide Ags emulsified in incomplete Freund’s adjuvant (IFA) are widely used to vaccinate cancer patients. While several clinical trials testing peptide/IFA-based vaccines have documented an increase in circulating Ag-specific T cells, objective therapeutic benefits are rare (31, 32). This may be due to the presence of large peptide deposits that are protected from degradation and can prime robust specific CD8+ T cell responses, which are detectable in circulation but not within tumor lesions. Thus, Ag persistence at the vaccination site can create a T cell graveyard in which specific T cells accumulate and compete with the tumor site (33). It is clear that other adjuvants need to be evaluated to improve peptide delivery and immune cell stimulation.
Therapeutic vaccines comprised of whole cells or cell lysates allow the immune system to interact with a broad antigenic repertoire. This is an important consideration, since tumors are heterogeneous and CM is not an exception (34). A meta-analysis combining results from 173 immunotherapy clinical trials of several cancers found that patients immunized with whole-tumor Ag, including irradiated tumor cells, modified tumor cells, tumor extracts, tumor mRNAs, and DC pulsed with tumor extracts, showed an 8.1% clinical response compared with 3.6% in patients immunized with synthetic or recombinant peptides and proteins (35). Whole-cell vaccines are still in clinical trials. The GVAX vaccine (36), consisting of irradiated genetically modified cell lines that secrete granulocyte–macrophage colony stimulating factor (GM-CSF, Molgramostim), is currently being assayed in stage IIB-IV melanoma patients in a phase I study (NCT01435499). We have tested a vaccine comprised of allogeneic irradiated CM cells (VACCIMEL) plus BCG as an adjuvant, combined with i.d. injection of GM-CSF to promote the local attraction of monocytes and their subsequent differentiation into DC. In the Phase I study, VACCIMEL shown to be safe, and with a maximum follow-up of 139 months, mean DFS and OS have not yet been reached for stage II/III vaccinated patients (DFS and OS 58.3%) (José Mordoh, personal communication 2014); all stage IV patients progressed (37). Currently, the CSF-470 vaccine, which adds a fourth irradiated melanoma cell line to the VACCIMEL formulation, is being tested with BCG and GM-CSF against medium dose IFN-alpha 2b in a Phase II–III clinical trial in stage IIB, IIC, and III CM patients (NCT01729663).
Dendritic cell-vaccines consist of autologous treatments, in which DC precursors are purified from patients and are loaded in vitro with tumor Ag (irradiated cells), matured ex vivo, and then re-infused into the patient (38). In 2010, the FDA approved Sipuleucel-T, the first DC-based vaccine for the treatment of metastatic castrate resistant, hormone refractory prostate cancer (39). Sipuleucel-T (Provenge) is made of an autologous DC-vaccine preparation loaded with Prostatic acid phosphatase Ag and GM-CSF. In the case of CM, a phase I–II trial was conducted for stage IV patients in which autologous DC were pulsed with cocktail of synthetic five melanoma-associated peptides (gp100, tyrosinase, MAGE-A2, MAGE-A3, and MART-1 or MAGE-A1) restricted to HLA-A2 or HLA-A24, and KLH as adjuvant (40). Clinical responses were observed and OS increased: 13.6 vs. 7.3 months (vaccinated vs. non-vaccinated), and 21.9 vs. 8.1 months (high vs. low ELISPOT). Additionally, the presence of the MAGE-A1 auto-antibody before vaccination was determined as a positive prognostic factor. Other clinical studies have used autologous DC loaded with peptides, tumor cell lysates (41), killed melanoma cells (42), or tumor Ag mRNA electroporation (43), but overall clinical responses remained disappointingly low. However, the induction of specific immune responses to tumor Ags has been described. Several years ago, we demonstrated that autologous DC can capture apoptotic-necrotic, gamma-irradiated, allogeneic melanoma cells, inducing the maturation and subsequent migration of DC in response to the lymph node homing CCL19 chemokine, both in a murine model (44) and with DC isolated from peripheral blood monocytes from healthy donors (45). We conducted a Phase I clinical trial for 16 CM patients. In that study, we observed gp100 and MART-1 specific CD8+T lymphocytes through ELISPOT and tetramer analysis in five HLA-A*0201 patients before and after vaccination (46). In an updated analysis with a maximum follow-up of 132 months, 88.9% stage II–III patients still show no evidence of melanoma recurrence (José Mordoh, personal communication 2014).
Dendritic cell-based vaccine design has been variable, it is difficult to assess the best inoculation site, the optimal number of DCs to elicit effective immune responses, the Ag-loading strategies and how to monitor immune effector recruitment in the tumor microenvironment. A recent review indicated that DC-based cancer vaccines can elicit adaptive and innate anti-tumor immunity in at least half of all patients; however, clinical responses remain disappointingly scarce (47). The observed dissociation between clinical objective response and the prolongation of OS in some patients indicates that alternative surrogate endpoints should be used to assess the therapeutic effectiveness of DC-based immunotherapy. DC-based vaccines can positively affect the clinical outcome in the form of increased patient survival rather than the induction of tumor objective responses. The clinical benefit of DC-based immunotherapy is real but small. With 8.5% of melanoma patients achieving an objective response, DC therapy has comparable efficacy to Dacarbazine, the standard-of-care chemotherapy, and to ipilimumab, to which 5–15% of patients have an objective response (47). Today, there are no surrogate markers indicative of relevant anti-tumor immunization to evaluate vaccine efficacy, meaning that DFS and OS are the parameters that define vaccine success. Furthermore, there are no predictive markers indicating anti-tumor immune competence that could identify the patients most likely to benefit (47).
Adoptive Cell Therapy
Adoptive Cell Therapy involves the administration of autologous ex vivo-expanded tumor-reactive T lymphocytes to properly preconditioned recipients. T lymphocytes can be expanded from tumor infiltrating lymphocytes (TIL) ex vivo from a patient’s own metastasis after IL-2 and anti-CD3 antibody stimulation. The expanded T cells are then re-administered after chemotherapy-induced lymphodepletion of the patient. Such melanoma TILs recognize their autologous tumor in >75% of patients. Although adoptive cell therapy (ACT) is typically administered only once, clinical responses can endure for years. The largest published description of ACT with significant follow-up showed an ORR of 56% in 93 patients who had followed a variety of lymphodepleting regimens, 19 patients (21%) had ongoing complete responses (CR) at 5–9 years of follow-up (48). Other investigators have published their experiences with ACT and reported a preliminary ORR of approximately 50%, including some CR (49–51).
Other Immunotherapies
The first immunotherapeutic drug approved for treatment of metastatic melanoma was recombinant Interleukin-2 or aldesleukin (Proleukin, Prometheus Laboratories), which promotes activation of T, B, and NK-cells. Studies indicate that high doses of IL-2 (HD IL-2) induced long-term responses in 16% of patients and a CR in 6% of cases (52). IL-2 provided the first “proof of principle” that a drug targeting the immune system can control melanoma at long-term. However, HD IL-2 is very toxic. Serious AEs can occur, such as capillary leak syndrome, breathing problems, serious infections, seizures, allergic reactions, and heart problems among other possible complications (53).
Later on, Interferon alpha 2b (IFN-α 2b) was approved by the FDA as a post-surgical adjuvant treatment. Previous studies have shown that adjuvant treatment with IFN-α2b at intermediate doses for 2 years increased DFS of melanoma patients with high risk of recurrence (stages IIb, IIC and III) in 10%, with no significant effect on OS (54). IFN-α 2b is associated with severe side effects. Based on the results of the EORTC 18991 trial, the FDA approved PEG-interferon α-2b (PEG-IFN) (Sylatron) as adjuvant therapy for high-risk melanoma. The median PFS of the PEG-IFN group was significantly longer than the observation group, while OS, a secondary endpoint, was not significantly different between the two groups. One-third of the patients receiving PEG-IFN discontinued treatment because of toxicity. PEG-IFN is characterized by a longer half-life and can be administered subcutaneously. Much progress has been made in unraveling the mechanisms of action of IFN’s anti-tumor activity. These include cell cycle inhibition by G1 arrest and anti-proliferative activity (55), the induction of apoptosis (56–58), and the reduction of angiogenesis in some tumors by reducing basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) production (59, 60). Several indirect immunomodulatory effects of interferon treatment have been identified, including a role in T cell differentiation and B cell development, which may aid in immune surveillance by increasing Ag processing and upregulating MHC-I expression, thus facilitating Ag recognition by cytotoxic CD8+ T cells. Besides, IFN-α produces strong activation of monocytes and macrophages. Activated macrophages produce reactive oxygen species and reactive nitrogen intermediates, which have cytotoxic effects on target cells (61). Additionally, activated monocytes produce cytokines that initiate a Th1 response. More recently, IFN-α has been shown to induce activation of NK-cells through the increase of NKG2D and CD161 stimulatory signaling, thus enhancing NK-cell killing of tumor cells (62, 63).
Lights and Shadows of Therapeutic Approaches for Melanoma Treatment
The phenomenon of oncogene-addicted tumors allowed the development of targeted therapies with impressive clinical results and, for the first time, rapid remissions in advanced metastatic melanoma patients. These results supported the approval of targeted therapies. However, targeted therapies are only useful for patients whose tumors possess specific mutations. There can be serious related AEs, and the emergence of escape mechanisms and drug-resistance may be difficult to overcome.
The unraveling of key immunomodulatory pathways involved in anti-tumor immune responses has allowed the development and approval of specific therapeutic approaches. For instance, immune checkpoint-specific antagonist mAbs allow the achievement of long-lasting clinical responses in a small proportion of advanced metastatic patients; these responses are often slow to manifest and take several months to achieve (5). Furthermore, there are concerning irAEs, primarily because these therapies are directed toward targets that are also involved in physiological processes. Additionally, specific biomarkers are needed to implement these treatments in the patient populations that would benefit the most.
Melanoma therapeutic vaccines have shown to be safe and to preserve the quality of life of patients with cancer (64, 65). Specific anti-tumor Ag responses have been demonstrated in about half of vaccinated patients. A small number of patients documented clinical OR (~8.5%) (66, 67). The most rational application of therapeutic vaccines seems to be as an adjuvant therapy aimed at the control of micro-metastatic dissemination. However, there is insufficient evidence supporting melanoma vaccine efficacy, or any correlation between immune responses and clinical objective responses. Most clinical studies have tested these vaccines in metastatic patients, perhaps foreshadowing possible control in earlier stage melanoma recurrence. Immune monitoring in the tumor microenvironment is difficult to assess and relevant immune effectors have not yet been identified.
Long-term responses to cytokine therapies such as IL-2 and IFN-α have been documented, though in very few patients, highlighting once again the intimate relationship between the immune system and melanoma (52, 54). However, the impact of cytokine therapy on patients’ OS has not been demonstrated and the associated AEs remain a major problem. Additional research is needed to discover appropriate markers that allow the identification of the patients that are most likely to respond to these therapies.
The ORR of ACT is approximately 50% (51). Some CR have been reported, suggesting that this strategy may be an attractive alternative for some melanoma patients. However, this procedure is expensive, only 30–40% of biopsy specimens yield satisfactory T cell population recovery for TIL preparation, and the process is laborious and time intensive (49). Furthermore, prior host conditioning with chemotherapy is required to increase the response to ACT. This conditioning is associated with serious AEs, including opportunistic infections and the frequent induction of vitiligo and uveitis, presumably due to autoimmunity (49).
The absence of an association between objective responses and OS has been widely reported with immunotherapeutic drugs and targeted therapies. Immunotherapies often produce an atypical clinical response pattern that includes delayed initial increases in tumor burden, which is associated with inflammation or immune cell infiltration of the tumor lesions, followed by regression and changes in disease progression kinetics (68). Altogether, these data underscore the idea that cancer immunotherapies need alternative efficacy endpoints in addition to the traditional outcome parameters used in oncology clinical trials.
Immunotherapy and Oncogene-Targeted Drugs: The Power of Combination Strategies
As we have seen, new melanoma treatments, including oncogene-targeted therapies and immunotherapies, have been recently approved. Due to their different modes of action and possible interactions, a special interest has arisen in using combination therapies to improve melanoma control. Targeted therapies can achieve impressive and rapid tumor remissions, though these results are shadowed by the emergence of resistance mechanisms through selection in heterogeneous tumors, limiting clinical response to a relatively short duration. In contrast, immunotherapy can give rise to long-term melanoma control by eliciting active immune effectors that may lead to curative responses, but in a small, select group of patients.
Why combine oncogene-targeted therapies and immunotherapy? There is increasing evidence that oncogenes play a role in the modulation of the expression of immune regulatory genes and therefore interfere with the immune microenvironment. We will focus on BRAFV600 in melanoma, where there is the most evidence. Initial reports showed that BRAFV600E CM cell lines secreted immunosuppressive IL-10, VEGF, and IL-6 molecules and suppressed IL-12 and TNF-α production by DC in vitro, and that this could be reversed by MEK-inhibitors or specific siRNA for BRAFV600E or STAT-3 (69). Human BRAFV600E CM cell lines also induced the expression of IL-1α and IL-1β, which in turn induced COX-2 and PD ligand expression in tumor-associated fibroblasts, suppressing T cell function. Interestingly, IL-1α expression was down-regulated in human tumor biopsies following vemurafenib treatment (70).
Studies investigating the underlying mechanisms of action describe an increase in CD4+, CD8+/Treg, and NK-cell numbers following PLX-4720 activity in BRAFV600E-driven murine melanoma models, showing that host immunity greatly contributes to inhibitor activity. Furthermore, the combination of PLX-4720 and anti-CCL2 or agonistic anti-CD137 antibodies improved anti-tumor responses (71). Regarding innate immunity, NK-cells will play a critical role in the control of BRAFV600E metastatic melanoma tumors treated with PLX-4720 through a perforin-dependent pathway. In the context of IL-2, PLX-4720-induced ERK1/2 phosphorylation, CD69 expression and IFN-γ release post NKp30 ligation in human NK-cells. A corollary from this work was that simultaneous inhibition by BRAF and MEK might preclude the activation of BRAFWT NK-cells. A low dose of IL-2 improved the anti-metastatic efficacy of PLX-4720, supporting the combination of NK-cell stimulatory agents with BRAF-inhibitors (72). With regards to adaptive immunity, BRAFV600E inhibition reportedly increased the expression of melanocyte differentiation Ags on CM cells and enhanced the activation of Ag-specific T cells in vivo (73). Neither PLX-4720 nor PLX-4032 BRAFV600E specific inhibitors interfered with the viability or the functionality of T cells, allowing the implementation of a combinatorial approach with immunotherapy (74). Other works reported that BRAF-inhibitors induced a paradoxical MAPK-dependent functional activation of T cells (75). This effect was dose-dependent on BMS-908662, a pan-RAF inhibitor (with activity against B-RAFV600, B-RAFwt, A-RAF, and C-RAF), and dose-independent on PLX-4720 inhibitor. The authors proposed that the paradoxical ERK activation in BRAFwt cells may be a common event with ATP-competitive inhibitors of the BRAF kinase domain and showed that MEK inhibition blocks this paradoxical activation. Interestingly, the combination of BMS-908662 with a CTLA-4 blockade improved anti-tumor action. In patients, reports indicated that BRAF-inhibitors induced tumor Ag expression and the infiltration of CD8+ and CD4+ T cells in metastases shortly after the initiation of treatment, which was followed by a reduction in tumor size. Progressing patients, however, showed a decrease in TILs. These results further support combinatorial strategies (76). Other analysis of tumor biopsies pre- and post-treatment with combinatorial dabrafenib/trametinib treatment showed a reduction of the immunosuppressive cytokines IL-6 and IL-8. However, CD8+ tumor infiltration, along with PD-1, Tim-3, and PD-L1 expression also increased following treatment, suggesting that these oncogene-targeted therapies might be limited and should be combined with immunotherapy to achieve long-term responses (77).
There remains much to learn about how the exposure of different classes of immune cells to vemurafenib modulates immune system activity. Interestingly, two ongoing clinical studies are currently analyzing the kinetics and effects of BRAF inhibition with vemurafenib (960 mg BID) on the innate and adaptive immune system in patients with unresectable melanoma expressing a BRAFV600 mutation (NCT01942993 and NCT01813214). These studies will evaluate changes in the immune cellular signature in blood circulation, comparing the baseline to different time points after the initiation of vemurafenib treatment through immunofluorescence and flow cytometry on blood samples. Additionally, the timeline of the vemurafenib-induced increase in T cell infiltration of tumors will be established through analysis of biopsies. As will other immune-related parameters, such as the activation state of TILs, expression of immune-inhibitory proteins (B7-H1/PD-L1, IDO, Arginase), changes in endothelial homing receptor ligands and tumor-associated chemokines, the presence of immune/inflammatory expression patterns, the presence of tissue-specific destruction and IFN-gamma upregulation, and in vitro determination of tumor cell lysis in comparison to allogeneic tumor cells. These studies will provide a systematic and valuable body of data that will aid in understanding the immune modulation induced by anti-BRAF therapy. And will help to develop more rationalized combination strategies with BRAF-targeted therapies and immunotherapy.
Currents Avenues of Research
In Table 1, we present a selected list of ongoing clinical studies that are exploring different combination therapies involving oncogene-targeted therapy with different immunotherapeutic approaches (current as of November 2014, source Clinical trials.gov and EMEA data base).
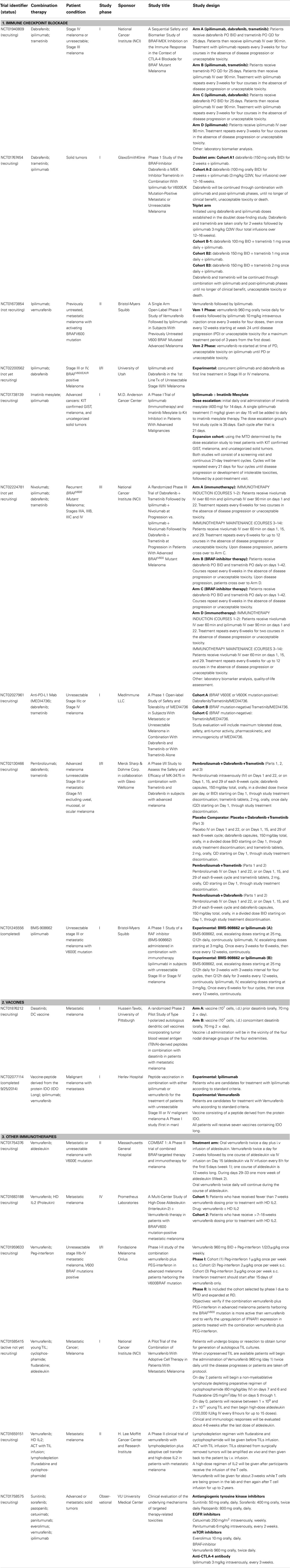
Table 1. Ongoing clinical studies combining immunotherapy and oncogene-targeted therapy.
Immune Checkpoint Blockade and Oncogene-Targeted Therapies
Expectations are high regarding immune checkpoint blockade and oncogene-targeted combination therapy, largely because of their individual success in the treatment of advanced melanoma per se, but also due to previous studies that have described oncogene modulation of the immune microenvironment. Several initial phase I/II trials are being conducted to evaluate the safety, quality of life, and immune functional status changes, in order to define an administration schedule and related biomarkers for further clinical development. These trials usually exclude patients with active autoimmune disease or known immune impairment; for example, patients receiving systemic immunosuppression for organ transplantation.
Surprisingly, a study with ipilimumab and vemurafenib in metastatic BRAFV600 mutated CM patients had to be terminated due to hepatic toxicities (NCT01400451) (78). Patients received a running period of oral vemurafenib, and then concomitant vemurafenib with intravenous ipilimumab. In both cohorts, most patients developed grade 2/3 hepatic AEs, including elevation in aminotransferase and total bilirubin levels. The AEs were asymptomatic and reversible with either temporary drug discontinuation or administration of glucocorticoids. A similar study terminated because of serious AEs was the combination of vemurafenib, ipilimumab, and DNE3 (a PI3K–AKT inhibitor) in advanced melanoma patients (NCT02095652). In order to analyze the underlying mechanisms of targeted therapy-related toxicities, an observational study is being conducted in patients with advanced solid tumors treated with standard palliative targeted therapies (NCT01758575). Monotherapies include antiangiogenic RTK inhibitors, EGFR inhibitors, mTOR inhibitors, BRAF-inhibitors, and ipilimumab. The study will investigate toxicity both at the systemic and local tissue level.
In spite of these reported toxicities, protocols designed with other drug combinations are currently being conducted. A phase II study assessing a sequential design that combines vemurafenib followed by ipilimumab has completed patient recruitment and results are expected for 2015 (NCT01673854). A new phase I/II trial studying concurrent treatment with dabrafenib and ipilimumab is ongoing (NCT02200562). Another phase I trial is assessing the co-administration of the BMS-908662 pan-RAF inhibitor and ipilimumab (NCT01245556). A combination of ipilimumab, dabrafenib, and trametinib is being assessed in advanced BRAFV600 CM patients. One such study is a four-arm, randomized trial with a sequential design that initiated treatment with dabrafenib and/or trametinib followed by ipilimumab (NCT01940809). Yet another open trial is testing a concomitant schedule; a doublet arm in which patients receive a running cycle of dabrafenib followed by co-administration with ipilimumab, and a triplet arm where patients receive dabrafenib, trametinib, and ipilimumab (NCT01767454).
An interesting option for c-KIT mutated melanoma and GIST patients is a dose escalation schedule with cycles of imatinib followed by ipilimumab, currently underway (NCT01738139). Regarding the PD-1–PD-L1 axis, pembrolizumab plus dabrafenib and Trametinib concomitant administration is being assessed in advanced melanoma patients. This study replaces pembolizumab with a placebo and conducts dabrafenib and trametinib monotherapy arms (NCT02130466). When nivolumab and ipilimumab are co-administered there are discernable induction and maintenance phases. Furthermore, MEDI4736 (PD-L1) in combination with oncogene-targeted therapies is being studied in advanced patients (NCT02027961). Patients with BRAFV600E/K tumors will receive dabrafenib, trametinib, and MEDI4736 combination therapy. Patients with BRAF mutation-negative tumors will only receive trametinib plus MEDI4736. A randomized trial in BRAFV600 stage III–IV melanoma patients is being conducted with multi-therapy combination (NCT02224781). The trial compares a group treated with dabrafenib plus trametinib followed by ipilimumab plus nivolumab in the case of progression, to a group treated with ipilimumab plus nivolumab followed by dabrafenib plus trametinib at progression. The primary outcome focuses on OS; the secondary outcome on PFS and toxicities. Additional outcomes include genetic characteristics and symptom burden.
Vaccines and Oncogene-Targeted Therapies
Besides inhibiting target kinases in cancer cells, dasatinib also inhibits a wide variety of kinases, such as src, tec, syk, and gck-families, which are essential for immune system function (79). Chronic myeloid leukemia patients have an increased proportion of granzyme B-expressing T cells at diagnosis, which is further increased by dasatinib therapy. Furthermore, dasatinib-treated CML patients have a higher proportion of effector CD4+ T cells that differentiate into Th1-type cytokine producers and are capable of producing IFN-γ, which is key for tumor control (80). Interestingly, although reports have indicated that dasatinib inhibits T cell activation via a blockade of lymphocyte-specific protein tyrosine kinase (Lck)-mediated proximal T cell receptor signaling in vitro (81), when administered in vivo dasatinib can profoundly enhance T effector cell activation, expansion, and function (82). In preclinical models, Yang et al. have recently reported that dasatinib treatment in BALB/c mice bearing P815 mastocytomas improved the recruitment of T cells into the tumor and surrounding stroma, an immunological response required to achieve the clinical benefits associated with therapeutic vaccines (83). Thus, a combinatorial protocol was tested in mice bearing established sub-cutaneous M05 (B16.OVA) melanoma. This protocol used specific vaccination with an OVA-peptide loaded DC-vaccine to activate and expand tumoricidal CD8+ T cells plus systemic administration of dasatinib to facilitate the refined targeting of vaccine-induced effector T cells to the tumor microenvironment. This approach provided a superior anti-tumor response to either single-agent modality alone. The increased efficacy of the combinatorial treatment included a reduction in the hypoxic-signaling associated with reduced levels of immunosuppressive CD11b+Gr1+ myeloid-derived suppressor cells and Treg populations in the melanoma microenvironment. Furthermore, dasatinib in combination with the DC-vaccine upregulated Type-1 T cell-recruiting CXCR3-ligand chemokines in the tumor stroma, which correlated with the activation and recruitment of Type-1, vaccine-induced CXCR3+CD8+ TILs and CD11c+ DC into the tumor. This combinatorial approach resulted in a profound “spreading” of the repertoire of melanoma-associated Ags recognized by CD8+ TILs (82). This evidence provided the rationale for a phase I clinical study of dasatinib in combination with a DC-cell based vaccine loaded with tumor blood vessel Ag (TBVA) in metastatic melanoma patients, which is currently ongoing (NCT01876212).
A newly discovered tumor cell escape mechanism is through the upregulation of the tryptophan-catabolizing enzyme known as IDO. Tryptophan is essential for the function of T cells; hence, the depletion of Trp leads to T cell anergy and apoptosis (84). IDO upregulation leads to cancer progression by suppressing T cell immunity, thereby elucidating IDO as a novel potential target for anticancer therapy. The IDO pathway is linked to Treg biology, since IDO-expressing DCs induce the differentiation of naïve CD4+ cells toward a FoxP3+ phenotype (84). Cancer patients possess spontaneous IDO-peptide specific T cells that are able to recognize and kill both IDO positive tumor cells and DCs (85). Targeting IDO is being explored in clinical trials for patients with metastatic solid tumors using IDO inhibitors such as DMT-1 and INCB024360 or IDO-peptide vaccines. The vaccine combinatorial approach may induce long-lasting IDO-specific memory T cells that could re-activate and be recruited to the tumor site when needed. Targeting IDO by a synthetic peptide vaccine [IDOlong; IDO (194–214) peptide sequence: DTLLKALLEIASCLEKALQVF] in combination with ipilimumab or vemurafenib was tested in a recently completed phase I clinical study for metastatic melanoma patients (NCT02077114). The study results have not yet been published, but will evaluate safety and tolerability as well as immune response. Reactivity to epitopes nested within the sequence of the peptide used for vaccination will be assessed in T cells from peripheral blood at different times. Reactivity will be assessed using ELISPOT (IFN-γ and TNF-α) and fluorochrome-conjugated HLA tetramers to enumerate the frequency of CD8+ T cell specific precursors for epitopes within the peptide sequence.
Other Immunotherapies and Oncogene-Targeted Therapies
IL-2 is an immunotherapy drug that increases the proliferation of the T lymphocytes and NK-cells responsible for targeting and eliminating cancer cells. Unfortunately, this elimination is accompanied by high toxicity, since IL-2 is used in high-dose regimen. As previously mentioned, targeting of mutated BRAFV600E with vemurafenib increases Ag expression on the surface of melanoma cells, which can make them easier targets for cytotoxic T lymphocytes and NK-cells. This suggests that combining BRAF-targeted therapy with IL-2 could contribute to increased tumor destruction. This hypothesis is being tested in a phase II clinical study (NCT01754376). The study design consists in primary treatment with oral vemurafenib BID for 2 weeks followed by one course of aldesleukin intravenous infusion (12 weeks long). Patients will also receive vemurafenib twice daily during the course of aldesleukin. Since this is a single arm study the efficacy of the vemurafenib/aldesleukin combination, measured by PFS, will be evaluated in comparison to an historic control of vemurafenib alone. The study will assess response rate, OS, safety, and toxicity. Preclinical data indicating that pharmacologic inhibition of mutated BRAF enhance the immunogenicity of melanoma without adversely affecting the cellular immune response will also be assessed. The study will explore biomarkers that may be relevant to predicting patient responsiveness to vemurafenib and aldesleukin, explaining primary or acquired resistance to vemurafenib, indicating the pharmacodynamic effects of vemurafenib and monitoring the disease. Another open-label, uncontrolled, two-arm, multi-center study has been designed to assess CR rate in BRAFV600 metastatic melanoma patients who have received vemurafenib plus HD IL-2 (NCT01683188). Patients will initially receive treatment with vemurafenib interspersed with two courses of HD IL-2. Eligible patients have been on vemurafenib therapy for 0–18 weeks, those patients previously taking vemurafenib must have responding or stable disease and all must meet the requirements for HD IL-2 dosing. Two cohorts will be enrolled, differing only in how they are characterized prior to HD IL-2 treatment: Cohort 1, patients naïve to vemurafenib and HD IL-2 therapy; Cohort 2, patients who have been on vemurafenib therapy for >7–18 weeks with stable or responding disease before starting HD IL-2. Patients in both cohorts will discontinue vemurafenib prior to each treatment with HD IL-2 and resume dosing after each discharge. Patients will receive up to two courses (four cycles) of HD IL-2.
IFN-α acts via engagement of a Type I IFN receptor consisting of two subunits (IFNAR1 and IFNAR2) followed by signaling through the JAK and the STAT family to activate the transcription of IFN-stimulated genes [reviewed in Ref. (58)]. A phase I/II trial has been launched to test this combination in advanced melanoma patients. This study is designed to evaluate the safety and the efficacy of the vemurafenib/PEG–interferon combination as well as the IFNAR1 upregulation induced by this treatment. Previous preclinical work reported by Kumar et al. demonstrated in a BRAFV600E melanoma cell line that oncogenic BRAF–MAPK signaling leads to the acceleration of IFNAR1 degradation by inducing βTrcp2 expression (86). Pharmacologic inhibition of either RAF or MEK1 stabilized IFNAR1 in melanoma cells and decreased their tumorigenicity. The destabilization and downregulation of IFNAR1 in BRAFV600E melanomas might account for the suboptimal anti-proliferative effects of IFN. Preclinical results obtained in murine models suggest that a combination treatment of IFN-α with RAF or MAPK inhibitors could decrease BRAF signaling and βTrcp2 expression and should prevent the rapid degradation of IFNAR1. This increase in the extent of IFN-α signaling could provide a promising strategy for treatment of melanoma. However, in vitro results also suggests that the BRAF-inhibitor BAY 43-9006 (Sorafenib) may decrease the extent of direct anti-proliferative responses to IFN-α by directly inhibiting the catalytic activity of JAK (86).
As previously mentioned, another immunotherapeutic strategy for the treatment of metastatic melanoma is ACT, with autologous TIL infusion after a lymphodepleting regimen, combined with IL-2 administration to support TIL growth. Furthermore, PLX-4720 significantly increased tumor infiltration by adoptively transferred T cells in vivo and enhanced the anti-tumor activity of ACT in a xenograft model with labeled T cells and human CM cell lines (87). This increase in tumor infiltration was primarily an effect of the inhibition of VEGF secretion by tumor cells. Currently, two open clinical studies are testing the combination of vemurafenib with ACT patients in BRAFV600 melanoma. In the study NCT01585415, the safety of vemurafenib administration will be evaluated both before and concurrent to autologous TIL infusion along with high HD-IL2, following a non-myeloablative lymphodepleting preparative regimen with Fluradabine. Furthermore, this study may provide information on how this combination of therapies mediates clinical tumor regression in patients, as well as the immunologic impact of BRAFV600E inhibition on the lymphoid infiltrate in melanoma deposits. In another study, the efficacy and side effects of this combination will also be analyzed (NCT01659151). This combination of therapies is designed to take advantage of the vemurafenib-induced upregulation of target Ags before TIL infusion. Furthermore, BRAF inhibition could contribute to the “autovaccination” of patients as tumor destruction leads to epitope exposition and immune effector elicitation. However, some questions will remain unanswered, since treatment with vemurafenib before harvesting the tumor for use in TIL could contribute to an increase in T cell infiltrate in the melanoma lesions. This could enrich TIL samples and potentially improve ACT outcome. These studies may elucidate whether the pre-ACT lymphodepleting regimen, necessary for ACT success, impairs vemurafenib treatment. Knight et al. have suggested that this may be the case if vemurafenib anti-tumor activity is mediated by resident immune cells (71). Thus, further research is necessary to assess the timing of TIL recovery, the lymphodepletion regimen and vemurafenib administration.
Future Directions and Perspectives
Combination therapies comprised of oncogene-targeted and immunotherapeutic strategies are a promising, emerging approach to melanoma treatment. Oncogenes impact not only tumor proliferation, but also other cancer hallmarks such as immune system evasion, which favors a tumor-permissive immune microenvironment. There is increasing evidence that oncogene-targeted inhibitors not only induce tumor cell death, but can also contribute to the restoration of an anti-tumor immune microenvironment. Oncogene-targeted therapies offer the possibility of rapid bulky tumor elimination, generate a vaccine-like boost and Ag tumor spreading and favor tumor infiltration by immune effectors, all of which may contribute to more extensive and durable tumor control.
Disease stage is a significant consideration in the design of combination strategies. In advanced metastatic melanoma, response kinetics has been a key factor in agent selection. Patients who have rapidly progressing tumors with druggable mutations can benefit from oncogene-targeted drugs first, allowing the elimination of important tumor masses in a short time. Sequential immunotherapy administration could cause tumor immune infiltration and therefore, promote durable immune control of disease dissemination. Concomitant administration of oncogene-targeted therapies with immunotherapy has produced serious AEs, though results are pending from additional clinical studies. Advanced patients with stable or slowly progressing disease might benefit from immunotherapies that act gradually but achieve long-lasting responses. Recent results with the antagonist mAb pembrolizumab are encouraging for patients who move on to immunotherapy or oncogene-targeted therapies. Instead, in the adjuvant setting, cancer vaccines could be the first choice to elicit a robust immune response. Their combination with an immune checkpoint blockade could enhance and prolong immune stimulation and effector numbers.
The choice of the best therapeutic design for combining immunotherapy with oncogene-targeted drugs (sequential vs. concurrent schemes), avoidance of toxicity and induction of long-lasting clinical responses are controversial and challenging issues. There is an urgent need for biomarker development through histologic and genomic analysis of the tumor and its microenvironment. Biomarkers will be able to predict which patients are likely to have a clinical benefit and which will have severe AEs. The use of biomarkers will give rise to a more personalized medicine for cancer treatment.
The development of more effective melanoma treatments based on oncogene-targeted therapy in combination with immunotherapy can emerge from novel hypothesis-testing and biomarker-driven clinical trials designed to optimize both drug dosing and immunotherapy scheduling. Ongoing and future clinical studies will certainly contribute to this future and illuminate these new roads for melanoma therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funds from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT); Instituto Nacional del Cáncer (INC); Fundación Cáncer-FUCA; Fundación Sales and Fundación Mosoteguy, Argentina. María Marcela Barrio is member of the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina. Mariana Aris is a Post-Doctoral fellow from the CONICET. We would like to thank María Luisa Pollack from the Library at the Instituto Alexander Fleming for her kind assistance.
Abbreviations
ACT, adoptive cell therapy; AE, adverse events; Ag, antigen/s; APC, antigen-presenting cells; CM, cutaneous melanoma; CR, complete response; DC, dendritic cells; DFS, disease-free survival; EMEA, European Medicines Agency; FDA, Federal Drug Administration; HD IL-2, high-dose interleukin-2; IFA, incomplete Freund’s adjuvant; irAEs, immune-related adverse events; mAbs, monoclonal antibodies; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; RTK, receptor tyrosine kinase; TIL, tumor infiltrating lymphocytes.
References
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin (2014) 64:9–29. doi:10.3322/caac.21208
2. Coit DG, Andtbacka R, Anker CJ, Bichakjian CK, Carson WE, Daud A, et al. Melanoma, version 2.2013: featured updates to the NCCN guidelines. J Natl Compr Canc Netw (2013) 11:395–407.
3. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364:2507–16. doi:10.1056/NEJMoa1103782
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (2012) 380:358–65. doi:10.1016/S0140-6736(12)60868-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
6. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet (2014) 6736:1–9. doi:10.1016/S0140-6736(14)60958-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Bis S, Tsao H. Melanoma genetics: the other side. Clin Dermatol (2013) 31:148–55. doi:10.1016/j.clindermatol.2012.08.003
8. Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature (2012) 485:502–6. doi:10.1038/nature11071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Siroy AE, Boland GM, Milton DR, Roszik J, Frankian S, Malke J, et al. Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J Invest Dermatol (2015) 135(2):508–15. doi:10.1038/jid.2014.366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417:949–54. doi:10.1038/nature00766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn (2013) 15:220–6. doi:10.1016/j.jmoldx.2012.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet (2012) 44:133–9. doi:10.1038/ng.1026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol (2011) 8:426–33. doi:10.1038/nrclinonc.2011.69
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer (2013) 49:1073–9. doi:10.1016/j.ejca.2012.11.004
15. Trudel S, Odolczyk N, Dremaux J, Toffin J, Regnier A, Sevestre H, et al. The clinical response to vemurafenib in a patient with a rare BRAFV600DK601del mutation-positive melanoma. BMC Cancer (2014) 14:727. doi:10.1186/1471-2407-14-727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol (2013) 31:1767–74. doi:10.1200/JCO.2012.44.7888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med (2012) 367:107–14. doi:10.1056/NEJMoa1203421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol (2013) 31:3182–90. doi:10.1200/JCO.2012.47.7836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich J, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet (2009) 41:1127–32. doi:10.1038/ng.438.Analysis
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol (2011) 29:2904–9. doi:10.1200/JCO.2010.33.9275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Kluger HM, Dudek AZ, McCann C, Ritacco J, Southard N, Jilaveanu LB, et al. A phase 2 trial of dasatinib in advanced melanoma. Cancer (2011) 117:2202–8. doi:10.1002/cncr.25766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi:10.1126/science.1203486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Aris M, Barrio MM, Mordoh J. Lessons from cancer immunoediting in cutaneous melanoma. Clin Dev Immunol (2012) 2012:192719. doi:10.1155/2012/192719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271:1734–6. doi:10.1126/science.271.5256.1734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol (2013) 31:616–22. doi:10.1200/JCO.2012.44.6112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28:3167–75. doi:10.1200/JCO.2009.26.7609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369:122–33. doi:10.1056/NEJMoa1302369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Hwu W, Topalian SL, Hwu P, Chen S, Salay TM, Alaparthy S, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Aris M, Tapia IJ, Mordoh J, Barrio MM. Cutaneous melanoma: molecular biology, risk factors and treatment options. Melanoma: Molecular Biology, Risk Factors and Treatment Options. Nova Publisher Group. p. 39–66.
31. Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol (2005) 175:6169–76. doi:10.4049/jimmunol.175.9.6169
32. Vacchelli E, Martins I, Eggermont A, Fridman WH, Galon J, Sautès-fridman C, et al. Peptide vaccines in cancer therapy trial watch. Oncoimmunology (2012) 1(9):1557–76. doi:10.4161/onci.22428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Hailemichael Y, Overwijk WW. Peptide-based anticancer vaccines: the making and unmaking of a T-cell graveyard. Oncoimmunology (2013) 2:e24743. doi:10.4161/onci.24743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Aris M, Zubieta MR, Colombo M, Arriaga JM, Bianchini M, Alperovich M, et al. MART-1- and gp100-expressing and -non-expressing melanoma cells are equally proliferative in tumors and clonogenic in vitro. J Invest Dermatol (2012) 132:365–74. doi:10.1038/jid.2011.312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Neller MA, López JA, Schmidt CW. Antigens for cancer immunotherapy. Semin Immunol (2008) 20:286–95. doi:10.1016/j.smim.2008.09.006
36. Nemunaitis J. Vaccines in cancer: GVAX, a GM-CSF gene vaccine. Expert Rev Vaccines (2005) 4:259–74. doi:10.1586/14760584.4.3.259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Barrio MM, de Motta PT, Kaplan J, von Euw EM, Bravo AI, Chacón RD, et al. A phase I study of an allogeneic cell vaccine (VACCIMEL) with GM-CSF in melanoma patients. J Immunother (2006) 29:444–54. doi:10.1097/01.cji.0000208258.79005.5f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature (2007) 449:419–26. doi:10.1038/nature06175
39. Beer TM, Bernstein GT, Corman JM, Glode LM, Hall SJ, Poll WL, et al. Randomized trial of autologous cellular immunotherapy with sipuleucel-T in androgen-dependent prostate cancer. Clin Cancer Res (2011) 17:4558–67. doi:10.1158/1078-0432.CCR-10-3223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Oshita C, Takikawa M, Kume A, Miyata H, Ashizawa T, Iizuka A, et al. Dendritic cell-based vaccination in metastatic melanoma patients: phase II clinical trial. Oncol Rep (2012) 28:1131–8. doi:10.3892/or.2012.1956
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Bercovici N, Haicheur N, Massicard S, Vernel-Pauillac F, Adotevi O, Landais D, et al. Analysis and characterization of antitumor T-cell response after administration of dendritic cells loaded with allogeneic tumor lysate to metastatic melanoma patients. J Immunother (2008) 31:101–12. doi:10.1097/CJI.0b013e318159f5ba
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Palucka AK, Ueno H, Connolly J, Kerneis-Norvell F, Blanck J-P, Johnston DA, et al. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J Immunother (2006) 29:545–57. doi:10.1097/01.cji.0000211309.90621.8b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Benteyn D, Van Nuffel AMT, Wilgenhof S, Corthals J, Heirman C, Neyns B, et al. Characterization of CD8+ T-cell responses in the peripheral blood and skin injection sites of melanoma patients treated with mRNA electroporated autologous dendritic cells (TriMixDC-MEL). Biomed Res Int (2013) 2013:976383. doi:10.1155/2013/976383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Goldszmid RS, Idoyaga J, Bravo AI, Steinman R, Mordoh J, Wainstok R. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol (2003) 171:5940–7. doi:10.4049/jimmunol.171.11.5940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Von Euw EM, Barrio MM, Furman D, Bianchini M, Levy EM, Yee C, et al. Monocyte-derived dendritic cells loaded with a mixture of apoptotic/necrotic melanoma cells efficiently cross-present gp100 and MART-1 antigens to specific CD8(+) T lymphocytes. J Transl Med (2007) 5:19. doi:10.1186/1479-5876-5-19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Von Euw EM, Barrio MM, Furman D, Levy EM, Bianchini M, Peguillet I, et al. A phase I clinical study of vaccination of melanoma patients with dendritic cells loaded with allogeneic apoptotic/necrotic melanoma cells. Analysis of toxicity and immune response to the vaccine and of IL-10 -1082 promoter genotype as predictor of diseas. J Transl Med (2008) 6:6. doi:10.1186/1479-5876-6-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol (2014) 15:e257–67. doi:10.1016/S1470-2045(13)70585-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi:10.1158/1078-0432.CCR-11-0116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol (2008) 26:5233–9. doi:10.1200/JCO.2008.16.5449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol (2005) 23:2346–57. doi:10.1200/JCO.2005.00.240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res (2010) 16:2646–55. doi:10.1158/1078-0432.CCR-10-0041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Tarhini AA, Kirkwood JM, Gooding WE, Cai C, Agarwala SS. Durable complete responses with high-dose bolus interleukin-2 in patients with metastatic melanoma who have experienced progression after biochemotherapy. J Clin Oncol (2007) 25:3802–7. doi:10.1200/JCO.2006.10.2822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Shaker MA, Younes HM. Interleukin-2: evaluation of routes of administration and current delivery systems in cancer therapy. J Pharm Sci (2009) 98:2268–98. doi:10.1002/jps.21596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Kirkwood JM, Manola J, Ibrahim J, Sondak V, Ernstoff MS, Rao U. A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin Cancer Res (2004) 10:1670–7. doi:10.1158/1078-0432.CCR-1103-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Sangfelt O, Erickson S, Grander D. Mechanisms of interferon-induced cell cycle arrest. Front Biosci (2000) 5:D479–87. doi:10.2741/Sangfelt
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Jewell AP, Worman CP, Lydyard PM, Yong KL, Giles FJ, Goldstone AH. Interferon-alpha up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br J Haematol (1994) 88:268–74. doi:10.1111/j.1365-2141.1994.tb05017.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Sangfelt O, Strander H. Apoptosis and cell growth inhibition as antitumor effector functions of interferons. Med Oncol (2001) 18:3–14. doi:10.1385/MO:18:1:3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Bekisz J, Baron S, Balinsky C, Morrow A, Zoon KC. Antiproliferative properties of type I and type II interferon. Pharmaceuticals (Basel) (2010) 3:994–1015. doi:10.3390/ph3040994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber H-P, et al. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell (2004) 6:485–95. doi:10.1016/j.ccr.2004.09.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Raig ET, Jones NB, Varker KA, Benniger K, Go MR, Biber JL, et al. VEGF secretion is inhibited by interferon-alpha in several melanoma cell lines. J Interferon Cytokine Res (2008) 28:553–61. doi:10.1089/jir.2008.0118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Prasanna SJ, Saha B, Nandi D. Involvement of oxidative and nitrosative stress in modulation of gene expression and functional responses by IFNgamma. Int Immunol (2007) 19:867–79. doi:10.1093/intimm/dxm058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Konjević G, Mirjačić Martinović K, Vuletić A, Babović N. In-vitro IL-2 or IFN-α-induced NKG2D and CD161 NK cell receptor expression indicates novel aspects of NK cell activation in metastatic melanoma patients. Melanoma Res (2010) 20:459–67. doi:10.1097/CMR.0b013e32833e3286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Konjevic G, Mirjacic-Martinovic K, Vuletic A, Babovic N. In vitro increased natural killer cell activity of metastatic melanoma patients with interferon-α alone as opposed to its combination with 13-cis retinoic acid is associated with modulation of NKG2D and CD161 activating receptor expression. J BUON (2012) 17:761–9. doi:10.1016/S0959-8049(12)71706-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Draube A, Klein-González N, Mattheus S, Brillant C, Hellmich M, Engert A, et al. Dendritic cell based tumor vaccination in prostate and renal cell cancer: a systematic review and meta-analysis. PLoS One (2011) 6:e18801. doi:10.1371/journal.pone.0018801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Leonhartsberger N, Ramoner R, Falkensammer C, Rahm A, Gander H, Höltl L, et al. Quality of life during dendritic cell vaccination against metastatic renal cell carcinoma. Cancer Immunol Immunother (2012) 61:1407–13. doi:10.1007/s00262-012-1207-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med (2004) 10:909–15. doi:10.1038/nm1100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Huber ML, Haynes L, Parker C, Iversen P. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J Natl Cancer Inst (2012) 104:273–9. doi:10.1093/jnci/djr514
68. Madan RA, Gulley JL, Fojo T, Dahut WL. Therapeutic cancer vaccines in prostate cancer: the paradox of improved survival without changes in time to progression. Oncologist (2010) 15:969–75. doi:10.1634/theoncologist.2010-0129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med (2006) 203:1651–6. doi:10.1084/jem.20051848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res (2012) 18:5329–40. doi:10.1158/1078-0432.CCR-12-1632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, et al. Host immunity contributes to the anti- melanoma activity of BRAF inhibitors. J Clin Invest (2013) 123:1371–81. doi:10.1172/JCI66236DS1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. De Andrade LF, Ngiow SF, Stannard K, Rusakiewicz S, Kalimutho M, Khanna KK, et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res (2014) 74(24):7298–308. doi:10.1158/0008-5472.CAN-14-1339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw C-NJ, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res (2010) 70:5213–9. doi:10.1158/0008-5472.CAN-10-0118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res (2010) 16:6040–8. doi:10.1158/1078-0432.CCR-10-1911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res (2014) 2:70–9. doi:10.1158/2326-6066.CIR-13-0160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Wilmott JS, Scolyer RA, Long GV, Hersey P. Combined targeted therapy and immunotherapy in the treatment of advanced melanoma. Oncoimmunology (2012) 1:997–9. doi:10.4161/onci.19865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res (2013) 19:1225–31. doi:10.1158/1078-0432.CCR-12-1630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Ribas A, Hodi FS, Callahan MK, Konto C, Wolchok JD. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med (2013) 368:1365–6. doi:10.1056/NEJMc1301264
79. Hantschel O, Rix U, Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk Lymphoma (2008) 49:615–9. doi:10.1080/10428190801896103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Kreutzman A, Ilander M, Porkka K, Vakkila J, Mustjoki S. Dasatinib promotes Th1-type responses in granzyme B expressing T-cells. Oncoimmunology (2014) 3:e28925. doi:10.4161/onci.28925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Schade AE, Schieven GL, Townsend R, Jankowska AM, Susulic V, Zhang R, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood (2008) 111:1366–77. doi:10.1182/blood-2007-04-084814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Lowe DB, Bose A, Taylor JL, Tawbi H, Lin Y, Kirkwood JM, et al. Dasatinib promotes the expansion of a therapeutically superior T-cell repertoire in response to dendritic cell vaccination against melanoma. Oncoimmunology (2014) 3:e27589. doi:10.4161/onci.27589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Yang Y, Liu C, Peng W, Lizée G, Overwijk WW, Liu Y, et al. Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood (2012) 120:4533–43. doi:10.1182/blood-2012-02-407163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117:1147–54. doi:10.1172/JCI31178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Sørensen RB, Berge-Hansen L, Junker N, Hansen CA, Hadrup SR, Schumacher TNM, et al. The immune system strikes back: cellular immune responses against indoleamine 2,3-dioxygenase. PLoS One (2009) 4:e6910. doi:10.1371/journal.pone.0006910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kumar KGS, Liu J, Li Y, Yu D, Thomas-Tikhonenko A, Herlyn M, et al. Raf inhibitor stabilizes receptor for the type I interferon but inhibits its anti-proliferative effects in human malignant melanoma cells. Cancer Biol Ther (2007) 6:1437–41. doi:10.4161/cbt.6.9.4569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res (2013) 19:393–403. doi:10.1158/1078-0432.CCR-12-1626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: melanoma, oncogene-targeted therapy, immunotherapy, combination therapy
Citation: Aris M and Barrio MM (2015) Combining immunotherapy with oncogene-targeted therapy: a new road for melanoma treatment. Front. Immunol. 6:46. doi: 10.3389/fimmu.2015.00046
Received: 02 December 2014; Accepted: 25 January 2015;
Published online: 09 February 2015.
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Viktor Umansky, German Cancer Research Center (DKFZ), GermanyGregory B. Lesinski, The Ohio State University Comprehensive Cancer Center, USA
Copyright: © 2015 Aris and Barrio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Marcela Barrio, Centro de Investigaciones Oncológicas – FUCA, Cramer 1180, Buenos Aires 1426, Argentina e-mail:YmFycmlvLm1hcmNlbGFAZ21haWwuY29t