 Romy E. Hoeppli
Romy E. Hoeppli Dan Wu
Dan Wu Laura Cook
Laura Cook Megan K. Levings
Megan K. Levings- Department of Surgery, Child and Family Research Institute, University of British Columbia, Vancouver, BC, Canada
Regulatory T cells (Tregs) are suppressive T cells that have an essential role in maintaining the balance between immune activation and tolerance. Their development, either in the thymus, periphery, or experimentally in vitro, and stability and function all depend on the right mix of environmental stimuli. This review focuses on the effects of cytokines, metabolites, and the microbiome on both human and mouse Treg biology. The role of cytokines secreted by innate and adaptive immune cells in directing Treg development and shaping their function is well established. New and emerging data suggest that metabolites, such as retinoic acid, and microbial products, such as short-chain fatty acids, also have a critical role in guiding the functional specialization of Tregs. Overall, the complex interaction between distinct environmental stimuli results in unique, and in some cases tissue-specific, tolerogenic environments. Understanding the conditions that favor Treg induction, accumulation, and function is critical to defining the pathophysiology of many immune-mediated diseases and to developing new therapeutic interventions.
Introduction
Regulatory T cells (Tregs) are a suppressive subset of CD4+ T helper (Th) cells important for the regulation of immune responses. The best-characterized Tregs are defined by expression of the transcription factor forkhead box protein 3 (FOXP3) and demethylation of the Treg-specific demethylated region (TSDR) in the FOXP3 locus. Demethylation of this element is thought to be crucial to maintain the stable, high expression of FOXP3 necessary for lineage stability and suppressive function (1, 2). Additional Treg markers include constitutive expression of the high-affinity IL-2Rα chain (CD25) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) (3), along with low expression of the IL-7Rα chain (CD127) (4, 5). CD4+CD25+FOXP3+ Tregs can be divided into two main types: thymically derived Tregs (tTregs) and peripherally derived Tregs (pTregs) (6). Although it is difficult to distinguish between tTregs and pTregs phenotypically, both are thought to have an essential role in immune regulation (7).
Because of their immunoregulatory function, Tregs are an attractive therapeutic target in many different immune-mediated diseases, including transplantation, autoimmunity, and autoinflammation (8). An emerging concept is that Tregs are functionally specialized to their local environments (9), with the local milieu of cytokines, metabolites, and catabolites having major effects on the phenotype and function of these cells. In this review, we discuss current knowledge on how environmental factors affect Treg development, maintenance, and function, focusing on key recent findings in the area of cytokines, metabolites, and the microbiome.
Cytokines
The Role of Cytokines in tTreg Development in the Thymus
Development of tTregs in the thymus is critically dependent on signals from the T cell receptor (TCR), CD28, and cytokines. Of particular importance are cytokines that signal via the common γ chain (γc), (10, 11), a topic that has been extensively reviewed (12, 13) (Table 1). Although most data suggest that IL-2 provides the essential signal to CD25+FOXP3− single positive tTreg precursors to differentiate into FOXP3+ cells, in the absence of IL-2, IL-15 provides a compensatory mechanism (14). In addition, a recent report found that CD25−FOXP3+ precursors have a specific requirement for IL-15 signaling to develop into tTregs in vitro and in vivo (15). Notably, while a complete absence of signaling from γc cytokines leads to a total lack of Tregs (16), in IL-2/IL-15-deficient mice a few Tregs remain (17). These findings suggest there other cytokines that signal through γc that can partially substitute for IL-2 and IL-15 in instructing tTreg development. An unanswered question is what cells in the thymus make IL-2, and/or other γc cytokines, and under what conditions? As dendritic cells (DCs) have been shown to make IL-2 (18) and are present in the human thymic medulla in close proximity to developing tTregs (19) they are an obvious candidate, but this has yet to be experimentally investigated.
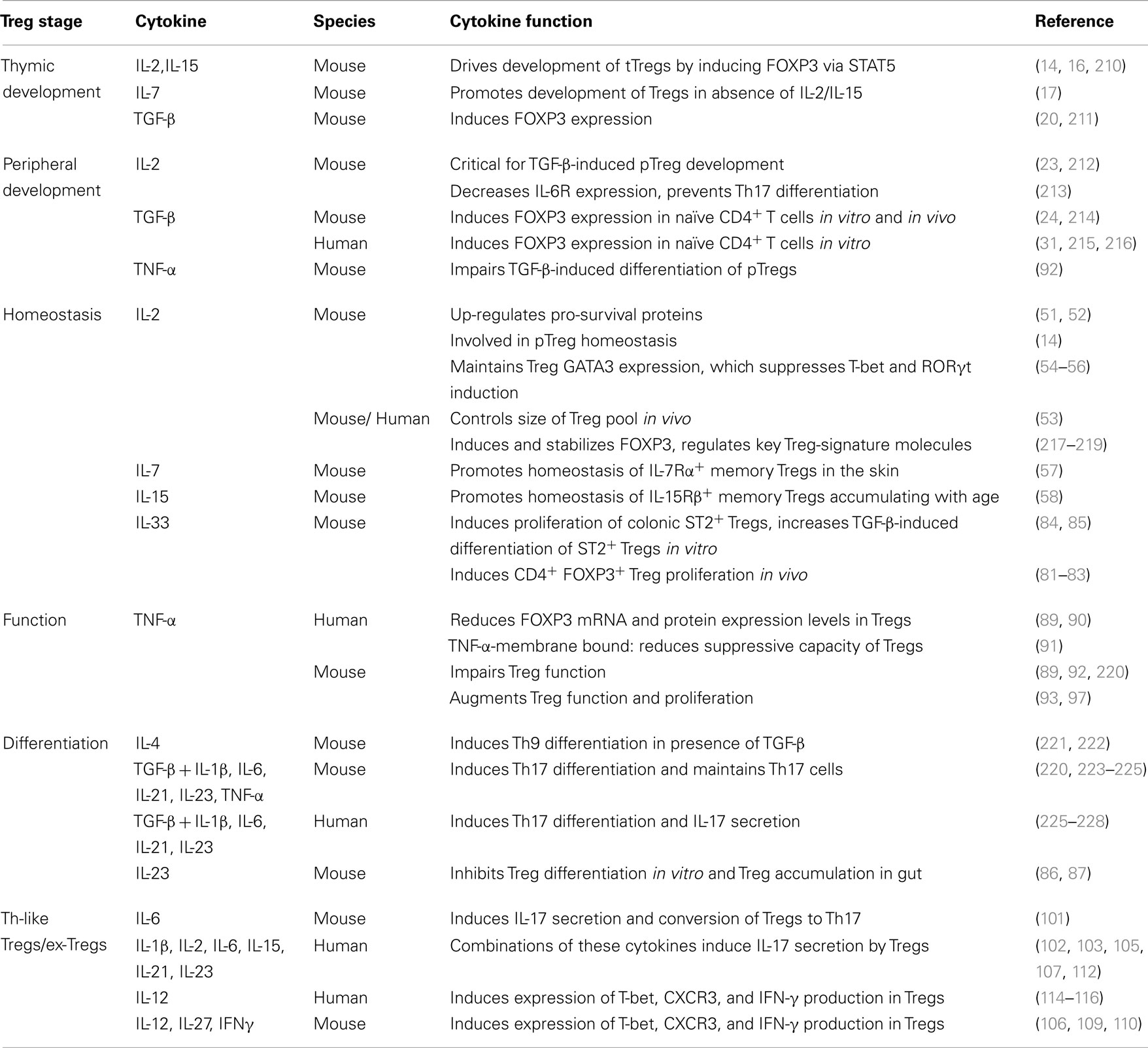
Table 1. Summary of cytokines that influence Tregs.
In addition to γc cytokines, TGF-β also has a critical role in tTreg development. In mice, thymocyte apoptosis leads to production of TGF-β by thymic macrophages, DCs, and epithelial cells, leading to TGF-β-induced FOXP3 expression and tTreg differentiation (20). Interestingly, in mice, this apoptosis in the thymus only occurs after birth, providing an explanation for the long-standing finding that murine tTregs only begin to develop 3 days after birth (21). How this finding relates to tTreg development in humans is unknown, but neonatal humans clearly have tTregs (22), so presumably this process occurs long before birth. Although the relative importance of IL-2 versus TGF-β in tTreg differentiation versus survival is a subject of much debate, both of these cytokines are clearly important for this lineage and understanding the biology of this system in humans will be key to developing therapies to boost tTreg development in vivo.
The Role of Cytokines in pTreg Development in the Periphery
The appropriate cytokine milieu is also a critical factor for the development of pTregs. In mice, both TGF-β and IL-2 are required to drive the conversion of CD4+CD25−FOXP3−naïve T cells into CD4+CD25+FOXP3+ pTregs (11, 23–25). However, the final outcome of TGF-β signaling is highly influenced by other surrounding cytokines. For example, anti-inflammatory conditions augment the effects of TGF-β, potentiating pTreg development (26). Conversely, pro-inflammatory cytokines (IL-1β, IL-6, IL-21, IL-23, and/or TNF-α) counteract TGF-β-induced FOXP3 expression and instead drive Th17 cell development by enhancing expression of retinoid-related orphan receptor γt (RORγt), the master Th17 lineage transcription factor (27).
Because of their potential application as a cell-based therapy, many groups have explored the cytokine combinations that can drive the differentiation of FOXP3+ Tregs in vitro (iTregs) from naïve human CD4+ T cells. Early evidence suggested that, as for mice, TCR stimulation in the presence of both TGF-β and IL-2 induced FOXP3 expression. However, the interpretation of these data became difficult when it was recognized that all activated human T cells transiently express FOXP3. Indeed, although TGF-β and IL-2-stimulated human CD4+ T cells express FOXP3, their TSDR remains methylated (2, 28, 29), a phenotype indicative of cells that are not stably committed to the Treg lineage. In addition, there are controversial findings on whether the resulting cells are suppressive, with some studies finding suppressive function (30), and others not (2, 28, 29, 31). It is important to note that human Treg suppression assays are particularly difficult to interpret when in vitro cultured cells are used due to non-specific effects mediated by media consumption and cell killing (32). Therefore, analysis of the TSDR status, and not functional assays, may be a more reliable way to measure human iTreg development. Collectively, these data suggest that while TGF-β may be necessary for differentiation of mouse and human pTregs in vivo, it is likely not sufficient, with other unknown environmental factors needed for their full development.
Interestingly, activated human Tregs express high levels of latent TGF-β coupled to latency-associated peptide and bound to the cell surface protein GARP (33–35). Therefore, Tregs themselves can drive the generation of new pTregs by providing a source of TGF-β (36, 37), offering a molecular explanation for a process termed “infectious tolerance” that has been observed for many years in animal models of transplantation (38–40). Mucosal DCs are also a rich source of TGF-β because they express integrin αvβ8, which converts extracellular latent TGF-β to its active form (41, 42). These cells may therefore be particularly important for the differentiation of intestinal pTregs that, as discussed in more detail below, are required for intestinal homeostasis.
Based on evidence that in humans TGF-β alone does not induce robust differentiation of stable Tregs, many studies have sought to define whether addition of other cytokines and/or compounds can enhance the effect (43). The most convincing evidence comes from addition of either the vitamin A metabolite all trans retinoic acid (ATRA, discussed further in the Section “Metabolites” below) or the mTOR inhibitor rapamycin. In mice, ATRA can be effectively generated by mucosal DCs and functions to enhance TGF-β-mediated pTreg generation, (44–46). In humans, suppressive iTregs can be generated with ATRA and TGF-β, but their stability based on the methylation of the TSDR is unknown (47–49). Similarly, addition of rapamycin enhances TGF-β-induced FOXP3 expression (49), and although the stability of these cells is unknown, there is an ongoing clinical trial to test their potential as a cellular therapy in hematopoietic stem cell transplantation (NCT01634217). Notably, rapamycin can also increase the stability of fully differentiated human Tregs in vitro (29), and of adoptively transferred non-human primate Tregs in vivo (50). These data provide a strong rationale to consider using rapamycin therapy to promote Treg function in vivo.
The Role of Cytokines in Treg Homeostasis
After development, naïve and memory Tregs in both mice and humans continue to rely heavily on IL-2 signaling for survival and homeostasis. IL-2 may also be important for facilitating Treg survival because it upregulates expression of pro-survival protein myeloid leukemia cell differentiation 1 (MCL1), which counter-regulates the FOXP3-induced pro-apoptotic protein BCL-2-interacting mediator of cell death (BIM) (51, 52). Indeed, administration of IL-2 to mice enhances Treg survival in vivo and reduces expression of the pro-apoptotic protein caspase 3 (53). In mice and humans, IL-2 also maintains Treg function by inducing FOXP3 mRNA, stabilizing FOXP3 protein expression, and regulating key Treg-signature molecules such as CTLA-4 and glucocorticoid-induced tumor necrosis factor receptor related protein (GITR) (11).
IL-2 is also essential to prevent the polarization of Tregs into pro-inflammatory effector cells (54, 55). For example, IL-2 signaling in Tregs is required to sustain expression of the GATA-binding protein 3 (GATA3) transcription factor (55, 56). Although this protein is commonly thought of as a Th2 cell lineage-defining protein, its expression is required for negative regulation of the TBX21 and RORC loci, which encode two transcription factors that feedback to diminish FOXP3 expression (55). It is currently not clear whether the role of GATA3 in Tregs is due to direct binding of GATA3 to regulatory regions in the TBX21 and RORC loci, or indirect via positive regulation of FOXP3 itself, which can then repress TBX21 and RORC transcription.
Whether or not other cytokines that signal via γc can substitute for IL-2 during pTreg development/survival in vivo remains unclear. Of note, some murine memory Tregs residing in the skin, or accumulating with age seem to preferentially rely on IL-7 or IL-15 for homeostasis (57, 58). Although human Tregs can definitely proliferate in response to IL-15 (59, 60), the relevance of IL-7 in humans is unclear as the lack of IL7Rα expression is a defining feature of human Tregs (4, 5).
Because of the essential role of exogenous IL-2 for keeping Tregs alive and maintaining FOXP3 expression, therapeutic approaches that deliver IL-2 signals specifically to Tregs are being actively explored. For example, delivery of IL-2/anti-IL-2-antibody complexes in pre-clinical studies stimulates Treg expansion and reduces disease in models of type 1 diabetes (T1D), experimentally induced autoimmune encephalomyelitis (EAE), collagen-induced arthritis, and angiotensin II-induced aortic stiffening (61–64). Similarly, in models of proteinuric kidney disease and renal ischemia-reperfusion injury, administration of IL-2/anti-IL-2-antibody complexes promotes Treg expansion, improves renal function, and reduces inflammation and disease symptoms (65, 66). In clinical trials, low-dose IL-2 therapy has been investigated for the treatment of graft versus host disease (GVHD) and T1D and appears to successfully expand the circulating Treg cell pool (67–70). A major caveat, however, is finding a dose regimen of IL-2 that only affects Tregs and does not activate CD8+ T cells and NK cells in parallel, as recently observed in a clinical trial of low dose IL-2 and rapamycin in T1D (71, 72). Another consideration is that IL-2-based therapies might not work in subjects who have genetic defects in IL-2R-signaling such as patients with a T1D-susceptibility IL-2RA haplotype (73) or whose Tregs have become IL-2-unresponsive (74).
A converse application of IL-2 targeted therapy is blockade of IL-2, which could theoretically be beneficial in the setting of cancer where depletion of Tregs could boost anti-tumor immunity (75). Interestingly anti-CD25 mAbs (basiliximab, daclizumab) were originally developed as immunosuppressive agents designed to deplete effector T cells and are still in common use today in transplantation. Investigation into whether daclizumab may also affect Tregs has revealed that it does indeed cause a reduction in Tregs by approximately 50%, both in the setting of autoimmunity (multiple sclerosis) and in cancer immunotherapy (76, 77). Basiliximab has similar effects in transplantation (78). However, post daclizumab therapy, the remaining 50% of Tregs are fully functional (77). These data suggest that, at least using current agents, IL-2 blockade is actually not a very effective way to deplete Tregs, possibly because of their elevated expression of the high affinity IL-2 receptor (i.e. CD25) and/or the ability of other cytokines to compensate in vivo.
Another cytokine that has recently gained interest as a regulator of Treg biology is IL-33, a member of the IL-1 cytokine family that signals via a heterodimeric receptor consisting of interleukin-1 receptor-related protein ST2 and the IL-1 receptor accessory protein IL1RAcP (79). Expressed by stromal and immune cells, IL-33 is well known to have a pathological role in airway inflammation and arthritis because it enhances and prolongs immune activation (80). Surprisingly, however, IL-33 treatment can actually protect against experimental colitis and rejection of HLA-mismatched cardiac allografts in mice by promoting Th2 cells and FOXP3+ Tregs (81–83). This anti-inflammatory effect of IL-33 on Tregs seems to be mediated in part via DCs, as IL-33-dependent expansion of murine ST2+FOXP3+ Tregs requires secretion of IL-2 by ST2+ DCs exposed to IL-33 (84).
In some tissues, however, there may be direct effects of IL-33 on Tregs. For example, more than 50% of colonic Tregs express ST2 enabling them to quickly respond to IL-33 released by epithelial cells upon tissue damage (85). Functionally, IL-33 can increase TGF-β-induced proliferation of colonic ST2+ Tregs in vitro and stabilize FOXP3 expression in inflamed tissues in vivo (85). Notably, IL-23, which is known to inhibit pTreg differentiation (86, 87), reduces expression of ST2 on Tregs (85), resulting in abrogation of the IL-33-mediated increase in pTreg induction and stabilization. Therefore, the balance between IL-33 and IL-23 may be an important factor in determining the outcome of tissue localized immune responses. In humans, IL-33 was previously thought to be an attractive target for therapeutic blocking (88), as a variety of inflammatory diseases feature elevated serum levels of IL-33. However, in light of its newly discovered function in promoting Treg expansion and function, inhibition of IL-33 could also have deleterious effects in some settings.
Control of Treg Function by Cytokines
The function of Tregs is also controlled by local the cytokine milieu, with mounting evidence that the presence of pro-inflammatory cytokines affect Treg suppression both directly and indirectly. Cytokines with direct effects on Tregs, such as tumor necrosis factor alpha (TNF-α), provide possible therapeutic targets for modulating Treg function. TNF-α is a pleiotropic cytokine that can act on a wide range of cells. Tregs express the TNF receptor, and there is evidence for both positive and negative effects of TNF-α on their function. Recent evidence shows that TNF-α induces expression of protein phosphatase 1 (PP1), which de-phosphorylates the C-terminal DNA-binding domain of FOXP3, resulting in a reduction in its function as a transcription factor (89). Notably, treatment of rheumatoid arthritis subjects with TNF-α-antibodies restores Treg function, decreases PP1 expression, and increases FOXP3 phosphorylation. These data are consistent with previous studies showing that TNF-α impairs Treg function in rheumatoid arthritis by reducing FOXP3 expression (90), and that Tregs expressing membrane-bound TNF-α are less suppressive than TNF-α negative Tregs (91). TNF-α also impairs TGF-β-induced pTreg development in EAE by reducing FOXP3 transcription (92).
Data reporting negative effects of TNF-α on Tregs contrast to a series of reports showing that TNF-α signaling through the TNF receptor 2, which is expressed by a subset of mouse and human effector and memory Tregs, enhances Treg proliferation and suppressive activity (93, 94). Notably, one of the common side effects of TNF-α therapy is psoriasis (95, 96) and data from mouse models suggest this may be due to an anti-TNF-α-mediated decrease in Treg frequency in the skin (97). Therefore, environmental TNF-α may actually bolster Treg function. Understanding how the local tissues define whether TNF-α has a negative or positive effect on Treg function will be key to understanding the side effects of this very common therapy.
Similar to conventional CD4+ T cells, Tregs respond to lineage-defining cytokines, resulting in differentiation into subsets that seem to mirror classical Th1, Th2, and Th17 cells (98, 99). Th1-like, Th2-like, and Th17-like peripheral CD4+CD45RO+CD127lowCD25high memory Tregs can be identified in human peripheral blood on the basis of differential expression of the chemokine receptors CXCR3, CCR4, and CCR6, respectively (100). A major question is whether these subsets of Th-like Tregs are protective or pathogenic. Evidence for the former comes from studies showing that Th-like Tregs remain suppressive and are necessary to provide protection from various diseases (101–114). On the other hand, in humans with autoimmunity and/or inflammation, Th1-like FOXP3+ Tregs that express T-bet, CXCR3 and produce IFN-γ appear to lose their suppressive function (115, 116), and multiple reports have shown that Th17-like Tregs are enriched at inflammatory sites, indicating a potential role in disease pathogenesis (116–120). We have also recently shown the first evidence for IL-13+ Th2-like Tregs, which are significantly increased in the skin, but not the blood, of subjects with systemic sclerosis (121). IL-13 is a pro-fibrotic cytokine that drives tissue fibrosis in this disease and in vitro experiments revealed that IL-33 increases the proportion of IL-13-producing Tregs in cultures of skin biopsies from healthy controls. Therefore, in addition to promoting Treg survival as described above, in some cases, IL-33 may cause detrimental changes to Treg function.
Metabolites
Dietary metabolites are another important environmental factor that influence Treg differentiation and function, especially in the gut. Research on the effect of metabolites on Tregs has particularly focused on vitamins A, D, and tryptophan. Understanding the effect of these and other metabolites on Tregs could identify dietary supplements that enhance Treg-based therapies and novel compounds that enhance in vitro expansion of stable Tregs.
Vitamin A
All trans retinoic acid (ATRA) is the main bioactive metabolite of vitamin A and, as briefly discussed above, is well known to have an important role in the differentiation of pTregs (122, 123). In vivo, a major source of ATRA appears to be mucosal DCs which in mice characteristically express CD103 (integrin αεβ7), (44–46, 124–126). Since mucosal DCs also express integrin αvβ8, which converts extracellular latent TGF-β to its active form (41), these cells can drive the synergistic induction of FOXP3+ pTregs, which specifically express gut homing markers, including CCR9 and integrin α4β7 (46). In humans, it has recently been demonstrated that ATRA acts on DCs and gives them the ability to preferentially drive the induction of gut homing Tr1 cells, an IL-10-producing FOXP3− Treg subset (127). The Tr1 cells produced by ATRA-producing DCs in this study displayed in vitro suppressive function, expressed gut homing markers CCR9 and integrin α4β7 and also produced IFN-γ.
As described in the Section “Cytokines,” Tregs can convert into Th-like cells in response to different inflammatory cytokines, a phenomenon, which may prove to be an obstacle for their use as immunotherapy. Recent work by Lu et al. has demonstrated that pre-treatment of human CD4+CD25highCD127low Tregs with ATRA almost completely prevents IL-1β/IL-6-driven conversion to Th1/Th17-like cells (128). Upon in vitro expansion in the presence of IL-1β and IL-6, ATRA-primed Tregs maintained high FOPX3 expression, suppressive function and were superior to untreated Tregs in preventing xenogeneic GVHD in mice. A similar effect of ATRA has also been observed in a study of individuals with autoimmune hepatitis type 2. Holder et al. demonstrated that the suppressive function of Tregs specific for liver enzyme cytochrome P450IID6 (the main disease autoantigen) was impaired following culture with IL-1β/IL-6; however, this was prevented by the addition of ATRA (129). Therefore, ATRA appears to be important for Tr1 cell differentiation in the gut and for stabilizing Tregs under inflammatory conditions, and has shown potential for therapeutic use in mouse models of colitis and periodontitis (130, 131).
Vitamin D
Vitamin D metabolites have long been recognized as important immunomodulators and exert their effects by binding to the vitamin D receptor, which is expressed on many immune cells including activated T cells (132). The active vitamin D metabolite calcitriol (1,25-dihydroxy vitamin D3) can be metabolized from vitamin D in the diet or synthesized in the skin following UV exposure. Calcitriol is known to promote the growth of both FOXP3+ and IL-10 producing Tregs, while inhibiting Th17 cells (133, 134). It has recently been shown that calcitriol also induces expression of skin (CCR10 and CLA) and inflamed tissue (CXCR6) homing receptors in Tregs (135), and that addition of TGF-β enhances calcitriol-driven expansion of FOXP3+ Tregs in vitro (136). Furthermore, calcidiol (25-hydroxy vitamin D3), a vitamin D metabolite similar to calcitriol, has been shown to prime mucosal DCs to induce suppressive IL-10 and IFN-γ producing Tr1 cells (127). A recent study has suggested that calcitriol could be a useful adjunct therapy with allergens in sublingual immunotherapy as it specifically enhanced Treg responses to allergens in vitro (137). It is interesting to note that, following treatment with UVB, MS patients had enhanced levels of serum calcitriol, which correlated with increased levels of circulating pTregs (138). This link between UVB exposure, serum levels of vitamin D metabolites, and Treg frequency might contribute to the observed epidemiological associations between environmental UVB exposure and incidence of autoimmune disease (139–141).
Metabolites That Activate Aryl Hydrocarbon Receptors
Numerous metabolites have been described that can activate the aryl hydrocarbon receptor (AHR), a transcription factor that alters the balance between Tregs and Th17 cells. The direction of this balance shift is though to be ligand-dependent, with some AHR ligands preferentially promoting Tregs and others promoting Th17 cells (142–144). For example, kynurenine, which is produced when tryptophan is catabolized by indoleamine 2,3-dioxygenase (IDO), is an AHR agonist that is important for generating Tregs and inhibiting Th17 cell development (145). Indeed, many tolerogenic cells, such as plasmacytoid DCs (146, 147) produce IDO, and through the production of tryptophan metabolites preferentially induce Tregs. Notably, both IDO and AHR are highly expressed in human placenta, implying that tryptophan metabolites acting via AHR also induces Tregs in pregnancy (148), a process critical for maternal/fetal tolerance (149).
Another tryptophan metabolite, cinnabarinic acid, has been identified as a novel AHR ligand (150) that is also an agonist of the type-4 metabotropic glutamate receptor. Cinnabarinic acid has been shown to prevent onset of EAE in mice following administration of myelin oligodendrocyte glycoprotein peptide through enhancing immune responses that were dominated by Tregs (151). Other dietary metabolites that can act as AHR ligands and promote Tregs include indole-3-carbanole (I3C) and 3,3’-diindolylmethane (DIM), derived from cruciferous vegetables. Treatment of EAE mice with either I3C or DIM completely protects against disease symptoms, significantly reduces immune cell infiltration into the CNS, increases Tregs, and reduces Th17 cells (152). These effects are AHR-dependent as treatment with an AHR antagonist reversed the protective effects of I3C and DIM. Similarly, a study of methionine–choline-deficient (MCD)-diet induced mouse non-alcoholic steatohepatitis (NASH) found that administering DIM reduced disease and shifted the immune dominance from Th17 cells toward Tregs using AHR-dependent mechanisms (153).
Studies of these natural metabolites have also led to the identification of novel AHR ligands, such as benzimidazoisoquinolines, which are not part of a normal diet. Administration of these compounds to mice increased Treg frequency and suppressed GVHD in an AHR-dependent manner (154). Understanding how the activity of AHR controls the balance between Tregs and inflammatory T cells will lead to new approaches to alter this balance therapeutically (155).
Purine Metabolism
Another important metabolic process is purine catabolism, which regulates the balance of pro-inflammatory adenosine 5’-triphosphate (ATP) and immunosuppressive adenosine. Through expression of adenosine receptors and the ecto-enzymes CD39 and CD73 that metabolize ATP, Tregs are able to both react to, and modulate, immune purinergic signals. CD39 and CD73 function to sequentially catabolize extracellular ATP: CD39 catalyzes the conversion of ATP into adenosine diphosphate (ADP) and adenosine monophosphate (AMP); and CD73 converts AMP into adenosine (156). Whereas ATP signals through type 2 purinergic (P2) receptors to initiate pro-inflammatory responses, adenosine, signals through type 1 purinergic (P1) receptors to suppress immune responses (157).
Mouse Tregs express both CD39 and CD73 and the production of adenosine by these enzymes is thought to be one of the Treg mechanisms of suppression (158). In contrast, most human CD39+ Tregs do not express CD73, and it is thought that human Tregs primarily generate adenosine when they are in proximity to CD73+ cells (159). Interestingly, expression of CD39 enables DCs and neutrophils to move along an ATP concentration gradient to sites of inflammation (160–162), and this may also be true for Tregs. Evidence using the EAE model showing that CD39 has an important role in directing migration of Tregs to lymphoid draining sites of the central nervous system (163) supports this hypothesis.
Regulatory T cells themselves can also respond to adenosine (157), which signals through the A2 class of P1 receptors to stimulate a positive feedback loop by increasing expression of CD73 mRNA via stimulation of cyclic AMP (cAMP) response elements in the CD73 locus (164). Adenosine can also act in an autocrine manner via A2A receptors expressed on Tregs to enhance their generation, CTLA-4 expression, and suppressive function (165).
Interestingly, in comparison to the negligible levels of cAMP levels in conventional T cells, human Tregs generate and maintain high intracellular levels of cAMP (166–168). CD39 may be important in this process, as intracellular production of cAMP is increased by extracellular adenosine signaling through A2 receptors. Of note, Tregs can mediate suppression by transferring cAMP through gap junctions into neighboring conventional T cells and DCs (166, 169). Intracellular cAMP also positively feedbacks on Tregs stimulating upregulation of both CTLA-4 (170) and CD39 expression (171).
In terms of the effects of cAMP on pTreg differentiation, in mice there may actually be negative effects as cAMP can suppress TGF-β-driven differentiation of pTregs in vitro (172). This negative effect of cAMP is likely due to cAMP-mediated activation of protein kinase A, which enhances TGF-β-mediated activation of mitogen-activated protein kinases ERK and JNK. In humans, however, the effect of cAMP on pTreg differentiation may be different as studies of prostaglandin E2 and vasoactive intestinal peptide, compounds that increase cAMP levels, result in increased pTreg generation and function (172, 173). As growing evidence shows that signaling through G-protein coupled receptors, which stimulate the cAMP pathway, has major effects on Treg differentiation and function (discussed in Section “Microbiome”), developing a full understanding of how cAMP affects Treg biology will be an important area of future research.
Microbiome
There are approximately 1000 species of different microbes colonizing the gut, with densities of 104-105 bacteria per millimeter of effluent in the proximal small intestine and 1011 bacteria per gram of luminal content in the colon (174). The high microbial content in the large intestine poses a large challenge to the mucosal immune system, as it needs to tolerate commensal microbiota and dietary antigens while maintaining the ability to eliminate pathogens. Induction of colonic Tregs is crucial in fostering this immune homeostasis.
It is now appreciated that a major site for development of pTregs is the colon, resulting in a large population of regulatory cells that have a distinct TCR repertoire and are critical for intestinal homeostasis (175). Since colonic pTregs are significantly reduced in germ-free mice, commensal microbiota has an essential role in inducing these cells (175–177). Similarly, the development of pTregs in the liver (178) and lungs (179) also requires the presence of commensal microbiota early in life. The exact mechanism behind the induction of colonic pTregs remains unknown, but several microbial components have been found to enhance their expansion and function, including short-chain fatty acids (SCFAs) (180–182), and the bacterial molecule polysaccharide A (PSA) of Bacteroides fragilis (183, 184) (Figure 1).
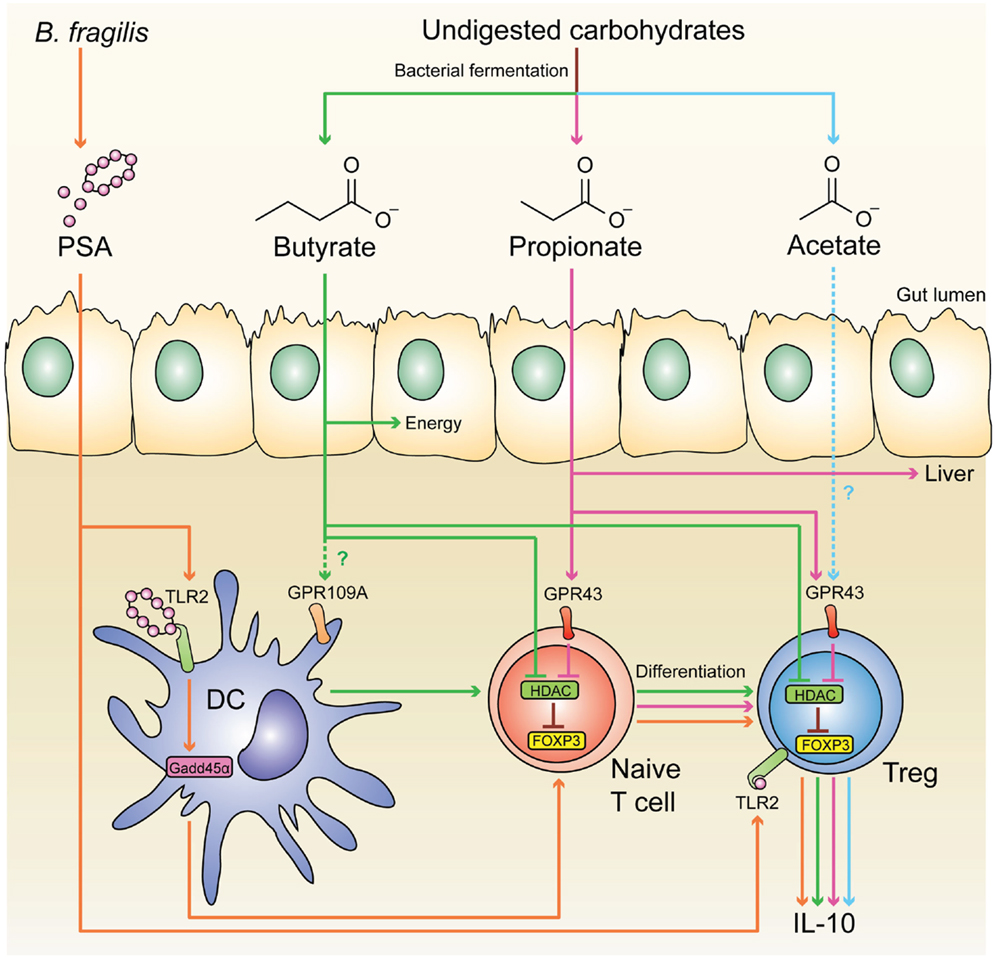
Figure 1. Microbial-derived molecules promote colonic Treg differentiation. Undigested dietary carbohydrates are fermented by gut commensal bacteria to produce the SCFAs acetate, propionate, and butyrate. Administration of acetate in drinking water results in the accumulation of IL-10+ colonic Tregs, and this effect is independent of HDAC inhibition and acetylation of the Foxp3 CNS1 region. Although acetate is a potent GPR43 ligand, it is not clear whether acetate mediates its effect through this receptor. GPR43 expression in colonic Tregs is required for propionate to inhibit HDAC function and enhance FOXP3 expression, thereby promoting Treg differentiation and IL-10 production, Butyrate has similar effects by either directly acting on Tregs or through modulating DC function to enhance their Treg-inducing ability; however, the role of GPR109A in these effects is controversial. Purified PSA derived from B. fragilis can also directly act on Tregs through TLR2 to promote Treg function by enhancing expression of effector molecules including IL-10, TGF-β2, and granzyme B. Membrane-bound PSA cannot act directly on Tregs, instead it interacts with TLR2 on DCs to promote Treg differentiation in a Gadd45α-dependent manner.
Short-Chain Fatty Acids
Despite substantial individual variation in the composition of the microbial community, in healthy adults most gut bacteria belong to two phyla: Firmicutes and Bacteroidetes (185). Because of the large anaerobic community and low oxygen availability in the colon, bacterial metabolism is dominated by fermentation and anaerobic respiration where nitrate, sulfate, and other compounds are used as electron acceptors (186). Undigested dietary carbohydrates are fermented to produce gases and organic acids, particularly the SCFAs acetate (C2), propionate (C3), and butyrate (C4), typically at a ratio of 3:1:1, respectively (187). Of these three main SCFAs, acetate can be produced by enteric bacteria and acetogens; propionate is a by-product of the succinate pathway in Bacteroidetes; and butyrate is formed from two acetyl-CoA molecules in Firmicutes (188). Specific species that have been recognized by their high levels of butyrate production include Faecalibacterium prausnitzii and the cluster IV and XIVa of genus Clostridium (189). SCFAs are the most abundant (50–100 mM) in the proximal colon, where most fermentation occurs (190). However, in the peripheral blood, only acetate remains in relatively high concentrations, since butyrate is preferentially utilized by colonocytes as an energy source and propionate is metabolized by hepatocytes (190).
Recent work has revealed that SCFAs are important in promoting the differentiation of colonic Tregs. An early study reported that germ-free mice have reduced colonic Treg numbers, and that colonization by bacterial strains belonging to the cluster IV and XIVa of the genus Clostridium rescues the deficiency and protects mice from colitis (176). Administration of acetate, propionate, and butyrate in drinking water mimics the effect of Clostridium colonization in germ-free mice, resulting in an elevated Treg frequency in the colonic lamina propria and increased IL-10 production by these Tregs (180, 182).
Of the three main SCFAs, butyrate has been found to be the most potent inducer of colonic Tregs. Mice fed a diet enriched in butyrylated starches have more colonic Tregs than those fed a diet containing propinylated or acetylated starches (181). Arpaia et al. tested an array of SCFAs purified from commensal bacteria and confirmed butyrate was the strongest SCFA-inducer of Tregs in vitro (180). Mechanistically, it has been proposed that butyrate, and possibly propionate, promote Tregs through inhibiting histone deacetylase (HDAC), causing increased acetylation of histone H3 in the Foxp3 CNS1 region, and thereby enhancing FOXP3 expression (180, 181).
Short-chain fatty acids partially mediate their effects through G-protein coupled receptors (GPR), including GPR41, GPR43, and GPR109A. GPR41 and GPR43 are stimulated by all three major SCFAs (191), whereas GPR109A only interacts with butyrate (192). In mice, colonic and small intestinal Tregs express GPR43, and expression of this receptor is required for propionate-mediated HDAC inhibition and Treg expansion (182). There are conflicting results as to whether GPR109A is required for butyrate to mediate its pro-Treg effect. In both mice and humans, GPR109A is only expressed on DCs and macrophages, but not on T cells (192). Singh et al. found that splenic DCs from Gpr109a−/− mice were not able to induce Treg differentiation in response to butyrate (192). Additionally, the study found that butyrate treatment increases transcription of Aldha1 (aldehyde dehydrogenase) in a GPR109A-dependent manner (192). As discussed above, this enzyme is important in vitamin A metabolism, so these data suggest that GPR109A signaling may increase ATRA production by APCs and indirectly promote Treg differentiation and function. In contrast, Arpaia et al. reported that butyrate-pre-treated Gpr109a−/− DCs are not defective in in vitro generation of Tregs (180). The reason for these discrepant findings is not clear, but overall the emerging data demonstrating that SCFAs can have both direct and indirect effects on Tregs and have opened up an exciting new area of research.
Polysaccharide A
Another microbial component capable of enhancing Treg function is PSA from the commensal gut bacterial strain B. fragilis. The initial study reported that in germ-free mice either colonization by B. fragilis or administration of purified PSA induces IL-10 secretion by CD4+ T cells and reduces gut inflammation (193). A subsequent study confirmed that the IL-10-expressing CD4+ T cells were FOXP3+ Tregs and that PSA treatment increases Treg frequency and their expression of effector molecules including IL-10, TGF-β2, granzyme B, and CCR6 (184). Notably, the authors found that in the absence of APCs, PSA acts directly on Tregs through toll-like receptor 2 (TLR2) to induce the observed effects (194). It has also been demonstrated that administration of PSA protects against induction of EAE in mice through TLR2-mediated expansion of CD39+ Tregs (195).
It remains unknown how PSA is recognized by the mucosal immune system. Since the genome of B. fragilis does not contain genes for any known bacterial secretion system (196) and PSA is a large capsular polysaccharide (197), it has been proposed that B. fragilis delivers PSA by secreting outer membrane vesicles (OMVs) (198). Shen et al. observed that oral administration of PSA-containing OMVs purified from B. fragilis is sufficient to protect mice from experimental colitis and that TLR2 expression on DCs, but not T cells, is required to promote IL-10 production by Tregs (198). Subsequent work identified that PSA-treated plasmacytoid DCs, but not conventional DCs, are responsible for inducing IL-10-secreting Tregs (199). Therefore, while Tregs can directly respond to purified PSA, immune responses to membrane-bound PSA require TLR2+ DCs. It is worth noting that many other TLR ligands can also directly or indirectly impact Treg function [reviewed in Ref. (200)]. In contrast to PSA, other TLR2 ligands, such as Pam3CSK4 and FSL-1, inhibit the function of both mouse and human Tregs (201–204). How multiple TLR signals are integrated in the mucosal environment is unknown.
Probiotics
Long before the direct effects of microbial products on Tregs were understood at the molecular levels, many groups have been exploring the potential therapeutic use of bacteria in the form of probiotics to modulate Tregs. For example, administration of a five-strain probiotic mixture (designated IRT5, including Lactobacillus acidophilus, Lactobacillus casei, Lactobacillus reuteri, Bifidobacterium bifidum, and Streptococcus thermophilus) in mice increases the proportion of Tregs in the mesenteric lymph nodes (205). CD11c+ DCs purified from these treated mice also had higher expression of IL-10, TGF-β, and IDO, and were more capable of inducing Treg differentiation compared to DCs from control mice (205). A more recent study demonstrated that administration of L. reuteri to mice was sufficient to prevent high-fat-diet-induced adipose inflammation and obesity, an effect that was associated with enhanced Treg induction and IL-10 expression (206). In vitro, L. casei and L. reuteri can also prime human monocyte-derived DCs to stimulate IL-10-producing Tregs through the adhesion molecule DC-SIGN (207). Lopez et al. found that DCs exposed to B. bifidum membrane vesicles strongly induced Treg differentiation in vitro, suggesting that the potential use of the membrane vesicle as a safe adjunct therapy (208).
Although much work is still needed to elucidate the details of how commensal microbiota induce Tregs, numerous randomized trials in the past decade using Lactobacillus and Bifidobacterium to treat inflammatory disorders have already demonstrated the clinical benefit of this approach (209). Indeed, while delivery of purified PSA or SCFAs may represent an effective, transient therapy, the use of probiotics may offer a well-tolerated long-term therapeutic solution to enhancing intestinal immunoregulatory cells.
Conclusion
Environmental stimuli influence all aspects of Treg biology: from development and differentiation to migration and function. As well as refining our understanding of how well-described cytokines affect Tregs, we are also discovering new cytokines, such as IL-33, which have a critical role in Treg function (Table 1). Other key factors influencing Tregs, particularly in the gut, are dietary metabolites, catabolites, and bacterial components from the microbiome. There is emerging evidence that retinoic acid is a key metabolite for expanding a stable population of Tregs, data that have clear implications for developing therapeutic approaches. Furthermore, aspects of the microbiome clearly help determine which commensal antigens the immune system is educated against and have a previously unappreciated role in influencing T cell differentiation in the gut. Further studies in this area will expand our knowledge of T cell biology and hopefully uncover new elements of disease pathogenesis and guide the development of Treg-based therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol (2007) 37:2378–89. doi:10.1002/eji.200737594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5:e38. doi:10.1371/journal.pbio.0050038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10:490–500. doi:10.1038/nri2785
4. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203:1701–11. doi:10.1084/jem.20060772
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med (2006) 203:1693–700. doi:10.1084/jem.20060468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14:307–8. doi:10.1038/ni.2554
7. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity (2011) 35:109–22. doi:10.1016/j.immuni.2011.03.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Singer BD, King LS, D’alessio FR. Regulatory T cells as immunotherapy. Front Immunol (2014) 5:46. doi:10.3389/fimmu.2014.00046
9. Pesenacker AM, Broady R, Levings MK. Control of tissue-localized immune responses by human regulatory T cells. Eur J Immunol (2014). doi:10.1002/eji.201344205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Burchill MA, Yang J, Vang KB, Farrar MA. Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol Lett (2007) 114:1–8. doi:10.1016/j.imlet.2007.08.005
11. Goldstein JD, Pérol L, Zaragoza B, Baeyens A, Marodon G, Piaggio E. Role of cytokines in thymus- versus peripherally derived-regulatory T cell differentiation and function. Front Immunol (2013) 4:155. doi:10.3389/fimmu.2013.00155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev (2011) 241:63–76. doi:10.1111/j.1600-065X.2011.01004.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Mahmud SA, Manlove LS, Farrar MA. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT (2013) 2:e23154. doi:10.4161/jkst.23154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor-dependent STAT5 activation is required for the development of Foxp3+ regulatory T Cells. J Immunol (2007) 178:280–90. doi:10.4049/jimmunol.178.1.280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Marshall D, Sinclair C, Tung S, Seddon B. Differential requirement for IL-2 and IL-15 during bifurcated development of thymic regulatory T cells. J Immunol (2014) 193:5525–33. doi:10.4049/jimmunol.1402144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6:1142–51. doi:10.1038/ni1263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Bayer AL, Lee JY, De La Barrera A, Surh CD, Malek TR. A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J Immunol (2008) 181:225–34. doi:10.4049/jimmunol.181.1.225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Granucci F, Vizzardelli C, Pavelka N, Feau S, Persico M, Virzi E, et al. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat Immunol (2001) 2:882–8. doi:10.1038/ni0901-882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, et al. Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature (2005) 436:1181–5. doi:10.1038/nature03886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Konkel JE, Jin W, Abbatiello B, Grainger JR, Chen W. Thymocyte apoptosis drives the intrathymic generation of regulatory T cells. Proc Natl Acad Sci U S A (2014) 111:E465–73. doi:10.1073/pnas.1320319111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in post-thymectomy autoimmune oophoritis in mice. II. Requirement of Lyt-1 cells in normal female mice for the prevention of oophoritis. J Exp Med (1982) 156:1577–86. doi:10.1084/jem.156.6.1565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Ndure J, Flanagan KL. Targeting regulatory T cells to improve vaccine immunogenicity in early life. Front Microbiol (2014) 5:477. doi:10.3389/fmicb.2014.00477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Davidson TS, Dipaolo RJ, Andersson J, Shevach EM. Cutting edge: IL-2 is essential for TGF-mediated induction of Foxp3+ T regulatory cells. J Immunol (2007) 178:4022–6. doi:10.4049/jimmunol.178.7.4022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol (2008) 9:194–202. doi:10.1038/ni1549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Chen W, Konkel JE. TGF-beta and ‘adaptive’ Foxp3(+) regulatory T cells. J Mol Cell Biol (2010) 2:30–6. doi:10.1093/jmcb/mjp004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Karman J, Jiang JL, Gumlaw N, Zhao H, Campos-Rivera J, Sancho J, et al. Ligation of cytotoxic T lymphocyte antigen-4 to T cell receptor inhibits T cell activation and directs differentiation into Foxp3+ regulatory T cells. J Biol Chem (2012) 287:11098–107. doi:10.1074/jbc.M111.283705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Sakaguchi S, Vignali DAA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol (2013) 13:461–7. doi:10.1038/nri3464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38:1654–63. doi:10.1002/eji.200838105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Rossetti M, Spreafico R, Saidin S, Chua C, Moshref M, Leong JY, et al. Ex vivo-expanded but not in vitro-induced human regulatory T cells are candidates for cell therapy in autoimmune diseases thanks to stable demethylation of the FOXP3 regulatory T cell-specific demethylated region. J Immunol (2014) 194:11–124. doi:10.4049/jimmunol.1401145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med (2011) 3:83ra41. doi:10.1126/scitranslmed.3001809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood (2007) 110:2983–90. doi:10.1182/blood-2007-06-094656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. McMurchy AN, Levings MK. Suppression assays with human T regulatory cells: a technical guide. Eur J Immunol (2012) 42:27–34. doi:10.1002/eji.201141651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS One (2008) 3:e2705. doi:10.1371/journal.pone.0002705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol (2009) 39:3315–22. doi:10.1002/eji.200939684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A (2009) 106:13445–50. doi:10.1073/pnas.0901944106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med (2002) 196:255–60. doi:10.1084/jem.20020394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ, et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med (2008) 205:1975–81. doi:10.1084/jem.20080308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Waldmann H, Cobbold S. How do monoclonal antibodies induce tolerance? A role for infectious tolerance? Annu Rev Immunol (1998) 16:619–44. doi:10.1146/annurev.immunol.16.1.619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Kendal AR, Waldmann H. Infectious tolerance: therapeutic potential. Curr Opin Immunol (2010) 22:560–5. doi:10.1016/j.coi.2010.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kendal AR, Chen Y, Regateiro FS, Ma J, Adams E, Cobbold SP, et al. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J Exp Med (2011) 208:2043–53. doi:10.1084/jem.20110767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature (2007) 449:361–5. doi:10.1038/nature06110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Worthington JJ, Fenton TM, Czajkowska BI, Klementowicz JE, Travis MA. Regulation of TGFbeta in the immune system: an emerging role for integrins and dendritic cells. Immunobiology (2012) 217:1259–65. doi:10.1016/j.imbio.2012.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Dons EM, Raimondi G, Cooper DK, Thomson AW. Induced regulatory T cells: mechanisms of conversion and suppressive potential. Hum Immunol (2012) 73:328–34. doi:10.1016/j.humimm.2011.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204:1757–64. doi:10.1084/jem.20070590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317:256–60. doi:10.1126/science.1145697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med (2007) 204:1775–85. doi:10.1084/jem.20070602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Wang J, Huizinga TW, Toes RE. De novo generation and enhanced suppression of human CD4+CD25+ regulatory T cells by retinoic acid. J Immunol (2009) 183:4119–26. doi:10.4049/jimmunol.0901065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS One (2011) 6:e15868. doi:10.1371/journal.pone.0015868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL, et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplant (2011) 11:1148–57. doi:10.1111/j.1600-6143.2011.03558.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Singh K, Stempora L, Harvey RD, Kirk AD, Larsen CP, Blazar BR, et al. Superiority of rapamycin over tacrolimus in preserving nonhuman primate treg half-life and phenotype after adoptive transfer. Am J Transplant (2014) 14:2691–703. doi:10.1111/ajt.12934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat Immunol (2013) 14:959–65. doi:10.1038/ni.2649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, et al. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity (2013) 38:1116–28. doi:10.1016/j.immuni.2013.02.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Amado IF, Berges J, Luther RJ, Mailhé M-P, Garcia S, Bandeira A, et al. IL-2 coordinates IL-2-producing and regulatory T cell interplay. J Exp Med (2013) 210:2707–20. doi:10.1084/jem.20122759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity (2011) 35:337–48. doi:10.1016/j.immuni.2011.08.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Invest (2011) 121:4503–15. doi:10.1172/JCI57456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Rudra D, Deroos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol (2012) 13:1010–9. doi:10.1038/ni.2402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Gratz IK, Truong H-A, Yang SH-Y, Maurano MM, Lee K, Abbas AK, et al. Cutting edge: memory regulatory T cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol (2013) 190:4483–7. doi:10.4049/jimmunol.1300212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Raynor J, Sholl A, Plas DR, Bouillet P, Chougnet CA, Hildeman DA. IL-15 fosters age-driven regulatory T cell accrual in the face of declining IL-2 levels. Front Immunol (2013) 4:161. doi:10.3389/fimmu.2013.00161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Clark RA, Kupper TS. IL-15 and dermal fibroblasts induce proliferation of natural regulatory T cells isolated from human skin. Blood (2007) 109:194–202. doi:10.1182/blood-2006-02-002873
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Xu S, Sun Z, Sun Y, Zhu J, Li X, Zhang X, et al. IL-15 and dendritic cells induce proliferation of CD4+CD25+ regulatory T cells from peripheral blood. Immunol Lett (2011) 140:59–67. doi:10.1016/j.imlet.2011.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28:687–97. doi:10.1016/j.immuni.2008.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med (2009) 206:751–60. doi:10.1084/jem.20082824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Lee SY, Cho ML, Oh HJ, Ryu JG, Park MJ, Jhun JY, et al. Interleukin-2/anti-interleukin-2 monoclonal antibody immune complex suppresses collagen-induced arthritis in mice by fortifying interleukin-2/STAT5 signalling pathways. Immunology (2012) 137:305–16. doi:10.1111/imm.12008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Majeed B, Tawinwung S, Eberson LS, Secomb TW, Larmonier N, Larson DF. Interleukin-2/anti-interleukin-2 immune complex expands regulatory T Cells and reduces angiotensin II-induced aortic stiffening. Int J Hypertens (2014) 2014:126365. doi:10.1155/2014/126365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Polhill T, Zhang GY, Hu M, Sawyer A, Zhou JJ, Saito M, et al. IL-2/IL-2Ab complexes induce regulatory T cell expansion and protect against proteinuric CKD. J Am Soc Nephrol (2012) 23:1303–8. doi:10.1681/ASN.2011111130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Kim MG, Koo TY, Yan JJ, Lee E, Han KH, Jeong JC, et al. IL-2/anti-IL-2 complex attenuates renal ischemia-reperfusion injury through expansion of regulatory T cells. J Am Soc Nephrol (2013) 24:1529–36. doi:10.1681/ASN.2012080784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Bayer AL, Pugliese A, Malek TR. The IL-2/IL-2R system: from basic science to therapeutic applications to enhance immune regulation. Immunol Res (2013) 57:197–209. doi:10.1007/s12026-013-8452-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1:295–305. doi:10.1016/S2213-8587(13)70113-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Matsuoka K-I, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5:ra143–79. doi:10.1126/scitranslmed.3005265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu M-F, Liu H, et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res (2014) 20:2215–25. doi:10.1158/1078-0432.CCR-13-3205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12:180–90. doi:10.1038/nri3156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes (2012) 61:2340–8. doi:10.2337/db12-0049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Garg G, Tyler JR, Yang JHM, Cutler AJ, Downes K, Pekalski M, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol (2012) 188:4644–53. doi:10.4049/jimmunol.1100272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Bending D, Pesenacker AM, Ursu S, Wu Q, Lom H, Thirugnanabalan B, et al. Hypomethylation at the regulatory T cell-specific demethylated region in CD25hi T cells is decoupled from FOXP3 expression at the inflamed site in childhood arthritis. J Immunol (2014) 193:2699–708. doi:10.4049/jimmunol.1400599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer (2010) 127:759–67. doi:10.1002/ijc.25429
76. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med (2012) 4:134ra162. doi:10.1126/scitranslmed.3003330
77. Huss DJ, Mehta DS, Sharma A, You X, Riester KA, Sheridan JP, et al. In vivo maintenance of human regulatory T cells during CD25 blockade. J Immunol (2014) 194:84–92. doi:10.4049/jimmunol.1402140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Bouvy AP, Klepper M, Kho MM, Boer K, Betjes MG, Weimar W, et al. The impact of induction therapy on the homeostasis and function of regulatory T cells in kidney transplant patients. Nephrol Dial Transplant (2014) 29:1587–97. doi:10.1093/ndt/gfu079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Palmer G, Lipsky BP, Smithgall MD, Meininger D, Siu S, Talabot-Ayer D, et al. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine (2008) 42:358–64. doi:10.1016/j.cyto.2008.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Mirchandani AS, Salmond RJ, Liew FY. Interleukin-33 and the function of innate lymphoid cells. Trends Immunol (2012) 33:389–96. doi:10.1016/j.it.2012.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, Fichtner-Feigl S. Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int (2011) 24:1027–39. doi:10.1111/j.1432-2277.2011.01306.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol (2011) 187:4598–610. doi:10.4049/jimmunol.1100519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Duan L, Chen J, Zhang H, Yang H, Zhu P, Xiong A, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol Med (2012) 18:753–61. doi:10.2119/molmed.2011.00428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol (2014) 193:4010–20. doi:10.4049/jimmunol.1400481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513:564–8. doi:10.1038/nature13577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwakura Y, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity (2008) 28:559–70. doi:10.1016/j.immuni.2008.02.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity (2010) 33:279–88. doi:10.1016/j.immuni.2010.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nat Rev Rheumatol (2011) 7:321–9. doi:10.1038/nrrheum.2011.53
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat Med (2013) 19:322–8. doi:10.1038/nm.3085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood (2006) 108:253–61. doi:10.1182/blood-2005-11-4567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Wang J, Van Dongen H, Scherer HU, Huizinga TWJ, Toes REM. Suppressor activity among CD4+,CD25++ T cells is discriminated by membrane-bound tumor necrosis factor alpha. Arthritis Rheum (2008) 58:1609–18. doi:10.1002/art.23460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Zhang Q, Cui F, Fang L, Hong J, Zheng B, Zhang JZ. TNF-α impairs differentiation and function of TGF-β-induced Treg cells in autoimmune diseases through Akt and Smad3 signaling pathway. J Mol Cell Biol (2013) 5:85–98. doi:10.1093/jmcb/mjs063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol (2007) 179:154–61. doi:10.4049/jimmunol.179.1.154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Chen X, Oppenheim JJ. TNF-alpha: an activator of CD4+FoxP3+TNFR2+ regulatory T cells. Curr Dir Autoimmun (2010) 11:119–34. doi:10.1159/000289201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Suryaprasad AG, Prindiville T. The biology of TNF blockade. Autoimmun Rev (2003) 2:346–57. doi:10.1016/S1568-9972(03)00048-X
96. Nguyen K, Vleugels RA, Velez NF, Merola JF, Qureshi AA. Psoriasiform reactions to anti-tumor necrosis factor α therapy. J Clin Rheumatol (2013) 19:377–81. doi:10.1097/RHU.0b013e3182a702e8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Ma HL, Napierata L, Stedman N, Benoit S, Collins M, Nickerson-Nutter C, et al. Tumor necrosis factor alpha blockade exacerbates murine psoriasis-like disease by enhancing Th17 function and decreasing expansion of Treg cells. Arthritis Rheum (2010) 62:430–40. doi:10.1002/art.27203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev (2014) 259:159–72. doi:10.1111/imr.12175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev (2014) 259:173–91. doi:10.1111/imr.12173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood (2012) 119:4430–40. doi:10.1182/blood-2011-11-392324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-. J Immunol (2007) 178:6725–9. doi:10.4049/jimmunol.178.11.6725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Koenen HJPM, Smeets RL, Vink PM, Van Rijssen E, Boots AMH, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood (2008) 112:2340–52. doi:10.1182/blood-2008-01-133967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113:4240–9. doi:10.1182/blood-2008-10-183251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science (2009) 326:986–91. doi:10.1126/science.1172702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1beta and IL-2 convert human Treg into T(H)17 cells. Clin Immunol (2009) 131:298–307. doi:10.1016/j.clim.2008.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Koch MA, Tucker-Heard GS, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10:595–602. doi:10.1038/ni.1731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Voo KS, Wang Y-H, Santori FR, Boggiano C, Wang Y-H, Arima K, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A (2009) 106:4793–8. doi:10.1073/pnas.0900408106
108. Zheng Y, Chaudhry A, Kas A, Deroos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458:351–6. doi:10.1038/nature07674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Feng T, Cao AT, Weaver CT, Elson CO, Cong Y. Interleukin-12 converts Foxp3+ regulatory T cells to interferon-γ-producing Foxp3+ T cells that inhibit colitis. Gastroenterology (2011) 140:2031–43. doi:10.1053/j.gastro.2011.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Hall AOHH, Beiting DP, Tato CM, John B, Lombana CG, Pritchard GH, et al. The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity (2012) 37:511–23. doi:10.1016/j.immuni.2012.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Koenecke C, Lee C-W, Thamm K, Föhse L, Schafferus M, Mittrücker H-W, et al. IFN-γ production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol (2012) 189:2890–6. doi:10.4049/jimmunol.1200413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Afzali B, Mitchell PJ, Edozie FC, Povoleri GAM, Dowson SE, Demandt L, et al. CD161 expression characterizes a subpopulation of human regulatory T cells that produces IL-17 in a STAT3-dependent manner. Eur J Immunol (2013) 43:2043–54. doi:10.1002/eji.201243296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Chaudhry A, Rudensky AY. Control of inflammation by integration of environmental cues by regulatory T cells. J Clin Invest (2013) 123:939–44. doi:10.1172/JCI57175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Pesenacker AM, Bending D, Ursu S, Wu Q, Nistala K, Wedderburn LR. CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood (2013) 121:2647–58. doi:10.1182/blood-2012-08-443473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med (2011) 17:673–5. doi:10.1038/nm.2389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186:3918–26. doi:10.4049/jimmunol.1003099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity (2009) 31:772–86. doi:10.1016/j.immuni.2009.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol (2011) 186:4388–95. doi:10.4049/jimmunol.1003251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Pesenacker AM, Wedderburn LR. T regulatory cells in childhood arthritis – novel insights. Expert Rev Mol Med (2013) 15:e13–13. doi:10.1017/erm.2013.14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20:62–8. doi:10.1038/nm.3432
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. MacDonald KM, Dawson NAJ, Huang Q, Dunne JV, Levings MK, Broady R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol (2015). doi:10.1016/j.jaci.2014.12.1932
122. Chen N, Onisko B, Napoli JL. The nuclear transcription factor RARalpha associates with neuronal RNA granules and suppresses translation. J Biol Chem (2008) 283:20841–7. doi:10.1074/jbc.M802314200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Raverdeau M, Mills KH. Modulation of T cell and innate immune responses by retinoic Acid. J Immunol (2014) 192:2953–8. doi:10.4049/jimmunol.1303245
124. Belkaid Y, Oldenhove G. Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity (2008) 29:362–71. doi:10.1016/j.immuni.2008.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med (2008) 205:2139–49. doi:10.1084/jem.20080414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, et al. Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J Exp Med (2013) 210:1961–76. doi:10.1084/jem.20122508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Bakdash G, Vogelpoel LT, Van Capel TM, Kapsenberg ML, De Jong EC. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol (2014). doi:10.1038/mi.2014.64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A (2014) 111:E3432–40. doi:10.1073/pnas.1408780111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Holder BS, Grant CR, Liberal R, Ma Y, Heneghan MA, Mieli-Vergani G, et al. Retinoic acid stabilizes antigen-specific regulatory T-cell function in autoimmune hepatitis type 2. J Autoimmun (2014) 53:26–32. doi:10.1016/j.jaut.2014.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Hong K, Zhang Y, Guo Y, Xie J, Wang J, He X, et al. All-trans retinoic acid attenuates experimental colitis through inhibition of NF-kappaB signaling. Immunol Lett (2014) 162:34–40. doi:10.1016/j.imlet.2014.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Wang L, Wang J, Jin Y, Gao H, Lin X. Oral administration of all-trans retinoic acid suppresses experimental periodontitis by modulating the Th17/Treg imbalance. J Periodontol (2014) 85:740–50. doi:10.1902/jop.2013.130132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. von Essen MR, Kongsbak M, Schjerling P, Olgaard K, Odum N, Geisler C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol (2010) 11:344–9. doi:10.1038/ni.1851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Joshi S, Pantalena LC, Liu XK, Gaffen SL, Liu H, Rohowsky-Kochan C, et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol (2011) 31:3653–69. doi:10.1128/MCB.05020-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Urry Z, Chambers ES, Xystrakis E, Dimeloe S, Richards DF, Gabrysova L, et al. The role of 1alpha,25-dihydroxyvitamin D3 and cytokines in the promotion of distinct Foxp3+ and IL-10+ CD4+ T cells. Eur J Immunol (2012) 42:2697–708. doi:10.1002/eji.201242370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Van Belle TL, Vanherwegen AS, Feyaerts D, De Clercq P, Verstuyf A, Korf H, et al. 1,25-dihydroxyvitamin D3 and its analog TX527 promote a stable regulatory T cell phenotype in T cells from Type 1 diabetes patients. PLoS One (2014) 9:e109194. doi:10.1371/journal.pone.0109194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Chambers ES, Suwannasaen D, Mann EH, Urry Z, Richards DF, Lertmemongkolchai G, et al. 1alpha,25-dihydroxyvitamin D3 in combination with transforming growth factor-beta increases the frequency of Foxp3(+) regulatory T cells through preferential expansion and usage of interleukin-2. Immunology (2014) 143:52–60. doi:10.1111/imm.12289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Urry ZL, Richards DF, Black C, Morales M, Carnes J, Hawrylowicz CM, et al. Depigmented-polymerised allergoids favour regulatory over effector T cells: enhancement by 1alpha, 25-dihydroxyvitamin D3. BMC Immunol (2014) 15:21. doi:10.1186/1471-2172-15-21
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Breuer J, Schwab N, Schneider-Hohendorf T, Marziniak M, Mohan H, Bhatia U, et al. Ultraviolet B light attenuates the systemic immune response in central nervous system autoimmunity. Ann Neurol (2014) 75:739–58. doi:10.1002/ana.24165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Cantorna MT. Vitamin D and its role in immunology: multiple sclerosis, and inflammatory bowel disease. Prog Biophys Mol Biol (2006) 92:60–4. doi:10.1016/j.pbiomolbio.2006.02.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Dobson R, Giovannoni G, Ramagopalan S. The month of birth effect in multiple sclerosis: systematic review, meta-analysis and effect of latitude. J Neurol Neurosurg Psychiatry (2013) 84:427–32. doi:10.1136/jnnp-2012-303934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Mouli VP, Ananthakrishnan AN. Review article: vitamin D and inflammatory bowel diseases. Aliment Pharmacol Ther (2014) 39:125–36. doi:10.1111/apt.12553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453:65–71. doi:10.1038/nature06880
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol (2010) 11:846–53. doi:10.1038/ni.1915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Duarte JH, Di Meglio P, Hirota K, Ahlfors H, Stockinger B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS One (2013) 8:e79819. doi:10.1371/journal.pone.0079819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol (2009) 183:2475–83. doi:10.4049/jimmunol.0900986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol (2010) 185:5953–61. doi:10.4049/jimmunol.1001628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Curran TA, Jalili RB, Farrokhi A, Ghahary A. IDO expressing fibroblasts promote the expansion of antigen specific regulatory T cells. Immunobiology (2014) 219:17–24. doi:10.1016/j.imbio.2013.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Hao K, Zhou Q, Chen W, Jia W, Zheng J, Kang J, et al. Possible role of the ‘IDO-AhR axis’ in maternal-foetal tolerance. Cell Biol Int (2013) 37:105–8. doi:10.1002/cbin.10023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol (2004) 5:266–71. doi:10.1038/ni1037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Lowe MM, Mold JE, Kanwar B, Huang Y, Louie A, Pollastri MP, et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS One (2014) 9:e87877. doi:10.1371/journal.pone.0087877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Fazio F, Zappulla C, Notartomaso S, Busceti C, Bessede A, Scarselli P, et al. Cinnabarinic acid, an endogenous agonist of type-4 metabotropic glutamate receptor, suppresses experimental autoimmune encephalomyelitis in mice. Neuropharmacology (2014) 81:237–43. doi:10.1016/j.neuropharm.2014.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Rouse M, Singh NP, Nagarkatti PS, Nagarkatti M. Indoles mitigate the development of experimental autoimmune encephalomyelitis by induction of reciprocal differentiation of regulatory T cells and Th17 cells. Br J Pharmacol (2013) 169:1305–21. doi:10.1111/bph.12205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Liu Y, She W, Wang F, Li J, Wang J, Jiang W. 3, 3’-diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int Immunopharmacol (2014) 23:489–98. doi:10.1016/j.intimp.2014.09.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Punj S, Kopparapu P, Jang HS, Phillips JL, Pennington J, Rohlman D, et al. Benzimidazoisoquinolines: a new class of rapidly metabolized aryl hydrocarbon receptor (AhR) ligands that induce AhR-dependent Tregs and prevent murine graft-versus-host disease. PLoS One (2014) 9:e88726. doi:10.1371/journal.pone.0088726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev (2014) 13:668–77. doi:10.1016/j.autrev.2013.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta (2008) 1783:673–94. doi:10.1016/j.bbamcr.2008.01.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther (2006) 112:358–404. doi:10.1016/j.pharmthera.2005.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D, et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol (2014) 177:531–43. doi:10.1111/cei.12354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Mizumoto N, Kumamoto T, Robson SC, Sevigny J, Matsue H, Enjyoji K, et al. CD39 is the dominant Langerhans cell associated ecto-NTPDase: modulatory roles in inflammation and immune responsiveness. Nat Med (2002) 8:358–65. doi:10.1038/nm0402-358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Salmi M, Jalkanen S. Cell-surface enzymes in control of leukocyte trafficking. Nat Rev Immunol (2005) 5:760–71. doi:10.1038/nri1705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Corriden R, Chen Y, Inoue Y, Beldi G, Robson SC, Insel PA, et al. Ecto-nucleoside triphosphate diphosphohydrolase 1 (E-NTPDase1/CD39) regulates neutrophil chemotaxis by hydrolyzing released ATP to adenosine. J Biol Chem (2008) 283:28480–6. doi:10.1074/jbc.M800039200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Wang Y, Begum-Haque S, Telesford KM, Ochoa-Reparaz J, Christy M, Kasper EJ, et al. A commensal bacterial product elicits and modulates migratory capacity of CD39(+) CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes (2014) 5:552–61. doi:10.4161/gmic.29797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Narravula S, Lennon PF, Mueller BU, Colgan SP. Regulation of endothelial CD73 by adenosine: paracrine pathway for enhanced endothelial barrier function. J Immunol (2000) 165:5262–8. doi:10.4049/jimmunol.165.9.5262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi:10.3389/fimmu.2012.00190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med (2007) 204:1303–10. doi:10.1084/jem.20062129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, et al. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol (2009) 182:4017–24. doi:10.4049/jimmunol.0803310
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Klein M, Vaeth M, Scheel T, Grabbe S, Baumgrass R, Berberich-Siebelt F, et al. Repression of cyclic adenosine monophosphate upregulation disarms and expands human regulatory T cells. J Immunol (2012) 188:1091–7. doi:10.4049/jimmunol.1102045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Fassbender M, Gerlitzki B, Ullrich N, Lupp C, Klein M, Radsak MP, et al. Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell Immunol (2010) 265:91–6. doi:10.1016/j.cellimm.2010.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Bodor J, Bopp T, Vaeth M, Klein M, Serfling E, Hunig T, et al. Cyclic AMP underpins suppression by regulatory T cells. Eur J Immunol (2012) 42:1375–84. doi:10.1002/eji.201141578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Liao H, Hyman MC, Baek AE, Fukase K, Pinsky DJ. cAMP/CREB-mediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) expression. J Biol Chem (2010) 285:14791–805. doi:10.1074/jbc.M110.116905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Cao J, Zhang X, Wang Q, Wang X, Jin J, Zhu T, et al. Cyclic AMP suppresses TGF-beta-mediated adaptive Tregs differentiation through inhibiting the activation of ERK and JNK. Cell Immunol (2013) 285:42–8. doi:10.1016/j.cellimm.2013.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Pozo D, Anderson P, Gonzalez-Rey E. Induction of alloantigen-specific human T regulatory cells by vasoactive intestinal peptide. J Immunol (2009) 183:4346–59. doi:10.4049/jimmunol.0900400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Walter J, Ley R. The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol (2011) 65:411–29. doi:10.1146/annurev-micro-090110-102830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature (2011) 478:250–4. doi:10.1038/nature10434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331:337–41. doi:10.1126/science.1198469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity (2011) 34:794–806. doi:10.1016/j.immuni.2011.03.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Maria A, English KA, Gorham JD. Appropriate development of the liver Treg compartment is modulated by the microbiota and requires TGF-beta and MyD88. J Immunol Res (2014) 2014:279736. doi:10.1155/2014/279736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med (2014) 20:642–7. doi:10.1038/nm.3568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi:10.1038/nature12726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi:10.1038/nature12721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi:10.1126/science.1241165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Ochoa-Reparaz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, et al. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol (2010) 3:487–95. doi:10.1038/mi.2010.29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A (2010) 107:12204–9. doi:10.1073/pnas.0909122107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science (2005) 308:1635–8. doi:10.1126/science.1110591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Sieber JR, McInerney MJ, Gunsalus RP. Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol (2012) 66:429–52. doi:10.1146/annurev-micro-090110-102844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Cummings JH, Hill MJ, Bone ES, Branch WJ, Jenkins DJ. The effect of meat protein and dietary fiber on colonic function and metabolism. II. Bacterial metabolites in feces and urine. Am J Clin Nutr (1979) 32:2094–101.
188. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol (2014) 12:661–72. doi:10.1038/nrmicro3344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Guilloteau P, Martin L, Eeckhaut V, Ducatelle R, Zabielski R, Van Immerseel F. From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr Res Rev (2010) 23:366–84. doi:10.1017/S0954422410000247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut (1987) 28:1221–7. doi:10.1136/gut.28.10.1221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Sleeth ML, Thompson EL, Ford HE, Zac-Varghese SE, Frost G. Free fatty acid receptor 2 and nutrient sensing: a proposed role for fibre, fermentable carbohydrates and short-chain fatty acids in appetite regulation. Nutr Res Rev (2010) 23:135–45. doi:10.1017/S0954422410000089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40:128–39. doi:10.1016/j.immuni.2013.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature (2008) 453:620–5. doi:10.1038/nature07008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science (2011) 332:974–7. doi:10.1126/science.1206095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Wang Y, Telesford KM, Ochoa-Reparaz J, Haque-Begum S, Christy M, Kasper EJ, et al. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun (2014) 5:4432. doi:10.1038/ncomms5432
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Cerdeno-Tarraga AM, Patrick S, Crossman LC, Blakely G, Abratt V, Lennard N, et al. Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science (2005) 307:1463–5. doi:10.1126/science.1107008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell (2005) 122:107–18. doi:10.1016/j.cell.2005.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Shen Y, Giardino Torchia ML, Lawson GW, Karp CL, Ashwell JD, Mazmanian SK. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe (2012) 12:509–20. doi:10.1016/j.chom.2012.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker HC, Kasper DL. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe (2014) 15:413–23. doi:10.1016/j.chom.2014.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Himmel ME, Hardenberg G, Piccirillo CA, Steiner TS, Levings MK. The role of T-regulatory cells and Toll-like receptors in the pathogenesis of human inflammatory bowel disease. Immunol (2008) 125(2):145–53. doi:10.1111/j.1365-2567.2008.02939.x
201. Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A (2006) 103:7048–53. doi:10.1073/pnas.0601554103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Sutmuller RP, Den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest (2006) 116:485–94. doi:10.1172/JCI25439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Oberg HH, Ly TT, Ussat S, Meyer T, Kabelitz D, Wesch D. Differential but direct abolishment of human regulatory T cell suppressive capacity by various TLR2 ligands. J Immunol (2010) 184:4733–40. doi:10.4049/jimmunol.0804279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Nyirenda MH, Sanvito L, Darlington PJ, O’brien K, Zhang GX, Constantinescu CS, et al. TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J Immunol (2011) 187:2278–90. doi:10.4049/jimmunol.1003715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Kwon HK, Lee CG, So JS, Chae CS, Hwang JS, Sahoo A, et al. Generation of regulatory dendritic cells and CD4+Foxp3+ T cells by probiotics administration suppresses immune disorders. Proc Natl Acad Sci U S A (2010) 107:2159–64. doi:10.1073/pnas.0904055107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Poutahidis T, Kleinewietfeld M, Smillie C, Levkovich T, Perrotta A, Bhela S, et al. Microbial reprogramming inhibits western diet-associated obesity. PLoS One (2013) 8:e68596. doi:10.1371/journal.pone.0068596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Smits HH, Engering A, Van Der Kleij D, De Jong EC, Schipper K, Van Capel TM, et al. Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin. J Allergy Clin Immunol (2005) 115:1260–7. doi:10.1016/j.jaci.2005.03.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Lopez P, Gonzalez-Rodriguez I, Sanchez B, Ruas-Madiedo P, Suarez A, Margolles A, et al. Interaction of Bifidobacterium bifidum LMG13195 with HT29 cells influences regulatory-T-cell-associated chemokine receptor expression. Appl Environ Microbiol (2012) 78:2850–7. doi:10.1128/AEM.07581-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Vitaliti G, Pavone P, Guglielmo F, Spataro G, Falsaperla R. The immunomodulatory effect of probiotics beyond atopy: an update. J Asthma (2014) 51:320–32. doi:10.3109/02770903.2013.862259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108:1571–9. doi:10.1182/blood-2006-02-004747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol (2008) 9:632–40. doi:10.1038/ni.1607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF- to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol (2007) 178:2018–27. doi:10.4049/jimmunol.178.4.2018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Liao W, Lin J-X, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol (2011) 12:551–9. doi:10.1038/ni.2030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J Clin Invest (2003) 112:1437–43. doi:10.1172/JCI200319441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Amarnath S, Dong L, Li J, Wu Y, Chen W. Endogenous TGF-beta activation by reactive oxygen species is key to Foxp3 induction in TCR-stimulated and HIV-1-infected human CD4+CD25- T cells. Retrovirology (2007) 4:57–57. doi:10.1186/1742-4690-4-57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity (2008) 28:100–11. doi:10.1016/j.immuni.2007.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Passerini L, Allan SE, Battaglia M, Di Nunzio S, Alstad AN, Levings MK, et al. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25- effector T cells. Int Immunol (2008) 20:421–31. doi:10.1093/intimm/dxn002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
219. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (2010) 329:1667–71. doi:10.1126/science.1191996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med (2007) 13:423–31. doi:10.1038/nm1564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol (2008) 9:1347–55. doi:10.1038/ni.1677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Goswami R, Jabeen R, Yagi R, Pham D, Zhu J, Goenka S, et al. STAT6-dependent regulation of Th9 development. J Immunol (2012) 188:968–75. doi:10.4049/jimmunol.1102840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
223. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
224. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
225. Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol (2012) 181:8–18. doi:10.1016/j.ajpath.2012.03.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol (2008) 9:641–9. doi:10.1038/ni.1610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol (2008) 9:650–7. doi:10.1038/ni.1613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature (2008) 454:350–2. doi:10.1038/nature07021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: regulatory T cells, FOXP3, cytokines, metabolites, microbiome, plasticity, environment, immune regulation
Citation: Hoeppli RE, Wu D, Cook L and Levings MK (2015) The environment of regulatory T cell biology: cytokines, metabolites, and the microbiome. Front. Immunol. 6:61. doi: 10.3389/fimmu.2015.00061
Received: 16 December 2014; Paper pending published: 15 January 2015;
Accepted: 30 January 2015; Published online: 18 February 2015.
Edited by:
David A. Hafler, Yale School of Medicine, USAReviewed by:
António Gil Castro, University of Minho, PortugalMargarita Dominguez-Villar, Yale School of Medicine, USA
Copyright: © 2015 Hoeppli, Wu, Cook and Levings. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan K. Levings, Department of Surgery, Child and Family Research Institute, University of British Columbia, A4-186, 950 West 28th Avenue, Vancouver, BC V5Z 4H4, Canada e-mail:bWxldmluZ3NAbWFpbC51YmMuY2E=
†Romy E. Hoeppli, Dan Wu and Laura Cook have contributed equally to this work.