 Linda Ferrante
Linda Ferrante Siri Hauge Opdal
Siri Hauge Opdal- 1Department of Research in Forensic Pathology, Norwegian Institute of Public Health, Oslo, Norway
- 2Department of Pathology, Oslo University Hospital, Oslo, Norway
Several studies report signs of slight infection prior to death in cases of sudden infant death syndrome (SIDS). Based on this, a hypothesis of an altered immunological homeostasis has been postulated. The cytokines are important cellular mediators that are crucial for infant health by regulating cell activity during the inflammatory process. The pro-inflammatory cytokines favor inflammation; the most important of these are IL-1α, IL-1β, IL-6, IL-8, IL-12, IL-18, TNF-α, and IFN-γ. These cytokines are controlled by the anti-inflammatory cytokines. This is accomplished by reducing the pro-inflammatory cytokine production, and thus counteracts their biological effect. The major anti-inflammatory cytokines are interleukin-1 receptor antagonist (IL-1ra), IL-4, IL-10, IL-11, and IL-13. The last decade there has been focused on genetic studies within genes that are important for the immune system, for SIDS with a special interest of the genes encoding the cytokines. This is because the cytokine genes are considered to be the genes most likely to explain the vulnerability to infection, and several studies have investigated these genes in an attempt to uncover associations between SIDS and different genetic variants. So far, the genes encoding IL-1, IL-6, IL-10, and TNF-α are the most investigated within SIDS research, and several studies indicate associations between specific variants of these genes and SIDS. Taken together, this may indicate that in at least a subset of SIDS predisposing genetic variants of the immune genes are involved. However, the immune system and the cytokine network are complex, and more studies are needed in order to better understand the interplay between different genetic variations and how this may contribute to an unfavorable immunological response.
Introduction
Sudden infant death syndrome (SIDS) is defined as the sudden unexpected death of an infant <1 year of age, with onset of the fatal episode apparently occurring during sleep, that remains unexplained after a thorough investigation, including performance of a complete autopsy and review of the circumstances of death and the clinical history (1). Even though there have been numerous studies trying to understand the pathophysiological mechanisms of SIDS, we still not fully understand what causes these deaths, or how to prevent them. The fatal triangle developed by Rognum et al. (2) suggest that SIDS occur when an infant at the same time is in a vulnerable developmental stage, with a rapid development of both the central nervous system (CNS) and the immune system, has predisposing factors such as an unfortunate genetic “make-up” or brainstem astrogliosis, and that trigger events such as slight infection, prone sleeping, maternal smoking, or overheating are present (Figure 1) (2).
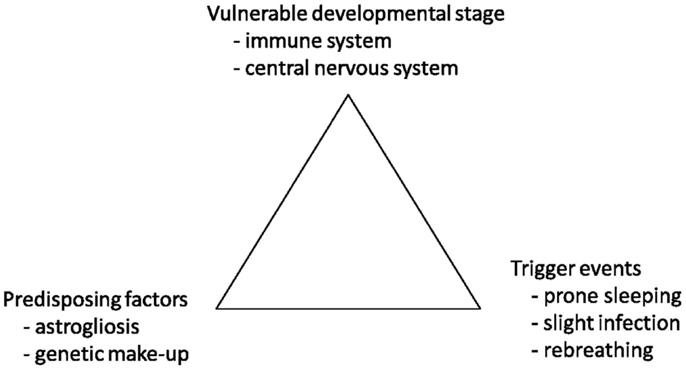
Figure 1. The hypothesis of a fatal triangle in SIDS. Modified with permission from Rognum and Saugstad (2).
SIDS and Infection
There is compelling evidence for a dysfunctional immune response in SIDS, and already in 1947, Werne et al. suggested that respiratory infection was the cause of death in an otherwise healthy infant (3). Since then, there have been numerous reports and papers describing signs of slight infection in SIDS infants (4–7). An immunological “overreaction” has been postulated since about half of the SIDS victims have had symptoms of slight infection in the days before death (8, 9).
Arnon et al. have hypothesized that some infants might suffer respiratory arrest due to botulinum toxin produced by Clostridium botulinum (10). From a cohort of 280 infants, they showed that botulinum toxin was present in 10 infants, of which 9 had been diagnosed as SIDS (10). Stoltenberg et al. have reported immune stimulation in both the upper airways and intestines, showing that SIDS had higher number of IgM immunocytes in the tracheal wall than controls, but significantly lower numbers of IgA and IgM immunocytes than cases of infectious death (11). In the duodenal mucosa, the number of IgA immunocytes was higher in SIDS cases than in controls. These findings indicate that the mucosal immune system is activated in a large proportion of SIDS (11). It is also shown that SIDS has higher IgG and IgA immunocyte density in the palatine tonsillar compartments than controls (12). Furthermore, salivary glands have a higher number of CD45+ stromal leukocytes, as well as intensified epithelial expression of HLA-DR and secretory component, and increased endothelial expression of HLA class I and II (13). These observations confirm that the immune system is activated in SIDS, probably with release of certain cytokines that are known to up-regulate epithelial expression of HLA-DR and secretory component (13).
A real breakthrough for the immunological overreaction theory was the demonstration by Vege et al. (8, 14), who showed that SIDS victims who have had signs of slight infection prior to death had both increased number of IgA immunocytes and HLA-DR expression in their laryngeal mucosa, as well as increased levels of IL-6 in their cerebrospinal fluid (CSF). In fact, half of the SIDS victims had CSF IL-6 concentrations in the same range as victims of meningitis and septicemia (8). A further support for the infection theory is a study performed on registry data from Norway and Sweden, which suggests that there is a co-variation between epidemics of whooping cough and SIDS (15). The association was stronger in Sweden than in Norway, which may reflect that Swedish infants are not vaccinated against Bordetella pertussis while the Norwegians infants are (15).
Stray-Pedersen et al. showed that SIDS victims with positive Helicobacter pylori stool antigen (HpSA) immunoassay had elevated IL-6 in the CSF compared to SIDS victims with negative HpSA test (16). Furthermore, detection of Helicobacter pylori antigen in stool was found associated with SIDS and death due to infection, indicating that this bacteria may represent a contributing factor to sudden death during the first months of life (16).
Surfactant protein A (SP-A) is a protein produced in the lungs, with a major purpose to reduce the surface tension at the alveolar air–liquid interface. Furthermore, it takes part in regulation of the inflammatory process. Interestingly, with regard to SIDS, there is a drop in alveolar SP-A expression in the first months after birth (17), corresponding to the classical age peak of SIDS. Thus, it may be hypothesized that this transient low expression of SP-A may be a part of the increased vulnerability for SIDS at that age (17).
It is also suggested that Staphylococcus aureus (S. aureus) are involved in events leading to SIDS (18). Based on observations from samples collected from the intestinal tract in SIDS compared with samples from feces from a group of healthy controls, it was shown that S. aureus and staphylococcal enterotoxins were more prevalent in SIDS. However, as much as 40% of the controls were positive for S. aureus, indicating that this bacteria is common in infants, and that the detection may not be seen as a support for the diagnosis of SIDS (18).
Another study investigated pyrogenic toxins of S. aureus in SIDS infants from different countries (19). The study reported these pyrogenic toxins in >50% of SIDS infants from three different countries; Scotland, France, and Australia, and suggest that further investigation into the effect of the toxins may be important (19). A study by Blackwell et al. found that the prevalence of S. aureus in nasopharyngeal flora was significantly higher in SIDS cases compared to age-matched healthy controls (20). Furthermore, SIDS found in a prone sleeping position more often had symptoms of slight infection prior to death than the babies put to sleep on their back.
The relationship between laryngeal immune stimulation, clinical signs of slight infection prior to death, and high levels of IL-6 in CSF may indicate an interaction between the immune system and the CNS (14). The assumption of such a relationship is strengthened by the recently reported increased IL-6 receptor expression on serotonergic cells in brain stem nuclei involved in respiratory regulation in SIDS cases compared to controls (21). Almost half of the investigated SIDS cases had signs of mild infection prior to death, and the study provides evidence for aberrant interactions in SIDS infants between IL-6 and the area of the brain stem involved in protective responses to hypercapnia, potentially induced by the combined effect of prone position and mild infection.
Complement Component C4
The first gene involved in the immune system to be investigated with regard to SIDS was the gene encoding complement component C4. This gene consists of two loci, C4A and C4B, and is highly polymorphic. Partial deletions of the C4 gene are common and found in 5–20% of Caucasians (22). The C4 gene has been investigated in both German and Norwegian SIDS victims, but none of these studies detected any differences between SIDS cases and controls with regard to allele frequencies (23–25). However, two of the studies revealed an association between slight infections prior to death and partial deletions of either the C4A or the C4B gene, which may indicate that this combination represents increased risk of sudden infant death (Table 1) (23, 25).
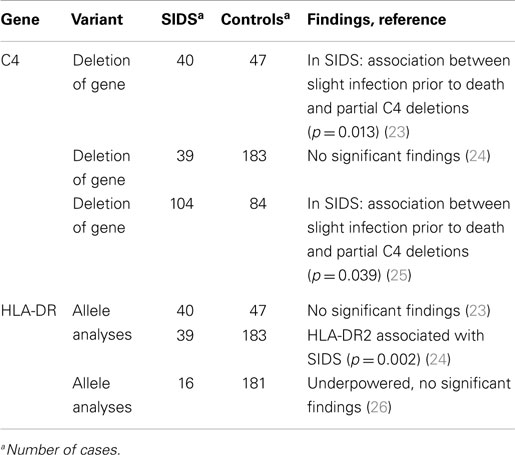
Table 1. C4 and HLA-DR gene variants investigated in cases of SIDS.
HLA-DR
Numerous diseases are associated with different alleles of the major histocompatibility complex, and HLA-DR has been investigated in a few SIDS victims (Table 1). There has been reported a significant decreased frequency of HLA-DR2 in a study including 39 SIDS cases and 183 controls (24). However, Schneider et al. (23) investigated 40 SIDS cases and found no significant difference in the HLA-DR gene frequencies between the SIDS cases and the controls, an observation, which was later confirmed in a study of 16 Norwegian SIDS cases (26).
IL-10
The genes most likely to explain the vulnerability to infection seen in SIDS are the cytokine genes. Several studies have investigated these genes in an attempt to uncover associations between SIDS and different genetic variants, and one of the most investigated is IL-10.
IL-10 is an important immune regulatory cytokine that downregulates the production of pro-inflammatory cytokines, such as IL-6 and TNF-α. Levels of IL-10 control the balance between inflammatory and humoral responses, and IL-10 therefore plays an important role in the development of infectious disease. Variability in IL-10 production has a hereditary component of approximately 75%, and the SNPs in the promoter region in position −1082, −819, and −592, as well as the microsatellite IL-10R and IL-10G, are collectively responsible for the production of the protein (27, 28).
The first study investigating the IL-10 gene in SIDS was by Summers et al., who investigated 23 SIDS cases and found that SIDS was associated with both the ATA haplotype and the −592A allele (29). However, according to the authors, babies who died of other causes might have been included in the SIDS group, which may have influenced the results (29). A Norwegian study of 214 SIDS cases was unable to confirm the association to SIDS, but found an association between the ATA haplotype and the ATA/ATA genotype and infectious death (Table 2) (30). The latter study also investigated the microsatellites, and found a higher percentage of the genotypes G21/G22 and G21/G23 in cases of infectious death compared to SIDS, and a higher percentage of G21/G22 in the SIDS cases compared to controls, while there were no differences between the groups for the IL-10R area (Table 2) (30). Subsequent studies investigating this gene have reported conflicting results, a small study investigating that 38 SIDS cases found an association between −592A and ATA/ATA and SIDS (31), while other studies did not (Table 2) (32, 33). An Australian study investigated the −1082A/G polymorphism in SIDS cases and SIDS parents from different countries, but did not find any differences in genotype frequency between this combined SIDS population and controls, neither between SIDS parents and controls (Table 2) (34). This study also investigated the influence of genotype and smoking on IL-10 responses. They found that the pooled data from smokers had significantly lower levels of IL-10 responses to TSTT, but there were no significant differences for smokers compared with non-smokers for the three genotypes (34). The lowest levels of IL-10 responses were observed among smokers who were homozygous for the −1082A allele, which is most prevalent among Aboriginal Australians and Bangladeshis. This is interesting, as the major difference between the risk factors for SIDS in these two groups is the level of exposure of infants to maternal smoking.
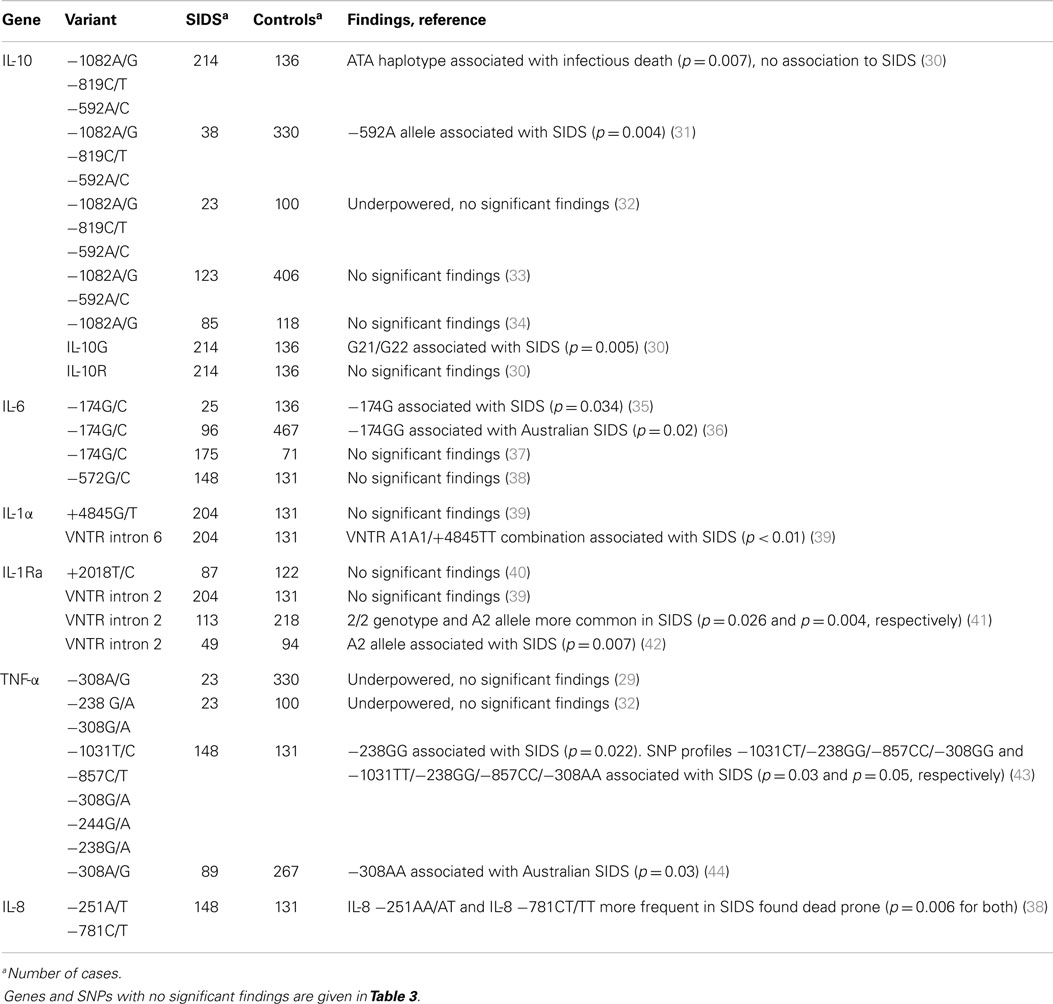
Table 2. Interleukin and cytokine gene polymorphisms investigated in cases of SIDS.
Based on the findings in these studies, one might speculate that in some situations an infant with an unfavorable IL-10 genotype may exhibit aberrant IL-10 production, which in turn leads to a disturbed immunological homeostasis and an increased risk for sudden death. This may be especially unfavorable if exposed to smoking, in particular in utero or if a smoking mother is breast-feeding.
IL-6
IL-6 is an acute phase protein that induces B- and T-cell growth and differentiation. IL-6 is also an important mediator of fever, and influences the effect of other cytokines. The first study of the IL-6 gene in cases of SIDS was a British study that included common polymorphisms in the genes encoding IL-4, IL-6, IFN-γ, TGF, and VEGF (35). They found significant differences for the genes encoding IL-6 and VEGF: the genotypes IL-6 −174GG, and VEGF −1154AA were more frequent in SIDS cases than in controls (Table 2) (35). Even though only a small number of SIDS cases were included, the authors suggest that the causation of SIDS is related to both fetal lung development and an infant’s innate ability to mount an inflammatory response to infection (35). The findings in the IL-6 gene have been confirmed in a study of Australian SIDS cases (36), but not in a Norwegian study (Table 2) (37). In the Australian study, it was in addition found a relationship between IL-6 responses to endotoxin, IL-6 genotype, and smoking status (36). A study by Ferrante et al., investigated the −572G/C polymorphism in the IL-6 gene in 148 SIDS cases, but it did not find any association between this SNP and SIDS (Table 2) (38). A study evaluating the correlation between HLA-DR expression in laryngeal mucosa and interleukin gene variation found that 12 of 13 SIDS cases (92%) with high HLA-DR expression, prone sleeping position, and sighs of infection prior to death had the IL-6 −176 CG/CC genotypes (p = 0.01) (45).
IL-1
IL-1 is a pro-inflammatory cytokine that induces the synthesis of acute phase proteins, and also induces fever. There are two structurally distinct forms of IL-1: IL-1α, which is the acidic form and IL-1β, which is the neutral form. IL-1 is regulated by the competitive antagonist IL-1Ra. The polymorphisms IL-1β −511C/T and IL-1Ra +2018T/C have a significant effect on the IL-1β levels. An Australian study investigated a combined SIDS group with cases from different countries with European controls, but did not find any association between those SNPs and SIDS (Table 2) (40). It was, however, shown that smoking had a significant effect on both IL-1β and IL-1ra responses to endotoxin, and that this effect differed according to genotype (40). This finding is highly interesting, since maternal smoking is one of the most well-known risk factors for SIDS (9, 46, 47).
A Norwegian study investigated a variable number of tandem repeat (VNTR) in intron 6 and the SNP in +4845G/T in the IL-1α gene, as well as the −511C/T polymorphism in the gene encoding IL-1β and a VNTR in intron 2 of the gene encoding IL-1ra (Tables 2 and 3) (39). When investigating each polymorphism separately, no association to SIDS was found. However, when combining VNTR and SNP genotypes, an association between the gene combination IL-1α VNTR A1A1/+4845TT and SIDS was disclosed, 16% of the SIDS cases had this combination compared to 1.8% of the controls (p < 0.01) (Table 2). In the SIDS group, it was also found that the genotypes IL-1β −511CC/CT were significantly more frequent in the SIDS victims who found dead in a prone sleeping position compared with SIDS victims who found dead in other sleeping positions (p = 0.004) (39).
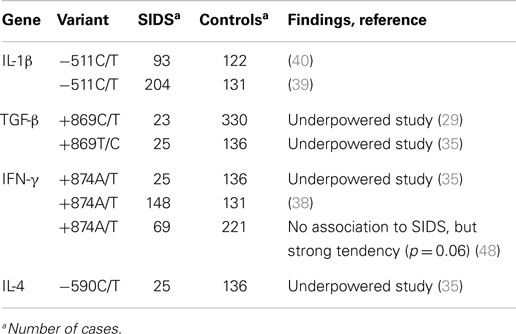
Table 3. Genes and SNPs with no significant findings with regard to SIDS.
The 89bp VNTR in the IL-1Ra gene have also been investigated in Australian SIDS cases, where it was found that carriage of the 2/2 genotype increased the risk for SIDS compared with the predominant 1/1 genotype (Table 2) (41). Homozygous carriers of allele 2 show a more severe and also prolonged pro-inflammatory immune response compared to other IL-1Ra genotypes (49), which may contribute to the vulnerability to infection seen in SIDS. A smaller study investigating the IL-1ra VNTR in 13 SIDS cases and 103 controls found an association between A2A2 and SIDS (Table 1) (42).
Tumor Necrosis Factor Alpha
Tumor necrosis factor alpha (TNF-α) is a transmembrane protein produced as a result of the presence of bacterial toxins. TNF-α is an important regulator of immune cells, in addition to stimulate inflammation and controlling viral replication. Two smaller studies have investigated the TNF-α polymorphisms −238A/G and −308A/G, but did not find any association to SIDS (Table 2) (29, 32). Five SNPs in the promoter region of the TNF-α gene have been investigated in a Norwegian SIDS population (43). The study found an association between the genotype −238GG and SIDS (Table 2). In addition, SNP profiles −1031CT/−238GG/−857CC/−308GG and −1031TT/−238GG/−857CC/−308AA found more often in SIDS; this may therefore be unfavorable SNP combinations (43). An Australian study investigated the −308G/A polymorphism in SIDS cases from different countries, and found a significantly higher proportion of the AA genotype among Australian SIDS cases compared to controls (Table 2) (44).
Other Cytokines
In addition to the genes mentioned above have also SNPs in the genes encoding IL-4 IL-8, IL-12, IL-13, IL-16, IL-18, TGF-β1, and INF-γ been investigated in SIDS (29, 35, 38). A Norwegian study found that the genotypes IL-8 −251AA/AT and IL-8 −781CT/TT were significantly more frequent in SIDS cases found dead in a prone sleeping position compared with SIDS cases found dead in other sleeping positions (Table 2) (38). Further, the IL-8 genotypes −251AA/AT and −781CT/TT were more often observed in SIDS cases with positive HLA-DR and one or more risk factors compared with SIDS cases with negative HLA-DR, no infection, and supine sleeping (45). An Australian study investigating SIDS cases from different countries found a marginal association with the IFN-γ +874AA genotype and SIDS (Table 3) (48).
Other Immune Genes
An increased vulnerability to infection may also be due to genetic variation in the genes encoding G-proteins. The most investigated polymorphism in the Gβ3 gene is C825T, and it is shown that T-allele results in increased G-protein-mediated signal transduction compared to the C-allele (50). Most interleukin-receptors are G-protein coupled, and an association between the Gβ3 825T allele and increased cell function has been reported (51). A study looking at the C825T polymorphism in SIDS victims, cases of infectious death, and live infant controls, revealed no difference in genotype frequency between SIDS cases and controls (52), but an association between the CC genotype and infectious death was found (p = 0.016). This observation may indicate that the presence of the 825T allele exerts a protective effect toward serious infection, perhaps through enhanced G-protein signaling.
Surfactant protein A and surfactant protein D (SP-D) are humoral molecules involved in the innate host defense against various bacterial and viral pathogens. Ten common SNPs that might influence expression of the genes encoding these two surfactants have been investigated in SIDS cases and controls (p = 0.08) (53). No difference in genotype distribution was found, even though there was a tendency for the most common SP-A haplotype, 6A2/1A0, to be overrepresented in cases with low immunohistochemical SP-A expression (53). None of the other SP-A haplotypes was associated with high or low SP-A expression, and the same was true for the two investigated SP-D SNPs (53).
Polymorphisms that influence the expression of toxin receptors could contribute to SIDS, at least in the cases where there is evidence of toxin involvement. One gene that influences the expression of receptors for staphylococcal enterotoxin B and C in humans are the TCRBV3S1 gene, and a C–T SNP in a recombination signal sequence (RSS) gene region is shown to influence the expression of the gene (54). This SNP have been investigated in 48 Australian SIDS cases and 96 controls, but no differences were found between SIDS cases and controls (55).
Another protein that might be of importance with regard to endotoxins in SIDS is CD14. The TT genotype of the CD14 −260C/T polymorphism causes a significantly higher density of CD14 receptor expression in monocytes, which makes the individual more sensitive to endotoxin than those with the CC genotype (56). This polymorphism has been investigated in an Australian cohort of 116 SIDS cases and 228 controls (57). No differences were found in genotype frequencies between SIDS cases and controls, and the authors conclude that the CD14 −260C/T polymorphism is unlikely to be involved in SIDS (57).
Discussion
The many genetic studies within immune genes in SIDS strongly suggest that these infants do have a combination of genotypes making them vulnerable to infections. The predispositions reported so far are mostly common SNPs with association to SIDS, but genetic variants with strong dominance in SIDS still remains to be uncovered. However, the challenges within genetic studies of SIDS are many. First and foremost is the fact that the SIDS diagnosis is an exclusion diagnosis and important challenge, and even though a great effort has been done in order to standardize is still different diagnostic criteria used in different countries. This might result in that infant with other causes of death, such as accidental asphyxia, or interstitial pneumonia might be included in the SIDS group, and thus camouflage a true genetic association.
Another challenge to genetic studies is the genetic variation between different ethnic groups, which makes it difficult to compare results between different studies without including a potential error. This must be taken into account when investigating SIDS. In addition, the investigated SIDS populations are often small, which make the genetic studies more difficult to interpret. The number of SIDS cases a year in, for instance, Norway is at the moment about 20, making it difficult and time consuming to be able to collect a large number of cases.
The fact that the number of cases and controls in each study often is small in relation to the number of statistical tests that is undertaken increases the risk of false negatives. Several studies include <25 SIDS cases, which makes it difficult to draw any firm conclusions. Even so, to our best of knowledge are all studies investigating immune genes in SIDS included in Tables 1–3, in order to give a comprehensive survey of the research done regarding this topic so far. A large scale GWAS study might be an important next step. The drawback of this method is, however, that important information may be lost due to the corrections that have to be made when investigating such a vast number of polymorphisms, in addition to the problem with limited number of cases. Even so, one might find polymorphisms that could be expected to have a small effect on the SIDS risk. This may be important as these gene variants may point directly to the underlying cause, such as for instance a sub-optimal immune reaction.
Despite the limitations, association studies are still able to report several genetic variations within many immune genes that might be associated with SIDS (Tables 1 and 2). This strongly suggest that these genes are of importance and hopefully in the future can provide even better and more accurate understanding of the role of the immune system in SIDS. However, immunology is a complicated biological field, and perhaps the most interesting frontiers in the genetics of immunity arise in the interaction between immune activity and other physiological or developmental processes. A better understanding of how genetic variation might influence the functional effect of the encoded proteins and how this can lead to a fatal outcome is important, and hopefully, with the new possibilities present within genetic research, this can be elucidated. Further would an in-depth analysis of the correlation between genotypes, interleukin response, and risk factors, such as smoking exposure, in different SIDS populations be interesting and might shed light on the involvement of the immune response in these deaths.
Conclusion
The studies so far suggest that some SIDS infants may have a genetic vulnerability in the regulation of the immune system. An unfortunate combination of polymorphisms in genes involved in the immune system, in particular in the cytokine genes, may lead to an imbalance in the immune response and render the infant unable to cope with an infection. However, the immune system and the cytokine network are complex, and more studies are needed in order to better understand the interplay between different genetic variations and how this may contribute to an unfavorable immunological response. The associations observed so far between different polymorphisms and SIDS and environmental factors for SIDS most likely represent only a small part of genetic patterns that may result in an unfortunate immunological reaction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, et al. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics (2004) 114:234–8. doi: 10.1542/peds.114.1.234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Rognum TO, Saugstad OD. Biochemical and immunological studies in SIDS victims. Clues to understanding the death mechanism. Acta Paediatr Suppl (1993) 82(Suppl 389):82–5. doi:10.1111/j.1651-2227.1993.tb12886.x
3. Werne J, Garrow I. Sudden deaths of infants allegedly due to mechanical suffocation. Am J Public Health Nations Health (1947) 37:675–87. doi:10.2105/AJPH.37.6.675
4. Blackwell CC, Weir DM. The role of infection in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25:1–6. doi:10.1111/j.1574-695X.1999.tb01320.x
5. Blood-Siegfried J. The role of infection and inflammation in sudden infant death syndrome. Immunopharmacol Immunotoxicol (2009) 31:516–23. doi:10.3109/08923970902814137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Highet AR. An infectious aetiology of sudden infant death syndrome. J Appl Microbiol (2008) 105:625–35. doi:10.1111/j.1365-2672.2008.03747.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Vege A, Rognum TO. Sudden infant death syndrome, infection and inflammatory responses. FEMS Immunol Med Microbiol (2004) 42:3–10. doi:10.1016/j.femsim.2004.06.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Vege A, Rognum TO, Scott H, Aasen AO, Saugstad OD. SIDS cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr (1995) 84:193–6. doi:10.1111/j.1651-2227.1995.tb13608.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Arnestad M, Andersen M, Vege A, Rognum TO. Changes in the epidemiological pattern of sudden infant death syndrome in southeast Norway, 1984-1998: implications for future prevention and research. Arch Dis Child (2001) 85:108–15. doi:10.1136/adc.85.2.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Arnon SS, Midura TF, Damus K, Wood RM, Chin J. Intestinal infection and toxin production by Clostridium botulinum as one cause of sudden infant death syndrome. Lancet (1978) 1:1273–7. doi:10.1016/S0140-6736(78)91264-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Stoltenberg L, Saugstad OD, Rognum TO. Sudden infant death syndrome victims show local immunoglobulin M response in tracheal wall and immunoglobulin A response in duodenal mucosa. Pediatr Res (1992) 31:372–5. doi:10.1203/00006450-199204000-00013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Stoltenberg L, Vege A, Saugstad OD, Rognum TO. Changes in the concentration and distribution of immunoglobulin-producing cells in SIDS palatine tonsils. Pediatr Allergy Immunol (1995) 6:48–55. doi:10.1111/j.1399-3038.1995.tb00258.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Thrane PS, Rognum TO, Brandtzaeg P. Up-regulated epithelial expression of HLA-DR and secretory component in salivary glands: reflection of mucosal immunostimulation in sudden infant death syndrome. Pediatr Res (1994) 35:625–8. doi:10.1203/00006450-199405000-00017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Vege A, Rognum TO, Anestad G. IL-6 cerebrospinal fluid levels are related to laryngeal IgA and epithelial HLA-DR response in sudden infant death syndrome. Pediatr Res (1999) 45:803–9. doi:10.1203/00006450-199906000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Lindgren C, Milerad J, Lagercrantz H. Sudden infant death and prevalence of whooping cough in the Swedish and Norwegian communities. Eur J Pediatr (1997) 156:405–9. doi:10.1007/s004310050626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Stray-Pedersen A, Vege A, Rognum TO. Helicobacter pylori antigen in stool is associated with SIDS and sudden infant deaths due to infectious disease. Pediatr Res (2008) 64:405–10. doi:10.1203/PDR.0b013e31818095f7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Stray-Pedersen A, Vege A, Stray-Pedersen A, Holmskov U, Rognum TO. Post-neonatal drop in alveolar SP-A expression: biological significance for increased vulnerability to SIDS? Pediatr Pulmonol (2008) 43:160–8. doi:10.1002/ppul.20750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Highet AR, Goldwater PN. Staphylococcal enterotoxin genes are common in Staphylococcus aureus intestinal flora in sudden infant death syndrome (SIDS) and live comparison infants. FEMS Immunol Med Microbiol (2009) 57:151–5. doi:10.1111/j.1574-695X.2009.00592.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Zorgani A, Essery SD, Madani OA, Bentley AJ, James VS, MacKenzie DA, et al. Detection of pyrogenic toxins of Staphylococcus aureus in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25:103–8. doi:10.1111/j.1574-695X.1999.tb01332.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Blackwell CC, MacKenzie DA, James VS, Elton RA, Zorgani AA, Weir DM, et al. Toxigenic bacteria and sudden infant death syndrome (SIDS): nasopharyngeal flora during the first year of life. FEMS Immunol Med Microbiol (1999) 25:51–8. doi:10.1111/j.1574-695X.1999.tb01326.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Rognum IJ, Haynes RL, Vege A, Yang M, Rognum TO, Kinney HC. Interleukin-6 and the serotonergic system of the medulla oblongata in the sudden infant death syndrome. Acta Neuropathol (2009) 118:519–30. doi:10.1007/s00401-009-0535-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Campbell RD, Dunham I, Kendall E, Sargent CA. Polymorphism of the human complement component C4. Exp Clin Immunogenet (1990) 7:69–84.
23. Schneider PM, Wendler C, Riepert T, Braun L, Schacker U, Horn M, et al. Possible association of sudden infant death with partial complement C4 deficiency revealed by post-mortem DNA typing of HLA class II and III genes. Eur J Pediatr (1989) 149:170–4. doi:10.1007/BF01958273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Keller E, Andreas A, Teifel-Greding J, Baur C, Josephi E, Beer G, et al. DNA analysis of HLA class II and III genes in sudden infant death (SIDS). Beitr Gerichtl Med (1990) 48:285–90.
25. Opdal SH, Vege A, Stave AK, Rognum TO. The complement component C4 in sudden infant death. Eur J Pediatr (1999) 158:210–2. doi:10.1007/s004310051051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Kaada B, Spurkland A. No association between HLA-DR2 and the sudden infant death syndrome. Acta Paediatr (1992) 81:283. doi:10.1111/j.1651-2227.1992.tb12226.x
27. Eskdale J, Gallagher G, Verweij CL, Keijsers V, Westendorp RG, Huizinga TW. Interleukin 10 secretion in relation to human IL-10 locus haplotypes. Proc Natl Acad Sci U S A (1998) 95:9465–70. doi:10.1073/pnas.95.16.9465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Eskdale J, Keijsers V, Huizinga T, Gallagher G. Microsatellite alleles and single nucleotide polymorphisms (SNP) combine to form four major haplotype families at the human interleukin-10 (IL-10) locus. Genes Immun (1999) 1:151–5. doi:10.1038/sj.gene.6363656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Summers AM, Summers CW, Drucker DB, Hajeer AH, Barson A, Hutchinson IV. Association of IL-10 genotype with sudden infant death syndrome. Hum Immunol (2000) 61:1270–3. doi:10.1016/S0198-8859(00)00183-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Opdal SH, Opstad A, Vege A, Rognum TO. IL-10 gene polymorphisms are associated with infectious cause of sudden infant death. Hum Immunol (2003) 64:1183–9. doi:10.1016/j.humimm.2003.08.359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Korachi M, Pravica V, Barson AJ, Hutchinson IV, Drucker DB. Interleukin 10 genotype as a risk factor for sudden infant death syndrome: determination of IL-10 genotype from wax-embedded postmortem samples. FEMS Immunol Med Microbiol (2004) 42:125–9. doi:10.1016/j.femsim.2004.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Perskvist N, Skoglund K, Edston E, Backstrom G, Lodestad I, Palm U. TNF-alpha and IL-10 gene polymorphisms versus cardioimmunological responses in sudden infant death. Fetal Pediatr Pathol (2008) 27:149–65. doi:10.1080/15513810802077651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Courts C, Madea B. No association of IL-10 promoter SNP −592 and −1082 and SIDS. Forensic Sci Int (2011) 204:179–81. doi:10.1016/j.forsciint.2010.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, et al. Interleukin-10 and sudden infant death syndrome. FEMS Immunol Med Microbiol (2004) 42:130–8. doi:10.1016/j.femsim.2004.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Dashash M, Pravica V, Hutchinson IV, Barson AJ, Drucker DB. Association of sudden infant death syndrome with VEGF and IL-6 gene polymorphisms. Hum Immunol (2006) 67:627–33. doi:10.1016/j.humimm.2006.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, et al. IL6 G-174C associated with sudden infant death syndrome in a Caucasian Australian cohort. Hum Immunol (2006) 67:819–25. doi:10.1016/j.humimm.2006.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Opdal SH, Rognum TO. The IL6 −174G/C polymorphism and sudden infant death syndrome. Hum Immunol (2007) 68:541–3. doi:10.1016/j.humimm.2007.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Ferrante L, Opdal SH, Vege A, Rognum T. Cytokine gene polymorphisms and sudden infant death syndrome. Acta Paediatr (2010) 99:384–8. doi:10.1111/j.1651-2227.2009.01611.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Ferrante L, Opdal SH, Vege A, Rognum TO. IL-1 gene cluster polymorphisms and sudden infant death syndrome. Hum Immunol (2010) 71:402–6. doi:10.1016/j.humimm.2010.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Moscovis SM, Gordon AE, Hall ST, Gleeson M, Scott RJ, Roberts-Thomson J, et al. Interleukin 1-beta responses to bacterial toxins and sudden infant death syndrome. FEMS Immunol Med Microbiol (2004) 42:139–45. doi:10.1016/j.femsim.2004.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Highet AR, Gibson CS, Goldwater PN. Variant interleukin 1 receptor antagonist gene alleles in sudden infant death syndrome. Arch Dis Child (2010) 95:1009–12. doi:10.1136/adc.2010.188268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Highet AR, Berry AM, Goldwater PN. Distribution of interleukin-1 receptor antagonist genotypes in sudden unexpected death in infancy (SUDI); unexplained SUDI have a higher frequency of allele 2. Ann Med (2010) 42:64–9. doi:10.3109/07853890903325360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ferrante L, Opdal SH, Vege A, Rognum TO. TNF-alpha promoter polymorphisms in sudden infant death. Hum Immunol (2008) 69:368–73. doi:10.1016/j.humimm.2008.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Moscovis SM, Scott RJ, Hall ST, Burns CJ, Blackwell CC. Genetic and environmental factors affecting TNF-a responses i relation to sudden infant death syndrome. Front Immunol Res Top (2014).
45. Ferrante L, Opdal SH, Vege A, Rognum TO. Is there any correlation between HLA-DR expression in laryngeal mucosa and interleukin gene variation in sudden infant death syndrome? Acta Paediatr (2013) 102:308–13. doi:10.1111/apa.12107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Schoendorf KC, Kiely JL. Relationship of sudden infant death syndrome to maternal smoking during and after pregnancy. Pediatrics (1992) 90:905–8.
47. Blair PS, Sidebotham P, Berry PJ, Evans M, Fleming PJ. Major epidemiological changes in sudden infant death syndrome: a 20-year population-based study in the UK. Lancet (2006) 367:314–9. doi:10.1016/S0140-6736(06)67968-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Moscovis SM, Scott RJ, Hall ST, Burns CJ, Blackwell CC. Virus infection and sudden death in infancy: the role of interferon-gamma. Front Immunol Res Top (2014).
49. Witkin SS, Gerber S, Ledger WJ. Influence of interleukin-1 receptor antagonist gene polymorphism on disease. Clin Infect Dis (2002) 34:204–9. doi:10.1086/338261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Rosskopf D, Koch K, Habich C, Geerdes J, Ludwig A, Wilhelms S, et al. Interaction of Gbeta3s, a splice variant of the G-protein Gbeta3, with Ggamma- and Galpha-proteins. Cell Signal (2003) 15:479–88. doi:10.1016/S0898-6568(02)00140-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lindemann M, Virchow S, Ramann F, Barsegian V, Kreuzfelder E, Siffert W, et al. The G protein beta3 subunit 825T allele is a genetic marker for enhanced T cell response. FEBS Lett (2001) 495:82–6. doi:10.1016/S0014-5793(01)02339-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Opdal SH, Melien O, Rootwelt H, Vege A, Arnestad M, Ole RT. The G protein beta3 subunit 825C allele is associated with sudden infant death due to infection. Acta Paediatr (2006) 95:1129–32. doi:10.1080/08035250600580529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Stray-Pedersen A, Vege A, Opdal SH, Moberg S, Rognum TO. Surfactant protein A and D gene polymorphisms and protein expression in victims of sudden infant death. Acta Paediatr (2009) 98:62–8. doi:10.1111/j.1651-2227.2008.01090.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Dresch C, Nardi NB, Chies JA. TCRBV3S1 and TCRBV18 gene segment polymorphisms in Brazilian Caucasoid and Black populations. Eur J Immunogenet (2002) 29:11–5. doi:10.1046/j.1365-2370.2002.00267.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Highet AR, Gibson CS, Goldwater PN. A polymorphism in a staphylococcal enterotoxin receptor gene (T cell receptor BV3 recombination signal sequence) is not associated with unexplained sudden unexpected death in infancy in an Australian cohort. Microb Pathog (2010) 49:51–3. doi:10.1016/j.micpath.2010.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Hubacek JA, Rothe G, Pit’ha J, Skodova Z, Stanek V, Poledne R, et al. C(-260)→T polymorphism in the promoter of the CD14 monocyte receptor gene as a risk factor for myocardial infarction. Circulation (1999) 99:3218–20. doi:10.1161/01.CIR.99.25.3218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Highet AR, Gibson CS, Goldwater PN. CD14 (C-260T) polymorphism is not associated with sudden infant death syndrome (SIDS) in a large South Australian cohort. Innate Immun (2011) 17:321–6. doi:10.1177/1753425910369272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: genetics, immune system, interleukins, infection, SIDS
Citation: Ferrante L and Opdal SH (2015) Sudden infant death syndrome and the genetics of inflammation. Front. Immunol. 6:63. doi: 10.3389/fimmu.2015.00063
Received: 18 November 2014; Accepted: 01 February 2015;
Published online: 20 February 2015.
Edited by:
Caroline Blackwell, University of Newcastle, AustraliaReviewed by:
Francesco Saverio Di Giovine, University of Sheffield, UKJames Alfred Morris, University Hospitals of Morecambe Bay NHS Foundation Trust, UK
Copyright: © 2015 Ferrante and Opdal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siri Hauge Opdal, Department of Research in Forensic Pathology, Norwegian Institute of Public Health, P. O. Box 4404, Nydalen, Oslo 0304, Norway e-mail:c2lyaS5oYXVnZS5vcGRhbEBmaGkubm8=