 Todd V. Brennan
Todd V. Brennan Victoria R. Rendell
Victoria R. Rendell Yiping Yang
Yiping Yang- 1Department of Surgery, Duke University, Durham, NC, USA
- 2Department of Medicine, Duke University, Durham, NC, USA
- 3Department of Immunology, Duke University, Durham, NC, USA
Allogeneic hematopoietic stem cell transplantation (Allo-HSCT) with donor lymphocyte infusion is the mainstay of treatment for many types of hematological malignancies, but the therapeutic effect and prevention of relapse is complicated by donor T-cell recognition and attack of host tissue in a process known as graft-versus-host disease (GvHD). Cytotoxic myeloablative conditioning regimens used prior to Allo-HSCT result in the release of endogenous innate immune activators that are increasingly recognized for their role in creating a pro-inflammatory milieu. This increased inflammatory state promotes allogeneic T-cell activation and the induction and perpetuation of GvHD. Here, we review the processes of cellular response to injury and cell death that are relevant following Allo-HSCT and present the current evidence for a causative role of a variety of endogenous innate immune activators in the mediation of sterile inflammation following Allo-HSCT. Finally, we discuss the potential therapeutic strategies that target the endogenous pathways of innate immune activation to decrease the incidence and severity of GvHD following Allo-HSCT.
Introduction
Allogeneic hematopoietic stem cell transplantation (Allo-HSCT) with donor lymphocyte infusion is currently the treatment of choice for several types of hematological malignancies associated with high rates of relapse, including chronic myeloid leukemia (CML) in adults and acute myeloid leukemia (AML) and acute lymphoid leukemia (ALL) in children and adults (1). While donor lymphocytes are critical for the prevention of tumor relapse, they also cause graft-versus-host disease (GvHD), the major complication of Allo-HSCT (2). Clinical manifestations of acute GvHD include rash, diarrhea, gastrointestinal hemorrhage, and jaundice, which result from immune-mediated injury to the epithelial cells of the skin, gastrointestinal tract, and liver, respectively. In severe cases, GvHD can be fatal and is the leading cause of mortality of Allo-HSCT recipients.
Allogeneic hematopoietic stem cell transplantation requires myeloablation by total body irradiation (TBI) and/or chemotherapy in order to provide space in the hematopoietic compartment for the engraftment of donor hematopoietic stem cells. TBI and chemotherapy also provide lymphocyte suppression necessary to prevent immunological rejection of the Allo-HSCT graft, which can be mediated by even small amounts of host immune defense. These myeloablative “conditioning” regimens result in cellular damage and death that produce sterile inflammatory immune responses through the release of endogenous innate immune-stimulatory molecules referred to as damage-associated molecular patterns, or “DAMPs” (3–5) (Figure 1). Endogenous molecules such as uric acid (5), adenosine triphosphate (ATP) (6), deoxyribonucleic acid (DNA) (7), N-formyl peptides (NFPs) (8), and heparan sulfate (9) activate specific pattern recognition receptors (PRRs) on innate immune cells. An increasing number of PRRs have been identified; of these some of the best studied include toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), the ATP receptor (P2X7R), and the formyl peptide receptors (FPR-1 and FPR-2). The downstream responses of these receptors on antigen presenting cells (APCs), such as dendritic cells (DCs), include increased expression of inflammatory cytokines and the upregulation of co-stimulation and antigen presenting molecules that play a critical role in initiating and orchestrating the resulting immune response.
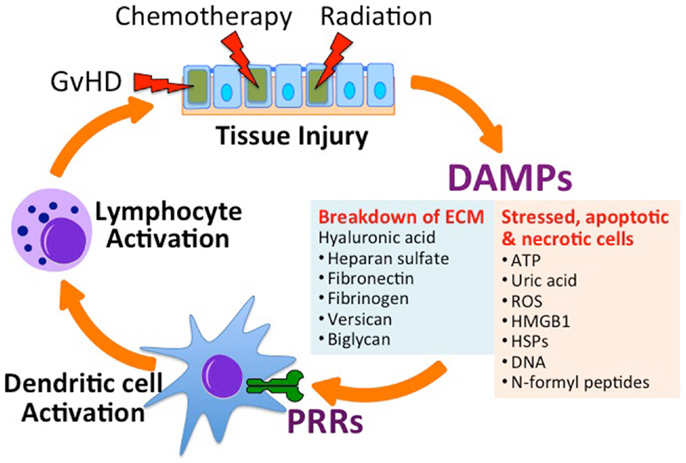
Figure 1. Tissue injury in the setting of allogeneic hematopoietic stem cell transplantation (Allo-HSCT) promotes graft-versus-host disease (GvHD). Conditioning regimens used in HSCT, which includes total body irradiation and chemotherapy, lead to tissue injury and the subsequent release of danger-associated molecular patterns (DAMPs) from the extracellular matrix (ECM) and from stressed, apoptotic and necrotic cells. Acting on specific receptors on DAMPs activate host antigen presenting cells (e.g., dendritic cells) through specific pattern recognition receptors (PRRs), leading to the activation of donor-derived lymphocytes. Activated allo-specific lymphocytes can provoke further tissue injury from GvHD. ATP, adenosine triphosphate; ROS, reactive oxygen species; HMGB1, high-mobility group box 1 protein; HSP, heat shock protein.
Many PRRs play a dual role in the recognition of DAMPs and pathogen-associated molecular patterns, or “PAMPs,” derived from microbial infection such as lipopolysaccharide (LPS) from bacteria, single-stranded RNA from viruses, and unmethylated CpG DNA motifs from bacteria and viral sources (4, 10–13). Similar to DAMPs, PAMPs also contribute to the inflammatory immune response following HSCT. For example, the loss of GI tract integrity due to epithelial barrier damage from conditioning regimens prior to allo-HSCT, as well as from GvHD following allo-HSCT, allows the translocation of gut commensal bacteria into the systemic circulation (14). In addition, the immune-compromised HSCT recipient is at high risk for viral, bacterial, and fungal infections, all potent sources of PAMPs. A complete discussion of the activation of the innate immune system by PAMPs is outside the scope of this review, but has been reviewed by others (15). The relative contribution of DAMPs versus PAMPs to the inflammatory response in the setting of Allo-HSCT is currently unclear.
In the setting of Allo-HSCT, DAMPs released following tissue damage and cell death caused by cytotoxic conditioning regimens and GvHD have the potential to enhance immune activation and worsen GvHD. A better understanding of their role in promoting immune activation will aid in the design of effective strategies to improve the outcome of Allo-HSCT. Here, we briefly review the mechanisms of cell injury involved in conditioning regimens for Allo-HSCT, highlight the potential cellular and molecular mediators of sterile inflammation in the setting of HSCT, and discuss potential therapeutic strategies of regulating these pathways to control GvHD in the setting of Allo-HSCT.
Tissue Injury from Conditioning Regimens
Multiple pathways of tissue injury occur following treatment with ionizing radiation (16, 17). Radioactive particles transfer their energy to tissues, resulting in the breaking of chemical bonds and the formation on unnatural chemical bonds. Radiation also creates free radicals and reactive oxidative species (ROS) that cause further direct tissue damage (18, 19). Following radiation, complex changes in cellular gene expression that vary between individuals, can lead to indirect cell damage (20). Tissues with high rates of proliferation, such as hematopoietic cells and enterocytes, are particularly susceptible to radiation injury as they become unable to meet their metabolic demands and unable to repair DNA damage prior to cell division (18).
Multiple chemotherapeutic regimens used in Allo-HSCT also injure host cells through a variety of cytotoxic mechanisms. While chemotherapeutic agents are dosed to be preferentially toxic to hematopoietic stem cells, they are capable of causing injury to most cell types (21). Efforts to decrease cell injury through the use of non-myeloablative conditioning regimens have been associated with decreased incidence and severity of GvHD compared to myeloablative regimen (22–24).
Cell responses to injury and stress following chemoradiation therapy include necrosis, apoptosis, and autophagy, all of which are pathways that can lead to cell death and subsequent innate immune activation (25). This response can be helpful in promoting engraftment of donor cells and in strengthening the alloimmune-dependent graft-versus-tumor (GvT) effect, or they can elicit detrimental effects by initiating GvHD. These responses to cell injury are described below.
Responses to Cell Injury
Necrosis
Necrosis is a cell death process involving cell membrane rupture that lacks the morphological and biochemical characteristics of autophagy or apoptosis. In the setting of HSCT, cell necrosis occurs following bone marrow myeloablation conditioning regimens and potentially from GvHD and infections resulting from immunosuppression. Although the initiating mechanisms are unclear, a characteristic set of changes occurs within the cell. There is a rapid loss of the cellular membrane ion potentials, cytoplasm, and cytoplasmic organelles swell, and there is a rise in cytosolic calcium levels, leading to protease and phospholipase activation. Mitochondrial dysfunction ensues and reactive oxygen species (ROS) are formed that further damage proteins, lipids, and DNA. Cell membrane integrity is eventually lost and membrane rupture occurs with release of intracellular contents into the extracellular space (26, 27). These released intracellular materials can act as DAMPs, alerting the innate immune system that an immune response is needed to either resolve a pathogen infection or initiate wound healing (28).
Apoptosis
Apoptosis is a process of programed cell death involving morphological cell changes of pyknosis, karyorrhexis, cell membrane blebbing, and loss of cell surface adhesions (25, 29). Recipient hematopoietic and stromal cells undergo apoptosis following conditioning regimens used in HSCT, and T-cells undergo apoptosis during GvHD (30).
The uptake of apoptotic cells by APCs can be anti-inflammatory and even potentially tolerogenic (31, 32). Injection of apoptotic cells at the time of HSCT has been shown to improve engraftment and decrease allo-immunization (33, 34). In a murine model of Allo-GvHD, the infusion of apoptotic APCs prior to Allo-HSCT decreased GvHD severity and improved survival (35, 36). However, if not phagocytized in a timely fashion, apoptotic cells will undergo necrosis, exposing previously hidden DAMPs. For example, apoptotic cells from monocytes, macrophages, and multiple cell lines cause the production of inflammatory cytokines and chemokines (37–39), and increased apoptosis of recipient stromal cells is linked to more severe cases of acute GvHD (40). Overall, the pro- and anti-inflammatory properties of apoptotic cells in the setting of HSCT and GvHD merits further investigation.
Autophagy
Cells under stress or undergoing starvation utilize autophagy to degrade and recycle organelles and intracellular macromolecules (41, 42). The role of autophagy in HSCT is unclear and is likely complex due to both pro- and anti-inflammatory properties of the autophagy process [reviewed by Leveque et al. (43)]. Treatment with radiation has been shown to upregulate autophagy in multiple cell types, particularly cells forming the gut barrier (44) and autophagy is theorized to limit inflammation by degrading inflammasomes (25, 45). In addition, the turnover of mitochondria by autophagy (mitophagy) is critical for removing damaged mitochondria that can be the source of multiple DAMPs (46–48).
Sources of Endogenous Innate Immune Activators
A variety of endogenous innate immune activators, or DAMPs, released during the cell death process have been identified. The following section will review the role of DAMPs in contributing to APC activation in the setting of HSCT, leading to GvHD.
Mitochondria
Mitochondria are a rich endogenous source of DAMPs, and there is increasing evidence that mitochondria play a central role in innate immune activation leading to regulation of the adaptive immune response (49–52) (Figure 2). Mitochondria evolved from an endosymbiotic relationship between eukaryotic cells and bacteria and retain structural features common to bacteria (53). Of particular relevance to their immune stimulating potential are their NFP-containing proteins and their circular DNA genome featuring unmethylated CpG motifs that resemble DNA of bacterial or viral origin. This is in contrast to mammalian genomic DNA, where between 60 and 90% of all CpG repeats are methylated (54).
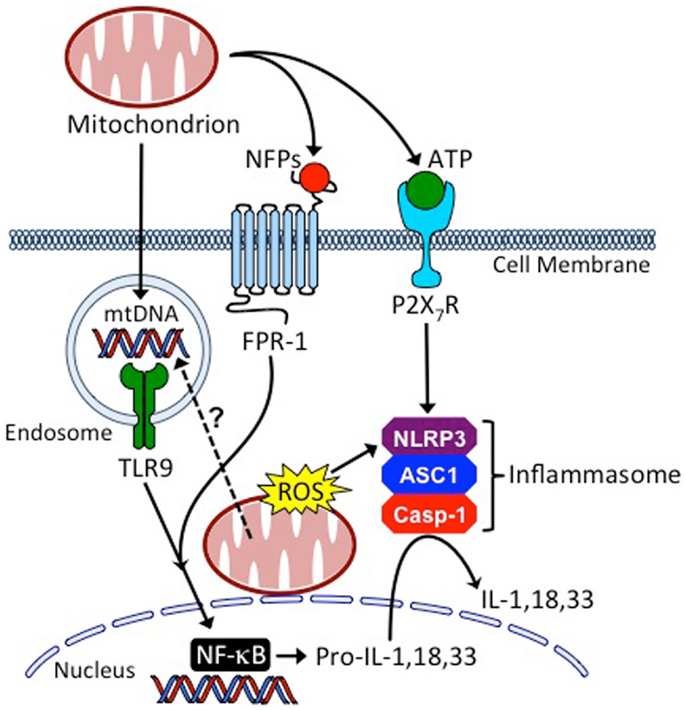
Figure 2. Mitochondria are central regulators of innate inflammatory responses. Cells injured from conditioning regimens used in HSCT, or as the result of GvHD, can leak mitochondria into the extracellular space where they release mitochondrial DNA (mtDNA), N-formyl peptides (NFPs), and adenosine triphosphate (ATP), causing host APC activation through binding to toll-like receptor 9 (TLR9), formyl peptide receptor 1 (FPR1) and the leukocyte purinergic receptor (P2X7R), respectively. Additionally, mitochondria of injured cells release reactive oxygen species (ROS) and their mtDNA may enter the endo-lysosomal compartment through the process of mitophagy to activate TLR9. These processes coordinate the production and maturation of inflammatory cytokines, such as interleukin (IL)-1, IL-18, and IL-33, through the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and the NLRP3 (NOD-, LRR-, and pyrin domain-containing 3 inflammasome). ASC1, apoptosis-associated speck-like protein containing a CARD; Casp-1, caspase-1.
There is growing evidence that mitochondrial DNA (mtDNA) is released in the setting of tissue injury. For example, plasma mtDNA is elevated in trauma patients (55), in patients with femur fracture (56), and in patients with acute liver injury (57–59). The release of mtDNA from injured tissue and cells may also play a key role in the pathogenesis of autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) (60–63).
Specific oligodeoxynucleotides (ODNs) derived from mtDNA have been identified that activate DCs in vitro based on their content of unmethylated CpG motifs (64). CpG ODNs are recognized by TLR9, leading to MyD88 activation and either an interferon (IFN) regulatory factor 3 (IRF3)- and IRF7-dependent type-1 IFN response or an NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)-initiated inflammatory response, depending on whether they localize to the endosomal or lysosomal compartment, respectively (65–68). In addition, mtDNA contains a high percentage of 8-hydroxy-2′-deoxyguanosine (8-OHG) residues that make mtDNA resistant to DNAses and increase their inflammatory potential (48, 69, 70). The high content of 8-OHG is the result of leaky oxidative machinery, the lack of efficient DNA repair mechanisms, and the absence of protective histones.
The administration of CpG ODNs at the time of Allo-HSCT accelerates GvHD in a host APC-TLR9-dependent manner and a host IFN-γ-dependent manner, but independent of host IL-6, IL-12, or natural killer (NK) cells (71). Interestingly, CpG administration also increased bone marrow rejection in a manner dependent on donor APC-TLR9 activation. In an MHC-mismatch murine model of HSCT using TLR9−/− recipient mice, the GvHD clinical score of TLR9−/− mice was significantly lower than that of wild-type mice, while no significant differences were observed when weight loss was considered alone (72).
Another GvHD study examining the role of MyD88, TRIF, TLR2/4, and TLR9 found that while deficiency in all these molecules decreased the intestinal immunopathology of GvHD, only TLR9 deficiency improved survival (73). Deficiencies in MyD88 and TLR9 also reduced the number of apoptotic cells in the gut in the same model of intestinal GvHD. In support of these findings, the administration of an inhibitory ODN that blocks TLR9 activation was shown to decrease severity of intestinal GvHD, measured by reduction in caspase-3 staining and decreased apoptotic cell counts (73). These results suggest a role of TLR9 activation by unmethylated CpG containing DNA, an endogenous source of which is mtDNA, in the inflammatory pathology of GvHD.
Mitochondrial-encoded proteins are initiated with N-formylmethionine residues similar to bacterial proteins (74). NFPs-derived from mitochondrial proteins are recognized by the formyl peptide receptors, FPR1 and FPR2, with high- and low-affinity, respectively. FPR1 and FPR2 are seven-transmembrane domain, G protein-coupled receptors, and their activation leads to neutrophil calcium mobilization, p38 and Erk1/2 activation, degranulation, oxidative burst, and chemotaxis (51). The role of NFPs in GvHD is unknown, but intravascular gradients of NFPs have been shown to guide neutrophils to sites of liver injury in mice (75).
Mitochondria also produce ATP, the main source of cellular energy, through oxidative phosphorylation. At high-extracellular concentrations, ATP activates the cell surface purinergic receptor, P2X7R, leading to activation of the NLRP3 inflammasome (6, 76, 77). Extracellular ATP has recently been shown to have a role in GvHD (78). Following TBI and during the development of acute GvHD, ATP level have been observed to be elevated in the peritoneal fluid of mice and humans, and GvHD survival in mice was improved by the breakdown of ATP with apyrase, with ATP receptor antagonism, and in mice genetically deficient for the P2X7 receptor (78).
Mitochondrial activation of the NLRP3 inflammasome also occurs through the release of ROS, resulting in the production of IL-1β and IL-18 through caspase-1 activation (79). ROS released from neutrophils at the site of gastrointestinal tract injury following radiation has been demonstrated. In irradiated mice, neutrophils strongly upregulate inducible phagocyte NADPH oxidase-1 (NOX1) (80). Deficiency in Cybb, the gene coding for NOX2, impairs ROS production, and irradiated Cybb-deficient allo-HSCT mouse recipients had protection from GVHD and improved survival (81).
In summary, as may be predicted from their bacterial origins, mitochondrial-derived molecules are capable of activating multiple inflammatory pathways used in host defense against microbial pathogens. There is increasing evidence that mitochondria are potent endogenous innate immune activators that can promote the pathogenesis of GvHD following Allo-HSCT.
Extracellular Matrix
Molecules from the extracellular matrix (ECM) also have been shown to become pro-inflammatory when they are fragmented and/or released. A number have been identified as having a role in immune activation including heparan sulfate, fibronectin, fibrinogen hyaluronate, biglycan, and tenascin (9, 82–88).
Hyaluronate and heparan sulfate induce DC maturation and alloimmune activation following recognition by TLR4 (9, 83). In a murine model examining skin manifestations of acute GvHD, hyaluronate interactions with CD44 promotes lymphocyte adherence to recipient endothelium (89). Serum heparan sulfate levels are significantly elevated in the setting of acute GvHD in a mouse model of Allo-HSCT (90). Increased serum heparan sulfate levels result in increased donor T-cell activation and proliferation along with GvHD severity. In human recipients of Allo-HSCT, similar elevations in serum heparan sulfate are detected and correlate with increasing GvHD grade (90).
High-Mobility Group Box Protein 1
High-mobility group box protein 1 (HMGB1) is a non-histone nuclear protein whose role is to bind DNA and regulate transcription. HMGB1 has been implicated in causing inflammation associated with ischemia–reperfusion injury, acute respiratory distress syndrome, multi-organ distress syndrome, trauma, and autoimmunity [reviewed by Bianchi et al. (91)]. Serum levels of HMGB1 are elevated in patients who developed acute GvHD following Allo-HSCT (92). In Allo-HSCT recipients conditioned with myeloablation, there is an association between polymorphisms in HMGB1 genotype and outcomes after Allo-HSCT (93). Interestingly, thrombomodulin, a natural neutralizer of HMGB1, has been demonstrated to treat refractory acute GvHD complicated by thrombotic microangiopathy (94).
Monosodium Urate Crystals
In a study identifying the immune-stimulatory products of cell damage released following treatment with ultraviolet (UV) radiation, uric acid was identified as a danger signal that causes priming of CD8+ T-cell responses. When added to DCs in vitro, uric acid rapidly upregulates expression of costimulatory molecules (5). Monosodium urate (MSU) is among several types of crystalline molecules that have been shown to initiate inflammatory immune responses through the activation of NLRs, including asbestos, silica, heme, and heme-like molecules (95–98). MSU crystals are potent activators of the NALP3 inflammasome, leading to release of IL-1β and IL-18, which act downstream to stimulate parenchymal cells to secrete chemokines and promote neutrophil infiltration (4, 99). In a mouse model of HSCT, uric acid levels in the peritoneal cavity were higher post TBI and chemotherapy pre-conditioning regimens. When uric acid depletion was induced with early uricase administration, treated mice had improved survival and lower IL-1β levels compared to control mice (100). A phase I study further explored the role of uric acid in GvHD by treating patients with recombinant urate oxidase for 5 days during their conditioning regimen for HSCT. The patients treated with urate oxidase indeed had lower serum levels of uric acid compared to matched controls, and, post-HSCT, these patients had a significantly decreased incidence of acute GvHD (101). A contradictory finding was shown in a retrospective study of 228 patients where low levels of uric acid at the time of HSCT was associated with significantly lower severity of GvHD (102). It was postulated that the antioxidant capacities of uric acid are also important for the prevention of GvHD and could outweigh its pro-inflammatory effects in some cases. Further investigation is needed to understand the competing roles of uric acid as an inflammatory mediator and an antioxidant and its contribution to GvHD pathogenesis.
Heat Shock Proteins
Heat shock proteins (HSPs) are molecular chaperones with a role in the formation of tertiary and quaternary protein structures and translocation within the cell. Numerous studies have reported on the immune-stimulatory potential of HSPs, which can elicit both innate and adaptive immune responses. In a human skin explant model, a high correlation was shown between the grade of GvHD and level of Hsp70 (103). In rats, Hsp70 expression in the spleen and lymph nodes is increased in the setting of GvHD (104). In human patients, homozygous Hsp70 gene polymorphisms are associated with a higher incidence and severity of GvHD (105).
Therapeutic Potential
Current therapeutic strategies for the prevention and management of acute GvHD following allogeneic HSCT rely on immunosuppressive medications such as corticosteroids, calcineurin inhibitors, mTOR inhibitors (e.g., sirolimus), and antimetabolites such as methotrexate and mycophenolate (106). Despite these aggressive approaches, GvHD is the principal cause of mortality among bone marrow transplant recipients. Additionally, the global immunosuppressive nature of these medications interferes with the beneficial properties of donor lymphocytes, such as the GvT response necessary for the prevention of tumor relapse (107) and immune reconstitution necessary for protection against microbial infections (108). Addressing inflammation derived from the endogenous innate immune agonists summarized above may provide mechanism-based treatments for GvHD.
One method to reduce endogenous inflammation derived from the release of DAMPs from injured, necrotic, or apoptotic cells is to reduce the severity of myeloablative conditioning before Allo-HSCT (23, 24) (Figure 3). A balance must be obtained between minimizing conditioning therapy and preventing rejection of the stem cell graft by host lymphocytes. One method for improving stem cell engraftment utilizes donor NK cells to aide in the elimination of host hematopoietic cells and augment the GvT response (109, 110). For this purpose, donor NK cell infusion is performed at the time of Allo-HSCT or cord blood transplantation in clinical trials for the treatment of hematologic malignancies (ClinicalTrials.gov Identifier: NCT01807611, NCT01621477, and NCT01619761). In this setting, donor NK function can be augmented using synthetic TLR9 agonists (e.g., CpG DNA), TLR3 agonists [e.g., Poly(I:C)], or IL-2 therapy.
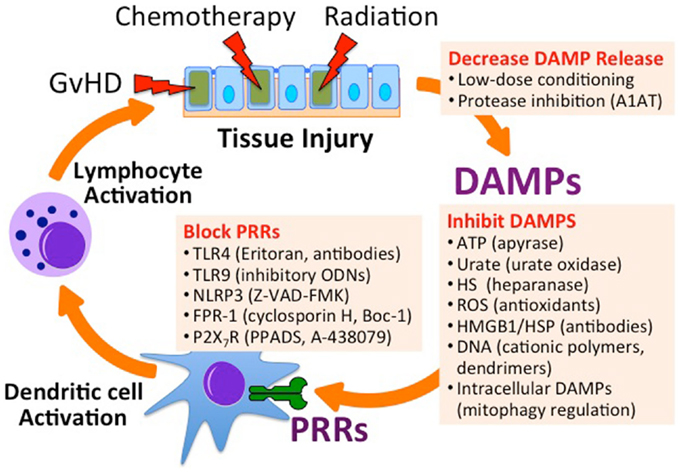
Figure 3. Inhibiting endogenous innate immune activation in the setting of Allo-HSCT. Utilizing low-dose chemoradiation conditioning regimens and protease inhibitors can minimize the release of DAMPs. Specific DAMPs can be targeted for degradation, inhibited by binders, or targeted for degradation. Antagonists can inhibit PRRs that recognize DAMPs. A1AT, alpha-1 antitrypsin; ODN, oligodeoxynucleotide; Z-VAD-FMK, carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone; Boc-1, tert-butyloxycarbonyl-methionyl-leucyl-phenylalanine; PPADS, pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid; A-438079, 3-((5-(2,3-dichlorophenyl)-1H-tetrazol-1-yl)methyl pyridine).
In addition to minimizing myeloablative pre-conditioning toxicity, there are a number of strategies focused on targeting specific DAMPs and subsequent innate immune activation. Dying and injured cells release ATP that activates its receptor, P2X7R, on innate immune cells contributing to inflammatory response to HSCT pre-conditioning therapy and GvHD (78). Wilhelm and colleagues have shown that metabolizing extracellular ATP with apyrase or P2X7R antagonism with pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS) is protective against GvHD in a murine model of allo-HSCT (78). As the authors also showed that ATP levels in the peritoneal cavity of human patients with GvHD following HSCT are dramatically elevated, methods of preventing the pathway of endogenous innate immune activation seem therapeutically promising.
Endogenous DNA, particularly hypomethylated CpG-rich mtDNA, is a TLR9-dependent innate immune activator, and methods to absorb extracellular DNA or blocking TLR9 are both potential avenues for decreasing GvHD severity. For example, using the synthetic TLR9 receptor inhibitory oligonucleotide, ODN-2088, Heimesaat and colleagues were able to decrease intestinal manifestation of GvHD in an irradiation-independent murine Allo-HSCT model (73). In a CpG-dependent model of acute liver failure, Lee and colleagues were able to use DNA binding polymers in vivo to prevent CpG related mortality in mice (111). Using these polymers at the time of either irradiation- or chemotherapy-based conditioning for Allo-HSCT, or at the onset of GvHD, has the potential to dampen the GvHD-immune response, but has not yet been tested.
Chemotherapy, irradiation, and GvHD result in tissue injury that can also lead to the degradation of ECM and the release of inflammatory glycosaminoglycans (GAGs), such as hyaluronate and heparan sulfate. GAGs can be released directly by glycolytic enzymes (e.g., heparanase), or by the proteolysis of extracellular or membrane bound proteoglycans to which they are attached. Alpha-1 antitrypsin (A1AT) is an abundant serum serine protease inhibitor critical for the prevention of neutrophil elastase-induced lung injury in the setting of chronic inflammation (112). The general immunosuppressive properties of A1AT have been observed in the prevention of acute myocardial ischemia–reperfusion injury (113) and ischemia–reperfusion-induced lung injury (114). A1AT has also been shown to prolong islet allograft survival in mice (115, 116). We and others have shown that administration of human A1AT decreases GvHD in murine models of Allo-HSCT (90, 117, 118), and it has recently been shown to preserve and enhance the NK-mediated GvT effect (119). A1AT is currently being studied in clinical trials for the treatment of steroid refractory GvHD (ClinicalTrials.gov Identifier: NCT01700036 and NCT01523821). Alternatively, the receptor for heparan sulfate, TLR4 (120), can be blocked by TLR4 antagonist antibodies (121) or TLR4 inhibitors such as Eritoran (122), TAK-242 (123), Ibudilast (124), and glucosamine dendrimers (125).
Future Perspectives
The pathogenesis of GvHD following Allo-HSCT is complex and multifactorial, resulting in significant obstacles for progress in this area of research. Successful attempts to address this critical problem for Allo-HSCT patients have focused on decreasing cytotoxic effects of chemoradiation pre-conditioning, decreasing microbial translocation across the damaged GI tract, and, more recently, targeting PRRs for both endogenous and exogenous sources of innate immune activation.
The potential for targeting DAMPs released following cell injury is appealing given the desire to directly target GvHD pathophysiology and minimize attenuation of the GvT effects. Intriguing future directions involve targeting all aspects of endogenous innate immune activation, including blocking the release of DAMPs, binding and inactivating extracellular DAMPs, blocking or inhibiting PRRs that recognize DAMPs, and blocking intracellular signal transduction molecules downstream of PRRs (Figure 3). As the role of intracellular DAMPs such as mitochondria and cytoplasmic DNA in the inflammatory response is being revealed, new insight into the pathogenesis of GvHD may be illuminated as well.
In summary, there is increasing evidence that innate immune activators released from damaged tissue potentiate GvHD and that targeting these endogenous activators and their receptors is a promising methodology for controlling GvHD while maintaining a competent immune system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to authors whose work has not been cited owing to space limitations. This work was supported by grants CA136934 (YY), CA047741 (YY), AI083000 (YY), and AI101263 (TB) from the National Institutes of Health.
References
1. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med (2006) 354:1813–26. doi: 10.1056/NEJMra052638
2. Truitt RL, Johnson BD. Principles of graft-vs.-leukemia reactivity. Biol Blood Marrow Transplant (1995) 1:61–8.
3. Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol (2008) 3:99–126. doi:10.1146/annurev.pathmechdis.3.121806.151456
4. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol (2010) 28:321–42. doi:10.1146/annurev-immunol-030409-101311
5. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature (2003) 425:516–21. doi:10.1038/nature01991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Haag F, Adriouch S, Braß A, Jung C, Möller S, Scheuplein F, et al. Extracellular NAD and ATP: partners in immune cell modulation. Purinergic Signal (2007) 3:71–81. doi:10.1007/s11302-006-9038-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Ishikawa K, Toyama-Sorimachi N, Nakada K, Morimoto M, Imanishi H, Yoshizaki M, et al. The innate immune system in host mice targets cells with allogenic mitochondrial DNA. J Exp Med (2010) 207:2297–305. doi:10.1084/jem.20092296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol (2002) 23:541–8. doi:10.1016/S1471-4906(02)02316-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by toll-like receptor 4. J Immunol (2002) 168:5233–9. doi:10.4049/jimmunol.168.10.5233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Pouwels SD, Heijink IH, Hacken ten NHT, Vandenabeele P, Krysko DV, Nawijn MC, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol (2014) 7:215–26. doi:10.1038/mi.2013.77
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol (2011) 22:416–25. doi:10.1681/ASN.2010040430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Brennan TV, Lunsford KE, Kuo PC. Innate pathways of immune activation in transplantation. J Transplant (2010) 2010:1–8. doi:10.1155/2010/826240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Beg AA. Endogenous ligands of toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol (2002) 23:509–12. doi:10.1016/S1471-4906(02)02317-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Lim J-Y, Choi M-S, Youn H, Choi EY, Min C-K. The influence of pretransplantation conditioning on graft-vs.-leukemia effect in mice. Exp Hematol (2011) 39:1018–29. doi:10.1016/j.exphem.2011.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Heidegger S, van den Brink MRM, Haas T, Poeck H. The role of pattern-recognition receptors in graft-versus-host disease and graft-versus-leukemia after allogeneic stem cell transplantation. Front Immunol (2014) 5:337. doi:10.3389/fimmu.2014.00337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Kim JH, Jenrow KA, Brown SL. Mechanisms of radiation-induced normal tissue toxicity and implications for future clinical trials. Radiat Oncol J (2014) 32:103–15. doi:10.3857/roj.2014.32.3.103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Shao L, Feng W, Li H, Gardner D, Luo Y, Wang Y, et al. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood (2014) 123:3105–15. doi:10.1182/blood-2013-07-515619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Chen Q, Xia X, Wu S, Wu A, Qi D, Liu W, et al. Apoptosis, necrosis, and autophagy in mouse intestinal damage after 15-Gy whole body irradiation. Cell Biochem Funct (2014) 32(8):647–56. doi:10.1002/cbf.3068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Gordon LI, Brown SG, Tallman MS, Rademaker AW, Weitzman SA, Lazarus HM, et al. Sequential changes in serum iron and ferritin in patients undergoing high-dose chemotherapy and radiation with autologous bone marrow transplantation: possible implications for treatment related toxicity. Free Radic Biol Med (1995) 18:383–9. doi:10.1016/0891-5849(94)E0145-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Smirnov DA, Morley M, Shin E, Spielman RS, Cheung VG. Genetic analysis of radiation-induced changes in human gene expression. Nature (2009) 459:587–91. doi:10.1038/nature07940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Mauch P, Constine L, Greenberger J, Knospe W, Sullivan J, Liesveld JL, et al. Hematopoietic stem cell compartment: acute and late effects of radiation therapy and chemotherapy. Int J Radiat Oncol Biol Phys (1995) 31:1319–39. doi:10.1016/0360-3016(94)00430-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Sorror ML, Maris MB, Storer B, Sandmaier BM, Diaconescu R, Flowers C, et al. Comparing morbidity and mortality of HLA-matched unrelated donor hematopoietic cell transplantation after nonmyeloablative and myeloablative conditioning: influence of pretransplantation comorbidities. Blood (2004) 104:961–8. doi:10.1182/blood-2004-02-0545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Mohty M, Malard F, Savani BN. High-dose total body irradiation and myeloablative conditioning before allogeneic stem cell transplantation: time to rethink? Biol Blood Marrow Transplant (2014). doi:10.1016/j.bbmt.2014.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Slavin S, Nagler A, Naparstek E, Kapelushnik Y, Aker M, Cividalli G, et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood (1998) 91:756–63.
25. Ouyang L, Shi Z, Zhao S, Wang F-T, Zhou T-T, Liu B, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif (2012) 45:487–98. doi:10.1111/j.1365-2184.2012.00845.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci (2007) 32:37–43. doi:10.1016/j.tibs.2006.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ (2009) 16:3–11. doi:10.1038/cdd.2008.150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Matzinger P. The danger model: a renewed sense of self. Science (2002) 296:301–5. doi:10.1126/science.1071059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26:239–57. doi:10.1038/bjc.1972.33
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Lin MT, Tseng LH, Frangoul H, Gooley T, Pei J, Barsoukov A, et al. Increased apoptosis of peripheral blood T cells following allogeneic hematopoietic cell transplantation. Blood (2000) 95:3832–9.
31. Kushwah R, Wu J, Oliver JR, Jiang G, Zhang J, Siminovitch KA, et al. Uptake of apoptotic DC converts immature DC into tolerogenic DC that induce differentiation of Foxp3+ Treg. Eur J Immunol (2010) 40:1022–35. doi:10.1002/eji.200939782
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Williams CA, Harry RA, McLeod JD. Apoptotic cells induce dendritic cell-mediated suppression via interferon-gamma-induced IDO. Immunology (2008) 124:89–101. doi:10.1111/j.1365-2567.2007.02743.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Perruche S, Kleinclauss F, Bittencourt Mde C, Paris D, Tiberghien P, Saas P. Intravenous infusion of apoptotic cells simultaneously with allogeneic hematopoietic grafts alters anti-donor humoral immune responses. Am J Transplant (2004) 4:1361–5. doi:10.1111/j.1600-6143.2004.00509.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Bittencourt MC, Perruche S, Contassot E, Fresnay S, Baron MH, Angonin R, et al. Intravenous injection of apoptotic leukocytes enhances bone marrow engraftment across major histocompatibility barriers. Blood (2001) 98:224–30. doi:10.1182/blood.V98.1.224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Gatza E, Rogers CE, Clouthier SG, Lowler KP, Tawara I, Liu C, et al. Extracorporeal photopheresis reverses experimental graft-versus-host disease through regulatory T cells. Blood (2008) 112:1515–21. doi:10.1182/blood-2007-11-125542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Florek M, Sega EI, Leveson-Gower DB, Baker J, Müller AMS, Schneidawind D, et al. Autologous apoptotic cells preceding transplantation enhance survival in lethal murine graft-versus-host models. Blood (2014) 124:1832–42. doi:10.1182/blood-2014-02-555128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Park DR, Thomsen AR, Frevert CW, Pham U, Skerrett SJ, Kiener PA, et al. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol (2003) 170:6209–16. doi:10.4049/jimmunol.170.12.6209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Altemeier WA, Zhu X, Berrington WR, Harlan JM, Liles WC. Fas (CD95) induces macrophage proinflammatory chemokine production via a MyD88-dependent, caspase-independent pathway. J Leukoc Biol (2007) 82:721–8. doi:10.1189/jlb.1006652
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell (2013) 49:1034–48. doi:10.1016/j.molcel.2013.01.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Wang L, Romero M, Ratajczak P, Lebœuf C, Belhadj S, Peffault de Latour R, et al. Increased apoptosis is linked to severe acute GVHD in patients with Fanconi anemia. Bone Marrow Transplant (2013) 48:849–53. doi:10.1038/bmt.2012.237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469:323–35. doi:10.1038/nature09782
42. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature (2008) 451:1069–75. doi:10.1038/nature06639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Leveque L, Le Texier L, Lineburg KE, Hill GR, MacDonald KP. Autophagy and haematopoietic stem cell transplantation. Immunol Cell Biol (2014) 93(1):43–50. doi:10.1038/icb.2014.95
44. Gorbunov NV, Kiang JG. Up-regulation of autophagy in small intestine Paneth cells in response to total-body gamma-irradiation. J Pathol (2009) 219:242–52. doi:10.1002/path.2591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Shi C-S, Shenderov K, Huang N-N, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13:255–63. doi:10.1038/ni.2215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi:10.1126/science.1232458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi:10.1016/j.immuni.2012.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Cloonan SM, Choi AMK. Mitochondria: commanders of innate immunity and disease? Curr Opin Immunol (2012) 24:32–40. doi:10.1016/j.coi.2011.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol (2011) 32:157–64. doi:10.1016/j.it.2011.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol (2011) 11:389–402. doi:10.1038/nri2975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep (2011) 12:901–10. doi:10.1038/embor.2011.157
53. Emelyanov VV. Mitochondrial connection to the origin of the eukaryotic cell. Eur J Biochem (2003) 270:1599–618. doi:10.1046/j.1432-1033.2003.03499.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Tucker KL. Methylated cytosine and the brain: a new base for neuroscience. Neuron (2001) 30:649–52. doi:10.1016/S0896-6273(01)00325-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi:10.1038/nature08780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J Orthop Trauma (2010) 24:534–8. doi:10.1097/BOT.0b013e3181ec4991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BHF, et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology (2012) 56:1971–82. doi:10.1002/hep.25801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H, Acute Liver Failure Study Group. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology (2014) 60:1336–45. doi:10.1002/hep.27265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest (2012) 122:1574–83. doi:10.1172/JCI59755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A (2009) 106:12061–6. doi:10.1073/pnas.0905441106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol (2007) 25:419–41. doi:10.1146/annurev.immunol.22.012703.104514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med (2005) 202:321–31. doi:10.1084/jem.20050338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Rodrigues HM, Juengel A, Gay RE, Gay S. Innate immunity, epigenetics and autoimmunity in rheumatoid arthritis. Mol Immunol (2009) 47:12–8. doi:10.1016/j.molimm.2009.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Ries M, Schuster P, Thomann S, Donhauser N, Vollmer J, Schmidt B. Identification of novel oligonucleotides from mitochondrial DNA that spontaneously induce plasmacytoid dendritic cell activation. J Leukoc Biol (2013) 94:123–35. doi:10.1189/jlb.0612278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature (2000) 408:740–5. doi:10.1038/35047123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Haas T, Schmitz F, Heit A, Wagner H. Sequence independent interferon-alpha induction by multimerized phosphodiester DNA depends on spatial regulation of toll-like receptor-9 activation in plasmacytoid dendritic cells. Immunology (2009) 126:290–8. doi:10.1111/j.1365-2567.2008.02897.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature (2005) 434:1035–40. doi:10.1038/nature03547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, et al. Cutting edge: role of toll-like receptor 9 in CpG DNA-induced activation of human cells. J Immunol (2001) 167:3555–8. doi:10.4049/jimmunol.167.7.3555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity (2013) 39:482–95. doi:10.1016/j.immuni.2013.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Collins LV, Hajizadeh S, Holme E, Jonsson I-M, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol (2004) 75:995–1000. doi:10.1189/jlb.0703328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Taylor PA, Ehrhardt MJ, Lees CJ, Panoskaltsis-Mortari A, Krieg AM, Sharpe AH, et al. TLR agonists regulate alloresponses and uncover a critical role for donor APCs in allogeneic bone marrow rejection. Blood (2008) 112:3508–16. doi:10.1182/blood-2007-09-113670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Calcaterra C, Sfondrini L, Rossini A, Sommariva M, Rumio C, Ménard S, et al. Critical role of TLR9 in acute graft-versus-host disease. J Immunol (2008) 181:6132–9. doi:10.4049/jimmunol.181.9.6132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Heimesaat MM, Nogai A, Bereswill S, Plickert R, Fischer A, Loddenkemper C, et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut (2010) 59:1079–87. doi:10.1136/gut.2009.197434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Marcker K, Sanger F. N-formyl-methionyl-S-RNA. J Mol Biol (1964) 8:835–40. doi:10.1016/S0022-2836(64)80164-9
75. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330:362–6. doi:10.1126/science.1195491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol (2006) 176:3877–83. doi:10.4049/jimmunol.176.7.3877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi:10.1038/nature04515
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Wilhelm K, Ganesan J, Müller T, Dürr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med (2010) 16:1434–8. doi:10.1038/nm.2242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi:10.1038/ni.1980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. François A, Milliat F, Guipaud O, Benderitter M. Inflammation and immunity in radiation damage to the gut mucosa. Biomed Res Int (2013) 2013:123241–9. doi:10.1155/2013/123241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat Med (2014) 20:648–54. doi:10.1038/nm.3517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kuhns DB, Nelson EL, Alvord WG, Gallin JI. Fibrinogen induces IL-8 synthesis in human neutrophils stimulated with formyl-methionyl-leucyl-phenylalanine or leukotriene B(4). J Immunol (2001) 167:2869–78. doi:10.4049/jimmunol.167.5.2869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Tesar BM, Jiang D, Liang J, Palmer SM, Noble PW, Goldstein DR. The role of hyaluronan degradation products as innate alloimmune agonists. Am J Transplant (2006) 6:2622–35. doi:10.1111/j.1600-6143.2006.01537.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Schaefer L, Babelova A, Kiss E, Hausser H-J, Baliova M, Krzyzankova M, et al. The matrix component biglycan is proinflammatory and signals through toll-like receptors 4 and 2 in macrophages. J Clin Invest (2005) 115:2223–33. doi:10.1172/JCI23755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature (2009) 457:102–6. doi:10.1038/nature07623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med (2009) 15:774–80. doi:10.1038/nm.1987
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates toll-like receptor 4. J Biol Chem (2001) 276:10229–33. doi:10.1074/jbc.M100099200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol (2001) 167:2887–94. doi:10.4049/jimmunol.167.5.2887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Milinkovic M, Antin JH, Hergrueter CA, Underhill CB, Sackstein R. CD44-hyaluronic acid interactions mediate shear-resistant binding of lymphocytes to dermal endothelium in acute cutaneous GVHD. Blood (2004) 103:740–2. doi:10.1182/blood-2003-05-1500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Brennan TV, Lin L, Huang X, Cardona DM, Li Z, Dredge K, et al. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD following allogeneic stem cell transplantation. Blood (2012) 120(14):2899–908. doi:10.1182/blood-2011-07-368720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev (2007) 220:35–46. doi:10.1111/j.1600-065X.2007.00574.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Yujiri T, Tagami K, Tanaka Y, Mitani N, Nakamura Y, Ariyoshi K, et al. Increased serum levels of high-mobility group box 1 protein in patients who developed acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Eur J Haematol (2010) 85:366–7. doi:10.1111/j.1600-0609.2010.01507.x
93. Kornblit B, Masmas T, Petersen SL, Madsen HO, Heilmann C, Schejbel L, et al. Association of HMGB1 polymorphisms with outcome after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2010) 16:239–52. doi:10.1016/j.bbmt.2009.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Inoue Y, Hatta Y, Takeuchi J, Kosugi S, Miura I. Successful treatment of refractory acute GVHD complicated by severe intestinal transplant-associated thrombotic microangiopathy using recombinant thrombomodulin. Thromb Res (2011) 127:603–4. doi:10.1016/j.thromres.2010.12.023
95. Dostert C, Pétrilli V, van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320:674–7. doi:10.1126/science.1156995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One (2009) 4:e6510. doi:10.1371/journal.pone.0006510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Chen C-J, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest (2006) 116:2262–71. doi:10.1172/JCI28075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukoc Biol (2008) 83:13–30. doi:10.1189/jlb.0607402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi:10.1038/nature04516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G, et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med (2013) 210:1899–910. doi:10.1084/jem.20130084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Yeh AC, Brunner AM, Spitzer TR, Chen Y-B, Coughlin E, McAfee S, et al. Phase I study of urate oxidase in the reduction of acute graft-versus-host disease after myeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20:730–4. doi:10.1016/j.bbmt.2014.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Ostendorf BN, Blau O, Uharek L, Blau IW, Penack O. Association between low uric acid levels and acute graft-versus-host disease. Ann Hematol (2015) 94(1):139–44. doi:10.1007/s00277-014-2180-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Jarvis M, Marzolini M, Wang XN, Jackson G, Sviland L, Dickinson AM. Heat shock protein 70: correlation of expression with degree of graft-versus-host response and clinical graft-versus-host disease. Transplantation (2003) 76:849–53. doi:10.1097/01.TP.0000083556.73555.78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Goral J, Mathews HL, Clancy J. Expression of 70-kDa heat-shock protein during acute graft-versus-host disease. Clin Immunol Immunopathol (1998) 86:252–8. doi:10.1006/clin.1997.4473
105. Bogunia-Kubik K, Lange A. HSP70-hom gene polymorphism in allogeneic hematopoietic stem-cell transplant recipients correlates with the development of acute graft-versus-host disease. Transplantation (2005) 79:815–20. doi:10.1097/01.TP.0000153157.97736.2C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. MacMillan ML, Weisdorf DJ, Wagner JE, Defor TE, Burns LJ, Ramsay NKC, et al. Response of 443 patients to steroids as primary therapy for acute graft-versus-host disease: comparison of grading systems. Biol Blood Marrow Transplant (2002) 8:387–94. doi:10.1053/bbmt.2002.v8.pm12171485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Zhang P, Chen BJ, Chao NJ. Prevention of GVHD without losing GVL effect: windows of opportunity. Immunol Res (2011) 49:49–55. doi:10.1007/s12026-010-8193-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. van Burik J-AH, Carter SL, Freifeld AG, High KP, Godder KT, Papanicolaou GA, et al. Higher risk of cytomegalovirus and Aspergillus infections in recipients of T cell-depleted unrelated bone marrow: analysis of infectious complications in patients treated with T cell depletion versus immunosuppressive therapy to prevent graft-versus-host disease. Biol Blood Marrow Transplant (2007) 13:1487–98. doi:10.1016/j.bbmt.2007.08.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Ruggeri L, Mancusi A, Burchielli E, Capanni M, Carotti A, Aloisi T, et al. NK cell alloreactivity and allogeneic hematopoietic stem cell transplantation. Blood Cells Mol Dis (2008) 40:84–90. doi:10.1016/j.bcmd.2007.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Ruggeri L, Aversa F, Martelli MF, Velardi A. Allogeneic hematopoietic transplantation and natural killer cell recognition of missing self. Immunol Rev (2006) 214:202–18. doi:10.1111/j.1600-065X.2006.00455.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Lee J, Sohn JW, Zhang Y, Leong KW, Pisetsky D, Sullenger BA. Nucleic acid-binding polymers as anti-inflammatory agents. Proc Natl Acad Sci U S A (2011) 108:14055–60. doi:10.1073/pnas.1105777108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Stockley RA. Alpha1-antitrypsin review. Clin Chest Med (2014) 35:39–50. doi:10.1016/j.ccm.2013.10.001
113. Toldo S, Seropian IM, Mezzaroma E, Van Tassell BW, Salloum FN, Lewis EC, et al. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J Mol Cell Cardiol (2011) 51:244–51. doi:10.1016/j.yjmcc.2011.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Gao W, Zhao J, Kim H, Xu S, Chen M, Bai X, et al. α1-Antitrypsin inhibits ischemia reperfusion-induced lung injury by reducing inflammatory response and cell death. J Heart Lung Transplant (2013) 33(3):309–15. doi:10.1016/j.healun.2013.10.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Lewis EC, Shapiro L, Bowers OJ, Dinarello CA. Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci U S A (2005) 102:12153–8. doi:10.1073/pnas.0505579102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Lewis EC, Mizrahi M, Toledano M, Defelice N, Wright JL, Churg A, et al. Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci U S A (2008) 105:16236–41. doi:10.1073/pnas.0807627105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Marcondes AM, Li X, Tabellini L, Bartenstein M, Kabacka J, Sale GE, et al. Inhibition of IL-32 activation by α-1 antitrypsin suppresses alloreactivity and increases survival in an allogeneic murine marrow transplantation model. Blood (2011) 118:5031–9. doi:10.1182/blood-2011-07-365247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A (2012) 109:564–9. doi:10.1073/pnas.1117665109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Marcondes AM, Karoopongse E, Lesnikova M, Margineantu D, Welte T, Dinarello CA, et al. α-1-antitrypsin (AAT)-modified donor cells suppress GVHD but enhance the GVL effect: a role for mitochondrial bioenergetics. Blood (2014) 124:2881–91. doi:10.1182/blood-2014-04-570440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through toll-like receptor 4. J Immunol (2004) 172:20–4. doi:10.4049/jimmunol.172.1.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Ungaro R, Fukata M, Hsu D, Hernandez Y, Breglio K, Chen A, et al. A novel toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am J Physiol Gastrointest Liver Physiol (2009) 296:G1167–79. doi:10.1152/ajpgi.90496.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Czeslick E, Struppert A, Simm A, Sablotzki A. E5564 (Eritoran) inhibits lipopolysaccharide-induced cytokine production in human blood monocytes. Inflamm Res (2006) 55:511–5. doi:10.1007/s00011-006-6057-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, et al. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl) sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol Pharmacol (2006) 69:1288–95. doi:10.1124/mol.105.019695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW. Ibudilast (AV-411): a new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin Investig Drugs (2007) 16:935–50. doi:10.1517/13543784.16.7.935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Shaunak S, Thomas S, Gianasi E, Godwin A, Jones E, Teo I, et al. Polyvalent dendrimer glucosamine conjugates prevent scar tissue formation. Nat Biotechnol (2004) 22:977–84. doi:10.1038/nbt995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: endogenous innate immune, graft-versus-host disease, hematopoietic stem cell transplantation, tissue injury
Citation: Brennan TV, Rendell VR and Yang Y (2015) Innate immune activation by tissue injury and cell death in the setting of hematopoietic stem cell transplantation. Front. Immunol. 6:101. doi: 10.3389/fimmu.2015.00101
Received: 10 December 2014; Accepted: 23 February 2015;
Published online: 16 March 2015.
Edited by:
Philippe Saas, Etablissement Français du Sang BFC, FranceReviewed by:
Reem Al-Daccak, Institut National de la Santé et de la Recherche Médicale, FranceNicolas Larmonier, University of Arizona, USA
Copyright: © 2015 Brennan, Rendell and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiping Yang, Departments of Medicine and Immunology, Duke University Medical Center, Box 103005, Durham, NC 27710, USA e-mail:eWFuZzAwMjlAbWMuZHVrZS5lZHU=