 Xinxin Wang
Xinxin Wang Leonard Benjamin Hills
Leonard Benjamin Hills Yina Hsing Huang
Yina Hsing Huang- 1California Institute for Biomedical Research, La Jolla, CA, USA
- 2Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Lebanon, NH, USA
- 3Department of Pathology, Geisel School of Medicine at Dartmouth, Lebanon, NH, USA
The phosphoinositide 3-kinase (PI 3-kinase, PI3K) pathway transduces signals critical for lymphocyte function. PI3K generates the phospholipid PIP3 at the plasma membrane to recruit proteins that contain pleckstrin homology (PH) domains – a conserved domain found in hundreds of mammalian proteins. PH domain–PIP3 interactions allow for rapid signal propagation and confer a spatial component to these signals. The kinases Akt and Itk are key PI3K effectors that bind PIP3 via their PH domains and mediate vital processes – such as survival, activation, and differentiation – in lymphocytes. Here, we review the roles and regulation of PI3K signaling in lymphocytes with a specific emphasis on Akt and Itk. We also discuss these and other PH domain-containing proteins as they relate more broadly to immune cell function. Finally, we highlight the emerging view of PH domains as multifunctional protein domains that often bind both lipid and protein substrates to exert their effects.
Lymphocyte Activation Receptors Signal through Class I PI3Ks
Phosphoinositide 3-kinase (PI3K) activation is important for lymphocyte survival, activation, differentiation, and migration. Many lymphocyte surface receptors activate class 1 PI3Ks, which phosphorylate phosphatidyl inositol 4,5-bisphosphate [PI(4,5)P2, PIP2] at the D-3 hydroxyl group of the myo-inositol ring to generate phosphatidyl inositol 3,4,5-trisphosphate [PI(3,4,5)P3, PIP3]. Two subclasses, 1A and 1B, are activated by distinct receptor types (Figure 1). Receptors or signaling adapters that are phosphorylated at YxxM sequence motifs signal though class IA PI3K, which includes p85α and p85β regulatory subunits and p110α, p110β, and p110δ catalytic subunits. These receptors include CD19, CD28, and ICOS co-receptors; IL-2, IL-7, IL-3, IL-15, and GM-CSF cytokine receptors (1–6); and receptors coupled to TRIM, DAP10, and MyD88 adapter proteins (7–11). Receptor ligation leads to tyrosine phosphorylation at the YxxM motif and subsequent recruitment of PI3K regulatory subunits through one or both Src homology 2 (SH2) domains. Regulatory subunits are then phosphorylated by Syk or Jak family tyrosine kinases to trigger activation of their constitutively associated catalytic subunits (3).
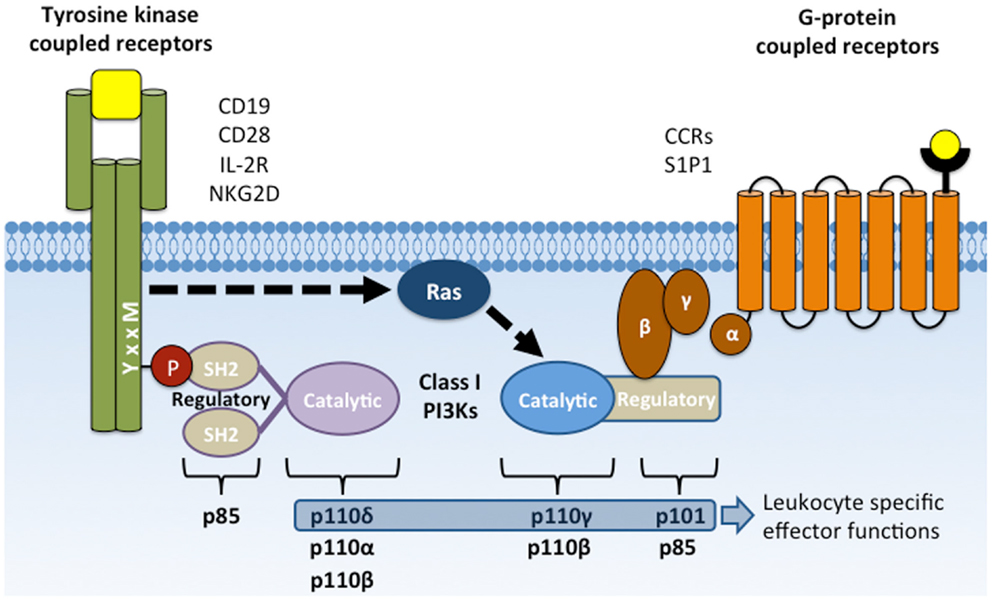
Figure 1. Activation of class I PI3Ks by YxxM signaling subunits and GPCRs. Membrane receptors that activate PI3K include CD19, CD28, and NKG2D co-receptors, cytokine receptors (e.g., IL-2R), G-protein-coupled receptors (chemokine receptors), and Fcγ receptor I and III. Class IA PI3Ks are recruited to the plasma membrane through SH2 domain interactions with phosphorylated YxxM motifs. Class IB PI3Ks are recruited and activated by direct interaction with the Gβγ subunit following GPCR activation. Activated PI3K phosphorylates the membrane lipid PI(4,5)P2 to form PI(3,4,5)P3.
G-protein-coupled receptors (GPCRs) signal through Class 1B PI3K, which includes p101 regulatory and p110γ catalytic subunits (12). These classic, seven transmembrane domain receptors include chemokine receptors and signal through heterotrimeric G proteins, Gα and Gβγ to promote cell migration. GPCR ligation dissociates the Gβγ dimer, allowing its binding to p101 regulatory subunits and subsequent activation of associated p110γ catalytic subunits. Activation of p110γ catalytic activity can also be induced by Ras activation (Ras-GTP) to promote migration of neutrophils (13).
Although many receptors activate class 1 PI3K, the magnitude and kinetics of PI3K activation differs greatly among receptors, depending on ligand binding kinetics and feedback circuitry that can either amplify or dampen PI3K signaling (14). Additionally, co-ligation of receptors, such as the T cell receptor (TCR) and the CD28 co-receptor, can cooperate to potentiate and sustain PI3K activation and PIP3 generation.
PIP3 Association with Pleckstrin Homology Domains
PI3K activation induces PIP3 accumulation, which comprises less than 5% of PIP2 levels and less than 1% of total membrane lipids (15). Despite its low overall abundance, super-resolution microscopy has revealed ~100 nm membrane clusters of PIP3 that create high local PIP3 concentrations (16). High affinity and specificity binding between PIP3 and pleckstrin homology (PH) domains of PI3K effectors helps to recruit and activate these effectors at the plasma membrane (Figure 2). Like protein–protein interactions that are induced by phosphorylation, PIP3 interactions with PH domains allow rapid transduction of downstream signals without new protein synthesis.
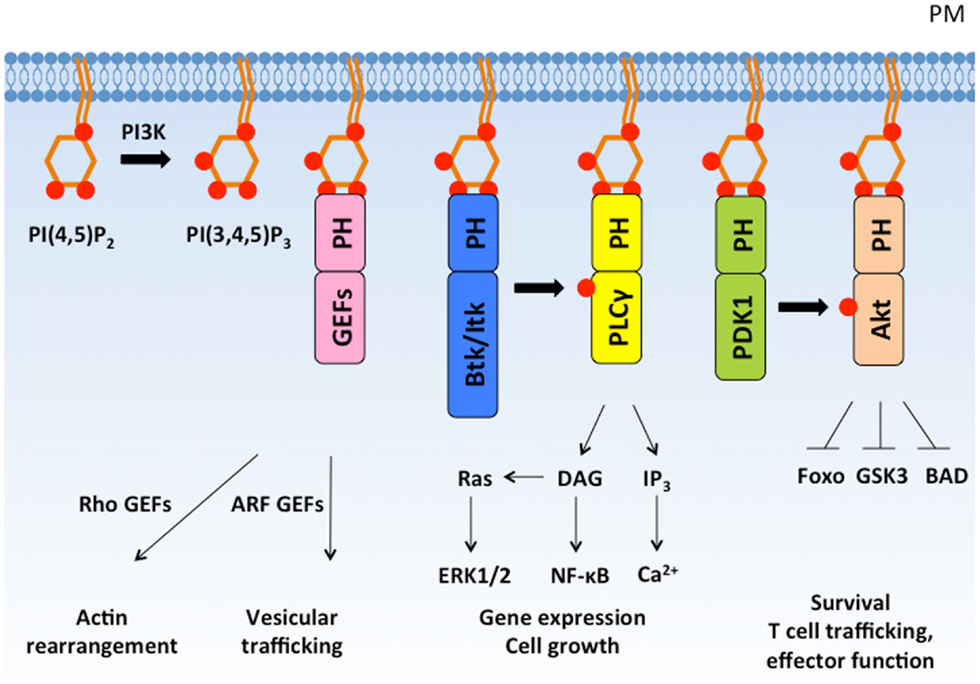
Figure 2. PI(3,4,5)P3 recruits PH domain-containing proteins to the plasma membrane and regulates diverse cellular responses. PI3K phosphorylates PI(4,5)P2 to form PI(3,4,5)P3, which recruits PH domain-containing signaling proteins to the plasma membrane. PH domain-containing proteins are activated at the plasma membrane and mediate important cellular responses such as cytoskeleton rearrangement, cell growth, proliferation, and survival. PM, plasma membrane; GEF, guanine nucleotide exchange factor.
The PH domain is an evolutionarily conserved structural fold found in proteins expressed in organisms ranging from yeast to mammals (17). The core of the PH domain is a seven-strand β-barrel that is encoded by approximately 120 amino acids and is composed of two anti-parallel β sheets and a C-terminal α helix (Figure 3). The mammalian genome contains roughly 300 PH domains found in proteins that perform diverse functions including cellular activation, cytoskeletal reorganization, vesicular trafficking, and cell cycle control. Approximately, 15% of PH domains, including Akt and Itk, bind to phosphoinositides with high specificity and affinity (Kd: nanomolar – low micromolar range). PH domains generally interact with phosphoinositides through positively charged lysine and arginine residues within the basic motif KXn(K/R)XR (18). However, not all PH domains bind to PIP3. Several PH domains interact with phosphoinositides that are selectively enriched in other membrane compartments, such as PI4P within the Golgi membrane (19) or PIP2 at the resting plasma membrane (17). Thus, conveying lipid specificity to PH domains constitutes a key mechanism for spatially sequestering distinct effector proteins within cells. Regulating the abundance of lipids either in resting or activated cells controls basal and induced effector activity. Additionally, regulated production of lipid ligands such as PIP3 within specific membrane nano-domains can induce polarized activation of downstream effectors in a robust but transient manner. This is because PIP3 abundance is not only spatial restricted but also finely controlled by receptor-induced PI3K-dependent PIP3 generation and by phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and SH2 domain-containing inositol 5′-phosphatase (SHIP) phosphatase-dependent PIP3 metabolism.
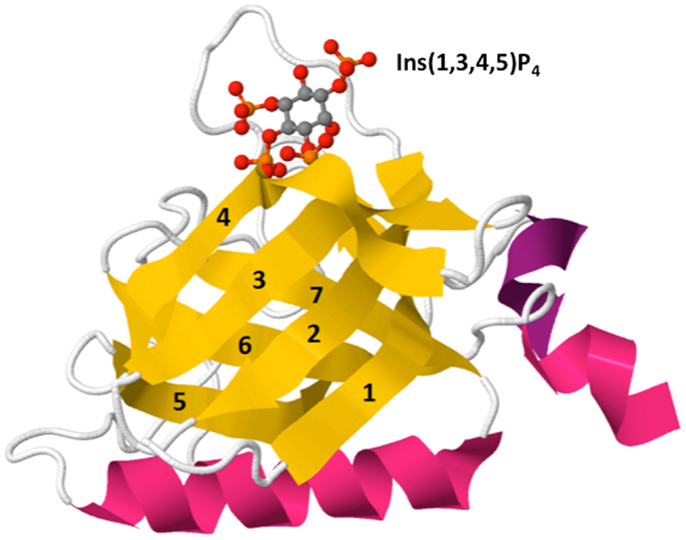
Figure 3. Crystal structure of Btk PH domain in complex with Ins(1,3,4,5)P4 (PDB ID: 2Z0P). The PH domain is comprised of a β-barrel formed by seven β-strands (yellow, 1–7) capped by an α-helix (pink). The hyper-variable loops of the β-barrel form the binding surface for lipid ligands such as Ins(1,3,4,5)P4 [top, shown by ball-and-stick model: red (oxygen), orange (phosphorus), and gray (carbon)].
Protein Phosphatases Inhibit PI3K Activation While Inositol Phosphatases Reduce PIP3 Levels
PI3K signaling is negatively regulated at distinct steps in its signaling cascade by both protein and lipid phosphatases. Protein tyrosine phosphatases SHP-1 and SHP-2 inhibit PI3K activation by preventing early receptor signaling and by directly down-regulating PI3K activity, the latter of which is accomplished by de-phosphorylation of phospho-tyrosine residues within signal adapter proteins and PI3K regulatory subunits (71). Inhibitory receptors that restrict lymphocyte activation through SHP-1 or SHP-2 include inhibitory killer-cell immunoglobulin-like receptors (KIR) on NK cells (72), CD22 on B cells (73), and CTLA-4 and PD-1 on T cells (74, 75). Phosphorylated immunoreceptor tyrosine-based inhibition motifs (ITIM) within the cytoplasmic domains of KIRs, CD22, and CTLA-4 recruit SHP-1 and SHP-2 to prevent activating signals at the plasma membrane (72, 74, 75). Persistent T cell activation signals can also be inhibited by SHP-1 and SHP-2 recruitment to the immunoreceptor tyrosine-based switch motif (ITSM) in PD-1, an inhibitory receptor expressed on chronically stimulated T cells (76, 77). For a detailed discussion regarding the requirements of SHP-1 and SHP-2 in T cell development, differentiation, and effector function, refer to Ref. (78).
In T cells, CTLA-4 can also directly repress Akt signaling by recruiting the Ser/Thr phosphatase PP2A (77), which dephosphorylates the T308 (79, 80) and possibly S473 (79), residues required for Akt activity. Thus, CTLA-4 utilizes a dual approach to antagonize CD28 and PI3K signaling: SHP-2-dependent inhibition of TCR signaling by CD3ε de-phosphorylation and PP2A-dependent de-phosphorylation of Akt (74, 77, 81).
Lipid and inositol phosphatases also prevent PI3K effector activation. PTEN and SHIP both dephosphorylate membrane PIP3. However, while PTEN converts PIP3 back to its lipid precursor PI(4,5)P2 to prevent further activation of PI3K effectors, SHIP converts PIP3 into PI(3,4)P2, a lipid that retains the ability to bind the Akt PH domain (82). In the latter case, subsequent de-phosphorylation of PI(3,4)P2 into PI(3)P by the inositol phosphatase, INPP4B is required to “turn off” Akt membrane recruitment (83). Inhibitory receptors including FcγIIB on B cells and mast cells and Ly49A and Ly49C on NK cells contain ITIM motifs that recruit SHIP through its SH2 domain (84, 85). Membrane receptors with cytosolic PDZ domains recruit PTEN to control PIP3 levels. Although the functional significance of PDZ domain-containing receptors on lymphocyte activation requires additional investigation, maintaining appropriate PTEN levels is crucial for the control of immune cell homeostasis and function (86).
General and Cell Type-Specific Akt Functions
Akt belongs to the AGC family of Serine/Threonine kinases. The three Akt isoforms are differentially expressed in various cell types but are 77–83% sequence identical. Akt activity prevents apoptosis, promotes protein expression, and regulates cellular metabolism (20–23). Akt mediates these general cellular functions through direct phosphorylation of RxRxxS*/T* motifs (24) found in a plethora of cellular targets including forkhead box transcription factors, TSC2, GSK3, and BAD, which are discussed in detail elsewhere (20). A somatic mutation in Akt that replaces glutamate with lysine at residue 17 (hereafter referred to as E17K) leads to cellular transformation and has been identified in human breast, colorectal, and ovarian cancer (25, 26). The E17K mutation is located in the lipid binding pocket of Akt’s PH domain and dramatically increases its affinity for membrane lipids, causing constitutive Akt signaling (27). Ectopic expression of E17K in hematopoietic stem cells is sufficient to induce development of lymphoblastic T cell lymphoma within 6–8 weeks following transfer into recipient mice (28). Similarly, conditional deletion of the Akt targets Foxo1/3/4 in mice leads to development of the same type of lymphomas 15–25 weeks after induction of Foxo deletion (29).
In lymphocytes, Foxo proteins regulate the gene expression of Rag recombinases, Ikaros, CCR7, IL-7R, TCF7, Eomes, and Foxp3, which are critical for controlling lymphocyte development, trafficking, and differentiation (30–37). Akt phosphorylation of Foxo1 and Foxo3 leads to their degradation and down-regulates Foxo-dependent gene expression (31, 38). Genetic ablation of both Foxo1 and Foxo3 causes a multi-focal autoimmune disease due to defective Foxp3 expression and T regulatory (Treg) cell specification and function (34). Similarly, retroviral expression of constitutively active myristoylated Akt (myrAkt) in CD4−CD8− thymocytes impairs Treg development in vivo following intrathymic transfer. Importantly, the inhibitory effect of myrAkt is on de novo but not established Foxp3 expression (39). In contrast, broad expression of myrAkt as a transgene under the control of the CD2 promoter leads to increased regulatory T cell numbers in vivo and enhanced suppressive activity (40). Interestingly, conventional CD4+ T cells expressing transgenic myrAkt are less responsive to TGFβ suppression and fail to differentiate into the Th17 lineage in response to TGFβ and IL-6 in vitro (40).
A proper balance of Akt activity is also required for appropriate CD8+ T cell maturation, effector function, and memory development (41). Uzel and colleagues recently published a study on patients with somatic dominant active p110δ (a catalytic subunit of PI3K) expression (42). T cell blasts from these patients have increased phosphorylation of AKT at T308 and S473, a decline in Foxo1, increased S6 activation, and glucose uptake. This hyperactive Akt/mTORC1 axis causes CD8 T cells to proliferate more vigorously, differentiate more readily into effector cells, and undergo cellular senescence. Sustained Akt activity in these patients also impairs development of CD8 memory T cells, which require a metabolic “switch” from glycolysis to fatty acid oxidation (41, 43). Furthermore, defective CD8 responses result in recurrent sinopulmonary infections and chronic viremia due to Epstein-Barr virus (EBV) and/or cytomegalovirus (CMV) infection (42). Cantrell and coworkers published a surprising finding demonstrating distinct roles for PDK1 and Akt in promoting cellular metabolism and effector responses of CD8 T cells, respectively (44). T cells expressing a catalytically inactive p110δ or treated with an Akt inhibitor are defective for Akt T308 phosphorylation. Akt-defective CD8 T cells proliferate normally in response to IL-2 but are unable to express proper lymphoid homing receptors and cytotoxic effector proteins (44). In contrast, conditional deletion of PDK1, the upstream activator of Akt, leads to defective glucose uptake and metabolism, resulting in reduced CD8 T cell proliferation. This indicates that PDK1 promotes proliferation in an Akt-independent manner (44). It remains to be determined whether PDK1 and Akt have distinct roles in cell types in which multiple functions have been attributed to Akt activity.
Tec Family Kinases Regulate Immune Cell Development and Function
The Tec family of non-receptor tyrosine kinases, including Tec, Btk, Itk/Emt/Tsk, Rlk/Txk, and Bmx/Etk, are differentially expressed in immune cells. Each Tec family member contains an N-terminal PIP3-binding PH domain except Rlk, which contains a cysteine-string motif that results in Rlk palmitoylation. In general, Tec kinases activate PLCγ to trigger Ca2+ and diacylglycerol (DAG) signaling. Mimicking Ca2+ and DAG activation with the addition of calcium ionophores and phorbol myristate acetate (PMA) is sufficient to induce many aspects of lymphocyte activation, differentiation, and effector responses in vitro. The requirement for Tec kinases in immune functions is apparent from the profound defects observed in human patients carrying mutations in Tec kinases and in mouse models of single and combined Tec kinase deficiencies.
In 1993, Btk was first identified in patients with X-linked agammaglobulinemia (XLA), an inherited immunodeficiency disease characterized by profound hypogammaglobulinemia due to severely decreased B cell numbers (45). XLA patients carry Btk mutations that prevent the maturation of pro-B cells into pre-B cells. Pre-B-cell receptor signaling at the pro-B to pre-B transition requires Btk activation by the Src kinase Lyn (46–48). A Btk mutation database generated from approximately 400 XLA patients indicates that the majority of missense mutations in the Btk PH domain are in the putative PIP3-binding pocket (49–51). The XLA missense mutants F25S, R28H, T33P, V64F, and V113D dramatically reduce Btk binding to PIP3 in vitro and disrupt Btk activation in B cells (52, 53). A similar mutation in mice, R28C also abolishes Btk binding to PIP3 and results in murine X-linked immunodeficiency (Xid) disease (53). These findings demonstrate the importance of PI3K-dependent PIP3 generation for the membrane recruitment and activation of Btk in promoting B cell receptor signaling during maturation and humoral immune responses.
While disruption of PIP3 association causes hypo-B-cell responses, enhanced PIP3 association also leads to B cell dysfunction. The Btk E41K mutant significantly increases Btk PH domain affinity for phosphoinositides and results in constitutive membrane localization when expressed ectopically in COS-7 cells (52, 53). Btk E41K expression allows cytokine-independent growth of the pro-B-cell line Y16 (54), demonstrating its gain-of-function activity. However, mice expressing a Btk E41K transgene controlled by the MHC class II locus are more severely B cell-deficient than even Xid mice (55). Lack of IgMhigh cells in the bone marrow suggest that constitutive Btk E41K activation leads to inappropriate deletion of immature B cells by mimicking strong BCR signals that promote apoptosis of auto-reactive B cells (55). Thus, appropriate levels of Btk activation are critical for developmental progression of B cells, productive B cell activation and differentiation, as well as deletion of auto-reactive cells.
The first patients identified with Itk mutations were initially diagnosed with Hodgkin’s lymphoma but subsequently characterized to have an underlying immunodeficiency disease that prevents control of EBV-induced B cell proliferation (56). Itk-deficient patients have decreased T cells (57), which are required to control EBV infection and prevent viral reactivation from latently infected B cells (58). Detailed characterization of Itk-deficient mice reveals multiple requirements for Itk during T cell development, differentiation, and function (59, 60). Like Btk in B cells, Itk participates in proximal antigen receptor signaling and is directly phosphorylated by a Src family kinase, in this case Lck (61). Activated Itk phosphorylates PLCγ1, which induces IP3-dependent increased intracellular Ca2+ levels as well as DAG-mediated signaling (59, 62, 63). Itk is required for efficient CD4+ T cell differentiation toward the Th2 and Th17 lineages (59). Itk-deficient mice cannot generate protective Th2 responses in multiple infection models, including Leishmania major, Nippostrongylus brasiliensis, and Schistosoma mansoni (59, 64). Defective Th2 differentiation is accompanied by substantially reduced production of the Th2 cytokines IL-4, IL-5, and IL-13 by Itk-deficient T cells (65, 66). Itk is also required for optimal production of the Th17 cytokine, IL-17A but not IL-17F (67). The selective requirement for Itk in IL-17A production is mechanistically linked to a requirement for the transcription factor nuclear factor of activated T cells (NFAT) in IL-17A transcription (64, 67, 68). Prolonged Itk activation maintains cytosolic Ca2+ levels to promote sustained calcineurin-dependent NFAT nuclear translocation. Itk deficiency or suboptimal TCR signaling restricts autoimmunity by biasing T cell differentiation from the Th17 toward the regulatory T cell lineage (69). In addition, autoimmune organ destruction can be limited by Itk-dependent control of transendothelial migration and tissue infiltration of effector T cells (70). Thus, mechanisms that regulate the magnitude and kinetics of Itk activity in T cells are important for induction of effector functions, specification of appropriate T cell lineages, and control of T cell trafficking.
Soluble Analogs of PIP3 Differentially Regulate PIP3 Effectors
Some PIP3-binding PH domains can associate with soluble PIP3 analogs. These include the cytosolic inositol phosphates Ins(1,3,4,5)P4 (IP4), Ins(1,2,3,4,5,6)P6 (IP6), and 5-PP-I(1,2,3,4,6)P5 (IP7) that are generated inducibly or constitutively by distinct inositol kinases (82). The effect of IP4, IP6, and IP7 binding is distinct for different PH domains and cell types (Figure 4).
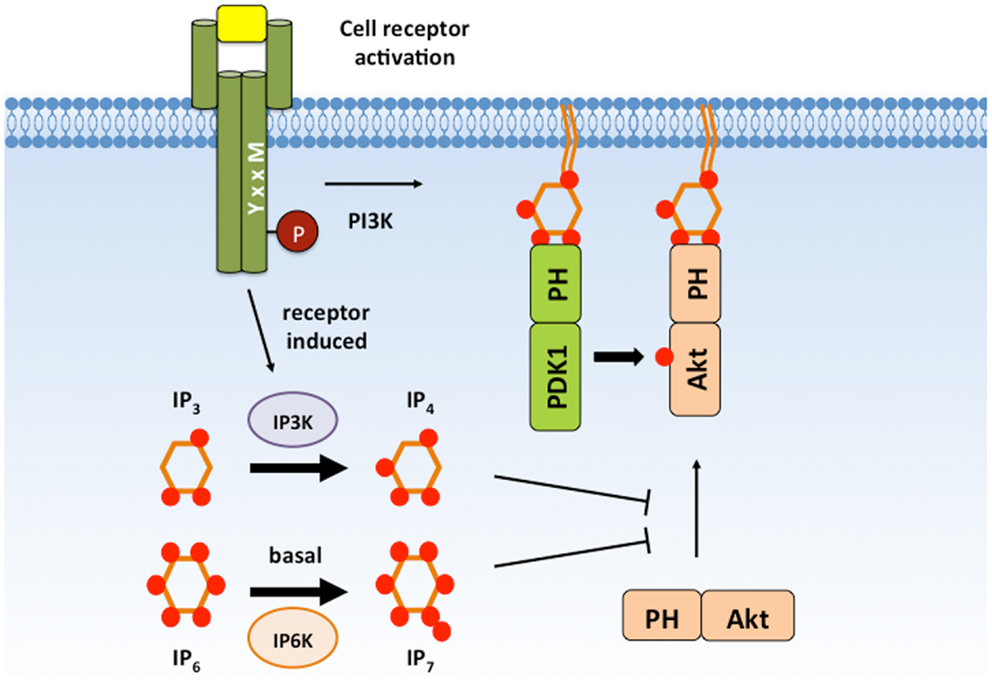
Figure 4. IP4 and IP7 negatively regulate Akt signaling. IP4 and IP7 are cytosolic PIP3 analogs that are able to associate with the Akt PH domain with high affinity and can compete with membrane PIP3. IP4 and IP7 binding has been proposed to dissociate Akt from the plasma membrane to prevent Akt activation and substrate accessibility. IP4, Ins(1,3,4,5)P4; IP7, 5-PP-(1,2,3,4,6)IP5; PIP3, PI(3,4,5)P3.
The inositol kinases IP3 kinase (Itpk) isoforms A, B, and C, and inositol polyphosphate multikinase (IPMK) can each generate IP4 by phosphorylating Ins(1,4,5)P3 (IP3) at the D-3 hydroxyl group [reviewed in Ref. (87)]. However, mice deficient in the ubiquitously expressed ItpkC or IPMK isoforms or in the neuronally enriched ItpkA isoform have no detectable immune abnormalities. In contrast, ItpkB expression is selectively enriched in hematopoietic cells and catalytically activated by the Ca2+-sensing protein calmodulin (CaM) following antigen receptor signaling. Analysis of ItpkB-deficient mice revealed a non-redundant requirement for ItpkB in lymphocyte development and activation (88–92). ItpkB deficiency results in severely reduced peripheral T cell numbers due to an absolute block in positive selection of CD4+CD8+ thymocytes (88). Defective activation of the Ras/MAP kinase pathway contributes to the T cell developmental defect (88, 89, 93). However, ItpkB-deficient CD4+CD8+ thymocytes are also defective in activation of Itk and its downstream effector PLC γ1 in response to TCR engagement (93). Itk fails to localize to the plasma membrane or assemble with the adapter protein LAT in the TCR signalosome of ItpkB-deficient thymocytes, indicating a requirement for IP4 in promoting Itk interactions (93). Interestingly, addition of IP4 increases binding of recombinant Itk PH domain to PIP3-coated beads in vitro, suggesting that IP4 may alter Itk PH domain conformation to enhance PIP3 accessibility (93).
Distinct from its effect on Itk, IP4 suppresses Akt activity by directly competing with PIP3 for binding to the Akt PH domain (94). ItpkB-deficient mice develop profound alterations in neutrophil and NK cell functions due to enhanced Akt activity during their development and activation (94, 95). Addition of membrane permeable IP4, but not an isomer, to the myeloid cell line HL-60 disrupts membrane localization of an Akt PH domain fused to GFP (94). In ItpkB-deficient neutrophils, Akt phosphorylation is enhanced in response to the bacterial peptide Formyl–Methionyl–Leucyl–Phenylalanine (fMLP). Enhanced Akt signaling in ItpkB-deficient neutrophils contributes to augmented anti-microbial and chemotaxis responses (94). The magnitude and kinetics of Akt phosphorylation are also increased in ItpkB-deficient NK cells (95). Elevated IFNγ secretion, granule exocytosis, and tumor cell lysis by ItpkB-deficient NK cells can be suppressed by Akt inhibition (95). Together, these studies indicate that IP4 dampens Akt activity in neutrophils and NK cells to restrict effector functions. Whether this occurs to shut-off innate functions during the resolution phase of an immune response or as a check to limit inflammatory damage remains unclear.
Similar to IP4, IP7 also competes with PIP3 for binding to the Akt PH domain and negatively regulates its activity (96). IP7 is generated by pyro-phosphorylation of IP6 at the 5-phosphate group by IP6 family kinases, IP6Ks (97, 98). While the importance of IP6K1 in lymphocyte function remains to be determined, analysis of IP6K1-deficient neutrophils demonstrates similar functional defects as ItpkB-deficient neutrophils. Both deficiencies result in enhanced fMLP-induced chemotaxis, superoxide production, and bacterial killing (94, 99). Akt membrane localization and activation are significantly increased in IP6K1-deficient neutrophils (99). Interestingly, IP7 is readily detectable in resting HL-60 cells but rapidly decreases upon fMLP stimulation (99). This suggests that IP7 may act to suppress initial Akt activation while IP4 regulates subsequent Akt activity following its induced production. Precise regulation of basal and induced IP4 and IP7 levels may act together to control the magnitude and kinetics of Akt activation in these innate immune cells. Future studies are required to determine the functional effects of IP4 and IP7 on Akt-dependent regulation of lymphocyte differentiation and effector responses. It also remains to be determined whether IP7 acts on other PIP3 effectors in immune cells as it does in Dictyostelium discoideum (100) or whether selective IP7 binding allows regulation of a particular subset of PIP3 effectors.
Recently, biochemical and structural analyses of Btk identified a new activating function for the inositol phosphate, IP6 (101). As with PIP3-containing liposomes, addition of soluble IP6 induces Btk trans-phosphorylation and activation. However, IP6 promotes Btk activation by an unconventional mechanism that is independent of the PIP3-binding pocket and membrane recruitment. Analysis of the co-crystal structure of IP6 with the Btk PH domain reveals an additional peripheral IP6 binding site sandwiched between two PH modules, termed the Saraste dimer. Molecular dynamics simulations suggest that IP6 neutralizes electrostatic forces in the monomer that oppose dimer formation. Mutation of the IP6 peripheral binding site disrupts transient dimerization and significantly abrogates IP6-dependent Btk trans-phosphorylation (101). IP6-induced Btk activation in solution represents a new PI3K-independent mechanism for controlling Btk activity. Considering that IP6 levels are basally high in lymphocytes, it will be important in future studies to determine whether IP6 contributes to tonic or B cell receptor-induced Btk function.
Proteins Interact with and Regulate the Activity of PH Domain-Containing Proteins
Although the Akt and Itk PH domains specifically bind to PIP3 with (nanomolar) affinities, only ~40 mammalian PH domains appear to be PIP3-regulated according to Teruel and colleagues, who developed a prediction algorithm based on experimental analyses of 130 mouse PH domains (102). The majority of PH domains do not interact with lipids or bind lipids promiscuously or with low affinity (Kd ≥ 10 μM). Furthermore, a growing number of PH domains have been reported to participate in inter- and/or intra-molecular protein interactions (discussed below). These findings support a revised view of PH domains as diverse, multifunctional domains that bind lipids, proteins, or both to regulate the activity of their parent proteins.
T and B cells induce Ca2+ and DAG-mediated signaling through PLCγ1- and PLCγ2-mediated cleavage of PIP2 (103, 104). T cell-specific ablation of PLCγ1 causes defects in thymocyte selection during T cell development, reduced T cell proliferation and cytokine secretion, and the development of autoimmunity resulting from defective regulatory T cells (104). PLCγ2 plays important roles in regulating B cells, neutrophils, mast cells, and dendritic cells (105–107). PLCγ1 and PLCγ2 both contain two PH domains. The conventional, N-terminal PH domain associates with PIP3 (108); however, the C-terminal PH domain is interrupted by an intervening amino acid sequence comprising two tandem SH2 domains and an SH3 domain (109, 110). This split PH domain is also critical for substrate binding (111). The C-terminal half of the PLCγ1 split PH domain associates with a partial PH domain in TRPC3 (112, 113), a Ca2+ channel that can mediate Ca2+ entry in T cells. The formation of this inter-molecular PH-like domain allows PLCγ1 to bind to its substrate PIP2 and is critical for TRPC3 membrane targeting and surface expression (113). Conversely, the split PH domain of PLCγ2 interacts with the small GTPase Rac2, which mediates PLCγ2 activation and localization to the plasma membrane (114–116).
Pleckstrin homology domains also participate in intra-molecular interactions. In resting cells, the Akt PH domain associates with the kinase domain (KD) to maintain a closed conformation in which the activation loop is blocked (117, 118). PIP3 binding to the Akt PH domain exposes the activation loop, allowing T308 and S473 to be accessed and phosphorylated by PDK1 and mTORC2, respectively (119). Phosphorylation of T308 and S473 fully activates Akt and keeps the activation loop “open” for substrate docking (117–119). PH domain mutations that disrupt PH–KD interaction (e.g., L52R and Q79K) result in constitutive Akt activation (119).
The Dbl family RhoGEF Vav is also regulated by lipid and intra-molecular interactions involving its PH domain (Figure 5). Vav plays crucial roles during T cell and B cell development (120, 121) and T cell, B cell, neutrophil, and NK cell activation (9, 107, 120–123). Vav contains a Dbl homology (DH) domain that promotes the activation of the small GTPase Rac in response to PI3K activation (124, 125). In quiescent cells, Vav1 adopts an auto-inhibitory conformation, which is stabilized by interactions between its PH, acidic (Ac), and calponin homology (CH) domains (126, 127). A truncation mutation of the Vav N-terminal CH domain was shown to have oncogenic potential (128), highlighting the importance of these intra-molecular interactions in limiting Vav activity. During T cell activation, Lck phosphorylates tyrosine residues within the Ac domain to release Vav1 from auto-inhibition (127). PIP3 binding to the PH domain significantly enhances Lck-dependent Vav1 phosphorylation in vitro (129) and promotes GEF activity (124, 129, 130) likely through the release of auto-inhibition (131). Interestingly, PIP2 binding to the Vav1 PH domain inhibits GEF activity (129). Thus, distinct lipids bind to the Vav1 PH domain to promote conformational changes that either reinforce or relieve its auto-inhibitory state.
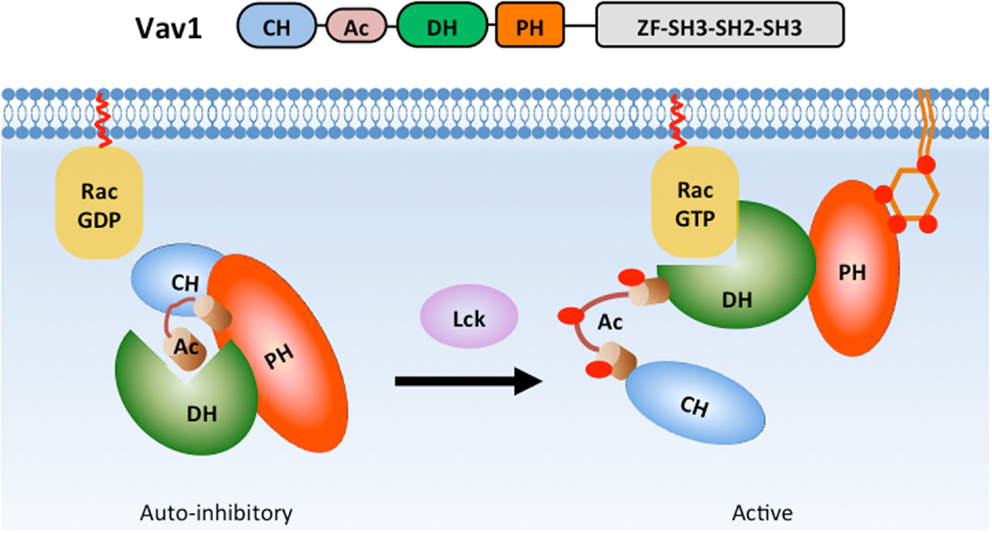
Figure 5. PH domain interactions stabilize Vav1 auto-inhibition in basal state. In the basal state, Vav1 adopts an auto-inhibitory conformation in which the substrate-docking site within the DH domain is blocked by interactions with a helix region from the Ac domain. The interactions between CH, PH, and Ac domains greatly strengthen the auto-inhibitory conformation (left). During T cell activation, phosphorylation of the Ac domain by Lck releases the substrate-docking site and allows GTPase binding (right).
Pleckstrin homology domains can also participate in inter-molecular interactions with other proteins. The PH domain of Dbs, a Cdc42/RhoGEF, associates with Cdc42 through the β3/β4 loop of its PH domain to improve substrate docking and catalysis (132). Interestingly, we recently identified the β3/β4 loop of the Itk PH domain as an important binding site for the ubiquitous Ca2+-sensing protein CaM (133). The CaM C-terminal EF hands bind to the β3/β4 loop of the Itk PH domain at basal intracellular Ca2+ levels while the CaM N-terminal EF hands engage the β5/β6 loop upon an increase in Ca2+ levels. CaM and PIP3 (but not IP4) reciprocally enhance binding of one another to the Itk PH domain in vitro, suggesting that CaM and PIP3 cooperate to regulate Itk signaling at the plasma membrane. Pharmacological inhibition of Ca2+/CaM activity or mutation of the CaM-binding β3/β4 loop disrupts Itk-dependent activation of PLCγ1 and downstream Ca2+ responses (133), indicating that CaM participates in a positive feedback loop whereby binding of CaM to the Itk PH domain enhances further Itk activation and downstream Ca2+ responses. Importantly, this positive feedback is required for optimal TCR-induced, NFAT-dependent production of the pro-inflammatory cytokine, IL-17A (133). Thus, CaM represents a novel protein-binding partner for the Itk PH domain that serves an important function in potentiating T cell pro-inflammatory responses (Figure 6). It remains to be determined how CaM, PIP3, and IP4 coordinate to regulate the kinetics and magnitude of Itk activation and whether they differentially participate in Itk-dependent T cell activation, differentiation, and effector responses.
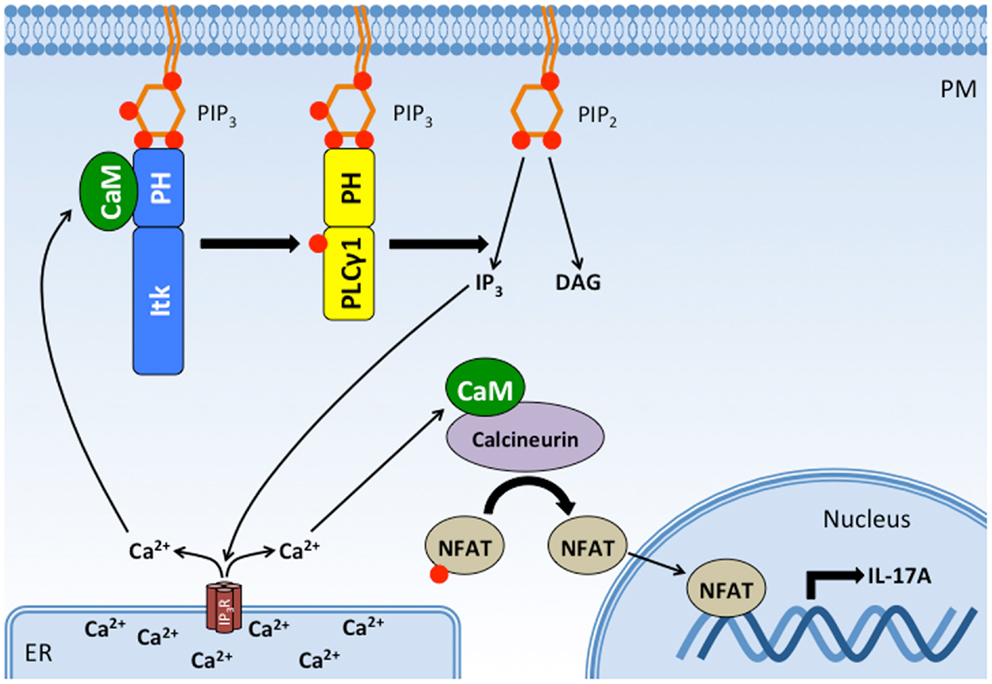
Figure 6. CaM binds the Itk PH domain in a positive feedback loop that potentiates Itk activity, intracellular Ca2+ release, and IL-17A production. Binding of Itk to PIP3 promotes Itk activation and the subsequent phosphorylation and activation of PLC γ1. PLCγ1 cleaves PIP2 to produce DAG and IP3, which binds IP3 receptors on the ER. The IP3 receptor is a ligand-gated Ca2+ channel, and its activation increases Ca2+ levels in the cytosol. Increased cytosolic Ca2+ activates CaM, which has at least two effects on T cell activation: (1) Ca2+/CaM binds to Itk’s PH domain, enhancing its interaction with PIP3 and Itk activity. (2) Ca2+/CaM binds to and activates calcineurin, a phophatase that dephosphorylates NFAT, allowing NFAT translocation to the nucleus where it drives the transcription of IL-17A. Thus, CaM binding to Itk’s PH domain completes a positive feedback loop that potentiates the downstream effects of Itk. PM, plasma membrane; ER, endoplasmic reticulum; Itk, IL-2-inducible tyrosine/T cell kinase; PLCγ1, phospholipase C gamma 1; CaM, calmodulin; NFAT, nuclear factor of activated T cells; IP3R, IP3 receptor.
Calmodulin has also been reported by Dong and colleagues to bind the PH domain of Akt family kinases (134). Using short peptide fragments of Akt1 in a pulldown assay, this interaction was further mapped to the first 42 residues of the Akt1 PH domain. Although CaM did not directly alter Akt kinase activity, CaM was reported to reduce the ability of PIP3 to co-precipitate Akt (134), suggesting that CaM competes with PIP3 to dampen Akt activity. However, this finding is inconsistent with other published data demonstrating a requirement for CaM in optimal Akt phosphorylation at T308 and S473 (135, 136). Thus, further investigation is warranted to clarify the functional significance of CaM binding to the AKT PH domain and to determine the precise role of this interaction in lymphocytes.
Conclusion
The studies discussed herein highlight the essential yet complex functions of PH domain-containing proteins in lymphocytes and other immune cells. It is well established that a subset of PH domains modulate the function of their parent proteins by binding to membrane-bound lipids as well as soluble lipid analogs. Furthermore, proteins regulated in this manner, such as the PI3K effector kinases Akt and Itk, are indispensable for immune cell function. Indeed, mutations that disrupt the lipid-binding capacity of PH domains are known to result in human disease, a phenomenon perhaps best demonstrated by the immunologic defects associated with mutations in Tec family kinases. Analogous and unique pathological processes observed in animal models and in vitro experiments reinforce the critical role of PH domain-containing proteins in the immune system. However, evidence increasingly shows that PH domains also interact with non-lipid substrates, and these interactions can be cooperative, antagonistic, or completely independent of lipid-binding capacity. The breadth of these interactions must be elucidated in order to fully understand role of PH domain-containing proteins in immune cell function. Thus, future work should investigate the capacity of PH domains to interact with multiple substrates, including both lipids and proteins, and should include careful evaluation of how binding of each substrate affects the binding of others.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grant AI089805 to YH.
References
1. Gadina M, Sudarshan C, Visconti R, Zhou YJ, Gu H, Neel BG, et al. The docking molecule gab2 is induced by lymphocyte activation and is involved in signaling by interleukin-2 and interleukin-15 but not other common gamma chain-using cytokines. J Biol Chem (2000) 275:26959–66. doi:10.1074/jbc.M004021200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Ward SG, Cantrell DA. Phosphoinositide 3-kinases in T lymphocyte activation. Curr Opin Immunol (2001) 13:332–8. doi:10.1016/S0952-7915(00)00223-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Koyasu S. The role of PI3K in immune cells. Nat Immunol (2003) 4:313–9. doi:10.1038/ni0403-313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Guthridge MA, Lopez AF. Phosphotyrosine/phosphoserine binary switches: a new paradigm for the regulation of PI3K signalling and growth factor pleiotropy? Biochem Soc Trans (2007) 35:250–2. doi:10.1042/BST0350250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Swainson L, Kinet S, Mongellaz C, Sourisseau M, Henriques T, Taylor N. IL-7-induced proliferation of recent thymic emigrants requires activation of the PI3K pathway. Blood (2007) 109:1034–42. doi:10.1182/blood-2006-06-027912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675–704. doi:10.1146/annurev-immunol-032712-095946
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol (2003) 4:557–64. doi:10.1038/ni929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Kolsch U, Arndt B, Reinhold D, Lindquist JA, Juling N, Kliche S, et al. Normal T cell development and immune functions in TRIM-deficient mice. Mol Cell Biol (2006) 26:3639–48. doi:10.1128/MCB.26.9.3639-3648.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Upshaw JL, Arneson LN, Schoon RA, Dick CJ, Billadeau DD, Leibson PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol (2006) 7:524–32. doi:10.1038/ni1325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Koelsch U, Schraven B, Simeoni L. SIT and TRIM determine T cell fate in the thymus. J Immunol (2008) 181:5930–9. doi:10.4049/jimmunol.181.9.5930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, et al. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol (2009) 85:966–77. doi:10.1189/jlb.1208763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol (2010) 11:329–41. doi:10.1038/nrm2882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Andrews S, Stephens LR, Hawkins PT. PI3K class IB pathway in neutrophils. Sci STKE (2007) 2007:cm3. doi:10.1126/stke.4072007cm2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene (2008) 27:5527–41. doi:10.1038/onc.2008.247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Insall RH, Weiner OD. PIP3, PIP2, and cell movement – similar messages, different meanings? Dev Cell (2001) 1:743–7. doi:10.1016/S1534-5807(01)00086-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Wang J, Richards DA. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol Open (2012) 1:857–62. doi:10.1242/bio.20122071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol (2008) 9:99–111. doi:10.1038/nrm2328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Isakoff SJ, Cardozo T, Andreev J, Li Z, Ferguson KM, Abagyan R, et al. Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO J (1998) 17:5374–87. doi:10.1093/emboj/17.18.5374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. De Matteis MA, Di Campli A, Godi A. The role of the phosphoinositides at the golgi complex. Biochim Biophys Acta (2005) 1744:396–405. doi:10.1016/j.bbamcr.2005.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi:10.1016/j.cell.2007.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle (2009) 8:2502–8. doi:10.4161/cc.8.16.9335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol (2012) 4:a011189. doi:10.1101/cshperspect.a011189
23. Limon JJ, Fruman DA. Akt and mTOR in B cell activation and differentiation. Front Immunol (2012) 3:228. doi:10.3389/fimmu.2012.00228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwag A, et al. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem (2000) 285:36108–15. doi:10.1074/jbc.M005497200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature (2007) 448:439–44. doi:10.1038/nature05933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature (2012) 486:405–9. doi:10.1038/nature11154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Landgraf KE, Pilling C, Falke JJ. Molecular mechanism of an oncogenic mutation that alters membrane targeting: glu17lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry (2008) 47:12260–9. doi:10.1021/bi801683k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood (2010) 115:1406–15. doi:10.1182/blood-2009-06-229443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell (2007) 128:309–23. doi:10.1016/j.cell.2006.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Fabre S, Carrette F, Chen J, Lang V, Semichon M, Denoyelle C, et al. FOXO1 regulates L-selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J Immunol (2008) 181:2980–9. doi:10.4049/jimmunol.181.5.2980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, Depinho RA, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol (2009) 10:176–84. doi:10.1038/ni.1689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity (2009) 30:358–71. doi:10.1016/j.immuni.2009.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Merkenschlager M, von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering foxo factors. J Exp Med (2010) 207:1347–50. doi:10.1084/jem.20101156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Ouyang W, Beckett O, Ma Q, Paik JH, Depinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol (2010) 11:618–27. doi:10.1038/ni.1884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Alkhatib A, Werner M, Hug E, Herzog S, Eschbach C, Faraidun H, et al. FoxO1 induces Ikaros splicing to promote immunoglobulin gene recombination. J Exp Med (2012) 209:395–406. doi:10.1084/jem.20110216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity (2012) 36:374–87. doi:10.1016/j.immuni.2012.01.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Michelini RH, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med (2013) 210:1189–200. doi:10.1084/jem.20130392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Hedrick SM. The cunning little vixen: foxo and the cycle of life and death. Nat Immunol (2009) 10:1057–63. doi:10.1038/ni.1784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med (2008) 205:565–74. doi:10.1084/jem.20071477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Pierau M, Engelmann S, Reinhold D, Lapp T, Schraven B, Bommhardt UH. Protein kinase B/Akt signals impair Th17 differentiation and support natural regulatory T cell function and induced regulatory T cell formation. J Immunol (2009) 183:6124–34. doi:10.4049/jimmunol.0900246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol (2012) 188:4305–14. doi:10.4049/jimmunol.1103568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15:88–97. doi:10.1038/ni.2771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123:4479–88. doi:10.1172/JCI69589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity (2011) 34:224–36. doi:10.1016/j.immuni.2011.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell (1993) 72:279–90. doi:10.1016/0092-8674(93)90667-F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Afar DE, Park H, Howell BW, Rawlings DJ, Cooper J, Witte ON. Regulation of Btk by Src family tyrosine kinases. Mol Cell Biol (1996) 16:3465–71.
47. Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science (1996) 271:822–5. doi:10.1126/science.271.5250.822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Niiro H, Clark EA. Regulation of B cell fate by antigen-receptor signals. Nat Rev Immunol (2002) 2:945–56. doi:10.1038/nri955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Vihinen M, Brandau O, Branden LJ, Kwan SP, Lappalainen I, Lester T, et al. BTKbase, mutation database for X-linked agammaglobulinemia (XLA). Nucleic Acids Res (1998) 26:242–7. doi:10.1093/nar/26.1.242
50. Vihinen M, Kwan S-P, Lester T, Ochs HD, Resnick I, Väliaho J, et al. Mutations of the human BTK gene coding for Bruton tyrosine kinase in X-linked agammaglobulinemia. Hum Mutat (1999) 13:280–5. doi:10.1002/(SICI)1098-1004(1999)13:4<280::AID-HUMU3>3.0.CO;2-L
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Nomura K, Kanegane H, Karasuyama H, Tsukada S, Agematsu K, Murakami G, et al. Genetic defect in human X-linked agammaglobulinemia impedes a maturational evolution of pro-B cells into a later stage of pre-B cells in the B cell differentiation pathway. Blood (2000) 96:610–7.
52. Fukuda M, Kojima T, Kabayama H, Mikoshiba K. Mutation of the pleckstrin homology domain of Bruton’s tyrosine kinase in immunodeficiency impaired inositol 1,3,4,5-tetrakisphosphate binding capacity. J Biol Chem (1996) 271:30303–6. doi:10.1074/jbc.271.48.30303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Varnai P, Rother KI, Balla T. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton’s tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem (1999) 274:10983–9. doi:10.1074/jbc.274.16.10983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Li T, Tsukada S, Satterthwaite A, Havlik MH, Park H, Takatsu K, et al. Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity (1995) 2:451–60. doi:10.1016/1074-7613(95)90026-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Maas A, Dingjan GM, Grosveld F, Hendriks RW. Early arrest in B cell development in transgenic mice that express the E41K Bruton’s tyrosine kinase mutant under the control of the CD19 promoter region. J Immunol (1999) 162:6526–33.
56. Huck K, Feyen O, Niehues T, Rüschendorf F, Hübner N, Laws H-J, et al. Girls homozygous for an IL-2–inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest (2009) 119:1350–8. doi:10.1172/JCI37901
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Serwas NK, Cagdas D, Ban SA, Bienemann K, Salzer E, Tezcan I, et al. Identification of ITK deficiency as a novel genetic cause of idiopathic CD4+ T cell lymphopenia. Blood (2014) 124:655–7. doi:10.1182/blood-2014-03-564930
58. Barton E, Mandal P, Speck SH. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol (2011) 29:351–97. doi:10.1146/annurev-immunol-072710-081639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol (2010) 2:a002287. doi:10.1101/cshperspect.a002287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Grasis JA, Tsoukas CD. Itk: the rheostat of the T cell response. J Signal Transduct (2011) 2011:297868. doi:10.1155/2011/297868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Heyeck SD, Wilcox HM, Bunnell SC, Berg LJ. Lck phosphorylates the activation loop tyrosine of the Itk kinase domain and activates Itk kinase activity. J Biol Chem (1997) 272:25401–8. doi:10.1074/jbc.272.40.25401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Perez-Villar JJ, Kanner SB. Regulated association between the tyrosine kinase Emt/Itk/Tsk and phospholipase-C gamma 1 in human T lymphocytes. J Immunol (1999) 163:6435–41.
63. Takesono A, Finkelstein LD, Schwartzberg PL. Beyond calcium: new signaling pathways for Tec family kinases. J Cell Sci (2002) 115:3039–48.
64. Fowell DJ, Shinkai K, Liao XC, Beebe AM, Coffman RL, Littman DR, et al. Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity (1999) 11:399–409. doi:10.1016/S1074-7613(00)80115-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Miller AT, Wilcox HM, Lai Z, Berg LJ. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity (2004) 21:67–80. doi:10.1016/j.immuni.2004.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Au-Yeung BB, Katzman SD, Fowell DJ. Cutting edge: Itk-dependent signals required for CD4+ T cells to exert, but not gain, Th2 effector function. J Immunol (2006) 176:3895–9. doi:10.4049/jimmunol.176.7.3895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, et al. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity (2009) 31:587–97. doi:10.1016/j.immuni.2009.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Liao XC, Littman DR. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity (1995) 3:757–69. doi:10.1016/1074-7613(95)90065-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med (2014) 211(3):529–43. doi:10.1084/jem.20131459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Jain N, Miu B, Jiang JK, Mckinstry KK, Prince A, Swain SL, et al. CD28 and ITK signals regulate autoreactive T cell trafficking. Nat Med (2013) 19:1632–7. doi:10.1038/nm.3393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Cuevas B, Lu Y, Watt S, Kumar R, Zhang J, Siminovitch KA, et al. SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J Biol Chem (1999) 274:27583–9. doi:10.1074/jbc.274.39.27583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Bryceson YT, Ljunggren HG. Arrestin NK cell cytotoxicity. Nat Immunol (2008) 9:835–6. doi:10.1038/ni0808-835
73. Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity (1998) 8:497–508. doi:10.1016/S1074-7613(00)80554-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Alegre ML, Frauwirth KA, Thompson CB. T cell regulation by CD28 and CTLA-4. Nat Rev Immunol (2001) 1:220–8. doi:10.1038/35105024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol (2013) 14:1212–8. doi:10.1038/ni.2762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173:945–54. doi:10.4049/jimmunol.173.2.945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev (2009) 228:342–59. doi:10.1111/j.1600-065X.2008.00760.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Ugi S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, et al. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol Cell Biol (2004) 24:8778–89. doi:10.1128/MCB.24.19.8778-8789.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem (2008) 283:1882–92. doi:10.1074/jbc.M709585200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T cell inactivation by CTLA-4. Science (1998) 282:2263–6. doi:10.1126/science.282.5397.2263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Huang YH, Sauer K. Lipid signaling in T cell development and function. Cold Spring Harb Perspect Biol (2010) 2:a002428. doi:10.1101/cshperspect.a002428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Fedele CG, Ooms LM, Ho M, Vieusseux J, O’toole SA, Millar EK, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A (2010) 107:22231–6. doi:10.1073/pnas.1015245107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature (1996) 383:263–6. doi:10.1038/383263a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Wang JW, Howson JM, Ghansah T, Desponts C, Ninos JM, May SL, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science (2002) 295:2094–7. doi:10.1126/science.1068438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Newton RH, Turka LA. Regulation of T cell homeostasis and responses by pten. Front Immunol (2012) 3:151. doi:10.3389/fimmu.2012.00151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Sauer K, Cooke MP. Regulation of immune cell development through soluble inositol-1,3,4,5-tetrakisphosphate. Nat Rev Immunol (2010) 10:257–71. doi:10.1038/nri2745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Pouillon V, Hascakova-Bartova R, Pajak B, Adam E, Bex F, Dewaste V, et al. Inositol 1,3,4,5-tetrakisphosphate is essential for T lymphocyte development. Nat Immunol (2003) 4:1136–43. doi:10.1038/ni980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Wen BG, Pletcher MT, Warashina M, Choe SH, Ziaee N, Wiltshire T, et al. Inositol (1,4,5) trisphosphate 3 kinase B controls positive selection of T cells and modulates Erk activity. Proc Natl Acad Sci U S A (2004) 101:5604–9. doi:10.1073/pnas.0306907101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Marechal Y, Pesesse X, Jia Y, Pouillon V, Perez-Morga D, Daniel J, et al. Inositol 1,3,4,5-tetrakisphosphate controls proapoptotic Bim gene expression and survival in B cells. Proc Natl Acad Sci U S A (2007) 104:13978–83. doi:10.1073/pnas.0704312104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Miller AT, Sandberg M, Huang YH, Young M, Sutton S, Sauer K, et al. Production of Ins(1,3,4,5)P4 mediated by the kinase Itpkb inhibits store-operated calcium channels and regulates B cell selection and activation. Nat Immunol (2007) 8:514–21. doi:10.1038/ni1458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Miller AT, Beisner DR, Liu D, Cooke MP. Inositol 1,4,5-trisphosphate 3-kinase B is a negative regulator of BCR signaling that controls B cell selection and tolerance induction. J Immunol (2009) 182:4696–704. doi:10.4049/jimmunol.0802850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Huang YH, Grasis JA, Miller AT, Xu R, Soonthornvacharin S, Andreotti AH, et al. Positive regulation of Itk PH domain function by soluble IP4. Science (2007) 316:886–9. doi:10.1126/science.1138684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Jia Y, Subramanian KK, Erneux C, Pouillon V, Hattori H, Jo H, et al. Inositol 1,3,4,5-tetrakisphosphate negatively regulates phosphatidylinositol-3,4,5-trisphosphate signaling in neutrophils. Immunity (2007) 27:453–67. doi:10.1016/j.immuni.2007.07.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Sauer K, Park E, Siegemund S, French AR, Wahle JA, Sternberg L, et al. Inositol tetrakisphosphate limits NK cell effector functions by controlling PI3K signaling. Blood (2013) 121:286–97. doi:10.1182/blood-2012-05-429241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell (2010) 143:897–910. doi:10.1016/j.cell.2010.11.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol (1999) 9:1323–6. doi:10.1016/S0960-9822(00)80055-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Manning BD. Insulin signaling: inositol phosphates get into the Akt. Cell (2010) 143:861–3. doi:10.1016/j.cell.2010.11.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Prasad A, Jia Y, Chakraborty A, Li Y, Jain SK, Zhong J, et al. Inositol hexakisphosphate kinase 1 regulates neutrophil function in innate immunity by inhibiting phosphatidylinositol-(3,4,5)-trisphosphate signaling. Nat Immunol (2011) 12:752–60. doi:10.1038/ni.2052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, et al. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell (2003) 114:559–72. doi:10.1016/S0092-8674(03)00640-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Wang Q, Vogan EM, Nocka LM, Rosen CE, Zorn JA, Harrison SC, et al. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife (2015). doi:10.7554/eLife.06074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Park WS, Heo WD, Whalen JH, O’rourke NA, Bryan HM, Meyer T, et al. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell (2008) 30:381–92. doi:10.1016/j.molcel.2008.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol (2007) 7:690–702. doi:10.1038/nri2152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Fu G, Chen Y, Yu M, Podd A, Schuman J, He Y, et al. Phospholipase C{gamma}1 is essential for T cell development, activation, and tolerance. J Exp Med (2010) 207:309–18. doi:10.1084/jem.20090880
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, et al. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity (2000) 13:25–35. doi:10.1016/S1074-7613(00)00005-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Wen R, Jou ST, Chen Y, Hoffmeyer A, Wang D. Phospholipase C gamma 2 is essential for specific functions of Fc epsilon R and Fc gamma R. J Immunol (2002) 169:6743–52. doi:10.4049/jimmunol.169.12.6743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Graham DB, Robertson CM, Bautista J, Mascarenhas F, Diacovo MJ, Montgrain V, et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J Clin Invest (2007) 117:3445–52. doi:10.1172/JCI32729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J (1998) 17:414–22. doi:10.1093/emboj/17.2.414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem (1997) 272:15045–8. doi:10.1074/jbc.272.24.15045
110. Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem (2001) 70:281–312. doi:10.1146/annurev.biochem.70.1.281
111. Kim SK, Wee SM, Chang JS, Kwon TK, Min DS, Lee YH, et al. Point mutations in the split PLC-gamma1 PH domain modulate phosphoinositide binding. J Biochem Mol Biol (2004) 37:720–5. doi:10.5483/BMBRep.2004.37.6.720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Lemmon MA. Pleckstrin homology domains: two halves make a hole? Cell (2005) 120:574–6. doi:10.1016/j.cell.2005.02.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. van Rossum DB, Patterson RL, Sharma S, Barrow RK, Kornberg M, Gill DL, et al. Phospholipase Cgamma1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature (2005) 434:99–104. doi:10.1038/nature03340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Piechulek T, Rehlen T, Walliser C, Vatter P, Moepps B, Gierschik P. Isozyme-specific stimulation of phospholipase C-gamma2 by Rac GTPases. J Biol Chem (2005) 280:38923–31. doi:10.1074/jbc.M509396200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Walliser C, Retlich M, Harris R, Everett KL, Josephs MB, Vatter P, et al. Rac regulates its effector phospholipase Cgamma2 through interaction with a split pleckstrin homology domain. J Biol Chem (2008) 283:30351–62. doi:10.1074/jbc.M803316200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Everett KL, Buehler A, Bunney TD, Margineanu A, Baxendale RW, Vatter P, et al. Membrane environment exerts an important influence on Rac-mediated activation of phospholipase Cgamma2. Mol Cell Biol (2011) 31:1240–51. doi:10.1128/MCB.01408-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol (2009) 7:e17. doi:10.1371/journal.pbio.1000017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, Brandhuber BJ. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One (2010) 5:e12913. doi:10.1371/journal.pone.0012913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Parikh C, Janakiraman V, Wu WI, Foo CK, Kljavin NM, Chaudhuri S, et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci U S A (2012) 109:19368–73. doi:10.1073/pnas.1204384109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Fischer KD, Zmuldzinas A, Gardner S, Barbacid M, Bernstein A, Guidos C. Defective T cell receptor signalling and positive selection of Vav-deficient CD4+ CD8+ thymocytes. Nature (1995) 374:474–7. doi:10.1038/374474a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Tedford K, Nitschke L, Girkontaite I, Charlesworth A, Chan G, Sakk V, et al. Compensation between Vav-1 and Vav-2 in B cell development and antigen receptor signaling. Nat Immunol (2001) 2:548–55. doi:10.1038/88756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol (2002) 2:476–86. doi:10.1038/nri840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Hall AB, Gakidis MA, Glogauer M, Wilsbacher JL, Gao S, Swat W, et al. Requirements for Vav guanine nucleotide exchange factors and Rho GTPases in FcgammaR- and complement-mediated phagocytosis. Immunity (2006) 24:305–16. doi:10.1016/j.immuni.2006.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Han J, Luby-Phelps K, Das B, Shu X, Xia Y, Mosteller RD, et al. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science (1998) 279:558–60. doi:10.1126/science.279.5350.558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Ma AD, Metjian A, Bagrodia S, Taylor S, Abrams CS. Cytoskeletal reorganization by G protein-coupled receptors is dependent on phosphoinositide 3-kinase gamma, a Rac guanosine exchange factor, and Rac. Mol Cell Biol (1998) 18:4744–51.
126. Li P, Martins IR, Amarasinghe GK, Rosen MK. Internal dynamics control activation and activity of the autoinhibited Vav DH domain. Nat Struct Mol Biol (2008) 15:613–8. doi:10.1038/nsmb.1428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Yu B, Martins IR, Li P, Amarasinghe GK, Umetani J, Fernandez-Zapico ME, et al. Structural and energetic mechanisms of cooperative autoinhibition and activation of Vav1. Cell (2010) 140:246–56. doi:10.1016/j.cell.2009.12.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Kranewitter WJ, Gimona M. N-terminally truncated Vav induces the formation of depolymerization-resistant actin filaments in NIH 3T3 cells. FEBS Lett (1999) 455:123–9. doi:10.1016/S0014-5793(99)00857-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Das B, Shu X, Day GJ, Han J, Krishna UM, Falck JR, et al. Control of intramolecular interactions between the pleckstrin homology and Dbl homology domains of Vav and Sos1 regulates Rac binding. J Biol Chem (2000) 275:15074–81. doi:10.1074/jbc.M907269199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol (2005) 6:167–80. doi:10.1038/nrm1587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Aghazadeh B, Lowry WE, Huang X-Y, Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell (2000) 102:625–33. doi:10.1016/S0092-8674(00)00085-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Rossman KL, Worthylake DK, Snyder JT, Siderovski DP, Campbell SL, Sondek J. A crystallographic view of interactions between Dbs and Cdc42: pH domain-assisted guanine nucleotide exchange. EMBO J (2002) 21:1315–26. doi:10.1093/emboj/21.6.1315
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Wang X, Boyken SE, Hu J, Xu X, Rimer RP, Shea MA, et al. Calmodulin and PI(3,4,5)P(3) cooperatively bind to the Itk pleckstrin homology domain to promote efficient calcium signaling and IL-17A production. Sci Signal (2014) 7:ra74. doi:10.1126/scisignal.2005147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Dong B, Valencia CA, Liu R. Ca(2+)/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J Biol Chem (2007) 282:25131–40. doi:10.1074/jbc.M702123200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Deb TB, Coticchia CM, Dickson RB. Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J Biol Chem (2004) 279:38903–11. doi:10.1074/jbc.M405314200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Coticchia CM, Revankar CM, Deb TB, Dickson RB, Johnson MD. Calmodulin modulates Akt activity in human breast cancer cell lines. Breast Cancer Res Treat (2009) 115:545–60. doi:10.1007/s10549-008-0097-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: PI3K, lymphocyte activation, pleckstrin homology domain, Akt signaling, Itk signaling
Citation: Wang X, Hills LB and Huang YH (2015) Lipid and protein co-regulation of PI3K effectors Akt and Itk in lymphocytes. Front. Immunol. 6:117. doi: 10.3389/fimmu.2015.00117
Received: 12 January 2015; Accepted: 02 March 2015;
Published online: 13 March 2015.
Edited by:
Klaus Okkenhaug, Babraham Institute, UKReviewed by:
M. Suresh, University of Wisconsin Madison, USALeslie J. Berg, University of Massachusetts Medical School, USA
Cosima T. Baldari, University of Siena, Italy
Copyright: © 2015 Wang, Hills and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yina Hsing Huang, Departments of Pathology and Microbiology and Immunology, The Geisel School of Medicine at Dartmouth, HB 7600, Borwell 650E, One Medical Center Drive, Lebanon, NH 03756, USA e-mail:eWluYS5oLmh1YW5nQGRhcnRtb3V0aC5lZHU=