 Steven K. Lundy
Steven K. Lundy Matthew W. Klinker
Matthew W. Klinker David A. Fox
David A. Fox- 1Department of Internal Medicine-Rheumatology, University of Michigan Medical School, Ann Arbor, MI, USA
- 2Graduate Training Program in Immunology, University of Michigan Medical School, Ann Arbor, MI, USA
Induction of immune tolerance is a key process by which the immune system is educated to modulate reactions against benign stimuli such as self-antigens and commensal microbes. Understanding and harnessing the natural mechanisms of immune tolerance may become an increasingly useful strategy for treating many types of allergic and autoimmune diseases, as well as for improving the acceptance of solid organ transplants. Our laboratory and others have been interested in the natural ability of some B lymphocytes to express the death-inducing molecule Fas ligand (FasL), and their ability to kill T helper (TH) lymphocytes. We have recently shown that experimental transformation of human B cells by a non-replicative variant of Epstein-Barr virus (EBV) consistently resulted in high expression of functional FasL protein. The production and release of FasL+ exosomes that co-expressed major histocompatibility complex (MHC) class II molecules and had the capacity to kill antigen-specific TH cells was also observed. Several lines of evidence indicate that FasL+ B cells and FasL+MHCII+ exosomes have important roles in natural immune tolerance and have a great deal of therapeutic potential. Taken together, these findings suggest that EBV-immortalized human B lymphoblastoid cell lines could be used as cellular factories for FasL+ exosomes, which would be employed to therapeutically establish and/or regain immune tolerance toward specific antigens. The goals of this review are to summarize current knowledge of the roles of FasL+ B cells and exosomes in immune regulation, and to suggest methods of manipulating killer B cells and FasL+ exosomes for clinical purposes.
Introduction
Fas ligand (FasL, CD178) is an important mediator of peripheral immune tolerance that is constitutively expressed by specialized epithelial cells at immune privileged sites of the body such as the eye and reproductive organs (1–3). Within the immune system, FasL expression is most often attributed to cytotoxic CD8+ T lymphocytes (CTL) and natural killer (NK) cells that are activated in response to viral infections or malignancies (4–8). A requirement for FasL expression by CD8+ veto T cells leading to deletion of effector CTL has been demonstrated to enhance tolerance of transplanted bone marrow cells (4, 9). Interaction of FasL with its receptor Fas (CD95) initiates a signaling cascade that leads to programmed cell death (apoptosis) in susceptible target cells (10, 11). The importance of FasL–Fas-mediated apoptosis to immune regulation was first highlighted by studies of mice with mutations affecting this pathway that developed lymphoproliferative disorders and produced autoantibodies similar to those found in systemic lupus erythematosus (SLE) patients (12, 13). In humans, defects in the FasL–Fas signaling pathway result in the autoimmune lymphoproliferative syndrome (ALPS), which most often manifests as autoimmune hemolytic anemia or other cytopenias caused by cell-specific autoantibody production (14). Thus, it is clear that FasL–Fas interactions play a significant role in regulating the production of pathogenic autoantibodies. It is well-established that mature B lymphocytes that have been activated through interactions with T helper (TH) lymphocytes are susceptible to FasL-mediated apoptosis, and presumably receive this signal from the interacting TH cell (15–17). What has often been overlooked is that some B lymphocytes express functional FasL constitutively, and that many types of stimuli can induce FasL expression in B cells (18). Therefore, it is important to consider that the reciprocal induction of apoptosis mediated by the B cell against the TH cell during these cognate interactions may also play a critical role in immune tolerance toward self-antigens.
KEY CONCEPT 1. Some B lymphocytes express functional FasL constitutively
In mice, these have been identified primarily within the CD5+ B-cell subset in spleen and lung. It remains to be determined how important they are in networks of mucosal and systemic immune tolerance, and which B-cell subset(s) in humans, if any, have similar functions.
FasL+ Killer B Cells
Expression of FasL by B cells was first reported in 1996 by Hahne and Tschopp, who demonstrated that activation of mouse B cells by mitogens led to FasL expression and killing of Fas-sensitive target cells by B cells from wild-type mice but not from FasL-mutant mice (19). Several other reports confirmed expression of FasL by both human and mouse B cells (20–30). Notably, it was found that human B-cell expression of FasL was induced during human immunodeficiency virus (HIV) and Epstein-Barr virus (EBV) infections, and that TH cells from infected individuals were very sensitive to Fas-mediated apoptosis (21, 27). While these data suggested that FasL expression by B cells might play a pathogenic role by inducing TH cell death in these viral infections, this was not explicitly tested. The first direct evidence that human B cells could induce TH cell apoptosis was demonstrated using FasL+ B-chronic lymphocytic leukemia (B-CLL) cells to kill a susceptible CD4+ T-cell leukemia line (23). FasL expression is common in the aggressive form of B-CLL, and has been reported in other human B-cell leukemias and lymphomas, most notably, multiple myeloma (31–34). Important questions that remain regarding human FasL+ B cells include: 1) are there specific human B-cell subsets that preferentially express FasL; 2) how is FasL expression regulated in human B cells; and 3) what are the normal functions of FasL+ B cells in humans? Studies performed in mice may provide some answers to these questions.
Analysis of TH cell apoptosis and the cellular distribution of FasL expression in the schistosome worm infection model led to our independent discovery of mouse FasL+ B cells (35). In that study, antigen-specific TH cell apoptosis was impaired when B cells but not CD8+ T cells were depleted from cell cultures, and was regained when TH cells were mixed with purified B cells from worm-infected mice. In a follow-up study, it was discovered that FasL expression was present on 1–2% of splenic B lymphocytes in uninfected mice, and was almost exclusively found within the CD5+ B-cell subset (36). The number of CD5+ and CD5neg B cells that expressed FasL increased significantly during the acute phase of schistosome infection, paralleling increases in TH cell apoptosis. Purified CD5+ B cells from both naïve and schistosome-infected mice had potent in vitro cytotoxic activity against TH cells isolated from schistosome-infected mice, but not naïve TH cells. In summary, mouse CD5+ B cells are constitutive and inducible expressers of functional FasL, and are efficient killer cells toward antigen-specific TH cells in vitro. It is yet to be determined if the human equivalents of CD5+ B cells, or other human B-cell subsets with reported regulatory functions, have the same constitutive expression of FasL and ability to kill antigen-specific TH cells in vivo (37).
Control of Killer B Lymphocyte Growth and Function
The schistosome model is an excellent system for studying the progression of the immune response. The initial reaction to worm egg deposition is an innate, pro-inflammatory reaction followed by acute TH1- and TH17-mediated inflammation that transitions to a strong TH2-mediated immune response, and which ultimately culminates in a chronic, fibrotic, and systemically immunosuppressive reaction (38). Peak FasL+ B-cell expansion and activation in the schistosome model occurred in the latter stages of the TH2 response and beginning of the chronic phase (35). B cells isolated from infected mice could be further induced to express surface FasL by treatment with interleukin 4 (IL-4) and IL-10 (36). More recently, we have shown that in vitro activation of purified splenic B cells from naïve mice with CD40 ligand (CD40L) and the TH2 cytokine IL-5 (but not IL-4) was sufficient to induce the proliferation of CD5+FasL+ B cells, and to increase the peptide-specific cytotoxic activity of B cells against TH cells (39). Naïve splenic B cells stimulated with the combination of CD40L and IL-5 and IL-4 were actually inhibited in their surface FasL expression and killer function, despite expressing similar levels of intracellular FasL. These data demonstrate novel differences in killer B-cell responses to the TH2 cytokine milieu and suggest that intracellular sequestration of FasL is a mechanism that regulates killer B-cell function.
KEY CONCEPT 2. Activation of purified splenic B cells from naïve mice with CD40 ligand (CD40L) and the TH2 cytokine IL-5 (but not IL-4) was sufficient to induce the proliferation of CD5+FasL+ B cells
This technique will be helpful for overcoming limitations in killer B-cell numbers and for studying the signaling requirements and in vivo effector functions of killer B cells in the future.
Until recently, IL-4 and IL-5 were generally accepted as cytokines produced by TH2 cells that have distinct but cooperative effects in driving TH2-mediated inflammation. However, a report by Islam et al. showed that IL-4 is an early activation product of TH2 cells and that chronically activated TH2 cells may switch to predominant production of IL-5 (40). It has also been reported that mucosal type 2 innate lymphoid cells (ILC2 cells) produce high levels of IL-5 compared to IL-4 when stimulated by IL-25 or IL-33, and are important contributors to TH2 inflammation. Interestingly, CD5+ B cells are more abundant in the mucosa, where they are commonly referred to as B-1a cells, and are sparse in the lymph nodes or circulation. It is quite likely that B-1a cells receive signals from ILC2 cells under homeostatic and inflammatory conditions. Although it remains to be formally proven, such an interaction would be expected to support mucosa-associated FasL+CD5+ B cells (Figure 1A). This may have important implications for protection from food allergies and local mucosal inflammation, and could play a role in the broader systemic immune tolerance mediated through the mucosal immune system.
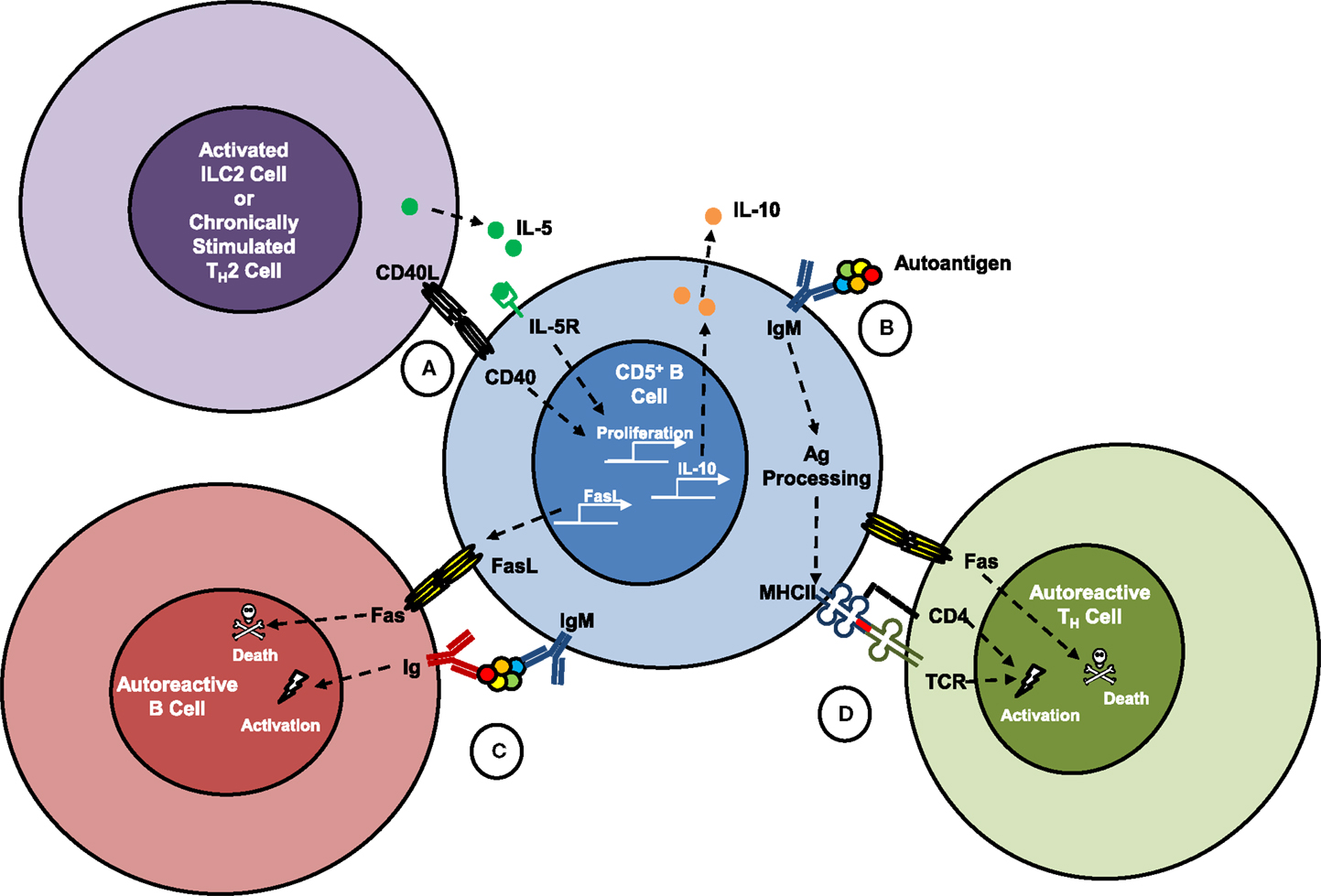
Figure 1. Hypothesized interactions of killer B cells with other lymphocytes. Fas ligand (FasL) expression is constitutive on mouse spleen and lung CD5+IgMhigh B cells, which have been shown to kill antigen-specific TH cells in vitro. Co-activation of mouse B cells through CD40 and the IL-5 receptor led to proliferation of CD5+ B cells, increased surface expression of FasL and killer function, and production and release of IL-10. (A) Interactions of killer B cells with mucosal type 2 innate lymphoid cells (ILC2), or with chronically stimulated TH2 cells in vivo would be expected to support their growth and functions, but has not been formally proven. (B) Surface immunoglobulins on CD5+ B cells are poly-reactive and are known to recognize autoantigens that once bound can be internalized and processed into peptides, which are then presented to TH cells on class II major histocompatibility (MHCII) molecules. (C) Binding of an autoantigen simultaneously by a killer B cell and an effector B cell could be a mechanism to explain B-cell fratricide, which has been described in several reports. (D) Killer B-cell uptake and presentation of autoantigens to TH cells in the context of FasL–Fas signaling in vivo could lead to activation-induced cell death and is hypothesized to be an important mechanism for maintaining peripheral tolerance and preventing autoimmune diseases. These processes may also play a role in tolerance to food antigens, other allergens, and commensal microbes.
Mice with an X-linked functional mutation of Bruton’s tyrosine kinase (Xid mice) or IL-5 deficiency have a severe impairment of mucosal B-1a cells (41–43). These mice have provided useful models to study the functions of CD5+ B cells in vivo. B-1a cells are a major source of natural antibodies, which are germline IgM immunoglobulins that are present at birth and which tend to have poly-reactive specificities for carbohydrates, lipids, and nucleic acids rather than protein components of antigens (44, 45). Interestingly, many of these germline-configured IgM antibodies also have a strong tendency to bind self-antigens (Figure 1B), and for this reason, B-1a cells have often been implicated as a source of autoantibodies and as contributors to autoimmune disease pathogenesis (46). The likelihood that self-reactive CD5+ B cells may also constitutively express FasL suggests that their more common role in autoimmunity may be to eliminate other self-reactive B and T cells (Figures 1C,D), and thus protect against the development of autoimmune diseases (47).
FasL+ B Cells and Tolerance
Immune tolerance is an active and complex process by which potentially harmful self-reactive lymphocytes are either eliminated, shunted into permanently inactive (anergic) states, or induced to become specialized immune regulatory cells. Breakdowns in tolerance that occur either centrally during lymphocyte development, or peripherally during inflammatory responses can lead to hyperactive immune reactions and severe pathological consequences (48). As evidenced by the autoimmune pathology associated with Fas or FasL-deficiencies, this death pathway is critical to normal immune tolerance (8). It is important to note that tolerance can also develop toward foreign antigens and is highly dependent on the location of antigen exposure and concurrent signals from the local environment. The fetal–maternal interface, gut mucosa, other sites of immune privilege, and the microenvironments created by tumors are especially well-suited to induction of tolerance (49). While the relative importance of FasL+ B cells in immune tolerance has not been studied extensively, several reports support an in vivo role for killer B cells in immune regulation and the induction of tolerance.
Minagawa et al. demonstrated that tolerance toward male H-Y antigen could be generated in mice through adoptive transfer of male splenic B cells into female recipients (50). The authors went on to show that tolerance was dependent on functional Fas receptor in the recipient mice, and that FasL on donor B cells was required (50). In separate studies, we showed that Xid mice, which lacked lung FasL+CD5+ B cells, failed to induce lung TH cell apoptosis in a chronic airway allergen exposure model (51). Low TH cell death in comparison to wild-type mice correlated with increased cytokine production, eosinophilia, and mucus production in the lungs despite high levels of IL-10 in the lung homogenates of the Xid mice (51). In the collagen-induced arthritis model, increased severity of joint inflammation correlated with decreased levels of FasL+ B cells in the spleen, and decreased killer function of B cells against antigen-specific TH cells (47). Montandon et al. demonstrated that activation of bone marrow-derived pro-B cells via toll-like receptor 9, induced pro-B-cell FasL expression, and that adoptive transfer of these cells into non-obese diabetic (NOD) mice resulted in protection from the spontaneous development of type 1 diabetes (52). An earlier study in NOD mice had also demonstrated a protective effect of B cells that were activated by bacterial lipopolysaccharide exposure, and expressed both FasL and the anti-inflammatory cytokine, transforming growth factor beta (TGFβ) (53). Many other studies have demonstrated the ability of transferred CD5+ B cells to suppress immune responses and induce therapeutic tolerance, however, the focus of those reports was on the ability of CD5+ B cells to produce IL-10, and unfortunately, FasL production and function were not addressed (54–59). It would be interesting to repeat some of the above studies using IL-10-deficient, TGFβ-deficient, and FasL-deficient B cells to measure the relative importance of each molecule to the therapeutic efficacy of B cells.
KEY CONCEPT 3. Tolerance toward male H-Y antigen could be generated in mice through adoptive transfer of male splenic B cells into female recipients
This study by Minagawa et al. demonstrated that the tolerizing effect of transferred B cells was Fas ligand-dependent. This report supports the concept that killer B cells could be targeted therapeutically to induce immune tolerance.
We have recently reported that B cells isolated from tumor-draining lymph nodes (TDLN) in a mouse breast tumor model were able to kill tumor cells in vitro and prevent metastases in vivo, and that B cells from IL-10-deficient mice were more potent killers of the tumor cells (60). FasL expression on TDLN B cells was not higher in the IL-10 knockout mice nor was there a difference in the total number of killer B cells. These data suggest that the lack of IL-10 from the adoptively transferred TDLN B cells may have had other inhibitory effects on B-cell killer functions, such as blocking critical interactions with the tumor cells and/or protective effects of IL-10 on the targeted tumor cells (60).
Restrictions on FasL Expression
Detection of FasL expression on the cell surface is usually not very robust, and most likely only represents a fraction of the total FasL protein produced by the cell. Cell types that do have measurable FasL on their surface, including B-1a cells, are often restricted in their distribution to specific niches where their interactions with Fas-sensitive target cells are more limited than if the FasL+ cells were allowed to circulate systemically. In cytotoxic T cells and NK cells, surface expression of FasL is transient due to the activity of metalloproteinases that cleave FasL and release the molecule in an inactive soluble form (61). Experimental systems in which FasL expression was forced to occur on cell surfaces, or in which cleavage of cell surface FasL was inhibited, most often resulted in the induction of target cell activation leading to inflammation rather than cell death (62–67). Moreover, the levels of transcription of the FasL gene in CTL and NK cells are very low in comparison to the amount of FasL protein found within the cells. This suggests that the FasL protein is fairly stable and is stored in the cell cytoplasm in a form that could be quickly transported to the cell surface when an appropriate target cell is identified.
In 1996, Martinez-Lorenzo and colleagues demonstrated that a preformed pool of FasL protein was sequestered within the cytoplasm of the immortalized human T-cell line, Jurkat (68). Upon stimulation, the FasL was rapidly released into the culture supernatant in a form that could induce cell death of other Jurkat T cells (68). In a subsequent study, these authors showed that bioactive FasL released by Jurkat was associated with microvesicles that could be removed by ultracentrifugation, and that similar FasL+ microvesicles were produced by non-transformed human T cells (69). It is now understood that these microvesicles were FasL+ exosomes and that this finding could explain conflicting data regarding soluble FasL and its role in mediating activation-induced cell death.
KEY CONCEPT 4. Bioactive FasL released by Jurkat was associated with microvesicles that could be removed by ultracentrifugation
This study and others support the concept that killer cells package much of their Fas ligand into exosomes, which limits the expression of the protein on the cell surface, yet provides a ready pool of FasL that can be quickly released upon activation.
Exosomes are extracellular vesicles that are approximately 30–100 nm in diameter, which are secreted by many types of cells, including B cells and other hematopoietic cells (70, 71). Exosomes can be found in many types of bodily fluids (blood, breast milk, urine), and ultracentrifugation is required to separate them from soluble proteins. Truly soluble FasL protein, removed from the cell surface by metalloproteinase-mediated cleavage, must now be considered separately from exosomal FasL that is membrane bound (66, 72–74). Many studies showing that soluble FasL was inhibitory of cell death were most likely influenced by the presence in the tested samples of cleaved FasL protein. Soluble FasL does not cause Fas aggregation and may block binding of membrane-associated FasL with death-inducing capacity. In contrast, exosomal FasL is very efficient at causing Fas aggregation, and may have been responsible for the results of those studies showing that “soluble” FasL was able to induce cell death. FasL+ exosomes are released by many types of FasL-producing cells, including NK cells, cytotoxic T cells, retinal pigment epithelial cells, and placental trophoblasts (75–77). FasL+ tumor cells can also produce FasL+ exosomes that target tumor-specific CD8+ T cells and aid in tumor immune escape (78). A direct demonstration that killer B cells can naturally produce FasL+ exosomes has not yet been reported, but it also has not been ruled out.
FasL+ exosomes circulating in normal human and mouse blood can co-express class II major histocompatibility complex (MHCII) molecules that are characteristic of professional antigen-presenting cells (APC) (79, 80). In APC, exosomes originate from the secretory lysosome where newly synthesized and recycled MHCII molecules are loaded with peptides derived from endocytosed proteins (Figure 2) (81). Thus, exosomes derived from APC contain peptide-loaded MHCII and co-stimulatory molecules, and are capable of activating antigen-specific TH cells (81). Unique to B cells is the fact that their APC function is restricted to antigens recognized by their surface immunoglobulins, which when bound to antigen are able to transfer the antigens directly to the secretory lysosomal compartment where MHCII+ exosomes are assembled (Figure 2) (82). In the case of an autoreactive B-1a cell that expresses FasL, we hypothesize that self-antigen uptake results in incorporation into MHCII+FasL+ exosomes (Figure 2). Subsequent binding of an autoreactive TH cell to these autoantigen/MHCII complexes and co-ligation by FasL either during cell–cell or cell–exosome interaction could then result in TH cell death and induction of immune tolerance toward that self-antigen. Similar processes could be envisioned if the B-1a cells were specific for food allergens, commensal bacteria, or other antigens toward which immune tolerance was desired. The unique positioning of B-1a cells at mucosal surfaces favors their involvement in maintaining local mucosal tolerance through surface expression of FasL and direct interaction with local cells. Beyond that, if the mucosal B-1a cells also produce FasL+MHCII+ exosomes, this could be an important yet overlooked natural mechanism by which systemic tolerance to mucosa-associated antigens is induced and maintained without the need for killer B-cell circulation outside the mucosa.
KEY CONCEPT 5. FasL+ exosomes circulating in normal human and mouse blood can co-express class II major histocompatibility complex (MHCII) molecules that are characteristic of professional antigen-presenting cells (APC)
The origin of these naturally occurring FasL+MHCII+ exosomes has not been positively identified and we hypothesize that killer B cells are one likely source.
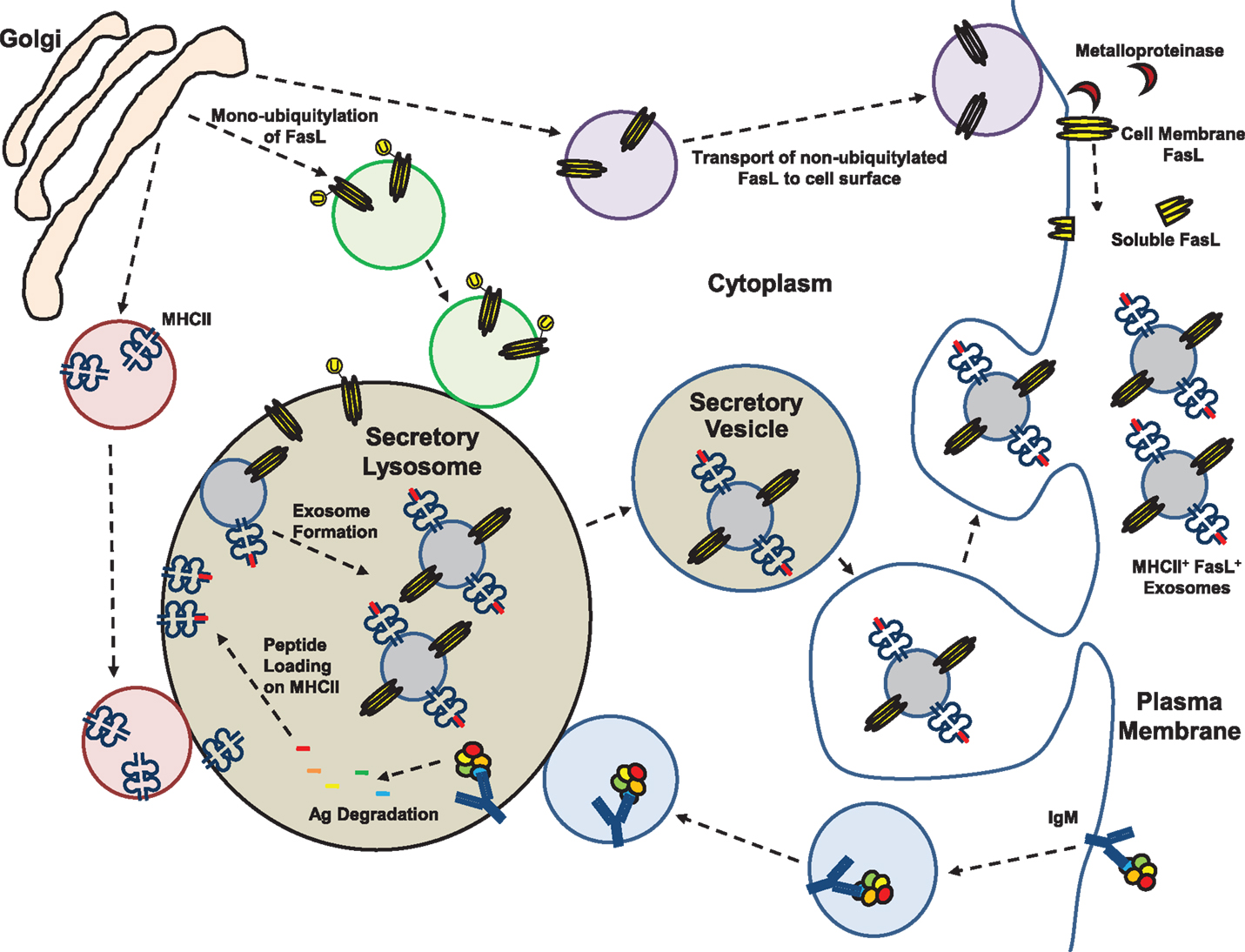
Figure 2. Membrane, exosome, and soluble forms of Fas ligand. Fas ligand (FasL) protein has been detected in several different forms in mouse and human cells and bodily fluids. Expression of FasL mRNA is generally low and the protein is assembled as a homotrimer in the trans-Golgi prior to transport to the cell surface. Plasma membrane expression of FasL is constitutive on a few specialized epithelial cell types at sites of immune privilege and on CD5+ B cells that have restricted localization. Cell surface expression of FasL is regulated by proteolytic cleavage by metalloproteinases, or by shunting of FasL protein into the secretory lysosomal compartment (SLC). Trafficking of FasL to the SLC is controlled by mono-ubiquitylation. The SLC is a site of antigen processing for molecules transported there from the cell surface by immunoglobulins, and processed peptides are loaded onto MHC class II (MHCII) molecules in the SLC. Through internal budding of the SLC membrane, FasL and MHCII are incorporated into exosomes that are stored in secretory vesicles of killer lymphocytes. Activation of the cell leads to their transport to the cell surface and release into the extracellular space.
FasL+ Exosomes and Immune Tolerance
The therapeutic application of exosomes has received increasing attention as a means to deliver signals to specific cell types, and as an alternative to adoptive immune cell therapy (83). Most of the efforts have been focused on using APC-derived exosomes to deliver activation signals for generating anti-tumor or anti-microbial immune responses (84–86). However, some investigators have focused instead on the immune-tolerizing capacity of exosomes, with particular interest placed on FasL+MHCII+ exosomes (70, 87). An early study showed that bone marrow-derived dendritic cells (BMDCs) genetically engineered to express FasL were able to produce FasL+MHCII+ exosomes that had direct killing effects on CD4+ TH cells (88). Further studies revealed the presence of naturally occurring FasL+MHCII+ exosomes in the blood of immunized mice, confirming the production of FasL+MHCII+ exosomes by non-transduced cells (80). Endogenously -produced FasL+MHCII+ exosomes mediated antigen-specific suppression of delayed-type hypersensitivity and experimental arthritis upon transfer to recipient mice (80). These naturally occurring FasL+MHCII+ exosomes did not express markers typical of BMDC-derived exosomes but did express CD11b, a surface molecule expressed by monocytes, macrophages, and mucosal B-1 cells. FasL+MHCII+ exosomes have also been found in human plasma, and had FasL-dependent suppressive activity against CD4+ T cells (79). To date, human FasL+ killer B cells have not been positively identified, making it difficult to define markers that would allow one to conclude whether or not they are the source of naturally -occurring human FasL+MHCII+ exosomes. However, one of our recent investigations demonstrated a very robust method for experimentally producing human B- cell-derived FasL+MHCII+ exosomes.
Human B Cells Transformed by EBV Produce FasL+ Exosomes
In attempting to identify human B cells that produce FasL, our laboratory screened a panel of 20 EBV-transformed B lymphoblastoid cell lines (B-LCL) that had been produced using the non-replicative laboratory B95-8 EBV strain (89). This virus has been used for decades to produce immortalized human B-LCL for studies of antibody rearrangement and other B-cell functions, and there are likely to be thousands of B-LCL distributed throughout the world. Although individual B-LCL had previously been demonstrated to express functional FasL, it was remarkable to find that every one of the lines we tested expressed a high amount of FasL protein. Despite easily detectable amounts of protein as measured by Western blots of whole cell lysates and intracellular staining, FasL protein was not present on the cell surface of the B-LCL (89). In keeping with what was noted above for other cell types that express FasL, we found that the B-LCL spontaneously produced and released FasL+MHCII+ exosomes that could be purified from the cell culture supernatant by ultracentrifugation (Figure 3) (89). The B-LCL exosomes were then used to induce death of autologous TH cells through antigen-dependent and FasL-mediated mechanisms as demonstrated by the use of bacterial superantigen or an exogenously loaded tetanus toxoid peptide in the presence or absence of FasL-neutralizing antibodies (Figure 3) (89). We believe that these findings open up the possibility that B-LCL could be used as cellular factories for producing FasL+MHCII+ exosomes for use in patient-customized treatments for many types of hyper-inflammatory conditions.
KEY CONCEPT 6. B-LCL spontaneously produced and released FasL+MHCII+ exosomes that could be purified from the cell culture supernatant by ultracentrifugation
These exosomes had potent killer function in vitro against human TH cells that was both antigen- and FasL-dependent. The fact that every B-LCL that was tested did this suggests that EBV could be used to create FasL+ exosomes from any individual’s B cells.
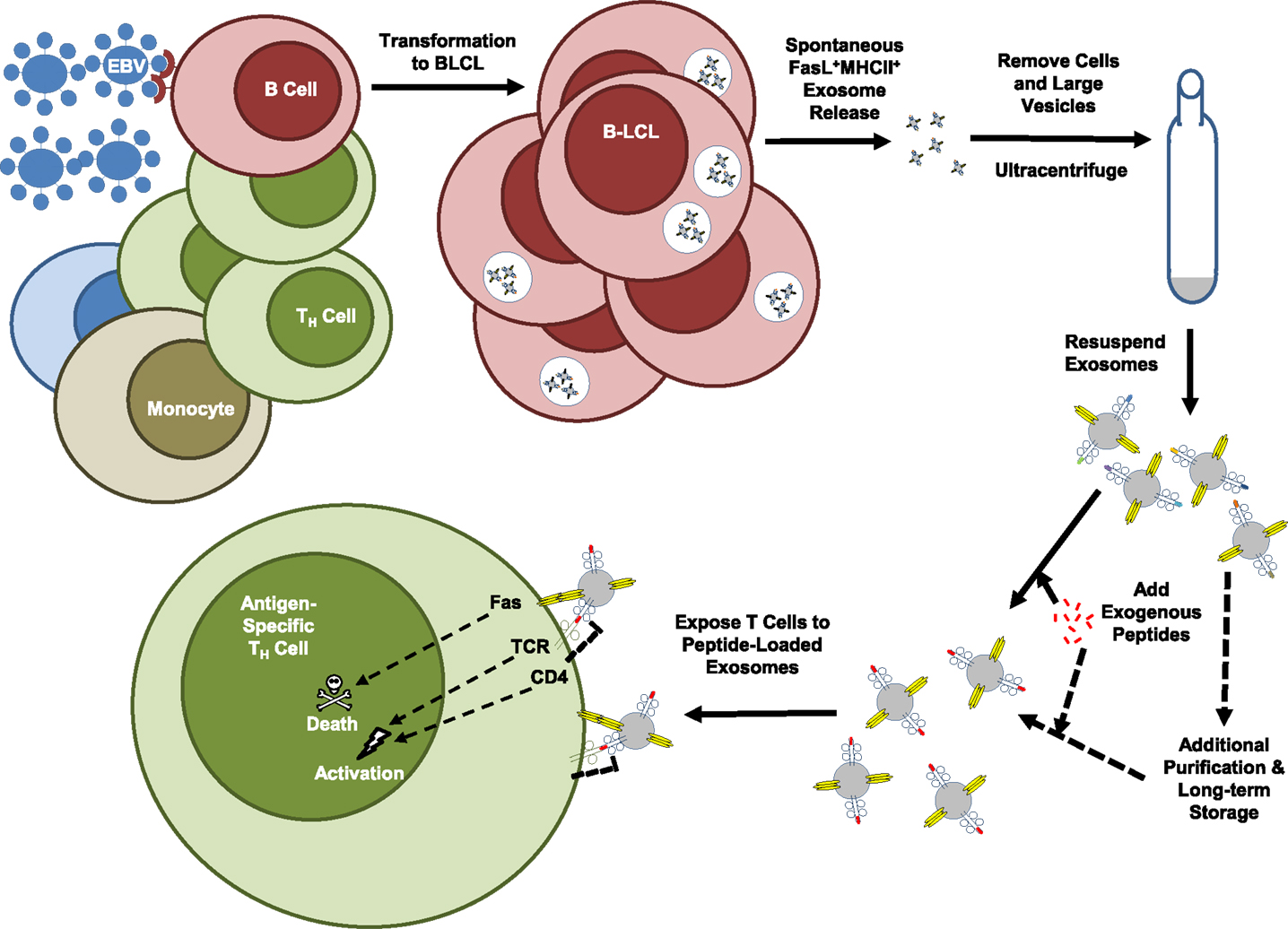
Figure 3. FasL+MHCII+ exosome production by EBV-transformed B-LCL. Epstein-Barr virus (EBV) has been used for many years to experimentally transform human B cells into immortalized B lymphoblastoid cell lines (B-LCL). The figure is a schematic of the methods recently employed to demonstrate the killer function of B-LCL-derived exosomes. Use of the non-replicative laboratory EBV strain, B95-8, resulted in consistent induction of FasL expression by transformed B-LCL. These B-LCL packaged FasL along with MHC class II molecules into exosomes that were then spontaneously released into supernatants of the B-LCL cultures grown in exosome-free media. Multiple rounds of centrifugation at increasing speeds removed cells, debris, and larger vesicles from the culture supernatant, which was then spun at 110,000 × g to pellet the exosome fraction. Resuspended exosomes were exposed to exogenous tetanus toxoid peptide or a bacterial superantigen, washed, and then added to cultures of autologous TH cells. The method resulted in TH cell death that was exosome and antigen-dependent, and FasL-mediated as demonstrated by the use of FasL-neutralizing antibodies.
Advantages of B-LCL Exosomes for Therapy
Cellular immunotherapy mediated by adoptive transfer of regulatory lymphocytes has been shown to suppress autoimmune diseases and allergic responses in many mouse models (90–92). These methods hold promise and have given us tools to dissect the importance of the different cell types and factors involved in immune regulation. However, many large hurdles remain in the translation of cellular immunotherapy into common clinical practice. Perhaps, the most important of these obstacles is that the transferred cells must be closely matched or identical to the MHC of the recipient in order to not be rejected. Ideally, this could be overcome if the transferred cells were autologous. The regulatory cells would have to be purified from patient blood, expanded in culture, quality-tested, and then reinfused in such a way that they would be viable and have the desired therapeutic effect. This form of treatment would be very expensive and time consuming. Notably, a defect in the function of regulatory T or B cells may be central to the cause of the inflammatory condition being treated, and therefore, infusion of more of these flawed cells may not be therapeutically helpful. Live regulatory cells may also turn off regulatory functions after transfer, or worse, change phenotype and participate in the inflammatory reaction. Much important work is being done and should continue in order to solve these problems and make adoptive cellular immunotherapy a better therapeutic option.
As subcellular alternatives to regulatory cell transfers, FasL+MHCII+ exosomes may hold some distinct advantages. First among these is that the killing of target TH cells by exosomes is not dependent on the presence of other cells. They also can be tailored to be antigen-specific and would be expected to only target a small subset of TH cells, while sparing desirable TH cells and protective immunity toward pathogenic microbes. Cell death induction by its nature is also likely to have more permanent or long-lasting effects in comparison to transfer of cells that express IL-10 or TGFβ which might only transiently modify the immune response. Exosomes lack the cellular machinery to switch their functions after transfer, and therefore, are much less likely to be turned off or to switch to a pathogenic role than are transferred regulatory cells (93). Exosomes have been reported to be functional even after long-term storage in low temperature freezers. This would suggest that once a batch of FasL+MHCII+ exosomes has been produced, isolated, and quality-tested, they could be banked in repositories until needed.
The EBV-transformation system that we utilized may provide some additional improvements over other methods of producing FasL+MHCII+ exosomes. The non-replicating, laboratory strain of the virus seems to be very efficient at transforming human B cells from any donor and activating their natural ability to express FasL. In our experience, these cells will grow indefinitely in cell culture and require relatively little labor to keep them healthy and producing exosomes. B-LCL can be frozen and stored in liquid nitrogen for decades without losing the ability to grow in cell culture. Transformation of the patient’s own B cells would allow for long-term propagation of the cells, scaling up of the culture as needed, and thus a potentially unlimited supply of FasL+ exosomes that would be 100% MHC-matched, and therefore, tolerated upon reinfusion into the patient. The plethora of lines that have already been transformed, frozen, and stored also provides a large potential repository of B-LCL that may closely match the MHC of the recipient patients, which could circumvent unexpected obstacles that might occur with transformation of a specific individual’s B cells.
KEY CONCEPT 7. A potentially unlimited supply of FasL+ exosomes that would be 100% MHC-matched, and therefore, tolerated upon reinfusion into the patient
Clinical translation of adoptive immunotherapy, whether it be cellular or vesicular, would be streamlined if mismatched MHC could be avoided. The exosome production method we describe may overcome this and other important obstacles.
Unlike the genetic manipulations that were required to force FasL expression in dendritic cells, the B-LCL spontaneously produce FasL+MHCII+ exosomes. By taking advantage of the natural ability of B cells to express functional FasL, EBV may promote its own survival and ability to persist in infected individuals. The fact that the non-replicating B95-8 laboratory strain is capable of driving FasL+MHCII+ exosome production is especially important since there are no active virions in the B-LCL culture, and therefore, little to no danger of transferring replicating EBV along with the exosome infusion. Although the direct application of B-LCL-generated FasL+ exosomes for use as immune-tolerizing agents in humans has not been tested, and is likely to be far off, the promising results from mouse models and their advantages over cellular immunotherapy as outlined above suggest that such a strategy may work and is worth further exploration.
Engineering EBV for Exosome Therapies
We envision that someday B-LCL-derived exosomes, such as those employed in our previous study, could be useful in several clinical applications. First, using the existing methods and the B95-8 strain of EBV, we would propose transforming B cells from the patients to be treated, isolating FasL+MHCII+ exosomes from cell cultures of these B-LCL and then loading them with exogenous peptides specific to the disease being targeted prior to infusion of autologous exosomes back into the patient (Figure 4A). Such a strategy, if successful, may be useful in treating allergies or autoimmune diseases in which the dominant antigens are identifiable. A similar strategy might also be used to tolerize a transplant recipient toward tissue from an allogeneic organ donor. In this approach, FasL+MHCII+ exosomes from B-LCL of a potential organ donor would be loaded with tissue peptides and infused into the recipient in order to induce death of donor MHC-specific TH cells (Figure 4A).
KEY CONCEPT 8. Someday B-LCL-derived exosomes, such as those employed in our previous study, could be useful in several clinical applications
The use of the non-replicative EBV strain to manufacture exosomes may mitigate some of the safety issues associated with this type of therapy, and also provide interesting opportunities to customize the exosomes through genetic engineering of the virus.
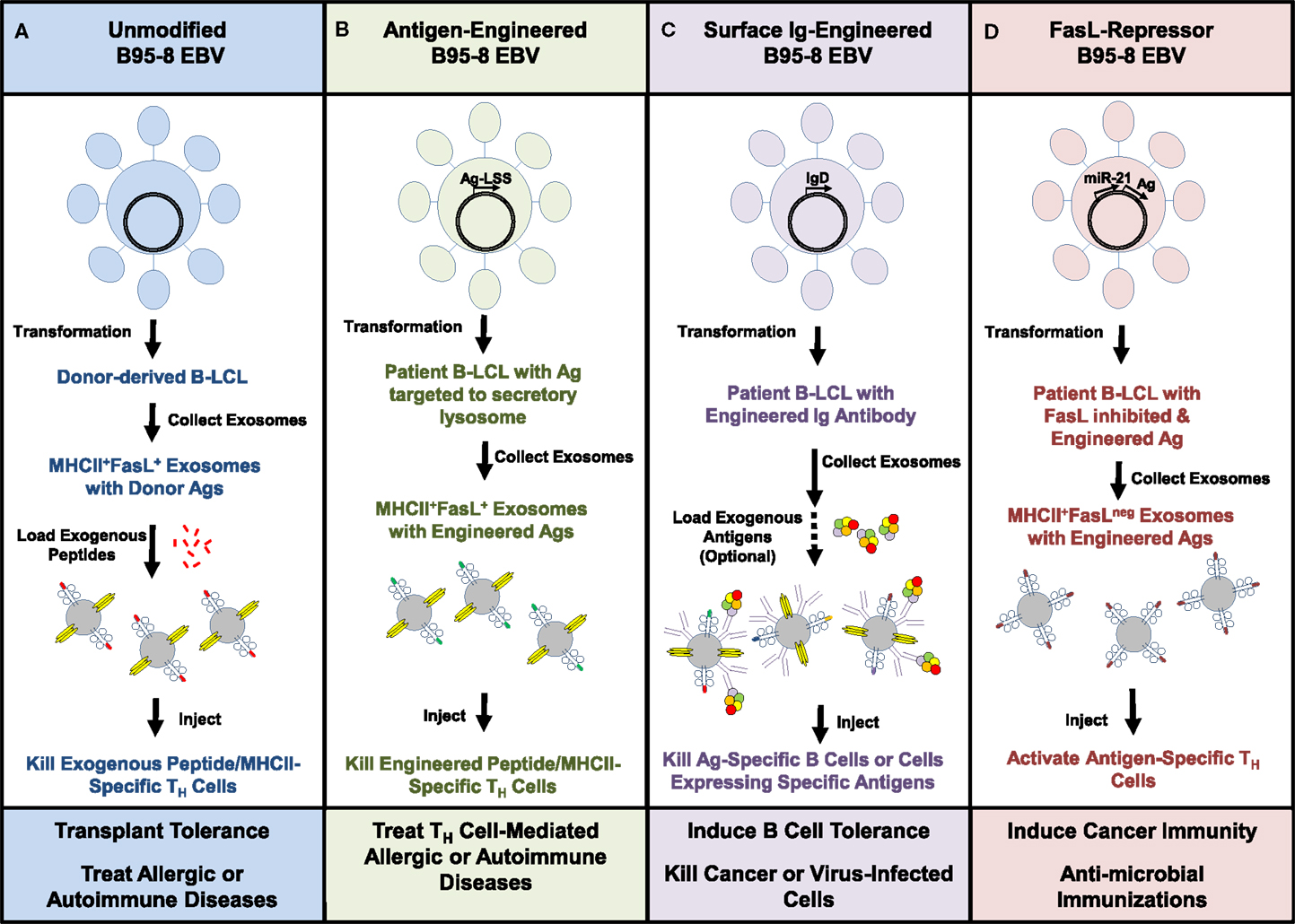
Figure 4. Potential therapeutic uses of B-LCL exosomes. Exosomes isolated from culture supernatants of EBV-transformed B-LCL were able to kill antigen-specific TH cells in vitro through a FasL-dependent mechanism. This schematic shows ways in which the B95-8 EBV strain might be used in its native form or genetically modified to manufacture customized B-LCL exosomes. (A) Unmodified virus could be used to generate autologous or allogeneic B-LCL that spontaneously produce FasL+MHCII+ exosomes. Replacement of MHC-bound peptides with exogenous peptides could be used to tailor the therapy toward specific antigens. (B) An alternative would be to modify the EBV genome to create B-LCL that express specific antigens linked to a lysosomal sorting sequence (LSS), which would target the proteins to the secretory lysosome where exosomes are assembled. (C) EBV engineered to express rearranged surface immunoglobulins (IgD or IgM) with known specificities onto exosomes may prove useful in targeting antigen-specific B cells, cancer, or virus-infected cells. (D) Inhibition of FasL expression in B-LCL through the repressor miR-21 or other molecules introduced into the EBV genome would be expected to yield FasLneg exosomes that could be used as vaccine adjuvants. Combining these genetic manipulations may further enhance the therapeutic potential of B-LCL-derived exosomes.
Beyond these uses of the existing EBV strain to produce therapeutic FasL+MHCII+ exosomes, we believe that there could be value in genetically modifying the B95-8 strain in order to further enhance the capabilities of B-LCL-derived exosomes. The genome of EBV is maintained in proliferating LCLs as a large episome (~167 kb), and techniques for engineering recombinant EBV are well-established (94, 95). Using a lysosomal sorting sequence fused to the protein of interest, the inserted gene products could be actively targeted into the secretory lysosomal compartment where FasL+MHCII+ exosomes are assembled and readied for transport to the cell surface (Figure 4B). Such a technique could be useful for targeting allergen proteins or autoantigens into the exosomal pathway as a potential improvement over loading cells or exosomes with exogenous peptides.
One means of improving the binding of exosomes to a desired target cell or tissue could be to engineer in a recombined immunoglobulin gene with a known specificity so that it is expressed on the exosome surface (Figure 4C). Expression of IgM and IgD has been found on the surface of naturally produced B-cell-derived exosomes (96). This technique may be especially useful for targeting other B lymphocytes with specificity for the same antigen, or for directing FasL+ exosomes to attack tumor cells or virus-infected cells. As described in a previous section, a recent study in mice demonstrated the ability of adoptively transferred FasL+ B cells to directly kill breast tumor cells and limit tumor metastases (60).
Finally, there are circumstances in which it might be desirable to be able to manufacture MHCII+ exosomes that do not express FasL. There is a great deal of interest in developing exosomes as vaccine adjuvants to induce T-cell responses against cancers and pathogenic microbes. We believe that the transformed B-LCL could provide a nearly unlimited source of individualized exosomes if the EBV virus could be engineered to repress instead of promote the expression of FasL (Figure 4D). One scenario would be to insert the coding sequence for micro RNA-21 (miR-21), a repressor of FasL expression, into the EBV genome (97, 98). The relative ease of creating and maintaining an immortalized B-LCL that naturally produces FasLnegMHCII+ exosomes could be a fundamental improvement over the use of non-transformed dendritic cells. Such an EBV virus could also be further manipulated genetically to express specific tumor antigens or microbial products to enhance the immunizing effects of the FasLneg B-LCL exosomes (Figure 4D).
Conclusion
There is still a great deal of basic and translational research to be done before we can attempt to manipulate killer B cells or their FasL+ exosomes in clinical trials. Some of the major questions that remain are: (1) is it safe to boost killer B-cell function in vivo and (2) what is the best approach toward achieving that goal. Fundamental to this pursuit will be better knowledge of how killer B cells are normally regulated in the body, their role in maintaining immune homeostasis and preventing disease, and the interactions they have with other regulatory or pro-inflammatory immune cells. It may be that many of the current therapies and medications used to treat autoimmune diseases or allergies have an effect on killer B cells that has gone unnoticed. In the case of development of B-LCL exosomes for therapy, it is likely that new techniques and equipment will have to be developed in order to reliably scale-up production and purify B-LCL-derived exosomes to the point that they can be tested for safety and efficacy in human subjects.
Research focused on immune regulatory pathways has yielded a wealth of knowledge and a number of potential targets for therapy. Beyond the established roles of regulatory T cells and their signature cytokines IL-10 and TGFβ in immune regulation, it has become increasingly clear that a larger network of cells and factors exists that induce and maintain immune tolerance. Increasing evidence suggests that killer B cells and FasL+MHCII+ exosomes play important and potentially irreplaceable roles in immune tolerance networks as specific regulators of TH cells. By increasing awareness of killer B cells and their therapeutic potential, we hope to foster interest in studying their biology and roles in human health and disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Edward T. and Ellen K. Dryer Foundation and grants from the National Institutes of Health: K01 AR053846, R03 AI0105029-01, and UM1 AI110557. SL was also supported by an Arthritis Investigator Award from the Arthritis Foundation. MK received stipend support from an NIH training grant award to the Immunology Graduate Program of the University of Michigan: T32 AI007413.
Authors Biography

References
1. French LE, Tschopp J. Constitutive Fas ligand expression in several non-lymphoid mouse tissues: implications for immune-protection and cell turnover. Behring Inst Mitt (1996) 97:156–60.
2. Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science (1995) 270:1189–92. doi: 10.1126/science.270.5239.1189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Uckan D, Steele A, Cherry, Wang BY, Chamizo W, Koutsonikolis A, et al. Trophoblasts express Fas ligand: a proposed mechanism for immune privilege in placenta and maternal invasion. Mol Hum Reprod (1997) 3:655–62. doi:10.1093/molehr/3.8.655
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Reich-Zeliger S, Zhao Y, Krauthgamer R, Bachar-Lustig E, Reisner Y. Anti-third party CD8+ CTLs as potent veto cells: coexpression of CD8 and FasL is a prerequisite. Immunity (2000) 13:507–15. doi:10.1016/S1074-7613(00)00050-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Rich RF, Green WR. Characterization of the Fas ligand/Fas-dependent apoptosis of antiretroviral, class I MHC tetramer-defined, CD8+ CTL by in vivo retrovirus-infected cells. J Immunol (2002) 168:2751–8. doi:10.4049/jimmunol.168.6.2751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Suda T, Okazaki T, Naito Y, Yokota T, Arai N, Ozaki S, et al. Expression of the Fas ligand in cells of T cell lineage. J Immunol (1995) 154:3806–13.
7. Oshimi Y, Oda S, Honda Y, Nagata S, Miyazaki S. Involvement of Fas ligand and Fas-mediated pathway in the cytotoxicity of human natural killer cells. J Immunol (1996) 157:2909–15.
8. Suda T, Nagata S. Why do defects in the Fas-Fas ligand system cause autoimmunity? J Allergy Clin Immunol (1997) 100:S97–101. doi:10.1016/S0091-6749(97)70013-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Reich-Zeliger S, Gan J, Bachar-Lustig E, Reisner Y. Tolerance induction by veto CTLs in the TCR transgenic 2C mouse model. II. Deletion of effector cells by Fas-Fas ligand apoptosis. J Immunol (2004) 173:6660–6. doi:10.4049/jimmunol.173.11.6660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity (1998) 8:615–23. doi:10.1016/S1074-7613(00)80566-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res (2007) 17:759–71. doi:10.1038/cr.2007.52
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell (1994) 76:969–76. doi:10.1016/0092-8674(94)90375-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today (1995) 16:39–43. doi:10.1016/0167-5699(95)80069-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Turbyville JC, Rao VK. The autoimmune lymphoproliferative syndrome: a rare disorder providing clues about normal tolerance. Autoimmun Rev (2010) 9:488–93. doi:10.1016/j.autrev.2010.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Kokkonen TS, Karttunen TJ. Fas/Fas ligand-mediated apoptosis in different cell lineages and functional compartments of human lymph nodes. J Histochem Cytochem (2010) 58:131–40. doi:10.1369/jhc.2009.954669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Eberl G, Jiang S, Yu Z, Schneider P, Corradin G, Mach JP. An anti-CD19 antibody coupled to a tetanus toxin peptide induces efficient Fas ligand (FasL)-mediated cytotoxicity of a transformed human B cell line by specific CD4+ T cells. Clin Exp Immunol (1998) 114:173–8. doi:10.1046/j.1365-2249.1998.00710.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Lundy SK. Killer B lymphocytes: the evidence and the potential. Inflamm Res (2009) 58(7):345–57. doi:10.1007/s00011-009-0014-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Hahne M, Renno T, Schroeter M, Irmler M, French L, Bornard T, et al. Activated B cells express functional Fas ligand. Eur J Immunol (1996) 26:721–4. doi:10.1002/eji.1830260332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Kondo E, Yoshino T, Nishiuchi R, Sakuma I, Nishizaki K, Kayagaki N, et al. Expression of Fas ligand mRNA in germinal centres of the human tonsil. J Pathol (1997) 183:75–9. doi:10.1002/(SICI)1096-9896(199709)183:1<75::AID-PATH1084>3.0.CO;2-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Samuelsson A, Sonnerborg A, Heuts N, Coster J, Chiodi F. Progressive B cell apoptosis and expression of Fas ligand during human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses (1997) 13:1031–8. doi:10.1089/aid.1997.13.1031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Mariani SM, Krammer PH. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur J Immunol (1998) 28:973–82. doi:10.1002/(SICI)1521-4141(199803)28:03<973::AID-IMMU973>3.0.CO;2-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Tinhofer I, Marschitz I, Kos M, Henn T, Egle A, Villunger A, et al. Differential sensitivity of CD4+ and CD8+ T lymphocytes to the killing efficacy of Fas (Apo-1/CD95) ligand+ tumor cells in B chronic lymphocytic leukemia. Blood (1998) 91:4273–81.
24. Bussing A, Stein GM, Pfuller U, Schietzel M. Induction of Fas ligand (CD95L) by the toxic mistletoe lectins in human lymphocytes. Anticancer Res (1999) 19:1785–90.
25. Sträter J, Mariani SM, Walczak H, Rücker FG, Leithäuser F, Krammer PH, et al. CD95 ligand (CD95L) in normal human lymphoid tissues: a subset of plasma cells are prominent producers of CD95L. Am J Pathol (1999) 154:193–201. doi:10.1016/S0002-9440(10)65265-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Mullauer L, Mosberger I, Chott A. Fas ligand expression in nodal non-Hodgkin’s lymphoma. Mod Pathol (1998) 11:369–75.
27. Tanner JE, Alfieri C. Epstein-Barr virus induces Fas (CD95) in T cells and Fas ligand in B cells leading to T-cell apoptosis. Blood (1999) 94:3439–47.
28. Verbeke CS, Wenthe U, Zentgraf H. Fas ligand expression in the germinal centre. J Pathol (1999) 189:155–60. doi:10.1002/(SICI)1096-9896(199910)189:2<155::AID-PATH442>3.0.CO;2-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Nilsson N, Ingvarsson S, Borrebaeck CA. Immature B cells in bone marrow express Fas/FasL. Scand J Immunol (2000) 51:279–84. doi:10.1046/j.1365-3083.2000.00701.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Rich RF, Cook WJ, Green WR. Spontaneous in vivo retrovirus-infected T and B cells, but not dendritic cells, mediate antigen-specific Fas ligand/Fas-dependent apoptosis of anti-retroviral CTL. Virology (2006) 346:287–300. doi:10.1016/j.virol.2005.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Sampalo A, Navas G, Medina F, Segundo C, Camara C, Brieva JA. Chronic lymphocytic leukemia B cells inhibit spontaneous Ig production by autologous bone marrow cells: role of CD95-CD95L interaction. Blood (2000) 96:3168–74. doi:10.1080/10428190290017033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hekimgil M, Cagirgan S, Pehlivan M, Doganavsargil B, Tombuloglu M, Soydan S. Immunohistochemical detection of CD 95 (Fas) & Fas ligand (Fas-L) in plasma cells of multiple myeloma and its correlation with survival. Leuk Lymphoma (2006) 47:271–80. doi:10.1080/10428190500286218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Silvestris F, Cafforio P, Grinello D, Dammacco F. Upregulation of erythroblast apoptosis by malignant plasma cells: a new pathogenetic mechanism of anemia in multiple myeloma. Rev Clin Exp Hematol (2002) (Suppl 1):39–46.
34. Silvestris F, Cafforio P, Tucci M, Grinello D, Dammacco F. Upregulation of osteoblast apoptosis by malignant plasma cells: a role in myeloma bone disease. Br J Haematol (2003) 122:39–52. doi:10.1046/j.1365-2141.2003.04374.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Lundy SK, Lerman SP, Boros DL. Soluble egg antigen-stimulated T helper lymphocyte apoptosis and evidence for cell death mediated by FasL (+) T and B cells during murine Schistosoma mansoni infection. Infect Immun (2001) 69:271–80. doi:10.1128/IAI.69.1.271-280.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Lundy SK, Boros DL. Fas ligand-expressing B-1a lymphocytes mediate CD4(+)-T-cell apoptosis during schistosomal infection: induction by interleukin 4 (IL-4) and IL-10. Infect Immun (2002) 70:812–9. doi:10.1128/IAI.70.2.812-819.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med (2011) 208:67–80. doi:10.1084/jem.20101499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Boros DL. T helper cell populations, cytokine dynamics, and pathology of the schistosome egg granuloma. Microbes Infect (1999) 1:511–6. doi:10.1016/S1286-4579(99)80090-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Klinker MW, Reed TJ, Fox DA, Lundy SK. Interleukin-5 supports the expansion of fas ligand-expressing killer B cells that induce antigen-specific apoptosis of CD4(+) T cells and secrete interleukin-10. PLoS One (2013) 8:e70131. doi:10.1371/journal.pone.0070131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, et al. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol (2011) 12:167–77. doi:10.1038/ni.1984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Khan WN. Colonel Bruton’s kinase defined the molecular basis of X-linked agammaglobulinemia, the first primary immunodeficiency. J Immunol (2012) 188:2933–5. doi:10.4049/jimmunol.1200490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, Dai WJ, et al. IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity (1996) 4:15–24. doi:10.1016/S1074-7613(00)80294-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Pecquet SS, Zazulak J, Simpson SD, Ernst PB. Reconstitution of xid mice with donor cells enriched for CD5+ B cells restores contrasuppression. Ann N Y Acad Sci (1992) 651:173–5. doi:10.1111/j.1749-6632.1992.tb24612.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B cell development and function in Btk-deficient mice. Immunity (1995) 3:283–99. doi:10.1016/1074-7613(95)90114-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev (2000) 176:154–70. doi:10.1034/j.1600-065X.2000.00607.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Steinberg BJ, Smathers PA, Frederiksen K, Steinberg AD. Ability of the xid gene to prevent autoimmunity in (NZB X NZW)F1 mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. J Clin Invest (1982) 70:587–97. doi:10.1172/JCI110651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lundy SK, Fox DA. Reduced Fas ligand-expressing splenic CD5+ B lymphocytes in severe collagen-induced arthritis. Arthritis Res Ther (2009) 11:R128. doi:10.1186/ar2795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Notarangelo LD, Gambineri E, Badolato R. Immunodeficiencies with autoimmune consequences. Adv Immunol (2006) 89:321–70. doi:10.1016/S0065-2776(05)89008-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Waldmann H. Tolerance: an overview and perspectives. Nat Rev Nephrol (2010) 6:569–76. doi:10.1038/nrneph.2010.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Minagawa R, Okano S, Tomita Y, Kishihara K, Yamada H, Nomoto K, et al. The critical role of Fas-Fas ligand interaction in donor-specific transfusion-induced tolerance to H-Y antigen. Transplantation (2004) 78:799–806. doi:10.1097/01.TP.0000129799.96439.6F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lundy SK, Berlin AA, Martens TF, Lukacs NW. Deficiency of regulatory B cells increases allergic airway inflammation. Inflamm Res (2005) 54:514–21. doi:10.1007/s00011-005-1387-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Montandon R, Korniotis S, Layseca-Espinosa E, Gras C, Mégret J, Ezine S, et al. Innate pro-B-cell progenitors protect against type 1 diabetes by regulating autoimmune effector T cells. Proc Natl Acad Sci U S A (2013) 110:E2199–208. doi:10.1073/pnas.1222446110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol (2001) 167:1081–9. doi:10.4049/jimmunol.167.2.1081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity (2008) 28:639–50. doi:10.1016/j.immuni.2008.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol (2010) 185:2240–52. doi:10.4049/jimmunol.1001307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, et al. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol (2010) 184:4801–9. doi:10.4049/jimmunol.0902385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491:264–8. doi:10.1038/nature11501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. van der Vlugt LE, Haeberlein S, de Graaf W, Martha TE, Smits HH. Toll-like receptor ligation for the induction of regulatory B cells. Methods Mol Biol (2014) 1190:127–41. doi:10.1007/978-1-4939-1161-5_10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. van der Vlugt LE, Zinsou JF, Ozir-Fazalalikhan A, Kremsner PG, Yazdanbakhsh M, Adegnika AA, et al. Interleukin 10 (IL-10)-producing CD1dhi regulatory B cells from Schistosoma haematobium-infected individuals induce IL-10-positive T cells and suppress effector T-cell cytokines. J Infect Dis (2014) 210:1207–16. doi:10.1093/infdis/jiu257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Tao H, Lu L, Xia Y, Dai F, Wang Y, Bao Y, et al. Antitumor effector B cells directly kill tumor cells via the Fas/FasL pathway and are regulated by IL-10. Eur J Immunol (2014). doi:10.1002/eji.201444625
61. Mariani SM, Matiba B, Baumler C, Krammer PH. Regulation of cell surface APO-1/Fas (CD95) ligand expression by metalloproteases. Eur J Immunol (1995) 25:2303–7. doi:10.1002/eji.1830250828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Askenasy N, Yolcu ES, Yaniv I, Shirwan H. Induction of tolerance using Fas ligand: a double-edged immunomodulator. Blood (2005) 105:1396–404. doi:10.1182/blood-2004-06-2364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Viard-Leveugle I, Bullani RR, Meda P, Micheau O, Limat A, Saurat JH, et al. Intracellular localization of keratinocyte Fas ligand explains lack of cytolytic activity under physiological conditions. J Biol Chem (2003) 278:16183–8. doi:10.1074/jbc.M212188200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Sano Y, Yamada J, Ishino Y, Adachi W, Kawasaki S, Suzuki T, et al. Non-cleavable mutant Fas ligand transfection of donor cornea abrogates ocular immune privilege. Exp Eye Res (2002) 75:475–83. doi:10.1006/exer.2002.2044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Gregory MS, Repp AC, Holhbaum AM, Saff RR, Marshak-Rothstein A, Ksander BR. Membrane Fas ligand activates innate immunity and terminates ocular immune privilege. J Immunol (2002) 169:2727–35. doi:10.4049/jimmunol.169.5.2727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Shudo K, Kinoshita K, Imamura R, Fan H, Hasumoto K, Tanaka M, et al. The membrane-bound but not the soluble form of human Fas ligand is responsible for its inflammatory activity. Eur J Immunol (2001) 31:2504–11. doi:10.1002/1521-4141(200108)31:8<2504::AID-IMMU2504>3.0.CO;2-C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. O’Connell J. Immune privilege or inflammation? The paradoxical effects of Fas ligand. Arch Immunol Ther Exp (Warsz) (2000) 48:73–9.
68. Martinez-Lorenzo MJ, Alava MA, Anel A, Pineiro A, Naval J. Release of preformed Fas ligand in soluble form is the major factor for activation-induced death of Jurkat T cells. Immunology (1996) 89:511–7. doi:10.1046/j.1365-2567.1996.d01-782.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Martínez-Lorenzo MJ, Anel A, Gamen S, Monle NI, Lasierra P, Larrad L, et al. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J Immunol (1999) 163:1274–81.
70. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol (2014) 14:195–208. doi:10.1038/nri3622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. McLellan AD. Exosome release by primary B cells. Crit Rev Immunol (2009) 29:203–17. doi:10.1615/CritRevImmunol.v29.i3.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Suda T, Hashimoto H, Tanaka M, Ochi T, Nagata S. Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J Exp Med (1997) 186:2045–50. doi:10.1084/jem.186.12.2045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, et al. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med (1998) 187:1205–13. doi:10.1084/jem.187.8.1205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Jang S, Krammer PH, Salgame P. Lack of proapoptotic activity of soluble CD95 ligand is due to its failure to induce CD95 oligomers. J Interferon Cytokine Res (2003) 23:441–7. doi:10.1089/107999003322277856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. McKechnie NM, King BC, Fletcher E, Braun G. Fas-ligand is stored in secretory lysosomes of ocular barrier epithelia and released with microvesicles. Exp Eye Res (2006) 83:304–14. doi:10.1016/j.exer.2005.11.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Stenqvist AC, Nagaeva O, Baranov V, Mincheva-Nilsson L. Exosomes secreted by human placenta carry functional Fas ligand and TRAIL molecules and convey apoptosis in activated immune cells, suggesting exosome-mediated immune privilege of the fetus. J Immunol (2013) 191:5515–23. doi:10.4049/jimmunol.1301885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol (2012) 189:2833–42. doi:10.4049/jimmunol.1101988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Abusamra AJ, Zhong Z, Zheng X, Li M, Ichim TE, Chin JL, et al. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol Dis (2005) 35:169–73. doi:10.1016/j.bcmd.2005.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Ren Y, Yang J, Xie R, Gao L, Yang Y, Fan H, et al. Exosomal-like vesicles with immune-modulatory features are present in human plasma and can induce CD4+ T-cell apoptosis in vitro. Transfusion (2011) 51:1002–11. doi:10.1111/j.1537-2995.2010.02909.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD. MHC class II+ exosomes in plasma suppress inflammation in an antigen-specific and Fas ligand/Fas-dependent manner. J Immunol (2007) 179:2235–41. doi:10.4049/jimmunol.179.4.2235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med (1996) 183:1161–72. doi:10.1084/jem.183.3.1161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Rialland P, Lankar D, Raposo G, Bonnerot C, Hubert P. BCR-bound antigen is targeted to exosomes in human follicular lymphoma B-cells. Biol Cell (2006) 98:491–501. doi:10.1042/BC20060027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Bastos-Amador P, Pérez-Cabezas B, Izquierdo-Useros N, Puertas MC, Martinez-Picado J, Pujol-Borrell R, et al. Capture of cell-derived microvesicles (exosomes and apoptotic bodies) by human plasmacytoid dendritic cells. J Leukoc Biol (2012) 91:751–8. doi:10.1189/jlb.0111054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Mignot G, Roux S, Thery C, Segura E, Zitvogel L. Prospects for exosomes in immunotherapy of cancer. J Cell Mol Med (2006) 10:376–88. doi:10.1111/j.1582-4934.2006.tb00406.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Altieri SL, Khan AN, Tomasi TB. Exosomes from plasmacytoma cells as a tumor vaccine. J Immunother (2004) 27:282–8. doi:10.1097/00002371-200407000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Taieb J, Chaput N, Zitvogel L. Dendritic cell-derived exosomes as cell-free peptide-based vaccines. Crit Rev Immunol (2005) 25:215–23. doi:10.1615/CritRevImmunol.v25.i3.30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Monguio-Tortajada M, Lauzurica-Valdemoros R, Borras FE. Tolerance in organ transplantation: from conventional immunosuppression to extracellular vesicles. Front Immunol (2014) 5:416. doi:10.3389/fimmu.2014.00416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Kim SH, Bianco N, Menon R, Lechman ER, Shufesky WJ, Morelli AE, et al. Exosomes derived from genetically modified DC expressing FasL are anti-inflammatory and immunosuppressive. Mol Ther (2006) 13:289–300. doi:10.1016/j.ymthe.2005.09.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Klinker MW, Lizzio V, Reed TJ, Fox DA, Lundy SK. Human B cell-derived lymphoblastoid cell lines constitutively produce fas ligand and secrete MHCII(+)FasL(+) killer exosomes. Front Immunol (2014) 5:144. doi:10.3389/fimmu.2014.00144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi:10.1146/annurev-immunol-020711-074934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Tanaka H, Zhang W, Yang GX, Ando Y, Tomiyama T, Tsuneyama K, et al. Successful immunotherapy of autoimmune cholangitis by adoptive transfer of forkhead box protein 3(+) regulatory T cells. Clin Exp Immunol (2014) 178:253–61. doi:10.1111/cei.12415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Wright GP, Ehrenstein MR, Stauss HJ. Regulatory T-cell adoptive immunotherapy: potential for treatment of autoimmunity. Expert Rev Clin Immunol (2011) 7:213–25. doi:10.1586/eci.10.96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity (2013) 38:414–23. doi:10.1016/j.immuni.2013.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Delecluse HJ, Hammerschmidt W. The genetic approach to the Epstein-Barr virus: from basic virology to gene therapy. Mol Pathol (2000) 53:270–9. doi:10.1136/mp.53.5.270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Yu Z, Lu J, Yu H, Yan Q, Zuo L, Li G. A precise excision of the complete Epstein-Barr virus genome in a plasmid based on a bacterial artificial chromosome. J Virol Methods (2011) 176:103–7. doi:10.1016/j.jviromet.2011.06.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Saunderson SC, Schuberth PC, Dunn AC, Miller L, Hock BD, MacKay PA, et al. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J Immunol (2008) 180:8146–52. doi:10.4049/jimmunol.180.12.8146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, et al. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem (2010) 285:20281–90. doi:10.1074/jbc.M110.109207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Zhang L, Dong LY, Li YJ, Hong Z, Wei WS. miR-21 represses FasL in microglia and protects against microglia-mediated neuronal cell death following hypoxia/ischemia. Glia (2012) 60:1888–95. doi:10.1002/glia.22404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: immune regulation, autoimmunity, allergy, transplantation, apoptosis, TH cells, EBV
Citation: Lundy SK, Klinker MW and Fox DA (2015) Killer B lymphocytes and their Fas ligand positive exosomes as inducers of immune tolerance. Front. Immunol. 6:122. doi: 10.3389/fimmu.2015.00122
Received: 30 January 2015; Paper pending published: 24 February 2015;
Accepted: 04 March 2015; Published online: 20 March 2015.
Edited by:
Yair Reisner, Weizmann Institute of Science, IsraelReviewed by:
Benny Chain, University College London, UKShlomit Reich-Zeliger, Weizmann Institute of Science, Israel
Copyright: © 2015 Lundy, Klinker and Fox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:c2tsdW5keUBtZWQudW1pY2guZWR1