 Jane Blood-Siegfried
Jane Blood-Siegfried- Duke University School of Nursing, Durham, NC, USA
Sudden infant death syndrome (SIDS) is still not well understood. It is defined as the sudden and unexpected death of an infant without a definitive cause. There are numerous hypotheses about the etiology of SIDS but the exact cause or causes have never been pinpointed. Examination of theoretical pathologies might only be possible in animal models. Development of these models requires consideration of the environmental and/or developmental risk factors often associated with SIDS, as they need to explain how the risk factors could contribute to the cause of death. These models were initially developed in common laboratory animals to test various hypotheses to explain these infant deaths – guinea pig, piglet, mouse, neonatal rabbit, and neonatal rat. Currently, there are growing numbers of researchers using genetically altered animals to examine specific areas of interest. This review describes the different systems and models developed to examine the diverse hypotheses for the cause of SIDS and their potential for defining a causal mechanism or mechanisms.
Introduction
Sudden infant death syndrome (SIDS) is the most common cause of infant mortality outside of the neonatal period in developed countries (1–3). It is a diagnosis made when every other possible cause can be excluded by the evaluation of all factors including an examination of the death scene, the review of the medical record, and a thorough autopsy (1, 4, 5). The incidence of SIDS is between 1 and 12 months of age with a peak between 2 and 4 months. Even though infants dying of SIDS appear normal and healthy, they often have had a recent mild viral illness that is not associated with the cause of death (1, 2, 6, 7).
Many of the risk factors associated with SIDS parallel those associated with susceptibility to infections. These include prone sleep position, maternal smoking, environmental tobacco smoke exposure, concurrent viral illness, ethnicity, gender, overheating, and drug or alcohol abuse during pregnancy (8–10). With the current decrease in SIDS due to the “Back to Sleep” campaign, the most significant risk factor continues to be maternal smoking during pregnancy (10–17).
Sudden infant death syndrome is most likely caused by several different mechanisms that trigger unexplained deaths. Animal models are important for evaluating the multiple hypotheses of causation and the relationships between: pathology related to sleep position; hyperthermia; hypoxia; reflux; anaphylaxis; infections; toxic exposures; substance abuse; and the role of neurotransmitters in regulation of basic physiologic functions. Animal models offer the ability to evaluate the interaction between different biologic systems in a single experiment.
This review will first outline the models used to assess the role of infection and inflammatory responses in these infant deaths. It will then review how the results of these studies might be applied to determine if inflammation could affect the physiological responses for other hypotheses proposed.
Inflammation and Infection
Several findings on autopsy of SIDS infants can be difficult to explain, particularly the finding of intrathoracic petechiae and liquid blood in the heart (18, 19). Evidence detected for inflammation has been summarized in the accompanying paper in this volume (Blackwell et al., this volume). These could be associated with efforts to overcome hypoxia, respiratory distress, suffocation, or resuscitation; however, they could also be due to coagulation problems associated with infection and inflammation (20, 21). There are often signs of inflammation and response to infection that are not due to the underlying, pre-existing symptoms. Thymic involution, is observed in more than half of SIDS cases and represents the response to severe stress (22). Thymic involution, intrathoracic petechiae, and liquid blood around the heart are also described in a developmental animal model of SIDS and are similar to the Rambaud results (23).
Anaphylaxis
Anaphylaxis is a potentially lethal inflammatory response. It was one of the first hypotheses proposed to explain SIDS and was examined by successful development of an animal model (24). Studies were conducted to evaluate allergy associated with anaphylaxis in conscious guinea pigs that had been previously sensitized to cow’s milk (Table 1). After challenge, these animals died quietly with some gasping and then appeared to fall asleep with the heart continuing to beat for a short time. These animals had two findings commonly found on SIDS autopsy, fluid blood around the heart and an empty bladder. Additionally, there was significant histamine release (24). Buckley showed that beta tryptases were elevated in SIDS infants indicating possible mast cell de-granulation prior to death (25). IgE levels were not affected but this is not a mandatory finding for anaphylaxis (25). Challenge from inhaled regurgitated milk in the lungs in a sensitized infant is a plausible scenario.
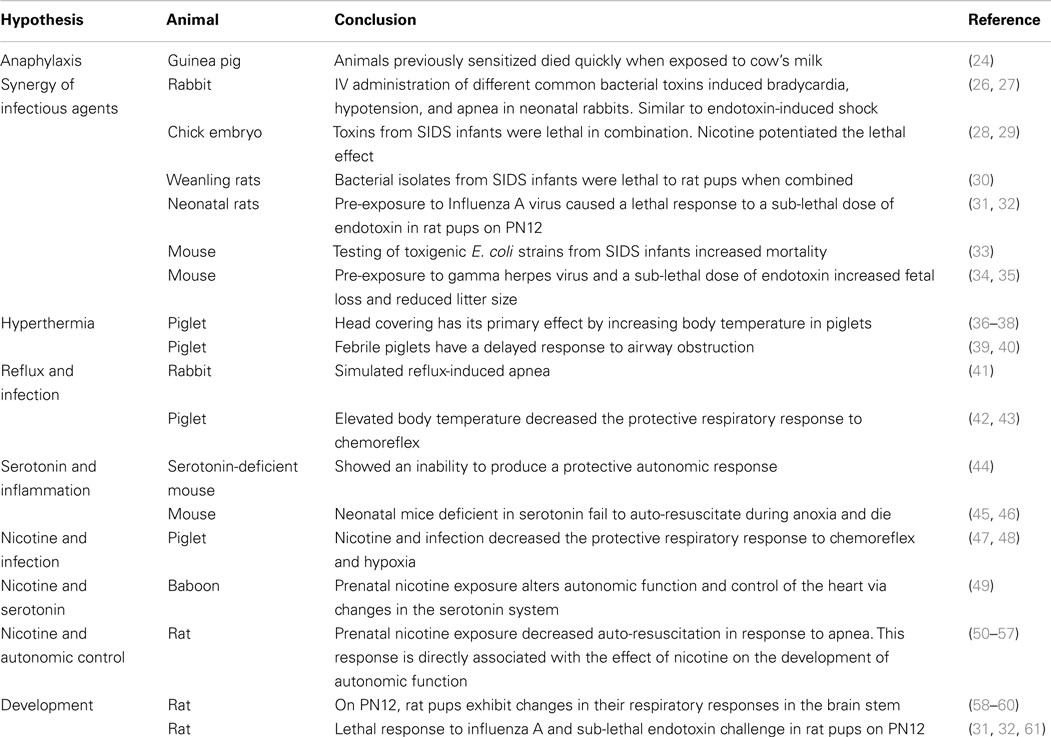
Table 1. Animal models examining hypotheses to explain SIDS.
Synergy of Infectious Agents
Assessment of the role of infectious agents and their toxins has employed several animal models. Each of these is considered below.
Rabbit
The response to intravenous injection of six common bacterial toxins was examined in 1–3 kg rabbits. The animals showed cardiac rate slowing, a drop in blood pressure, and apnea with sudden death. It was concluded that bacteria could produce toxins that cause inflammatory responses similar to those associated with endotoxin-induced shock (26) (Table 1). Catecholamine levels did not increase in these animals when the toxins were administered via the gastrointestinal tract; however, increasing doses administered intravenously demonstrated a dose-related increase in catecholamine levels and sudden death. A healthy gastrointestinal tract is not sensitive to these toxins (27).
Chick Embryo
The lethality of toxins obtained from nasopharyngeal preparations from SIDS infants were tested individually and in combination in chick embryos. Enterobacterial and staphylococcal toxins alone were only lethal at high dilutions, however when combined, these same toxins were lethal at much lower concentrations. Both of these strains are found together more often in the nasopharynx of SIDS victims than in healthy infants (28) (Table 1). When nicotine was added at very low concentrations, it further potentiated the lethal action of these bacterial toxins (29).
Weanling Rats
Weanling rats died rapidly without any symptoms prior to death following the injection of nasopharyngeal bacterial isolates obtained from SIDS. These animals had no signs of illness and negligible signs of inflammation in the heart, liver, and lungs (30) (Table 1). When E. coli and S. aureus were paired, the animals died more rapidly. Again this demonstrated the lethal synergy between different pathogens.
Neonatal Rat
Blood-Siegfried addressed susceptibility associated with developmental stage in relation to the timing of infectious insults. This model used a double insult with a non-lethal strain of influenza A virus and a sub-lethal dose of endotoxin. Animals were given an intranasal dose of the virus on postnatal (PN) day 10. When they were challenged with a sub-lethal dose of endotoxin 2 days after the viral exposure, 70% of the rat pups died within 8–10 h, quietly without significant symptoms. Animals displayed the characteristic intrathoracic petechiae, liquid blood around the heart, thymic involution, and other findings on pathology that have also been found in infants dying of SIDS (23). Older animals and younger animals did not die (31, 32). SIDS occurs between 2 and 4 months of age in human infants. The narrow window of lethality seen in this animal model on PN12 could be due to an increased susceptibility from normal developmental changes in the immune, endocrine system, or autonomic nervous system (31) (Table 1). These animals had an abnormal cytokine profile in response to that normally seen following endotoxin injection (62).
Pilot work suggests that rat pups exposed to nicotine in utero exhibited a similar pattern of mortality following endotoxin injection on PN11 or PN12. It was not necessary to pre-infect these animals with influenza. Prenatal nicotine exposure alone appears to be sufficient to alter the neonatal rat’s physiology to make the animal susceptible to a sub-lethal dose of endotoxin. Work in this model is continuing.
Mice
Pregnant mice were exposed to E. coli in the drinking water. One strain of E. coli was toxigenic, obtained from SIDS infants; three other strains were from normal infants. Dams exposed to the toxigenic strain had smaller litter sizes and some runting of the pups. Mortality was 18% for the SIDS E. coli strain compared to 9% for non-SIDS isolates (33) (Table 1). In a different mouse model, gamma herpes virus plus a non-lethal dose of endotoxin was responsible for a reduction of litter size and fetal loss (34) (Table 1). These studies point to a double hit hypothesis for fetal and neonatal death. A single toxin or infectious insult might not be lethal on its own, but in the right combination, and at the right developmental stage, they trigger a lethal event. The multi-hit hypothesis also explains why the spiral to death can often not be stopped once it has started, even with exhaustive medical care. The process begins long before it is noticeable and treatment can be started.
In all of these models, an infectious insult was lethal to young animals. The number of infectious insults increased lethality with nicotine exposure also acting synergistically with the infectious agent. These data support the premise that maternal smoking, both prenatal and PN, might increase the lethality of a presumably non-lethal infection in a susceptible infant. Inflammatory mediators can affect many of the mechanisms proposed to explain SIDS. The following section assesses these models and comments on how inflammation might contribute to the lethal processes investigated.
Respiration and Hyperthermia
Prone sleeping position plays a significant role in SIDS risk, not because of its effect on normal oxygen/carbon dioxide-exchange but rather on an increase in temperature. Elevated body temperature and increased toxin production by bacterial isolates in the nasal pharynx might be a contributing factor (33, 63, 64). Data suggest that prone position will exacerbate the consequences of a viral infection because it promotes an optimum temperature to increase the numbers and variety of bacterial species in upper respiratory secretions and stimulates bacterial toxin production (65).
It is well understood that infection affects the control of respiration and temperature. In studies of infants who died from SIDS while on heart monitors, the cardiac and respiratory tracings resembled those of infants with known infections rather than infants succumbing to asphyxia (66). Alteration of protective mechanisms, gasping, arousal, and efforts to restore normal blood pressure and heart rate are likely to be involved (67, 68). SIDS is more common in the winter months; therefore, these infants are usually well covered to keep them warm. Additionally, infants in a prone position tend to retain body temperature if overwrapped, even in cold weather, they risk becoming hyperthermic (69).
In a piglet model, Galland and colleagues (36, 37) tested the hypothesis that O2 consumption could be altered in a cold climate when the face was cold and the body remained hot. They found that artificially induced hyperthermia increased REM sleep, a period associated with risk for SIDS (37) (Table 1). This study promoted the hypothesis that the primary effect on mortality was an increased body temperature rather than any changes in O2 or CO2 gas exchange (38, 70).
In a separate experiment, piglets developed tachycardia with a drop in blood pressure when heated. Their respiratory rate increased to compensate but they developed hypocapnic alkalosis and a metabolic acidosis. In addition, these animals had necropsy findings similar to that seen in SIDS cases, excessive lung hemorrhage, and alveolar edema (39) (Table 1). Febrile piglets were shown to have a delayed protective response to airway obstruction (40). Hyperthermia resulting from infection or prone position might trigger event leading to SIDS.
Reflux and Infection
Reflux during sleep has been proposed to have a negative effect on the human infant’s ability of the human infant to auto-resuscitate. Simulated reflux in neonatal rabbits induced obstructive, central, and mixed apnea (41); however, the artificial instillation of a mildly acidic compound in a piglet model, stimulated normal protective responses in sedated and naturally sleeping animals (71, 72) (Table 1). Post-mortem lung changes in these piglets showed petechiae, characteristic of SIDS (73).
In a piglet model decerebrated, vagotomized animals were used to evaluate the effects of apnea and hyperthermia in sudden death. When body temperature was elevated, chemoreflex was prolonged (42) through a central nervous mechanism (43) (Table 1). Prenatal nicotine exposure decreased recovery from laryngeal chemoreflex in heated piglets (74). Again there was a connection with respiratory inhibition and hyperthermia.
Helicobacter pylori DNA and antigens have been found in post-mortem tissues of SIDS infants. A rat model has been used to determine if a fatal apnea could result from a response to gastroesophageal reflux (75, 76). The results were inconclusive that H. pylori are a primary cause of SIDS; however, this model is consistent with infectious challenge (77–79). Recent work by Highet and colleagues on samples from SIDS infants show that the gut microbiome are significantly different than found in control infants and should be explored further as a cause of inflammation (80).
Serotonin, Depression, and Inflammation
The serotonergic system of the brain stem, an area that controls heart rate and breathing, is a primary focus for the role of the central nervous system in the factors underlying SIDS. The neurotransmitter serotonin appears to be very sensitive to inflammation; in particular, the inflammatory cytokines often found in autopsy in the central nervous system of SIDS infants (81–83). A specific relationship between increased inflammation and decreased serotonin output has been established in studies on depression (84); the use of anti-inflammatory medications improves serotonin function as it improves depression (85). If SIDS is associated with low levels of serotonin in important structures of the brain stem, it is reasonable to examine the hypothesis that inflammation due to infection might be involved.
Genetically altered animals have been used to explore specific pathology in the serotonin system. An over-expression of serotonin auto-receptors in this mouse model leads to death associated with a decrease in serotonin, drop in heart rate, hypothermia, and a failure to initiate protective autonomic responses (44) (Table 1). Neonatal mice deficient in brainstem serotonin have spontaneous bradycardia in room air and fail to auto-resuscitate during episodic anoxia, ultimately dying from an inability to appropriately increase heart rate (45, 46) (Table 1). Inflammatory cytokines decrease levels of serotonin in the brain stem and might lead to death because of an inability to respond appropriately to key triggers SIDS such as hyperthermia, hypoxia, low blood pressure or decreased heart rate.
Nicotine
Smoking during pregnancy is a primary risk factor for SIDS (49, 86, 87). The risk is fairly low from environmental tobacco exposure (11, 13, 88–90); however, maternal smoking doubles the risk and that increases 3–4 times when the mother smokes more than 10 cigarettes per day (12, 86). Nicotine is considered a major neuroteratogen (91). Nicotine crosses the placenta during pregnancy and could explain why maternal smoking during pregnancy is more harmful on central respiratory mechanisms than PN exposure to environmental tobacco smoke (10, 13, 51). Prenatal nicotine exposure could result in an increase in susceptibility to other risk factors of SIDS in several ways that involve inflammation.
Nicotine and Infection
The synergy between infection and smoking has been examined in a piglet model. When animals are treated with nicotine and endotoxin then exposed to subglottic acidified saline solution are unable to auto-resuscitate and develop prolonged apnea. Nicotine and an inflammatory mediator interleukin-1β have a similar synergistic effect, decreasing the animal’s ability to respond to apnea (47). These experiments demonstrate the synergistic connection between nicotine and inflammation and lethal apnea (92) (Table 1). While Froen and colleagues did not directly examine the role of infection, inflammatory mediators are often used as proxy for inflammation and it is highly likely that infection is involved.
In vitro studies on the effects of infection and risk factors such as cigarette smoke on inflammatory responses have identified that these factors can increase inflammation and enhance pro-inflammatory responses (93, 94), these effects need to be tested in animal models in which they also change the autonomic and serotonergic response to infection.
Nicotine and Serotonin
Prenatal exposure of animals to nicotine has been shown to alter the response of the serotonergic system in the newborn by down-regulating receptors important for normal serotonin function (49, 95). This in turn alters normal function and control of the heart through its action on the autonomic nervous system (49) (Table 1). Any process that challenges the cardiovascular system such as overheating, infection, toxins, and low blood sugar levels, increases SIDS risk (64, 96, 97). Nicotine exposure might increase the risk of SIDS by altering how the infant responds to these challenges.
Nicotine and Autonomic Control
Investigators have used the rat model to evaluate the effect of prenatal nicotine exposure on the heart (55), brain (54), and respiration (56, 57) (Table 1). Nicotine stimulates the nicotinic acetylcholine receptor and mimics the effects of acetylcholine (98). Chronic exposure of these receptors during critical prenatal development could explain the abnormalities of receptor expression and acetylcholinesterase activity observed in tissues from SIDS infants. These mechanisms are critical for normal autonomic control of the heart.
A strain of rabbits was developed to examine the effect of cardiac muscarinic receptors on autonomic tone. Similar to infants dying from SIDS, these rabbits had over-expression of muscarinic receptors that was most pronounced between the fifth and the seventh week of life, a time of increased mortality in the rabbits (99). Abnormal autonomic development from maternal smoking during pregnancy might result in infants being unable to arouse and respond to a physiologic insult such as a drop in blood pressure, hypoxia, infection, or overheating.
Mechanisms that regulate inflammation also affect vagal nerve activity through the release of acetylcholine. Data suggest an association between heart rate variability, a prime example of normal vagal function, and inflammation (100). Stress can precipitate both depression and promote inflammatory responses through its action on the sympathetic and parasympathetic nervous system (101). All of these processes are so intertwined that evaluating them as a single response negates the complex sets of interactions in the human body and encourages the use of animal models that can mimic interactions between systems.
Developmental Susceptibility
One key factor in SIDS is the specific timing of susceptibility. By definition, SIDS infants are younger than 12 months of age with most SIDS occurring between 2 and 4 months. There is a peak around 3 months of age. Evaluation of a condition that occurs during such a specific time during the development of multiple interrelated physiologic systems can be quite difficult. Teasing out the combined effects requires the ability to measure multiple interactions at the same time in many organ systems.
Development of Inflammatory Responses
The peak age at which SIDS occurs corresponds to a time when infants are dependent on innate responses to infection. A dysregulation of innate inflammatory responses and the developmental change that mature the infant could contribute to the physiological changes leading to these sudden deaths (31, 32, 94, 102, 103). The immune system is beginning to recognize a large number of foreign antigens and developing its own antibody responses while the protective effect of maternal antibodies is waning (104). Small changes in the development of immune responses can turn a minor illness into a lethal event (62). This could explain why breastfeeding is protective for SIDS because breast milk contains biologically active antibodies that reduce colonization by potentially pathogenic bacteria as well as glycoproteins that reduce attachment of organisms to epithelial cells via lectin like adhesins (105, 106).
Male infants succumb to SIDS slightly more frequently than females. Peripheral blood monocytic cells from adult male donors exposed to cigarette smoke extract produced lower levels of some inflammatory cytokines than those from adult female donors. There were significant correlations between levels of pro-inflammatory cytokines and testosterone, but only in females. Testosterone levels in females correspond to those among male infants in the age range at greatest risk of SIDS. There is potentially an effect between cortisol levels and the testosterone surge in male infants during the time of SIDS risk but this needs to be evaluated further (94).
The normalization of the adult day–night temperature pattern occurs around the time of highest SIDS risk (107). Both human infants (108, 109) and rat pups (110, 111) have a nadir of cortisol production during this time of development that overlaps a time when the immune system is responding to new infections and the corresponding cortisol system might not be able to regulate the response. The levels of cortisol suppress pro-inflammatory responses to bacterial antigens during the day, just before and after the switch to diurnal circadian rhythm. The night-time levels before the switch have the same effect; however night-time levels of cortisol after the switch are much lower and do not have a suppressive effect on the immune system. In fact, they can enhance pro-inflammatory responses [(106); Blackwell et al., this volume]. Developmental immaturity at this age could lead to an inability to produce a protective response to an infection.
Development of the Neural System
The maturity of the autonomic nervous system and baroreflex control of the heart in human infants correspond to times of autonomic imbalance (112). This imbalance is influenced by stage of sleep, head covering, temperature, body position, and nicotine exposure, all known risk factors for SIDS (113–116). In 2- to 4-month olds, there is a significant difference in cardiac response to a hypotensive event in quiet sleep. This is in contrast to that seen in adults and newborns where heart rate elevates and is maintained in an elevated stage for several minutes (117). Ledwidge et al. (118) describes a case where a 5-month-old infant died during a sleep study because the heart rate failed to increase in response to a head-up tilt (decrease in blood pressure), even though all other parameters of the sleep study were normal.
Development and Respiration
Developmental changes in 12-day-old rats had an abrupt switch in brain stem expression of a receptor important for stimulating respiratory control. This period of imbalance is transitory but could leave the respiratory system vulnerable to insult such as infection that often causes apnea in infants (58–60, 119) (Table 1). These represent three very important areas of development that occur around the time of unexplained death that could be attributed to a SIDS-like event. This case supports the hypothesis that abnormal autonomic tone might contribute to SIDS deaths.
Conclusion
Any animal model used to mimic a human condition should reflect the pathophysiology and other parameters that are indicative of that condition. SIDS by definition is a sudden unexplained death that cannot be defined by symptoms or pathology commonly known to cause death. Because of this definition, there are very few clues to build a model on. The models presented here are an attempt to define possible clues on which to build a model with physical and epidemiologic findings of SIDS.
A developmental model like that described by Blood-Siegfried is critical to tease out the effect of inflammatory responses to infection on interactions among physiological systems and risk factors such as tobacco smoke (31, 32, 61, 120) (Table 1). This model not only causes pathological finding similar to those found on autopsy of SIDS infants but also reflects the narrow window of susceptibility not seen in many other models (23).
Further examination using this model could evaluate other possible mechanistic associations:
1) How does cigarette smoke extract affect 11- and 12-day-old rat pups?
2) How does the stress response as measured by catecholamine and corticosterone levels respond to the insult?
3) Do animals that are susceptible to death have specific changes in the circadian rhythms?
4) What is the normal development of autonomic tone and heart rate variability?
This developmental model allows the researcher to examine the effect of age within the context of the development of these systems and the interactions between systems.
We have only begun to tap into the multiple possibilities of connections involved in sudden and unexplained death. SIDS is a complex syndrome and the answer to its cause/s will require development of appropriate animal models to determine how all the parts fit together.
Conflict of Interest Statement
I have received no payment or services from a third party for any aspect of the submitted work. The developmental model was initially designed during a post-doctoral fellowship at the National Institute of Environmental Health Sciences. Further work was funded by a small grant from Duke University School of Nursing.
References
1. Willinger M, James LS, Catz C. Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediatr Pathol (1991) 11:677–84. doi: 10.3109/15513819109065465
2. Blackwell CC, Weir DM. The role of infection in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25:1–6. doi:10.1111/j.1574-695X.1999.tb01320.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Centers for Disease Control and Prevention. Sudden Infant Death Syndrome (SIDS) and Sudden Unexpected Infant Death (SUID): Home [Online]. (2008). Available from: http://www.cdc.gov/SIDS/index.htm
4. Rognum TO. Definition and pathologic features. In: Byard RW, Krouse HF, editors. Sudden Infant Death Syndrome: Problems, Progress and Possibilities. New York, NY: Oxford University Press (2001). p. 4–30.
5. Byard RW, Lee V. A re-audit of the use of definitions of sudden infant death syndrome (SIDS) in peer-reviewed literature. J Forensic Leg Med (2012) 19:455–6. doi:10.1016/j.jflm.2012.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Alvarez-Lafuente R, Aguilera B, Suarez-Mier MAP, Morentin B, Vallejo G, Gomez J, et al. Detection of human herpesvirus-6, Epstein-Barr virus and cytomegalovirus in formalin-fixed tissues from sudden infant death: a study with quantitative real-time PCR. Forensic Sci Int (2008) 178:106–11. doi:10.1016/j.forsciint.2008.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Dettmeyer R, Baasner A, Haag C, Bruch S, Schlamann M. Immunohistochemical and molecular-pathological diagnosis of myocarditis in cases of suspected sudden infant death syndrome (SIDS) – a multicenter study. Leg Med (2009) 11(Suppl 1):S124–7. doi:10.1016/j.legalmed.2009.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. American Academy of Pediatrics. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics (2005) 116:1245–55. doi:10.1542/peds.2005-1499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Mitchell EA. What is the mechanism of SIDS? Clues from epidemiology. Dev Psychobiol (2009) 51:215–22. doi:10.1002/dev.20369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Zhang K, Wang X. Maternal smoking and increased risk of sudden infant death syndrome: a meta-analysis. Leg Med (Tokyo) (2012) 15(3):115–21. doi:10.1016/j.legalmed.2012.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Klonoff-Cohen HS, Edelstein SL, Lefkowitz ES, Srinivasan IP, Kaegi D, Chang JC, et al. The effect of passive smoking and tobacco exposure through breast milk on sudden infant death syndrome. J Am Med Assoc (1995) 273:795–8. doi:10.1001/jama.273.10.795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. MacDorman MF, Cnattingius S, Hoffman HJ, Kramer MS, Haglund B. Sudden infant death syndrome and smoking in the United States and Sweden. Am J Epidemiol (1997) 146:249–57. doi:10.1093/oxfordjournals.aje.a009260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Alm B, Milerad J, Wennergren G, Skjaerven R, Oyen N, Norvenius G, et al. A case-control study of smoking and sudden infant death syndrome in the Scandinavian countries, 1992 to 1995. The Nordic Epidemiological SIDS Study. Arch Dis Child (1998) 78:329–34. doi:10.1136/adc.78.4.329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Chong DSY, Yip PSF, Karlberg J. Maternal smoking: an increasing unique risk factor for sudden infant death syndrome in Sweden. Acta Paediatr (2004) 93:471–8. doi:10.1080/08035250310023495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Anderson ME, Johnson DC, Batal HA. Sudden infant death syndrome and prenatal maternal smoking: rising attributed risk in the back to sleep era. BMC Med (2005) 3:4. doi:10.1186/1741-7015-3-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Mitchell EA, Milerad J. Smoking and the sudden infant death syndrome. Rev Environ Health (2006) 21:81–103. doi:10.1515/REVEH.2006.21.2.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Mitchell EA, Hutchison L, Stewart AW. The continuing decline in SIDS mortality. Arch Dis Child (2007) 92:625–6. doi:10.1136/adc.2007.116194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Krous HF, Haas EA, Chadwick AE, Masoumi H, Stanley C. Intrathoracic petechiae in SIDS: a retrospective population-based 15-year study. Forensic Sci Med Pathol (2008) 4:234–9. doi:10.1007/s12024-008-9054-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Harrison M, Curran C, Gillan JE. Mast-cell degranulation suggests nonimmune anaphylaxis as a cause of deaths in SIDS – an electron-microscopy study. Lab Invest (1992) 66:5.
21. Holgate ST, Walters C, Walls AF, Lawrence S, Shell DJ, Variend S, et al. The anaphylaxis hypothesis of sudden infant death syndrome (SIDS): mast cell degranulation in cot death revealed by elevated concentrations of tryptase in serum. Clin Exp Allergy (1994) 24:1115–22. doi:10.1111/j.1365-2222.1994.tb03316.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Rambaud C, Guibert M, Briand E, Grangeot-Keros L, Coulomb-L’hermin A, Dehan M. Microbiology in sudden infant death syndrome (SIDS) and other childhood deaths. FEMS Immunol Med Microbiol (1999) 25:59–66. doi:10.1111/j.1574-695X.1999.tb01327.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Blood-Siegfried J, Rambaud C, Nyska A, Germolec DR. Evidence for infection, inflammation and shock in sudden infant death: parallels between a neonatal rat model of sudden death and infants who died of sudden infant death syndrome. Innate Immun (2008) 14:145–52. doi:10.1177/1753425908090730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Coombs RR, Holgate ST. Allergy and cot death: with special focus on allergic sensitivity to cows’ milk and anaphylaxis. Clin Exp Allergy (1990) 20:359–66. doi:10.1111/j.1365-2222.1990.tb02794.x
25. Buckley MG, Variend S, Walls AF. Elevated serum concentrations of beta-tryptase, but not alpha-tryptase, in sudden infant death syndrome (SIDS). An investigation of anaphylactic mechanisms. Clin Exp Allergy (2001) 31:1696–704. doi:10.1046/j.1365-2222.2001.01213.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Siarakas S, Damas E, Murrell WG. Is cardiorespiratory failure induced by bacterial toxins the cause of sudden infant death syndrome? Studies with an animal model (the rabbit). Toxicon (1995) 33:635–49. doi:10.1016/0041-0101(95)00003-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Siarakas S, Damas E, Murrell WG. The effect of enteric bacterial toxins on the catecholamine levels of the rabbit. Pathology (1997) 29:278–85. doi:10.1080/00313029700169095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Sayers NM, Drucker DB, Morris JA, Telford DR. Lethal synergy between toxins of staphylococci and enterobacteria: implications for sudden infant death syndrome. J Clin Pathol (1995) 48:929–32. doi:10.1136/jcp.48.10.929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Sayers NM, Drucker DB, Morris JA, Telford DR. Significance of endotoxin in lethal synergy between bacteria associated with sudden infant death syndrome: follow up study. J Clin Pathol (1996) 49:365–8. doi:10.1136/jcp.49.5.365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Lee S, Barson AJ, Drucker DB, Morris JA, Telford DR. Lethal challenge of gnotobiotic weanling rats with bacterial isolates from cases of sudden infant death syndrome (SIDS). J Clin Pathol (1987) 40:1393–6. doi:10.1136/jcp.40.12.1393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Blood-Siegfried J, Nyska A, Lieder H, Joe M, Vega L, Patterson R, et al. Synergistic effect of influenza a virus on endotoxin-induced mortality in rat pups: a potential model for sudden infant death syndrome. Pediatr Res (2002) 52:481–90. doi:10.1203/00006450-200210000-00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Blood-Siegfried J, Nyska A, Geisenhoffer K, Lieder H, Moomaw C, Cobb K, et al. Alteration in regulation of inflammatory response to influenza a virus and endotoxin in suckling rat pups: a potential relationship to sudden infant death syndrome. FEMS Immunol Med Microbiol (2004) 42:85–93. doi:10.1016/j.femsim.2004.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Bettelheim KA, Luke RK, Johnston N, Pearce JL, Goldwater PN. A possible murine model for investigation of pathogenesis of sudden infant death syndrome. Curr Microbiol (2012) 64:276–82. doi:10.1007/s00284-011-0065-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Cardenas I, Mor G, Aldo P, Lang SM, Stabach P, Sharp A, et al. Placental viral infection sensitizes to endotoxin-induced pre-term labor: a double hit hypothesis. Am J Reprod Immunol (2011) 65:110–7. doi:10.1111/j.1600-0897.2010.00908.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Sarawar SR, Blackman MA, Doherty PC. Superantigen shock in mice with an inapparent viral infection. J Infect Dis (1994) 170(5):1189–94. doi:10.1093/infdis/170.5.1189
36. Galland BC, Peebles CM, Bolton DPG, Taylor BJ. Oxygen consumption in the newborn piglet during combined cold face hot body exposure. J Paediatr Child Health (1992) 28:S33–5. doi:10.1111/j.1440-1754.1992.tb02730.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Galland BC, Peebles CM, Bolton DPG, Taylor BJ. Sleep state organization in the developing piglet during exposure to different thermal stimuli. Sleep (1993) 16:610–9.
38. Galland BC, Peebles CM, Bolton DPG, Taylor BJ. The microenvironment of the sleeping newborn piglet covered by bedclothes: gas exchange and temperature. J Paediatr Child Health (1994) 30:144–50. doi:10.1111/j.1440-1754.1994.tb00599.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Elder DE, Bolton DP, Dempster AG, Taylor BJ, Broadbent RS. Pathophysiology of overheating in a piglet model: findings compared with sudden infant death syndrome. J Paediatr Child Health (1996) 32:113–9. doi:10.1111/j.1440-1754.1996.tb00906.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Voss LJ, Bolton DP, Galland BC, Taylor BJ. Effects of prior hypoxia exposure, endotoxin and sleep state on arousal ability to airway obstruction in piglets: implications for sudden infant death syndrome. Biol Neonate (2005) 88:145–55. doi:10.1159/000085896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Wetmore RF. Effects of acid on the larynx of the maturing rabbit and their possible significance to the sudden infant death syndrome. Laryngoscope (1993) 103:1242–54. doi:10.1288/00005537-199311000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Curran AK, Xia L, Leiter JC, Bartlett D. Elevated body temperature enhances the laryngeal chemoreflex in decerebrate piglets. J Appl Physiol (2005) 98:780–6. doi:10.1152/japplphysiol.00906.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Xia L, Leiter JC, Bartlett D. Laryngeal water receptors are insensitive to body temperature in neonatal piglets. Respir Physiol Neurobiol (2006) 150:82–6. doi:10.1016/j.resp.2005.05.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Audero E, Coppi E, Mlinar B, Rossetti T, Caprioli A, Banchaabouchi MA, et al. Sporadic autonomic dysregulation and death associated with excessive serotonin autoinhibition. Science (2008) 321:130–3. doi:10.1126/science.1157871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Cummings KJ, Li A, Deneris ES, Nattie EE. Bradycardia in serotonin-deficient Pet-1-/- mice: influence of respiratory dysfunction and hyperthermia over the first 2 postnatal weeks. Am J Physiol Regul Integr Comp Physiol (2010) 298:R1333–42. doi:10.1152/ajpregu.00110.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Cummings KJ, Hewitt JC, Li A, Daubenspeck JA, Nattie EE. Postnatal loss of brainstem serotonin neurons compromises the ability of neonatal rats to survive episodic severe hypoxia. J Physiol (2011) 589:5247–56. doi:10.1113/jphysiol.2011.214445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Froen JF, Akre H, Stray-Pedersen B, Saugstad OD. Adverse effects of nicotine and interleukin-1beta on autoresuscitation after apnea in piglets: implications for sudden infant death syndrome. Pediatrics (2000) 105:E52. doi:10.1542/peds.105.4.e52
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Froen JF, Akre H, Stray-Pedersen B, Saugstad OD. Prolonged apneas and hypoxia mediated by nicotine and endotoxin in piglets. Biol Neonate (2002) 81:119–25. doi:10.1159/000047196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Duncan JR, Garland M, Myers MM, Fifer WP, Yang M, Kinney HC, et al. Prenatal nicotine-exposure alters fetal autonomic activity and medullary neurotransmitter receptors: implications for sudden infant death syndrome. J Appl Physiol (2009) 107:1579–90. doi:10.1152/japplphysiol.91629.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Slotkin TA, Saleh JL, Mccook EC, Seidler FJ. Impaired cardiac function during postnatal hypoxia in rats exposed to nicotine prenatally: implications for perinatal morbidity and mortality, and for sudden infant death syndrome. Teratology (1997) 55:177–84. doi:10.1002/(SICI)1096-9926(199703)55:3<177::AID-TERA2>3.0.CO;2-#
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther (1998) 285:931–45.
52. St John WM, Leiter JC. Maternal nicotine depresses eupneic ventilation of neonatal rats. Neurosci Lett (1999) 267:206–8. doi:10.1016/S0304-3940(99)00364-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Fewell JE, Smith FG, Ng VKY. Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age-dependent manner. Respir Physiol (2001) 127:61–73. doi:10.1016/S0034-5687(01)00232-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Neff RA, Simmens SJ, Evans C, Mendelowitz D. Prenatal nicotine exposure alters central cardiorespiratory responses to hypoxia in rats: implications for sudden infant death syndrome. J Neurosci (2004) 24:9261–8. doi:10.1523/JNEUROSCI.1918-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Evans C, Wang J, Neff R, Mendelowitz D. Hypoxia recruits a respiratory-related excitatory pathway to brainstem premotor cardiac vagal neurons in animals exposed to prenatal nicotine. Neuroscience (2005) 133:1073–9. doi:10.1016/j.neuroscience.2005.03.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Huang ZG, Wang X, Dergacheva O, Mendelowitz D. Prenatal nicotine exposure recruits an excitatory pathway to brainstem parasympathetic cardioinhibitory neurons during hypoxia/hypercapnia in the rat: implications for sudden infant death syndrome. Pediatr Res (2005) 58:562–7. doi:10.1203/01.PDR.0000179380.41355.FC
57. Huang ZG, Griffioen KJS, Wang X, Dergacheva O, Kamendi H, Gorini C, et al. Differential control of central cardiorespiratory interactions by hypercapnia and the effect of prenatal nicotine. J Neurosci (2006) 26:21–9. doi:10.1523/JNEUROSCI.4221-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Liu QL, Wong-Riley MTT. Developmental changes in the expression of GABA(A) receptor subunits alpha 1, alpha 2, and alpha 3 in the rat pre-Botzinger complex. J Appl Physiol (2004) 96:1825–31. doi:10.1152/japplphysiol.01264.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Liu QL, Wong-Riley MTT. Postnatal developmental expressions of neurotransmitters and receptors in various brain stem nuclei of rats. J Appl Physiol (2005) 98:1442–57. doi:10.1152/japplphysiol.01301.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Wong-Riley MTT, Liu QL. Neurochemical development of brain stem nuclei involved in the control of respiration. Respir Physiol Neurobiol (2005) 149:83–98. doi:10.1016/j.resp.2005.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Blood-Siegfried J, Shelton B. Animal models of sudden unexplained death. FEMS Immunol Med Microbiol (2004) 42:34–41. doi:10.1016/j.femsim.2004.06.009
62. Gleeson M, Cripps AW. Development of mucosal immunity in the first year of life and relationship to sudden infant death syndrome. FEMS Immunol Med Microbiol (2004) 42:21–33. doi:10.1016/j.femsim.2004.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Molony N, Blackwell CC, Busuttil A. The effect of prone posture on nasal temperature in children in relation to induction of staphylococcal toxins implicated in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25:109–13. doi:10.1111/j.1574-695X.1999.tb01333.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Blackwell CC, Moscovis SM, Gordon AE, Al Madani OM, Hall ST, Gleeson M, et al. Cytokine responses and sudden infant death syndrome: genetic, developmental, and environmental risk factors. J Leukoc Biol (2005) 78:1242–54. doi:10.1189/jlb.0505253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Harrison LM, Morris JA, Telford DR, Brown SM, Jones K. The nasopharyngeal bacterial flora in infancy: effects of age, gender, season, viral upper respiratory tract infection and sleeping position. FEMS Immunol Med Microbiol (1999) 25:19–28. doi:10.1111/j.1574-695X.1999.tb01323.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Poets CF, Meny RG, Chobanian MR, Bonofiglo RE. Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res (1999) 45:350–4. doi:10.1203/00006450-199903000-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Harper RM. Sudden infant death syndrome: a failure of compensatory cerebellar mechanisms? Pediatr Res (2000) 48:140–2. doi:10.1203/00006450-200008000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Harper RM. Autonomic control during sleep and risk for sudden death in infancy. Arch Ital Biol (2001) 139:185–94.
69. Gilbert R, Rudd P, Berry PJ, Fleming PJ, Hall E, White DG, et al. Combined effect of infection and heavy wrapping on the risk of sudden unexpected infant death. Arch Dis Child (1992) 67:171–7. doi:10.1136/adc.67.2.171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Ponsonby AL, Dwyer T, Gibbons LE, Cochrane JA, Jones ME, Mccall MJ. Thermal environment and sudden infant death syndrome: case-control study. BMJ (1992) 304:277–82. doi:10.1136/bmj.304.6822.277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Jeffery HE, Page M, Post EJ, Wood AKW. Physiological studies of gastroesophageal reflux and airway protective responses in the young animal and human infant. Clin Exp Pharmacol Physiol (1995) 22:544–9. doi:10.1111/j.1440-1681.1995.tb02064.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. McKelvey GM, Post EJ, Wood AK, Jeffery HE. Airway protection following simulated gastro-oesophageal reflux in sedated and sleeping neonatal piglets during active sleep. Clin Exp Pharmacol Physiol (2001) 28:533–9. doi:10.1046/j.1440-1681.2001.03483.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Richardson MA, Adams J. Fatal apnea in piglets by way of laryngeal chemoreflex: postmortem findings as anatomic correlates of sudden infant death syndrome in the human infant. Laryngoscope (2005) 115:1163–9. doi:10.1097/01.MLG.0000165458.52991.1B
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Xia L, Leiter JC, Bartlett D Jr. Gestational nicotine exposure exaggerates hyperthermic enhancement of laryngeal chemoreflex in rat pups. Respir Physiol Neurobiol (2010) 171:17–21. doi:10.1016/j.resp.2010.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Pattison CP, Marshall BJ, Scott LW, Herndon B, Willsie SK. Proposed link between Helicobacter pylori (HP) and sudden infant death syndrome (SIDS): possible pathogenic mechanisms in an animal model. I. Effects of intratracheal urease. Gastroenterology (1998) 114:G3689. doi:10.1016/S0016-5085(98)83663-9
76. Orienstein DM. Aspiration pneumonias and gastroesophageal reflux-related respiratory disease. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson Textbook of Pediatrics. Philadelphia, PA: W. B. Saunders (2000). p. 1288–91.
77. Elitsur Y, Btriest W, Sabet Z, Neace C, Jiang C, Thomas E. Is sudden infant death syndrome associated with Helicobacter pylori infection in children? Helicobacter (2000) 5:227–31. doi:10.1046/j.1523-5378.2000.00035.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Ho GY, Windsor HM, Snowball B, Marshall BJ. Helicobacter pylori is not the cause of sudden infant death syndrome (SIDS). Am J Gastroenterol (2001) 96:3288–94. doi:10.1111/j.1572-0241.2001.05327.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Stray-Pedersen A, Vege A, Rognum TO. Helicobacter pylori antigen in stool is associated with SIDS and sudden infant deaths due to infectious disease. Pediatr Res (2008) 64:405–10. doi:10.1203/PDR.0b013e31818095f7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Highet AR, Berry AM, Bettelheim KA, Goldwater PN. Gut microbiome in sudden infant death syndrome (SIDS) differs from that in healthy comparison babies and offers an explanation for the risk factor of prone position. Int J Med Microbiol (2014) 304:735–41. doi:10.1016/j.ijmm.2014.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Vege A, Rognum TO, Scott H, Aasen AO, Saugstad OD. SIDS cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr (1995) 84:193–6. doi:10.1111/j.1651-2227.1995.tb13608.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Prandota J. Possible pathomechanisms of sudden infant death syndrome: key role of chronic hypoxia, infection/inflammation states, cytokine irregularities, and metabolic trauma in genetically predisposed infants. Am J Ther (2004) 11:517–46. doi:10.1097/01.mjt.0000140648.30948.bd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Rognum IJ, Haynes RL, Vege A, Yang M, Rognum TO, Kinney HC. Interleukin-6 and the serotonergic system of the medulla oblongata in the sudden infant death syndrome. Acta Neuropathol (2009) 118:519–30. doi:10.1007/s00401-009-0535-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Pomytkin IA, Cline BH, Anthony DC, Steinbusch HW, Lesch KP, Strekalova T. Endotoxemia resulting from decreased serotonin transporter (5-HTT) function: a reciprocal risk factor for depression and insulin resistance? Behav Brain Res (2014) 276:111–7. doi:10.1016/j.bbr.2014.04.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Hernandez ME, Mendieta D, Perez-Tapia M, Bojalil R, Estrada-Garcia I, Estrada-Parra S, et al. Effect of selective serotonin reuptake inhibitors and immunomodulator on cytokines levels: an alternative therapy for patients with major depressive disorder. Clin Dev Immunol (2013) 2013:267871. doi:10.1155/2013/267871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Hunt CE, Hauck FR. Sudden infant death syndrome. CMAJ (2006) 174:1861–9. doi:10.1503/cmaj.051671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Fleming P, Blair PS. Sudden infant death syndrome and parental smoking. Early Hum Dev (2007) 83:721–5. doi:10.1016/j.earlhumdev.2007.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Anderson HR, Cook DG. Passive smoking and sudden infant death syndrome: review of the epidemiological evidence. Thorax (1997) 52:1003–9. doi:10.1136/thx.52.11.1003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. McMartin KI, Platt MS, Hackman R, Klein J, Smialek JE, Vigorito R, et al. Lung tissue concentrations of nicotine in sudden infant death syndrome (SIDS). J Pediatr (2002) 140:205–9. doi:10.1067/mpd.2002.121937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Liebrechts-Akkerman G, Lao O, Liu F, Van Sleuwen BE, Engelberts AC, L’hoir MP, et al. Postnatal parental smoking: an important risk factor for SIDS. Eur J Pediatr (2011) 170:1281–91. doi:10.1007/s00431-011-1433-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Slotkin TA, Mackillop EA, Rudder CL, Ryde IT, Tate CA, Seidler FJ. Permanent, sex-selective effects of prenatal or adolescent nicotine exposure, separately or sequentially, in rat brain regions: indices of cholinergic and serotonergic synaptic function, cell signaling, and neural cell number and size at 6 months of age. Neuropsychopharmacology (2007) 32:1082–97. doi:10.1038/sj.npp.1301231
92. Froen JF, Amerio G, Stray-Pedersen B, Saugstad OD. Detrimental effects of nicotine and endotoxin in the newborn piglet brain during severe hypoxemia. Biol Neonate (2002) 82:188–96. doi:10.1159/000063610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Moscovis SM, Cox A, Hall ST, Burns CJ, Scott RJ, Blackwell CC. Effects of gender, cytokine gene polymorphisms and environmental factors on inflammatory responses. Innate Immun (2014). doi:10.1177/1753425914553645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Moscovis SM, Hall ST, Burns CJ, Scott RJ, Blackwell CC. The male excess in sudden infant deaths. Innate Immun (2014) 20:24–9. doi:10.1177/1753425913481071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Slotkin TA, Ryde IT, Tate CA, Seidler FJ. Lasting effects of nicotine treatment and withdrawal on serotonergic systems and cell signaling in rat brain regions: separate or sequential exposure during fetal development and adulthood. Brain Res Bull (2007) 73:259–72. doi:10.1016/j.brainresbull.2007.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Alkout AM, Blackwell CC, Weir DM, Poxton IR, Elton RA, Luman W, et al. Isolation of a cell surface component of Helicobacter pylori that binds H type 2, Lewis(a), and Lewis(b) antigens. Gastroenterology (1997) 112:1179–87. doi:10.1016/S0016-5085(97)70129-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, et al. IL6 G-174C associated with sudden infant death syndrome in a Caucasian Australian cohort. Hum Immunol (2006) 67:819–25. doi:10.1016/j.humimm.2006.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol (2000) 61:75–111. doi:10.1016/S0301-0082(99)00045-3
99. Livolsi A, Niederhoffer N, Dali-Youcef N, Rambaud C, Olexa C, Mokni W, et al. Cardiac muscarinic receptor overexpression in sudden infant death syndrome. PLoS One (2010) 5:e9464. doi:10.1371/journal.pone.0009464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Taylor L, Loerbroks A, Herr RM, Lane RD, Fischer JE, Thayer JF. Depression and smoking: mediating role of vagal tone and inflammation. Ann Behav Med (2011) 42:334–40. doi:10.1007/s12160-011-9288-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol (2006) 27:24–31. doi:10.1016/j.it.2005.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Weese-Mayer DE, Ackerman MJ, Marazita ML, Berry-Kravis EM. Sudden infant death syndrome: review of implicated genetic factors. Am J Med Genet (2007) 143A:771–88. doi:10.1002/ajmg.a.31722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Blood-Siegfried J. The role of infection and inflammation in sudden infant death syndrome. Immunopharmacol Immunotoxicol (2009) 31:516–23. doi:10.3109/08923970902814137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Butler JE. Immunologic aspects of breast feeding, antiinfectious activity of breast milk. Semin Perinatol (1979) 3:255–70.
106. Gordon AE, Al Madani O, Weir DM, Busuttil A, Blackwell C. Cortisol levels and control of inflammatory responses to toxic shock syndrome toxin-1 (TSST-1): the prevalence of night-time deaths in sudden infant death syndrome (SIDS). FEMS Immunol Med Microbiol (1999) 25:199–206. doi:10.1111/j.1574-695X.1999.tb01344.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Joseph DV, Jackson JA, Westaway J, Taub NA, Petersen SA, Wailoo MP. Effect of parental smoking on cotinine levels in newborns. Arch Dis Child Fetal Neonatal Ed (2007) 92:484–8. doi:10.1136/adc.2006.108506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Santiago LB, Jorge SM, Moreira AC. Longitudinal evaluation of the development of salivary cortisol circadian rhythm in infancy. Clin Endocrinol (1996) 44:157–61. doi:10.1046/j.1365-2265.1996.645466.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Mantagos S, Moustogiannis A, Vagenakis AG. Diurnal variation of plasma cortisol levels in infancy. J Pediatr Endocrinol Metab (1998) 11:549–53. doi:10.1515/JPEM.1998.11.4.549
110. Dent GW, Smith MA, Levine S. The ontogeny of the neuroendocrine response to endotoxin. Brain Res Dev Brain Res (1999) 117:21–9. doi:10.1016/S0165-3806(99)00091-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Yoshimura S, Sakamoto S, Kudo H, Sassa S, Kumai A, Okamoto R. Sex-differences in adrenocortical responsiveness during development in rats. Steroids (2003) 68:439–45. doi:10.1016/S0039-128X(03)00045-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Gournay V, Drouin E, Roze JC. Development of baroreflex control of heart rate in preterm and full term infants. Arch Dis Child (2002) 86:151–4. doi:10.1136/fn.86.3.F151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Galland BC, Reeves G, Taylor BJ, Bolton DPG. Sleep position, autonomic function, and arousal. Arch Dis Child (1998) 78:F189–94. doi:10.1136/fn.78.3.F189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Franco P, Chabanski S, Szliwowski H, Dramaix M, Kahn A. Influence of maternal smoking on autonomic nervous system in healthy infants. Pediatr Res (2000) 47:215–20. doi:10.1203/00006450-200002000-00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Franco P, Szliwowski H, Dramaix M, Kahn A. Influence of ambient temperature on sleep characteristics and autonomic nervous control in healthy infants. Sleep (2000) 23:401–7.
116. Trinder J, Kleiman J, Carrington M, Smith S, Breen S, Tan N, et al. Autonomic activity during human sleep as a function of time and sleep stage. J Sleep Res (2001) 10:253–64. doi:10.1046/j.1365-2869.2001.00263.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Myers MM, Gomez-Gribben E, Smith KS, Tseng A, Fifer WP. Developmental changes in infant heart rate responses to head-up tilting. Acta Paediatr (2006) 95:77–81. doi:10.1111/j.1651-2227.2006.tb02184.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Ledwidge M, Fox G, Matthews T. Neurocardiogenic syncope: a model for SIDS. Arch Dis Child (1998) 78:481–3. doi:10.1136/adc.78.5.481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Gao XP, Liu QS, Liu Q, Wong-Riley MT. Excitatory-inhibitory imbalance in hypoglossal neurons during the critical period of postnatal development in the rat. J Physiol (2011) 589:1991–2006. doi:10.1113/jphysiol.2010.198945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: anaphylaxis, development, hyperthermia, infection, inflammation, nicotine, serotonin, toxin
Citation: Blood-Siegfried J (2015) Animal models for assessment of infection and inflammation: contributions to elucidating the pathophysiology of sudden infant death syndrome. Front. Immunol. 6:137. doi: 10.3389/fimmu.2015.00137
Received: 06 January 2015; Paper pending published: 28 January 2015;
Accepted: 12 March 2015; Published online: 30 March 2015.
Edited by:
Caroline Blackwell, University of Newcastle, AustraliaReviewed by:
Paul Nathan Goldwater, University of Adelaide, AustraliaJan Matthias Braun, Leibniz Institute for Environmental Health, Germany
Copyright: © 2015 Blood-Siegfried. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jane Blood-Siegfried, Duke University School of Nursing, Box 3322, Durham, NC 27710, USA e-mail:Ymxvb2QwMDJAbWMuZHVrZS5lZHU=