 Vivek D. Gandhi
Vivek D. Gandhi Harissios Vliagoftis
Harissios Vliagoftis- Pulmonary Research Group, Department of Medicine, University of Alberta, Edmonton, AB, Canada
Airway epithelial cells are the first line of defense against the constituents of the inhaled air, which include allergens, pathogens, pollutants, and toxic compounds. The epithelium not only prevents the penetration of these foreign substances into the interstitium, but also senses their presence and informs the organism’s immune system of the impending assault. The epithelium accomplishes the latter through the release of inflammatory cytokines and chemokines that recruit and activate innate immune cells at the site of assault. These epithelial responses aim to eliminate the inhaled foreign substances and minimize their detrimental effects to the organism. Quite frequently, however, the innate immune responses of the epithelium to inhaled substances lead to chronic and high level release of pro-inflammatory mediators that may mediate the lung pathology seen in asthma. The interactions of airway epithelial cells with allergens will be discussed with particular focus on interactions-mediated epithelial release of cytokines and chemokines and their role in the immune response. As pollutants are other major constituents of inhaled air, we will also discuss how pollutants may alter the responses of airway epithelial cells to allergens.
Introduction
Allergic asthma is a complex disease that involves interactions of genetic and environmental factors (1). Genetic factors predispose to atopy, but the development of allergic sensitization is also dependent on environmental factors with exposure to a particular allergen being one of the most important. When sensitized individuals are exposed to the same allergen, they develop allergy symptoms from target organs; these symptoms can be organ specific in conditions like allergic rhinitis and asthma, or generalized, as in the case of anaphylaxis.
Aeroallergens are the major triggers for respiratory allergy (2), although foods, drugs, and other allergens can also trigger disease. The major mode of entry of aeroallergens in the body is through inhalation, although these allergens can also affect the immune system following skin exposure. The airway epithelium is the first line of defense against inhaled aeroallergens (3). The epithelium acts as a structural barrier to prevent invasion of inhaled particles carrying aeroallergens, but it is also the first innate immune cell type that interacts with antigens and other components of the inhaled particles. Studies on these interactions support the idea that epithelial innate immune functions may be instrumental for the development of an immune response through pro-inflammatory mucosal responses, following interactions with inhaled antigens and chemicals found in pathogens, allergens, and pollutants.
The theme of the review will be the role of epithelium as innate immune cells in allergic airway inflammation. Our discussion will focus primarily on cytokines and chemokines released upon interaction of the airway epithelium with major aeroallergens and how these mediators shape the immune response toward the inhaled allergen. We will also discuss the role of air pollutants in modifying the results of these interactions.
Airway Epithelium: An Immunologically Active Barrier
Airway epithelial cells express tight junction (TJ) proteins such as occludin, claudin, and zonula occludens, which give epithelial cell monolayers their barrier property. Apart from being a barrier, the airway epithelium plays multiple roles, such as maintaining airway surface liquid (ASL) levels, the mucociliary escalator, and also epithelium restitution upon injury (4, 5). To fulfill these roles, the airway epithelium is comprised of a number of specialized cell types that work in harmony to maintain homeostasis in the airways. The characteristics of the various epithelial cell types found in the lungs and their specific roles are summarized in Table 1.

Table 1. Different airway epithelial cell types and their characteristics.
Identification of pattern recognition receptors (PRRs) (6) and proteinase-activated receptors (PARs) (7) highlighted the potential of airway epithelial cells to sense/interact with allergens. Further secretion of immune mediators through activation of these receptors gave new insight to airway epithelial cells being an immunologically active innate immune cell. Similar to the airway epithelium, epithelium in other organs, such as gut (8, 9) and skin (10, 11), play the same multiple roles in tissue homeostasis and in the development of immune responses to foreign antigens.
Allergen–airway epithelium interactions and their effects on epithelial properties as well as on the immune system will be discussed below.
Allergen–Airway Epithelium Interactions
The airway epithelium and allergen–epithelium interactions play a pivotal role in airway immune responses. Allergen–airway interactions start with the recognition of an allergen by receptors present on the airway epithelium. These interactions result in partial loss of epithelial integrity and/or release of inflammatory mediators from epithelial cells. Inflammatory mediators can activate the innate immune system at the same time that the allergen can interact and activate dendritic cells (DCs) that are present below the epithelial monolayer. The default response of immune system to most of these interactions is the development of immune tolerance, which means that subsequent interactions of the organisms with the same allergen will not lead to pathology. In certain individuals, however, the same interactions can lead to the development of allergic sensitization. The exact circumstances and factors responsible for the decision between tolerance and allergic sensitization are not well understood. In mouse experimental models, it has been shown that activation of PRRs (12) or PAR-2 (13) can bias the system toward sensitization. It is also known that inflammatory mediators produced by epithelial cells after interactions with allergens may bias the immune response toward allergic sensitization through their effects on allergen-DC-T cell interactions (3). A subsequent exposure of an individual with an allergic sensitization to the same allergen results in IgE-mediated activation of mast cells (14). However, even in sensitized individuals, inflammatory mediators released from epithelial cells following interactions with allergens play an important role in recruiting inflammatory cells and mediating allergic inflammation. Since the aim of the review is to discuss the role of allergen–epithelium interactions and its immune outcomes, we will focus on the epithelial responses that play role in allergic sensitization, the first part of the process described above, and allergic inflammation, the second part of the process.
The airway epithelium–allergen interactions and the functional consequences of these interactions are affected by the structural and functional state of the epithelium at the time of these interactions. For example, decreased TJ protein expression in atopic asthmatics (15) could compromise the epithelial barrier function (16) allowing allergen invasion. Moreover, there is evidence that mucociliary clearance is not efficient in asthmatic airways (17, 18). It is not clear whether this impairment is the result of environmental factors, including the effects of inhaled allergens, or driven by genetics, but ineffective airway clearance may result in prolonged presence of the allergens in the airways allowing them to have protracted effects. In addition allergen can directly alter epithelial properties, as will be discussed below.
In vitro, ex vivo, and in vivo systems have been used to study the outcomes of direct allergen–epithelium interactions. In vitro approaches have generated interesting results about epithelial responses to allergens, but the biological relevance of these results is questionable as these interactions happen under artificial conditions and in the absence of other cells/factors, such as other immune cells that may modulate the effects. Ex vivo and in vivo approaches on the other hand suffer from the inability to determine that the effects seen are mediated by direct epithelium–allergen interactions and are not due to the indirect effects of other immune cells–allergens interactions on the airway epithelium. However, the two approaches give complementary and very important results and we will review them separately below.
In vitro research to understand allergen–epithelial interactions has been carried out using both alveolar and bronchial airway epithelial cells. A549 is the most utilized cell line to study interactions with alveolar epithelium while various cell lines, such as BEAS-2B, 16HBE14o-, Calu-3, NCI-H292, and primary airway epithelial cultures, from healthy and asthmatic patients, have been used to study interactions with the bronchial epithelium. In addition, both immersed cultures and air–liquid interface (ALI) cultures have been used in studies performed with bronchial epithelial cells. ALI cultures mimic a physiological airway epithelium and facilitate in vitro study of epithelial functions such as barrier property, mucus secretion, and mucociliary escalator, which is not possible with immersed cultures. ALI cultures also provide a unique opportunity to study cellular interactions between epithelial cells and other immune cells by using co-culture systems. However, this variety of experimental settings and cells used has generated often inconsistent and difficult-to-explain results.
In the following section, we will review studies focusing on interactions of airway epithelial cells with some of the most common aeroallergens including house dust mite (HDM), cockroach, fungal, and pollen allergens. A schematic of the mechanism of these interactions and the biological effects of interactions are shown in Figure 1. Allergens include proteins with very different structures and activities and can interact with epithelial cells through a variety of mechanisms. In vitro studies have shown the proteinase activity of allergens can alter epithelial cell morphology and cause epithelial cell detachment (19, 20). Moreover, proteinase activity of allergens has been implicated in allergic sensitization and allergic inflammation in animal models (21). In this review, we will classify the allergen–epithelial interactions into proteinase-dependent and proteinase-independent interactions.
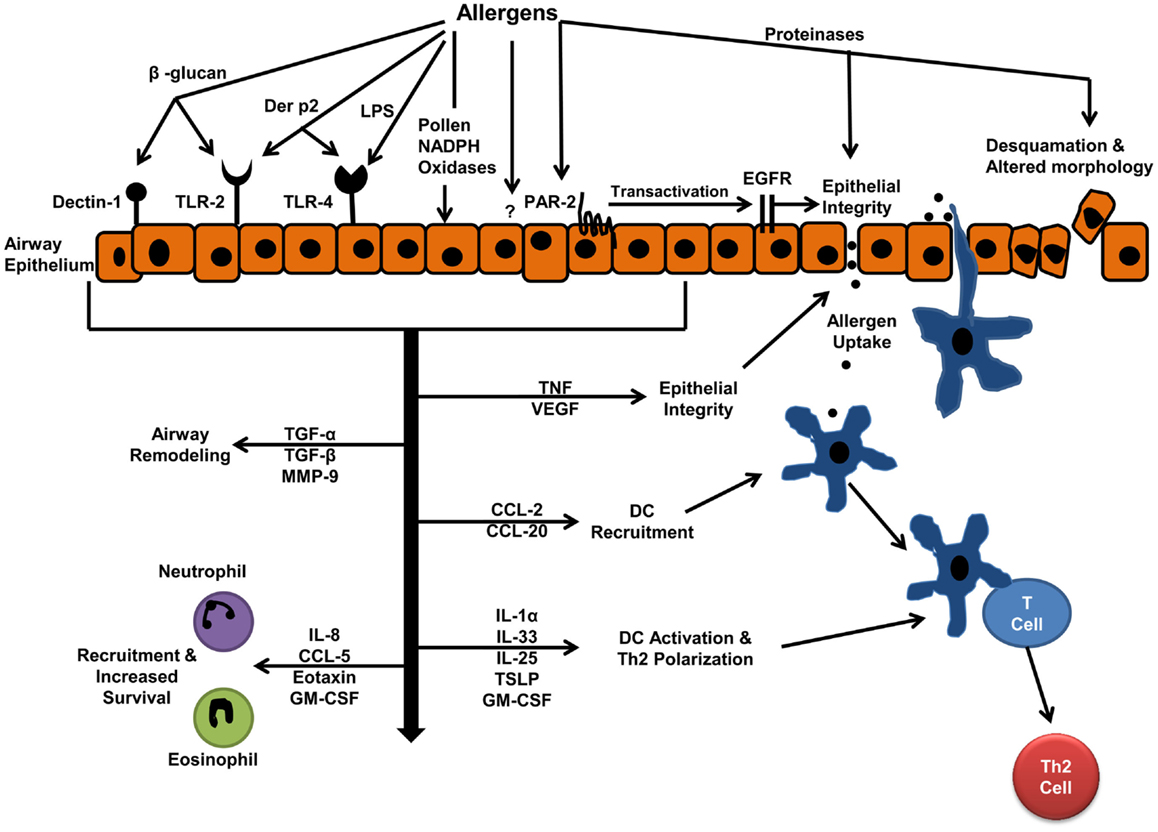
Figure 1. Allergen–epithelial interactions: effects on allergic sensitization, and allergic airway inflammation. Aeroallergen interactions with epithelium result in increased epithelial permeability, which facilitates allergen entry and uptake by dendritic cells (DCs). The mediators released by epithelium, upon allergen interactions, drive DC-T cell interactions toward Th2 immune response. These mediators also develop inflammation by recruiting inflammatory cells to the airways.
Epithelial Tight Junction Disruption
Most of the direct effects of allergens on the immune system require allergens to penetrate through the epithelium into deeper tissues. The main mechanism by which allergens cross the epithelial barrier is through the degradation of epithelial TJs. A HDM allergen with cysteine proteinase activity can degrade the adhesion protein occludin and allow allergens to penetrate through the epithelial monolayer (22). Similarly, cysteine and serine proteinases from pollen grains (23, 24) and fungi (25) can induce TJ degradation.
In addition to direct degradation of TJ, HDM proteinases can also have an indirect effect on TJ integrity. HDM proteinases activate PAR-2 (26), a pro-inflammatory receptor on epithelium, which in turn transactivates epidermal growth factor receptor leading to E-cadherin destabilization and loss of epithelial barrier integrity (27). Thus, although proteinases associated with allergens or allergen particles are prime candidates to mediate TJ degradation, this may not always be the case. Another study comparing various HDM extracts from different manufactures showed that the extract with the lowest proteinase activity caused maximum barrier dysfunction (28). The study also showed that this extract was the most potent inducer of inflammatory mediator release indicating that these mediators could contribute to epithelial barrier dysfunction. The idea was supported by the observation that cockroach allergens affect epithelial permeability indirectly through the release of vascular endothelial growth factor (VEGF) from epithelial cells (29). Chronic exposure to tumor necrosis factor-α (TNF) also causes barrier dysfunction by disrupting TJs (30). The observation that fungal allergens can induce TNF release from airway epithelial cells (31) indicates that this may be another indirect method utilized by allergens to increase airway epithelial permeability.
Finally, TJ degradation by allergens may also be specific to epithelium from asthmatics. For example, a study has shown that epithelium from only asthmatic individuals and not from healthy individuals was sensitive to fungal allergens-mediated increased epithelial permeability (31). However, it is not clear whether asthma-induced epigenetic changes make the airways of asthmatic individuals sensitive to allergens or it is genetic defects in the TJ proteins that make an individual more sensitive to allergen penetration, sensitization, and asthma development.
Epithelial Release of Inflammatory Mediators: Cytokines and Chemokines
Allergens interact directly with airway epithelial cells and induce the release of inflammatory mediators, including cytokines and chemokines. In most cases, however, the exact type of epithelial cell responsible for the release of mediators is not described. Many of the studies discussed below utilize immersed cultures of airway epithelial cells, which are comprised primarily of basal cells (32); basal cells have been shown to express receptors that can be activated by allergens (6, 33). Thus, we speculate that basal cells may be the main cell type releasing cytokines and chemokines in response to aeroallergen stimulations, although other epithelial cell types may also be involved. There is more information on the responses of epithelial cells, from different anatomical locations, to interactions with allergens and these will also be discussed below. We also discuss interactions of allergens with alveolar epithelial cells. Although allergen particles may not be able to reach the alveolar space, individual allergenic proteins may bind to other smaller particles [i.e., diesel exhaust particles (DEPs)] and reach this area.
House dust mite
House dust mite allergens induced the release of CCL20 from human airway epithelial cells, while cockroach allergens do not have the same effect (34); CCL20 release in this study was dependent on the interaction of β-glucan from HDM with dectin-1 on epithelial cells and independent of proteinase activity. β-glucan from HDM also induced CCL20 release from human nasal epithelial cells, but in this case through interactions with TLR-2 (35). Even though HDM-induced CCL20 release from human airway epithelial cells was proteinase-independent, in mouse models of allergic airway inflammation CCL20 release from airway epithelial cells was shown to be proteinase and PAR-2-dependent (36).
Der p2, an HDM allergen without proteinase activity, induced cytokine release from the airway epithelium through TLR-2 (37) and TLR-4 (38) activation. Der p2 is shown to induce reactive oxygen species (ROS) and ROS-mediated nerve growth factor (NGF) release from airway epithelium (39), and NGF was found to be important for the development of asthma features (40). An in vivo study, using a model of tissue specific TLR-4 knockout, showed that HDM-mediated allergic inflammation was dependent on the interaction between HDM and TLR-4 on airway structural cells (41). However, in this model, HDM-induced IL-25 and thymic stromal lymphopoietin (TSLP) were not TLR-4 dependent. IL-25 and TSLP are vital mediators for the development of allergic sensitization and are released primarily by epithelial cells. Thus the presence of these mediators, in the absence of epithelial TLR-4, shows a requirement of other receptors on airway epithelial cells for the development of inflammation.
In line with this argument, HDM has shown proteinase-dependent release of IL-25 from bronchial epithelial cells (42). Moreover, a study also demonstrated that the inflammatory property of HDM was mainly dependent on the proteinase activity of HDM extract (19). Der p3 and Der p9, HDM allergens with serine proteinase activity, induced chemotactic mediators CCL11 and granulocyte macrophage colony-stimulating factor (GM-CSF) release from alveolar cells through PAR-2 (43).
Cockroach allergens
Cockroach proteinases can also activate PAR-2 (44) and PAR-2-mediated release of CXCL8 from human alveolar (45) and bronchial epithelial cells (46) as well as CCL20 and GM-CSF from mouse tracheal epithelial cells (36). Cockroach allergens can also induce the release of IL-33 (47) and IL-25 (42) from human bronchial epithelial cells, but the role of proteinases in this instance is not clear.
These studies have primarily used HDM and cockroach whole body extracts to study their interactions with epithelial cells. However, humans are exposed to HDM and cockroach frass in their daily lives. It is true that majority of the allergens that are present in frass are also found in whole body extract (48, 49), allowing us to consider results from crude extracts. Antigen characteristics and abundance may also vary to some degree between species (50, 51) and the composition of allergen extracts varies from manufacturer to manufacturer (28, 49). These differences make at times the studies difficult to interpret and impossible to compare.
Fungal allergens
Alternaria alternata is the best studied fungal species for interactions with epithelial cells. It has been shown to induce proteinase-dependent IL-6 and CXCL8 release from alveolar (20) and bronchial epithelial cells (52); cytokine release from bronchial epithelial cells was shown to be PAR-2-mediated. Apart from these mediators, A. alternata has also been shown to induce PAR-2-mediated release of GM-CSF (52) and TSLP (53) from bronchial epithelial cells, as well as release of IL-33 (47) and IL-25 (42). Involvement of proteinases in the release of IL-33 and IL-25 is not clear. Aspergillus fumigatus (54) and Cladosporium herbarum (20) also induced proteinase-dependent release of IL-6 and CXCL8 from an alveolar cell line. Purified serine proteinase Pen c13 from Penicillium citrinum showed to induce CXCL8 in alveolar cell line and primary cells through PAR-1 and PAR-2 activation (55). Similarly, another purified serine proteinase Pen ch13 from Penicillium chrysogenum induced CXCL8, prostaglandin E2 (PGE2), and transforming growth factor-β (TGFβ) from A549 and primary human bronchial epithelial cells (25). Fungal extracts, similarly with HDM and cockroach extracts, possess chitin and it has been shown that chitin can induce CCL2 from a mouse airway epithelial cell line (56) but the effect of chitin on human airway epithelial cells have not been studied.
As discussed, allergens from HDM, cockroaches, and fungi are able to activate PAR-2 receptors, which are expressed throughout the airways. In vitro PAR-2 activation has been shown to release mediators from cells of different phenotypes, which are present in the airways, for example, IL-25 (42) and PGE2 from bronchial epithelial cells (7), matrix metallopeptidase 9 (MMP-9) from small airway epithelial cells (57), and CCL2 from alveolar epithelial cells (58).
Pollens
Pollen grains are complex structures containing proteins with and without enzymatic activity. We saw in the previous section that proteinases released from pollen grains can alter epithelial integrity; however, pollen-mediated release of mediators has been found to be proteinase-independent (19). Pollen extracts induced release of IL-6, CXCL8, GM-CSF (19), and TGF-β (59) from airway epithelial cells was independent of enzyme activity or lipopolysaccharide (LPS). Pollens also induced IL-25 from bronchial epithelial cells (42), but the mechanism of release is not known. Finally, pollen grains contain NADPH oxidases, which increase intracellular ROS in epithelial cells (60), and thus increase oxidative stress.
A number of the studies discussed here have been carried out with purified allergens and others with crude extracts that contain a great number of different allergens. An in vitro study compared inflammatory mediators induced by a purified grass pollen allergen Phl p1 (61) and a crude grass pollen extract (GPE) (62) in a human epithelial cell line. Both stimuli induced many common mediators; however, purified Phl p1 induced the release of granulocyte-colony stimulating factor (G-CSF) while GPE induced CCL3, CCL4, and CCL5. In addition to this differential regulation, the most important difference was that the purified Phl p1 upregulated CCL28 and downregulated TSLP, but the opposite regulation was evident for GPE (63). The authors suggested that the whole extract can have other inhibitory or stimulatory components which could be crucial to overcome/maintain the disease phenotype. This observation was supported by an in vivo study, which showed that a purified HDM allergen induced mild late asthmatic responses compared to complete allergen extracts that induced severe responses (64). Thus, it is advisable to use whole allergen extract for studies, which will mimic real life conditions.
Furthermore, the concentrations of allergens used for in vitro and/or in vivo studies may not be close to physiological concentrations encountered by human airway epithelial cells in vivo. Since biological responses are often dependent on the concentration of a protein in addition to the affinity of the ligand for its receptors, results seen in studies may not be similar with what would happen in vivo in humans.
Synergy with other inflammatory mediators
An important question is why aeroallergens do not induce inflammation in non-asthmatic individuals if they can directly activate epithelial cells. One possibility is that the tissue microenvironment where allergen–epithelial cell interactions take place influences the final outcome of these interactions. Asthmatic airways, in contrast to healthy airways, are characterized by the presence of chronic allergic inflammation. The levels of Th2 inflammatory cytokines such as IL-4 (65), IL-13 (66, 67), and fibrogenic mediators, such as TGF-β (68), are increased in asthmatic airways. There is evidence of synergy for HDM allergens with IL-4 and TGF-β to induce release of the Th2 chemoattractant CCL17 from airway epithelial cells (69). HDM also showed synergy with IL-4 and IL-13 for induction of CXCL8 and GM-CSF release from airway epithelial cells (70). Interestingly, IL-4 also enhanced TSLP release in response to A. alternata (53), while the Th1 cytokine interferon gamma (IFNγ) inhibited TSLP induction. TSLP plays an important role in allergic sensitization and thus this observation reflects the importance of the tissue microenvironment in the development of immune response. Cockroach allergens acted synergistically with TNF to induce PAR-2-mediated CXCL8 (71) and MMP-9 (72) release from airway epithelial cells. Finally, PAR-2 activation has also shown synergy with IL-4 for TSLP release (53) and with LPS for CXCL8 release and PAR-2 mRNA expression (73).
In addition, cytokines released by epithelial cells after direct activation by allergens may subsequently activate epithelial cells to release other mediators. For example, direct interaction of epithelial cells with allergens caused TSLP release (53), which could in turn activate epithelial cells to release IL-13 (74). Further, IL-13 acted on airway epithelium to induce periostin (75) and CCL11 (76) release. It was also shown that HDM allergens induced TLR4-mediated IL-1α release from human airway epithelial cells, which acted in an autocrine manner to induce dendritic cell (DC) chemoattractants IL-33 and GM-CSF from airway epithelial cells (77). Inflammation could therefore be perpetuated by this “loop effect” that allergens and cytokines/chemokines exert on the airway epithelium.
Asthmatic vs. non-asthmatic airway epithelial cells
A second important question is whether there is a difference in the allergen-induced response between asthmatic and healthy airway epithelia. Attempts to address this question have used cells isolated from the airways of asthmatic and non-asthmatic individuals; these cells retain their characteristics after culture in vitro (78). According to one study, untreated cells from HDM-allergic patients showed higher basal expression for genes regulating cytokines, chemokines, and growth factors compared to cells from non-allergic subjects (79). This upregulated expression in the absence of stimulus could be because of asthma-induced epigenetic changes in the epithelium, which could result in constant increased amounts of mediator release. Other studies have also shown that HDM allergen-mediated activation of airway epithelial cells from asthmatic patients, and not from healthy individuals, release CCL20 (80) and transforming growth factor-α (TGFα) (70), which can cause DC chemotaxis and cell proliferation, respectively. The latter manuscript has also shown that cells isolated from asthmatic individuals demonstrate a tendency for increased release of CXCL8 and GM-CSF upon HDM stimulation. These differences could be responsible for the altered immune response to allergens seen in the airways of asthma patients.
Effect of epithelial phenotype
We have already discussed the phenotypic and functional heterogeneity of airway epithelial cells. This heterogeneity may explain different epithelial responses to allergens depending on the part of the airways where the interaction takes place. There is substantial experimental evidence showing that cells from different compartments of the airways may respond differently to allergens. The HDM allergens Der p2 (81, 82) and Der f2 (82), which have no proteinase activity, induced IL-6, CXCL8, G-CSF, GM-CSF, CCL2, and CCL20 from bronchial epithelial cells, but not from alveolar epithelial cells, while HDM allergens with proteinase activity induced IL-6, CXCL8, and CCL2 release from alveolar epithelial cells (19) and CCL2 release from bronchial epithelial cells (80), but not from nasal epithelial cells (19). In addition, A. fumigatus (54) as well as timothy grass and birch pollens (19) showed CCL2 release from alveolar epithelial cells, but not from bronchial epithelial cells. This information may indicate that in the case of inhalation of multiple allergens, different allergens act on the upper and lower airway epithelial cells causing release of a variety of inflammatory mediators. Also receptors activated by allergens show heterogeneity in their pro-inflammatory responses. Small airway epithelial cells demonstrated PAR-2-mediated CCL11 release (83), while PAR-2-mediated activation of bronchial epithelial cells did not induce the release of CCL11 (52, 84).
In conclusion, interactions between aeroallergens and airway epithelial cells are complex events that are influenced by a number of factors as discussed above. A plethora of mediators are released from airway epithelium as a result of these interactions. In the subsequent sections, we will discuss the effects of these mediators on different immune cells, and thus on the immune response.
Role of Released Mediators in Shaping the Immune Response
As we mentioned above, allergen–airway epithelial cell interactions participate in both the development of allergic sensitization, and in the development of allergic airway inflammation in sensitized individuals. Here, we will describe the role of various epithelial-derived mediators in these two processes.
Role in Allergic Sensitization
Tight junction disruption induced by allergens, as has been discussed above, may facilitate antigen penetration and uptake by DCs, an important first step toward allergic sensitization. However, there is evidence regarding mechanisms, other than TJ disruption, which aid allergen uptake by DCs (85, 86). Another crucial factor required for the development of allergic sensitization is the presence of a favorable microenvironment for DCs. Allergen–airway epithelium interaction results in release of mediators, which provide a favorable environment that supports DC maturation, activation, and also directs DCs interaction with CD4 T cells toward Th2 phenotype, resulting in allergic sensitization and allergic inflammation (87). Most of these studies were done with HDM and cockroach allergens. However, these mechanisms could also be applied to pollen and fungal allergens as they also activate TLR-2, TLR-4, Dectin-1, and PAR-2 receptors that are important for HDM and cockroach effects. Some of the studies showing this effect will be reviewed here.
Allergen-mediated activation of PAR-2 on airway epithelial cells could induce CCL2 release (58), while dectin-1 (34) and TLR-2 (35) activation resulted in the epithelial release of CCL20; CCL2 (88) and CCL20 (89) are chemoattractants for immature DCs. This will result in the recruitment of the immature DCs to the airways, facilitating the first step toward the allergic sensitization, i.e., allergen–DC interaction and allergen uptake by DCs. CCL2 has been shown to play a role in the generation of Th2 responses in mouse models of allergy (90) and also of airway hyper-reactivity (91), a consequence of allergic sensitization. It is interesting that bronchial lavage from allergic asthmatics showed increased CCL20 (80), which indicates that this mechanism may be also important in vivo in humans.
Mediators released by the epithelium upon allergen interactions, such as TSLP (92), IL-25, and IL-33 (93), act on DCs to induce OX40L expression. This expression of OX40L on DCs is vital for the development of T cells into the Th2 phenotype (94). In mouse models, interaction of OX40L expressing DC with OX40 on T cells resulted in increased expression of IL-4 (95), which induces T cell differentiation to Th2 cells. Recent evidence showed the inability of DCs to induce Th2 responses in the absence of GM-CSF, another epithelial-derived mediator (96), suggesting a role of GM-CSF in the development of a DC phenotype that promotes Th2 response. In addition to its role in the development of antigen-specific Th2 cells, IL-33 may also play a more direct role in the development of humoral immune responses to inhaled aeroallergens, although the exact mechanism is not clear (97). Finally, IL-33 may also have other pro-inflammatory effects in allergic airway inflammation by supporting eosinophil survival (98) and IL-4 and IL-13 release from basophils (99).
In conclusion, proteinase activity of allergens may facilitate entry and uptake of allergens by DCs, while components, with and without proteinase activity, induce a cytokine/chemokine milieu that supports DCs maturation and polarizes their interactions with T cells toward development of Th2 responses.
Role in Allergic Airway Inflammation
Interactions of allergens with the airway epithelium are the first events that take place after allergen inhalation by an allergic individual. The mediators released from these interactions play an important role in the development of airway inflammation by acting as chemotactic or survival factors for inflammatory cells.
As mentioned previously, CCL2 (88, 100) and CCL20 (89) recruit monocytes or immature DCs to the site of inflammation. DCs can also act as inflammatory cells and promote inflammation in sensitized individuals (101). GM-CSF increases eosinophil survival (83), while CCL11 acts as chemoattractant for eosinophils to the airway (102). Airway epithelial cells also release neurotrophins under allergic inflammatory conditions that increase eosinophil survival (103).
Allergens mediate release of TGF-β (59). TGF-β is increased in the asthmatic airways and its release was further increased after allergen challenge (68). TGF-β has been shown to induce more extensive epithelial-mesenchymal transition (EMT) in vitro in airway epithelial cells isolated from asthmatic individuals compared to cells from healthy individuals (104). In addition, TGF-β-treated, but not untreated, airway epithelial cells undergo EMT upon HDM exposure in vitro (105). These observations raise the possibility of differential effect of allergens on airways of asthmatics vs. non-asthmatics but their significance has to be validated in vivo. Moreover, TGF-β has found to be involved in airway remodeling by inducing airway smooth muscle cell proliferation and increased mucus production (106). Other released mediators from the airway epithelium, such as IL-25 (107) and TSLP (108), are also involved in airway remodeling. Finally, a recent human study showed that neutralizing TSLP not only prevents the allergen-induced increase in exhaled nitric oxide and blood and sputum eosinophils, but also decreases their levels in mild allergic asthmatics (109).
Allergen-induced epithelial release of MMP-9 has not been reported; however, we have shown that activation of PAR-2, which is a target of allergen-proteinases, causes MMP-9 release from airway epithelial cells (57). Increased presence of MMP-9 in sputum has been observed in patients with severe asthma (110). The same study showed that after allergen challenge, MMP-9 activity was significantly increased in severe and mild asthmatics. MMP-9 could be responsible for airway remodeling by degrading extra cellular matrix (ECM).
As discussed, the activation of airway epithelial cells by allergens also releases CXCL8 and IL-6 (19, 20, 45, 81). CXCL8 may be a chemoattractant for eosinophils in allergic individuals (111), but it may also contribute to the neutrophilia seen in the airways in cases of acute asthma exacerbations or severe asthma (112). IL-6 has been found to be upregulated in severe asthma (113), but its role in asthma is uncertain as it possesses both pro- and anti-inflammatory properties (114).
The majority of these mediators released by the epithelium are also released by other immune cells in the airways. Thus, it would be an overstatement to conclude that the epithelial cell is the sole cell type responsible for the above mentioned responses. However, when HDM allergen-mediated epithelial NF-κB activation was inhibited in vivo, allergen-mediated inflammatory mediator release, inflammation, and remodeling was significantly reduced (115). This observation depicts that the mediators release by airway epithelium upon allergen-airway epithelium interactions contribute significantly in the development of allergic sensitization and allergic inflammation.
Interactions between Allergens and Pollutants
Pollutants are a major pro-inflammatory component of inhaled air and constitute a major health concern (116, 117). Pollutants are present in indoor and outdoor environments and can be gaseous such as ozone and nitrogen dioxide or particulate matter such as DEPs and cigarette smoke (CS). Direct interactions of these inhaled pollutants with airway epithelial cells have been discussed quite extensively elsewhere (118–121). More interestingly, the simultaneous presence of pollutants and aeroallergens in the air results in complex interactions between the two. Pollutants exert direct effects on aeroallergens but also alter the host responses to inhaled aeroallergens.
Pollutant Effects on Aeroallergens
Indoor and outdoor airborne particles carry aeroallergens. HDM and cockroach allergens are found in general on particles with a median diameter of 10–30 μm (122), while cat and dog allergens are found on particles with 5 μm mass median diameters (123, 124). Suspended particulate matter in homes can carry dog, pollen (125), and cat allergens (126). DEPs, a major outdoor particulate pollutant, have also been shown to bind pollen, dog, cat, and HDM allergens (125, 127). Because of their very fine size, DEPs can facilitate penetration of these allergens into the lungs and therefore increased numbers of pollution particles may increase the amount of allergen interacting with the epithelium.
It has also been shown that DEPs can disrupt pollen particles causing release of allergenic sub-pollen particles. Interestingly, detailed analysis of pollen obtained from areas with pollution showed increased presence of allergenic proteins (128, 129), which resulted in a higher allergenic property of the allergen (128). This may be another mechanism by which pollution increase allergen–epithelial interactions.
Finally, the effect of climate change and pollution on the allergenicity of pollen has been studied. Comparing recent and a decade-old pollen extract, the authors showed that the allergenic potency of the recent pollen extract was higher (130). It was further shown that the recent pollen extract harvested from the urban area had a higher allergenic potency than the recent pollen extract from suburb. Recently, the same authors also demonstrated that the recent pollen extract from an urban area, which has faced the climate change and increased pollution, was more effective at inducing transepithelial permeability and ROS production in the cultured airway epithelial cells (131).
Pollutant Effects on Aeroallergen-Induced Responses
The effects of various pollutants on the development of allergic sensitization and allergic airway inflammation have been an area of intense research in both animal and human systems. The first evidence 30 years ago showed that inhalation of ozone (132, 133) and DEPs (134) increase sensitization to inhaled allergens as measured by the presence of antigen-specific IgE (132) and subsequently resulted in increased anaphylactic sensitivity upon the allergen challenge. Inhalation of suspended particulate matter along with an allergen has shown to act as an adjuvant and increase IgE production (135). Similarly, in a human study, DEPs, when inhaled with allergen, promoted Th2 inflammation and allergen specific IgE (136).
The mechanisms of this priming/sensitization effect of pollutants have been studied in detail. Interestingly, mice exposed to the pollutant nitrogen dioxide prior to allergen exposure developed TLR2, MyD88, and NF-kB dependent sensitization to the allergen, resulting in Th2 inflammation and airway hyper-responsiveness (137). Pollutants such as ozone (138) and nitrogen dioxide (139) can induce maturation of CD11c+ myeloid DC and increase antigen uptake and antigen-presenting activity of DCs. DEPs and ambient particulate matter upregulated TSLP in bronchial epithelial cells (140) and DEP-induced TSLP can promote myeloid DCs maturation (141) and increase OX40L expression (142), favoring Th2 inflammation. These, and possibly others, are some of the mechanisms of increased allergic sensitization in the presence of pollutants.
The effects of pollutants on allergen-induced responses in sensitized individuals have also been studied in vivo. Motorcycle exhaust particles (143) and DEPs (144, 145) can increase airway hyper-responsiveness to allergen and allergen-mediated early and late inflammatory responses in different animal models of allergy. Similarly human studies have shown that prior exposure to pollutants such as ozone, nitric dioxide alone or in combination with sulfur dioxide increased bronchial responsiveness to pollen (146) and HDM allergens (147–149) and also amplified airway inflammation (150, 151).
The in vivo studies discussed above, however, do not allow us to identify the cells involved in pollutant–allergen interactions. The airway epithelium would be expected to play a significant role in these effects, especially since pollutants can directly activate airway epithelial cells. Nitrogen dioxide increased epithelial permeability and induced leukotriene C4 synthesis (152). Moreover, nitrogen dioxide (153), DEPs (154), and ozone (155) induced CXCL8, GM-CSF, and TNF from cultured airway epithelial cells. As was shown for allergen–epithelial cell interactions, ozone and nitrogen oxide released more inflammatory mediators from epithelial cells from asthmatics than those from healthy donors (156). This observation indicates that pollutants induce a different pro-inflammatory environment in the airways of asthmatics compared to non-asthmatics.
Limited data exist regarding the effect of pollutants on allergen–airway epithelium interactions. These studies have been performed primarily with alveolar cell line A549; because of the fine size of allergen-carrying pollutant particles, these particles can reach to alveolar space and therefore may affect alveolar epithelial cells. Studies have shown that exposure to submicron particles and allergen individually or in combination induced alterations in cellular morphology (increased microvilli), functions (increased lysozyme and surfactant-producing multilamellar bodies) (157), and metabolic activity (damage to mitochondria, tonofilaments, and rough endoplasmic reticulum) in lung epithelial cells (158). Sodium sulfite and HDM acted synergistically for detachment of the cells (159), which could cause inflammation and decrease epithelial barrier integrity. Further, exposure of these cells to the combination of pollen grain-Pb (160) and pollen allergens-DEPs (161) caused significant increase in IL-5 mRNA and Th2 cytokines release, respectively, from airway epithelial cells.
In summary, apart from having inflammatory effects on their own, pollutants can increase the concentration, exposure, and allergenic property of aeroallergens. Thus, pollutants exert a priming effect on immune system for allergens and also increase inflammatory responses to allergens.
Apart from these pollutants discussed so far, CS is a major factor influencing allergic sensitization and asthma. Smoking is a major source of indoor particles (162). Maternal smoking has been shown to increase the risk of asthma development in children (163). Using animal models, it has been shown that passive smoking acts as an adjuvant to increase allergen-mediated allergic immune responses (164) and airway remodeling (165). Effects of CS on airway epithelial cells are similar to other pollutants, i.e., increased allergen-mediated epithelial permeability and inflammatory properties, which have been reviewed in detail (166, 167).
Conclusion
Current literature establishes the airway epithelium as an innate immunity organ that senses inhaled allergens through an armory of receptors, and initiates innate and adaptive immunity. This potential has been established clearly through in vitro studies, although more detailed in vivo studies are still needed to validate these results.
Two approaches could improve our understanding regarding the role of airway epithelium in allergic inflammation. In vitro co-culture of epithelial cells grown in ALI with one or more of the other immune cells that may play a role in allergic inflammation, such as DCs and T cells, in the presence of particular allergens may clarify the sequence of events leading to the development of allergic airway inflammation. These studies should be coupled with in vivo models utilizing airway epithelium-specific strains of knockout mice. These studies should start with tissue-specific knockouts of epithelial receptors interacting with allergens and continue with similar knockout strains of signaling molecules. These studies may reveal common links between major allergens for their interactions with the airway epithelium and improve our understanding of the basic mechanisms leading to allergen-specific sensitization and inflammation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. von Mutius E. Gene-environment interactions in asthma. J Allergy Clin Immunol (2009) 123(1):3–11. doi:10.1016/j.jaci.2008.10.046
2. Craig TJ. Aeroallergen sensitization in asthma: prevalence and correlation with severity. Allergy Asthma Proc (2010) 31(2):96–102. doi:10.2500/aap.2010.31.3310
3. Lambrecht BN, Hammad H. Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol (2014) 134(3):499–507. doi:10.1016/j.jaci.2014.06.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology (2003) 8(4):432–46. doi:10.1046/j.1440-1843.2003.00493.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Tam A, Wadsworth S, Dorscheid D, Man SF, Sin DD. The airway epithelium: more than just a structural barrier. Therap Adv Respir Dis (2011) 5(4):255–73. doi:10.1177/1753465810396539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol (2004) 31(3):358–64. doi:10.1165/rcmb.2003-0388OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Asokananthan N, Graham PT, Fink J, Knight DA, Bakker AJ, McWilliam AS, et al. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J Immunol (2002) 168(7):3577–85. doi:10.4049/jimmunol.168.7.3577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14(3):141–53. doi:10.1038/nri3608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Stadnyk AW. Intestinal epithelial cells as a source of inflammatory cytokines and chemokines. Can J Gastroenterol (2002) 16(4):241–6.
10. Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol (2008) 17(12):1063–72. doi:10.1111/j.1600-0625.2008.00786.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol (2014) 14(5):289–301. doi:10.1038/nri3646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med (2002) 196(12):1645–51. doi:10.1084/jem.20021340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Davidson CE, Asaduzzaman M, Arizmendi NG, Polley D, Wu Y, Gordon JR, et al. Proteinase-activated receptor-2 activation participates in allergic sensitization to house dust mite allergens in a murine model. Clin Exp Allergy (2013) 43(11):1274–85. doi:10.1111/cea.12185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med (2012) 18(5):693–704. doi:10.1038/nm.2755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ. Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol (2008) 86(3):105–12. doi:10.1139/y08-004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol (2011) 128(3):.e1–12. doi:10.1016/j.jaci.2011.05.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Mezey RJ, Cohn MA, Fernandez RJ, Januszkiewicz AJ, Wanner A. Mucociliary transport in allergic patients with antigen-induced bronchospasm. Am Rev Respir Dis (1978) 118(4):677–84.
18. Bateman JR, Pavia D, Sheahan NF, Agnew JE, Clarke SW. Impaired tracheobronchial clearance in patients with mild stable asthma. Thorax (1983) 38(6):463–7. doi:10.1136/thx.38.6.463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Tomee JF, van Weissenbruch R, de Monchy JG, Kauffman HF. Interactions between inhalant allergen extracts and airway epithelial cells: effect on cytokine production and cell detachment. J Allergy Clin Immunol (1998) 102(1):75–85. doi:10.1016/S0091-6749(98)70057-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Kauffman HF, Tomee JF, van de Riet MA, Timmerman AJ, Borger P. Protease-dependent activation of epithelial cells by fungal allergens leads to morphologic changes and cytokine production. J Allergy Clin Immunol (2000) 105(6 Pt 1):1185–93. doi:10.1067/mai.2000.106210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Arizmendi NG, Abel M, Mihara K, Davidson C, Polley D, Nadeem A, et al. Mucosal allergic sensitization to cockroach allergens is dependent on proteinase activity and proteinase-activated receptor-2 activation. J Immunol (2011) 186(5):3164–72. doi:10.4049/jimmunol.0903812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Wan H, Winton HL, Soeller C, Tovey ER, Gruenert DC, Thompson PJ, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest (1999) 104(1):123–33. doi:10.1172/JCI5844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Vinhas R, Cortes L, Cardoso I, Mendes VM, Manadas B, Todo-Bom A, et al. Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy (2011) 66(8):1088–98. doi:10.1111/j.1398-9995.2011.02598.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Runswick S, Mitchell T, Davies P, Robinson C, Garrod DR. Pollen proteolytic enzymes degrade tight junctions. Respirology (2007) 12(6):834–42. doi:10.1111/j.1440-1843.2007.01175.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Tai HY, Tam MF, Chou H, Peng HJ, Su SN, Perng DW, et al. Pen ch 13 allergen induces secretion of mediators and degradation of occludin protein of human lung epithelial cells. Allergy (2006) 61(3):382–8. doi:10.1111/j.1398-9995.2005.00958.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Cho HJ, Choi JY, Yang YM, Hong JH, Kim CH, Gee HY, et al. House dust mite extract activates apical Cl(-) channels through protease-activated receptor 2 in human airway epithelia. J Cell Biochem (2010) 109(6):1254–63. doi:10.1002/jcb.22511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Heijink IH, van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J (2010) 36(5):1016–26. doi:10.1183/09031936.00125809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Post S, Nawijn MC, Hackett TL, Baranowska M, Gras R, van Oosterhout AJ, et al. The composition of house dust mite is critical for mucosal barrier dysfunction and allergic sensitisation. Thorax (2012) 67(6):488–95. doi:10.1136/thoraxjnl-2011-200606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Antony AB, Tepper RS, Mohammed KA. Cockroach extract antigen increases bronchial airway epithelial permeability. J Allergy Clin Immunol (2002) 110(4):589–95. doi:10.1067/mai.2002.127798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Hardyman MA, Wilkinson E, Martin E, Jayasekera NP, Blume C, Swindle EJ, et al. TNF-alpha-mediated bronchial barrier disruption and regulation by src-family kinase activation. J Allergy Clin Immunol (2013) 132(3):665.e–75.e. doi:10.1016/j.jaci.2013.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Leino MS, Loxham M, Blume C, Swindle EJ, Jayasekera NP, Dennison PW, et al. Barrier disrupting effects of alternaria alternata extract on bronchial epithelium from asthmatic donors. PLoS One (2013) 8(8):e71278. doi:10.1371/journal.pone.0071278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. de Jong PM, van Sterkenburg MA, Kempenaar JA, Dijkman JH, Ponec M. Serial culturing of human bronchial epithelial cells derived from biopsies. In vitro Cell Dev Biol Anim (1993) 29A(5):379–87. doi:10.1007/BF02633985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Ahmad S, Ahmad A, Rancourt RC, Neeves KB, Loader JE, Hendry-Hofer T, et al. Tissue factor signals airway epithelial basal cell survival via coagulation and protease-activated receptor isoforms 1 and 2. Am J Respir Cell Mol Biol (2013) 48(1):94–104. doi:10.1165/rcmb.2012-0189OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Nathan AT, Peterson EA, Chakir J, Wills-Karp M. Innate immune responses of airway epithelium to house dust mite are mediated through beta-glucan-dependent pathways. J Allergy Clin Immunol (2009) 123(3):612–8. doi:10.1016/j.jaci.2008.12.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ryu JH, Yoo JY, Kim MJ, Hwang SG, Ahn KC, Ryu JC, et al. Distinct TLR-mediated pathways regulate house dust mite-induced allergic disease in the upper and lower airways. J Allergy Clin Immunol (2013) 131(2):549–61. doi:10.1016/j.jaci.2012.07.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Day SB, Ledford JR, Zhou P, Lewkowich IP, Page K. German cockroach proteases and protease-activated receptor-2 regulate chemokine production and dendritic cell recruitment. J Innate Immun (2012) 4(1):100–10. doi:10.1159/000329132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Chiou YL, Lin CY. Der p2 activates airway smooth muscle cells in a TLR2/MyD88-dependent manner to induce an inflammatory response. J Cell Physiol (2009) 220(2):311–8. doi:10.1002/jcp.21764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, et al. Allergenicity resulting from functional mimicry of a toll-like receptor complex protein. Nature (2009) 457(7229):585–8. doi:10.1038/nature07548
39. Ye YL, Wu HT, Lin CF, Hsieh CY, Wang JY, Liu FH, et al. Dermatophagoides pteronyssinus 2 regulates nerve growth factor release to induce airway inflammation via a reactive oxygen species-dependent pathway. Am J Physiol Lung Cell Mol Physiol (2011) 300(2):216–24. doi:10.1152/ajplung.00165.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Ogawa H, Azuma M, Uehara H, Takahashi T, Nishioka Y, Sone S, et al. Nerve growth factor derived from bronchial epithelium after chronic mite antigen exposure contributes to airway hyperresponsiveness by inducing hyperinnervation, and is inhibited by in vivo siRNA. Clin Exp Allergy (2012) 42(3):460–70. doi:10.1111/j.1365-2222.2011.03918.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via toll-like receptor 4 triggering of airway structural cells. Nat Med (2009) 15(4):410–6. doi:10.1038/nm.1946
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kouzaki H, Tojima I, Kita H, Shimizu T. Transcription of interleukin-25 and extracellular release of the protein is regulated by allergen proteases in airway epithelial cells. Am J Respir Cell Mol Biol (2013) 49(5):741–50. doi:10.1165/rcmb.2012-0304OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Sun G, Stacey MA, Schmidt M, Mori L, Mattoli S. Interaction of mite allergens Der p3 and Der p9 with protease-activated receptor-2 expressed by lung epithelial cells. J Immunol (2001) 167(2):1014–21. doi:10.4049/jimmunol.167.2.1014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Hong JH, Lee SI, Kim KE, Yong TS, Seo JT, Sohn MH, et al. German cockroach extract activates protease-activated receptor 2 in human airway epithelial cells. J Allergy Clin Immunol (2004) 113(2):315–9. doi:10.1016/j.jaci.2003.11.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Lee MF, Wang NM, Liu SW, Lin SJ, Chen YH. Induction of interleukin 8 by American cockroach allergens from human airway epithelial cells via extracellular signal regulatory kinase and jun N-terminal kinase but not p38 mitogen-activated protein kinase. Ann Allergy Asthma Immunol (2010) 105(3):234–40. doi:10.1016/j.anai.2010.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Lee KE, Kim JW, Jeong KY, Kim KE, Yong TS, Sohn MH. Regulation of German cockroach extract-induced IL-8 expression in human airway epithelial cells. Clin Exp Allergy (2007) 37(9):1364–73. doi:10.1111/j.1365-2222.2007.02797.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol (2011) 186(7):4375–87. doi:10.4049/jimmunol.1003020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Arlian LG, Bernstein IL, Geis DP, Vyszenski-Moher DL, Gallagher JS, Martin B. Investigations of culture medium-free house dust mites. III. Antigens and allergens of body and fecal extract of Dermatophagoides farinae. J Allergy Clin Immunol (1987) 79(3):457–66. doi:10.1016/0091-6749(87)90364-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Patterson ML, Slater JE. Characterization and comparison of commercially available German and American cockroach allergen extracts. Clin Exp Allergy (2002) 32(5):721–7. doi:10.1046/j.1365-2222.2002.01397.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Helm RM, Squillace DL, Jones RT, Brenner RJ. Shared allergenic activity in Asian (Blattella asahinai), German (Blattella germanica), American (Periplaneta americana), and Oriental (Blatta orientalis) cockroach species. Int Arch Allergy Appl Immunol (1990) 92(2):154–61. doi:10.1159/000235207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Arlian LG, Bernstein IL, Vyszenski-Moher DL, Gallagher JS. Investigations of culture medium-free house dust mites. IV. Cross antigenicity and allergenicity between the house dust mites, Dermatophagoides farinae and D. pteronyssinus. J Allergy Clin Immunol (1987) 79(3):467–76. doi:10.1016/0091-6749(87)90364-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Matsuwaki Y, Wada K, White T, Moriyama H, Kita H. Alternaria fungus induces the production of GM-CSF, interleukin-6 and interleukin-8 and calcium signaling in human airway epithelium through protease-activated receptor 2. Int Arch Allergy Immunol (2012) 158(Suppl 1):19–29. doi:10.1159/000337756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Kouzaki H, O’Grady SM, Lawrence CB, Kita H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J Immunol (2009) 183(2):1427–34. doi:10.4049/jimmunol.0900904
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Tomee JF, Wierenga AT, Hiemstra PS, Kauffman HK. Proteases from Aspergillus fumigatus induce release of proinflammatory cytokines and cell detachment in airway epithelial cell lines. J Infect Dis (1997) 176(1):300–3. doi:10.1086/517272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Chiu LL, Perng DW, Yu CH, Su SN, Chow LP. Mold allergen, pen C 13, induces IL-8 expression in human airway epithelial cells by activating protease-activated receptor 1 and 2. J Immunol (2007) 178(8):5237–44. doi:10.4049/jimmunol.178.8.5237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Roy RM, Wuthrich M, Klein BS. Chitin elicits CCL2 from airway epithelial cells and induces CCR2-dependent innate allergic inflammation in the lung. J Immunol (2012) 189(5):2545–52. doi:10.4049/jimmunol.1200689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Vliagoftis H, Schwingshackl A, Milne CD, Duszyk M, Hollenberg MD, Wallace JL, et al. Proteinase-activated receptor-2-mediated matrix metalloproteinase-9 release from airway epithelial cells. J Allergy Clin Immunol (2000) 106(3):537–45. doi:10.1067/mai.2000.109058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Wang H, Yi T, Zheng Y, He S. Induction of monocyte chemoattractant protein-1 release from A549 cells by agonists of protease-activated receptor-1 and -2. Eur J Cell Biol (2007) 86(4):233–42. doi:10.1016/j.ejcb.2006.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Roschmann K, Farhat K, Konig P, Suck R, Ulmer AJ, Petersen A. Timothy grass pollen major allergen Phl p 1 activates respiratory epithelial cells by a non-protease mechanism. Clin Exp Allergy (2009) 39(9):1358–69. doi:10.1111/j.1365-2222.2009.03291.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Boldogh I, Bacsi A, Choudhury BK, Dharajiya N, Alam R, Hazra TK, et al. ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J Clin Invest (2005) 115(8):2169–79. doi:10.1172/JCI24422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Roschmann KI, van Kuijen AM, Luiten S, Jonker MJ, Breit TM, Fokkens WJ, et al. Purified timothy grass pollen major allergen Phl p 1 may contribute to the modulation of allergic responses through a pleiotropic induction of cytokines and chemokines from airway epithelial cells. Clin Exp Immunol (2012) 167(3):413–21. doi:10.1111/j.1365-2249.2011.04522.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Roschmann KI, Luiten S, Jonker MJ, Breit TM, Fokkens WJ, Petersen A, et al. Timothy grass pollen extract-induced gene expression and signalling pathways in airway epithelial cells. Clin Exp Allergy (2011) 41(6):830–41. doi:10.1111/j.1365-2222.2011.03713.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Roschmann KI, van Kuijen AM, Luiten S, Jonker MJ, Breit TM, Fokkens WJ, et al. Comparison of timothy grass pollen extract- and single major allergen-induced gene expression and mediator release in airway epithelial cells: a meta-analysis. Clin Exp Allergy (2012) 42(10):1479–90. doi:10.1111/j.1365-2222.2012.04033.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Van Der Veen MJ, Jansen HM, Aalberse RC, van der Zee JS. Der p 1 and Der p 2 induce less severe late asthmatic responses than native Dermatophagoides pteronyssinus extract after a similar early asthmatic response. Clin Exp Allergy (2001) 31(5):705–14. doi:10.1046/j.1365-2222.2001.01120.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Walker C, Bauer W, Braun RK, Menz G, Braun P, Schwarz F, et al. Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am J Respir Crit Care Med (1994) 150(4):1038–48. doi:10.1164/ajrccm.150.4.7921434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Berry MA, Parker D, Neale N, Woodman L, Morgan A, Monk P, et al. Sputum and bronchial submucosal IL-13 expression in asthma and eosinophilic bronchitis. J Allergy Clin Immunol (2004) 114(5):1106–9. doi:10.1016/j.jaci.2004.08.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Saha SK, Berry MA, Parker D, Siddiqui S, Morgan A, May R, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol (2008) 121(3):685–91. doi:10.1016/j.jaci.2008.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, Holgate ST, et al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med (1997) 156(2 Pt 1):642–7. doi:10.1164/ajrccm.156.2.9605065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Heijink IH, Marcel Kies P, van Oosterhout AJ, Postma DS, Kauffman HF, Vellenga E. Der p, IL-4, and TGF-beta cooperatively induce EGFR-dependent TARC expression in airway epithelium. Am J Respir Cell Mol Biol (2007) 36(3):351–9. doi:10.1165/rcmb.2006-0160OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Lordan JL, Bucchieri F, Richter A, Konstantinidis A, Holloway JW, Thornber M, et al. Cooperative effects of Th2 cytokines and allergen on normal and asthmatic bronchial epithelial cells. J Immunol (2002) 169(1):407–14. doi:10.4049/jimmunol.169.1.407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Page K, Hughes VS, Odoms KK, Dunsmore KE, Hershenson MB. German cockroach proteases regulate interleukin-8 expression via nuclear factor for interleukin-6 in human bronchial epithelial cells. Am J Respir Cell Mol Biol (2005) 32(3):225–31. doi:10.1165/rcmb.2004-0231OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Page K, Hughes VS, Bennett GW, Wong HR. German cockroach proteases regulate matrix metalloproteinase-9 in human bronchial epithelial cells. Allergy (2006) 61(8):988–95. doi:10.1111/j.1398-9995.2006.01103.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Ostrowska E, Sokolova E, Reiser G. PAR-2 activation and LPS synergistically enhance inflammatory signaling in airway epithelial cells by raising PAR expression level and interleukin-8 release. Am J Physiol Lung Cell Mol Physiol (2007) 293(5):1208–18. doi:10.1152/ajplung.00137.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Semlali A, Jacques E, Koussih L, Gounni AS, Chakir J. Thymic stromal lymphopoietin-induced human asthmatic airway epithelial cell proliferation through an IL-13-dependent pathway. J Allergy Clin Immunol (2010) 125(4):844–50. doi:10.1016/j.jaci.2010.01.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Yuyama N, Davies DE, Akaiwa M, Matsui K, Hamasaki Y, Suminami Y, et al. Analysis of novel disease-related genes in bronchial asthma. Cytokine (2002) 19(6):287–96. doi:10.1006/cyto.2002.1972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Matsukura S, Stellato C, Georas SN, Casolaro V, Plitt JR, Miura K, et al. Interleukin-13 upregulates eotaxin expression in airway epithelial cells by a STAT6-dependent mechanism. Am J Respir Cell Mol Biol (2001) 24(6):755–61. doi:10.1165/ajrcmb.24.6.4351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med (2012) 209(8):1505–17. doi:10.1084/jem.20112691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Gras D, Bourdin A, Vachier I, de Senneville L, Bonnans C, Chanez P. An ex vivo model of severe asthma using reconstituted human bronchial epithelium. J Allergy Clin Immunol (2012) 129(5):1259.e–66.e. doi:10.1016/j.jaci.2012.01.073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Vroling AB, Jonker MJ, Luiten S, Breit TM, Fokkens WJ, van Drunen CM. Primary nasal epithelium exposed to house dust mite extract shows activated expression in allergic individuals. Am J Respir Cell Mol Biol (2008) 38(3):293–9. doi:10.1165/rcmb.2007-0278OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Pichavant M, Charbonnier AS, Taront S, Brichet A, Wallaert B, Pestel J, et al. Asthmatic bronchial epithelium activated by the proteolytic allergen Der p 1 increases selective dendritic cell recruitment. J Allergy Clin Immunol (2005) 115(4):771–8. doi:10.1016/j.jaci.2004.11.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Osterlund C, Gronlund H, Polovic N, Sundstrom S, Gafvelin G, Bucht A. The non-proteolytic house dust mite allergen Der p 2 induce NF-kappaB and MAPK dependent activation of bronchial epithelial cells. Clin Exp Allergy (2009) 39(8):1199–208. doi:10.1111/j.1365-2222.2009.03284.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Osterlund C, Gronlund H, Gafvelin G, Bucht A. Non-proteolytic aeroallergens from mites, cat and dog exert adjuvant-like activation of bronchial epithelial cells. Int Arch Allergy Immunol (2011) 155(2):111–8. doi:10.1159/000318743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Vliagoftis H, Befus AD, Hollenberg MD, Moqbel R. Airway epithelial cells release eosinophil survival-promoting factors (GM-CSF) after stimulation of proteinase-activated receptor 2. J Allergy Clin Immunol (2001) 107(4):679–85. doi:10.1067/mai.2001.114245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Rudack C, Steinhoff M, Mooren F, Buddenkotte J, Becker K, von Eiff C, et al. PAR-2 activation regulates IL-8 and GRO-alpha synthesis by NF-kappaB, but not RANTES, IL-6, eotaxin or TARC expression in nasal epithelium. Clin Exp Allergy (2007) 37(7):1009–22. doi:10.1111/j.1365-2222.2007.02686.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Yang PC, Berin MC, Perdue MH. Enhanced antigen transport across rat tracheal epithelium induced by sensitization and mast cell activation. J Immunol (1999) 163(5):2769–76.
86. Takano K, Kojima T, Go M, Murata M, Ichimiya S, Himi T, et al. HLA-DR- and CD11c-positive dendritic cells penetrate beyond well-developed epithelial tight junctions in human nasal mucosa of allergic rhinitis. J Histochem Cytochem (2005) 53(5):611–9. doi:10.1369/jhc.4A6539.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol (1999) 17:255–81. doi:10.1146/annurev.immunol.17.1.255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Xu LL, Warren MK, Rose WL, Gong W, Wang JM. Human recombinant monocyte chemotactic protein and other C-C chemokines bind and induce directional migration of dendritic cells in vitro. J Leukoc Biol (1996) 60(3):365–71.
89. Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med (1998) 188(2):373–86. doi:10.1084/jem.188.2.373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature (2000) 404(6776):407–11. doi:10.1038/35006097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Campbell EM, Charo IF, Kunkel SL, Strieter RM, Boring L, Gosling J, et al. Monocyte chemoattractant protein-1 mediates cockroach allergen-induced bronchial hyperreactivity in normal but not CCR2-/- mice: the role of mast cells. J Immunol (1999) 163(4):2160–7.
92. Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med (2005) 202(9):1213–23. doi:10.1084/jem.20051135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Chu DK, Llop-Guevara A, Walker TD, Flader K, Goncharova S, Boudreau JE, et al. IL-33, but not thymic stromal lymphopoietin or IL-25, is central to mite and peanut allergic sensitization. J Allergy Clin Immunol (2013) 131(1):.e1–8. doi:10.1016/j.jaci.2012.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Hoshino A, Tanaka Y, Akiba H, Asakura Y, Mita Y, Sakurai T, et al. Critical role for OX40 ligand in the development of pathogenic Th2 cells in a murine model of asthma. Eur J Immunol (2003) 33(4):861–9. doi:10.1002/eji.200323455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Flynn S, Toellner KM, Raykundalia C, Goodall M, Lane P. CD4 T cell cytokine differentiation: the B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, Blr-1. J Exp Med (1998) 188(2):297–304. doi:10.1084/jem.188.2.297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Zhou Q, Ho AW, Schlitzer A, Tang Y, Wong KH, Wong FH, et al. GM-CSF-licensed CD11b+ lung dendritic cells orchestrate Th2 immunity to Blomia tropicalis. J Immunol (2014) 193(2):496–509. doi:10.4049/jimmunol.1303138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Canbaz D, Utsch L, Logiantara A, van Ree R, van Rijt LS. IL-33 promotes the induction immunoglobulin production after inhalation of house dust mite extract in mice. Allergy (2015). doi:10.1111/all.12594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol (2008) 121(6):1484–90. doi:10.1016/j.jaci.2008.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood (2009) 113(7):1526–34. doi:10.1182/blood-2008-05-157818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med (1989) 169(4):1485–90. doi:10.1084/jem.169.4.1485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med (2005) 201(6):981–91. doi:10.1084/jem.20042311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med (1996) 2(4):449–56. doi:10.1038/nm0496-449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Hahn C, Islamian AP, Renz H, Nockher WA. Airway epithelial cells produce neurotrophins and promote the survival of eosinophils during allergic airway inflammation. J Allergy Clin Immunol (2006) 117(4):787–94. doi:10.1016/j.jaci.2005.12.1339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med (2009) 180(2):122–33. doi:10.1164/rccm.200811-1730OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol (2010) 42(1):69–79. doi:10.1165/rcmb.2008-0449OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol (2005) 174(9):5774–80. doi:10.4049/jimmunol.174.9.5774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, et al. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax (2013) 68(1):82–90. doi:10.1136/thoraxjnl-2012-202003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Chen ZG, Zhang TT, Li HT, Chen FH, Zou XL, Ji JZ, et al. Neutralization of TSLP inhibits airway remodeling in a murine model of allergic asthma induced by chronic exposure to house dust mite. PLoS One (2013) 8(1):e51268. doi:10.1371/journal.pone.0051268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Gauvreau GM, O’Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med (2014) 370(22):2102–10. doi:10.1056/NEJMoa1402895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Mattos W, Lim S, Russell R, Jatakanon A, Chung KF, Barnes PJ. Matrix metalloproteinase-9 expression in asthma: effect of asthma severity, allergen challenge, and inhaled corticosteroids. Chest (2002) 122(5):1543–52. doi:10.1378/chest.122.5.1543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Sehmi R, Cromwell O, Wardlaw AJ, Moqbel R, Kay AB. Interleukin-8 is a chemo-attractant for eosinophils purified from subjects with a blood eosinophilia but not from normal healthy subjects. Clin Exp Allergy (1993) 23(12):1027–36. doi:10.1111/j.1365-2222.1993.tb00295.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Ordonez CL, Shaughnessy TE, Matthay MA, Fahy JV. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: clinical and biologic significance. Am J Respir Crit Care Med (2000) 161(4 Pt 1):1185–90. doi:10.1164/ajrccm.161.4.9812061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Morjaria JB, Babu KS, Vijayanand P, Chauhan AJ, Davies DE, Holgate ST. Sputum IL-6 concentrations in severe asthma and its relationship with FEV1. Thorax (2011) 66(6):537. doi:10.1136/thx.2010.136523
114. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta (2011) 1813(5):878–88. doi:10.1016/j.bbamcr.2011.01.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Tully JE, Hoffman SM, Lahue KG, Nolin JD, Anathy V, Lundblad LK, et al. Epithelial NF-B orchestrates house dust mite-induced airway inflammation, hyperresponsiveness, and fibrotic remodeling. J Immunol (2013) 191(12):5811–21. doi:10.4049/jimmunol.1301329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Anderson JO, Thundiyil JG, Stolbach A. Clearing the air: a review of the effects of particulate matter air pollution on human health. J Med Toxicol (2012) 8(2):166–75. doi:10.1007/s13181-011-0203-1
118. Devalia JL, Bayram H, Rusznak C, Calderon M, Sapsford RJ, Abdelaziz MA, et al. Mechanisms of pollution-induced airway disease: in vitro studies in the upper and lower airways. Allergy (1997) 52(38 Suppl):45–51.
119. Baeza-Squiban A, Bonvallot V, Boland S, Marano F. Airborne particles evoke an inflammatory response in human airway epithelium. Activation of transcription factors. Cell Biol Toxicol (1999) 15(6):375–80. doi:10.1023/A:1007653900063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Persinger RL, Poynter ME, Ckless K, Janssen-Heininger YM. Molecular mechanisms of nitrogen dioxide induced epithelial injury in the lung. Mol Cell Biochem (2002) 234-235(1–2):71–80.
121. Becker S, Mundandhara S, Devlin RB, Madden M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol Appl Pharmacol (2005) 207(2 Suppl):269–75.
122. Platts-Mills TA, Heymann PW, Longbottom JL, Wilkins SR. Airborne allergens associated with asthma: particle sizes carrying dust mite and rat allergens measured with a cascade impactor. J Allergy Clin Immunol (1986) 77(6):850–7. doi:10.1016/0091-6749(86)90383-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Wood RA, Laheri AN, Eggleston PA. The aerodynamic characteristics of cat allergen. Clin Exp Allergy (1993) 23(9):733–9. doi:10.1111/j.1365-2222.1993.tb00360.x
124. Custovic A, Green R, Fletcher A, Smith A, Pickering CA, Chapman MD, et al. Aerodynamic properties of the major dog allergen Can f 1: distribution in homes, concentration, and particle size of allergen in the air. Am J Respir Crit Care Med (1997) 155(1):94–8. doi:10.1164/ajrccm.155.1.9001295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Ormstad H, Johansen BV, Gaarder PI. Airborne house dust particles and diesel exhaust particles as allergen carriers. Clin Exp Allergy (1998) 28(6):702–8. doi:10.1046/j.1365-2222.1998.00302.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Ormstad H, Namork E, Gaarder PI, Johansen BV. Scanning electron microscopy of immunogold labeled cat allergens (Fel d 1) on the surface of airborne house dust particles. J Immunol Methods (1995) 187(2):245–51. doi:10.1016/0022-1759(95)00190-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Knox RB, Suphioglu C, Taylor P, Desai R, Watson HC, Peng JL, et al. Major grass pollen allergen Lol p 1 binds to diesel exhaust particles: implications for asthma and air pollution. Clin Exp Allergy (1997) 27(3):246–51. doi:10.1046/j.1365-2222.1997.d01-508.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Armentia A, Lombardero M, Callejo A, Barber D, Martin Gil FJ, Martin-Santos JM, et al. Is Lolium pollen from an urban environment more allergenic than rural pollen? Allergol Immunopathol (Madr) (2002) 30(4):218–24. doi:10.1016/S0301-0546(02)79124-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Cortegano I, Civantos E, Aceituno E, del Moral A, Lopez E, Lombardero M, et al. Cloning and expression of a major allergen from Cupressus arizonica pollen, Cup a 3, a PR-5 protein expressed under polluted environment. Allergy (2004) 59(5):485–90. doi:10.1046/j.1398-9995.2003.00363.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Jin HJ, Choi GS, Shin YS, Kim JH, Kim JE, Ye YM, et al. The allergenic potency of Japanese hop pollen is increasing with environmental changes in Korea. Allergy Asthma Immunol Res (2013) 5(5):309–14. doi:10.4168/aair.2013.5.5.309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Lee SI, Pham le D, Shin YS, Suh DH, Park HS. Environmental changes could enhance the biological effect of Hop J pollens on human airway epithelial cells. J Allergy Clin Immunol (2014) 134(2):470.e–2.e. doi:10.1016/j.jaci.2014.01.034
132. Gershwin LJ, Osebold JW, Zee YC. Immunoglobulin E-containing cells in mouse lung following allergen inhalation and ozone exposure. Int Arch Allergy Appl Immunol (1981) 65(3):266–77. doi:10.1159/000232766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Osebold JW, Zee YC, Gershwin LJ. Enhancement of allergic lung sensitization in mice by ozone inhalation. Proc Soc Exp Biol Med (1988) 188(3):259–64. doi:10.3181/00379727-188-42733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Muranaka M, Suzuki S, Koizumi K, Takafuji S, Miyamoto T, Ikemori R, et al. Adjuvant activity of diesel-exhaust particulates for the production of IgE antibody in mice. J Allergy Clin Immunol (1986) 77(4):616–23. doi:10.1016/0091-6749(86)90355-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Takafuji S, Suzuki S, Koizumi K, Tadokoro K, Ohashi H, Muranaka M, et al. Enhancing effect of suspended particulate matter on the IgE antibody production in mice. Int Arch Allergy Appl Immunol (1989) 90(1):1–7. doi:10.1159/000234990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Diaz-Sanchez D, Tsien A, Fleming J, Saxon A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J Immunol (1997) 158(5):2406–13.
137. Bevelander M, Mayette J, Whittaker LA, Paveglio SA, Jones CC, Robbins J, et al. Nitrogen dioxide promotes allergic sensitization to inhaled antigen. J Immunol (2007) 179(6):3680–8. doi:10.4049/jimmunol.179.6.3680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Koike E, Kobayashi T. Ozone exposure enhances antigen-presenting activity of interstitial lung cells in rats. Toxicology (2004) 196(3):217–27. doi:10.1016/j.tox.2003.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Hodgkins SR, Ather JL, Paveglio SA, Allard JL, LeClair LA, Suratt BT, et al. NO2 inhalation induces maturation of pulmonary CD11c+ cells that promote antigenspecific CD4+ T cell polarization. Respir Res (2010) 11:102. doi:10.1186/1465-9921-11-102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Bleck B, Grunig G, Chiu A, Liu M, Gordon T, Kazeros A, et al. MicroRNA-375 regulation of thymic stromal lymphopoietin by diesel exhaust particles and ambient particulate matter in human bronchial epithelial cells. J Immunol (2013) 190(7):3757–63. doi:10.4049/jimmunol.1201165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Bleck B, Tse DB, Curotto de Lafaille MA, Zhang F, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation and polarization via thymic stromal lymphopoietin. J Clin Immunol (2008) 28(2):147–56. doi:10.1007/s10875-007-9149-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Bleck B, Tse DB, Gordon T, Ahsan MR, Reibman J. Diesel exhaust particle-treated human bronchial epithelial cells upregulate Jagged-1 and OX40 ligand in myeloid dendritic cells via thymic stromal lymphopoietin. J Immunol (2010) 185(11):6636–45. doi:10.4049/jimmunol.1000719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Lee CC, Cheng YW, Liao JW, Chiang BL, Lai YL, Kang JJ. Motorcycle exhaust particles augment antigen-induced airway inflammation in BALB/c mice. J Toxicol Environ Health A (2008) 71(6):405–12. doi:10.1080/15287390701801687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Dong CC, Yin XJ, Ma JY, Millecchia L, Wu ZX, Barger MW, et al. Effect of diesel exhaust particles on allergic reactions and airway responsiveness in ovalbumin-sensitized brown Norway rats. Toxicol Sci (2005) 88(1):202–12. doi:10.1093/toxsci/kfi280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Hashimoto K, Ishii Y, Uchida Y, Kimura T, Masuyama K, Morishima Y, et al. Exposure to diesel exhaust exacerbates allergen-induced airway responses in guinea pigs. Am J Respir Crit Care Med (2001) 164(10 Pt 1):1957–63. doi:10.1164/ajrccm.164.10.2011070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Molfino NA, Wright SC, Katz I, Tarlo S, Silverman F, McClean PA, et al. Effect of low concentrations of ozone on inhaled allergen responses in asthmatic subjects. Lancet (1991) 338(8761):199–203. doi:10.1016/0140-6736(91)90346-Q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Devalia JL, Rusznak C, Herdman MJ, Trigg CJ, Tarraf H, Davies RJ. Effect of nitrogen dioxide and sulphur dioxide on airway response of mild asthmatic patients to allergen inhalation. Lancet (1994) 344(8938):1668–71. doi:10.1016/S0140-6736(94)90458-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Tunnicliffe WS, Burge PS, Ayres JG. Effect of domestic concentrations of nitrogen dioxide on airway responses to inhaled allergen in asthmatic patients. Lancet (1994) 344(8939–8940):1733–6. doi:10.1016/S0140-6736(94)92886-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Rusznak C, Devalia JL, Davies RJ. Airway response of asthmatic subjects to inhaled allergen after exposure to pollutants. Thorax (1996) 51(11):1105–8. doi:10.1136/thx.51.11.1105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Bascom R, Naclerio RM, Fitzgerald TK, Kagey-Sobotka A, Proud D. Effect of ozone inhalation on the response to nasal challenge with antigen of allergic subjects. Am Rev Respir Dis (1990) 142(3):594–601. doi:10.1164/ajrccm/142.3.594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Peden DB, Setzer RW Jr, Devlin RB. Ozone exposure has both a priming effect on allergen-induced responses and an intrinsic inflammatory action in the nasal airways of perennially allergic asthmatics. Am J Respir Crit Care Med (1995) 151(5):1336–45. doi:10.1164/ajrccm.151.5.7735583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Devalia JL, Sapsford RJ, Cundell DR, Rusznak C, Campbell AM, Davies RJ. Human bronchial epithelial cell dysfunction following in vitro exposure to nitrogen dioxide. Eur Respir J (1993) 6(9):1308–16.
153. Devalia JL, Campbell AM, Sapsford RJ, Rusznak C, Quint D, Godard P, et al. Effect of nitrogen dioxide on synthesis of inflammatory cytokines expressed by human bronchial epithelial cells in vitro. Am J Respir Cell Mol Biol (1993) 9(3):271–8. doi:10.1165/ajrcmb/9.3.271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Ohtoshi T, Takizawa H, Okazaki H, Kawasaki S, Takeuchi N, Ohta K, et al. Diesel exhaust particles stimulate human airway epithelial cells to produce cytokines relevant to airway inflammation in vitro. J Allergy Clin Immunol (1998) 101(6 Pt 1):778–85. doi:10.1016/S0091-6749(98)70307-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Rusznak C, Devalia JL, Sapsford RJ, Davies RJ. Ozone-induced mediator release from human bronchial epithelial cells in vitro and the influence of nedocromil sodium. Eur Respir J (1996) 9(11):2298–305. doi:10.1183/09031936.96.09112298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Bayram H, Sapsford RJ, Abdelaziz MM, Khair OA. Effect of ozone and nitrogen dioxide on the release of proinflammatory mediators from bronchial epithelial cells of nonatopic nonasthmatic subjects and atopic asthmatic patients in vitro. J Allergy Clin Immunol (2001) 107(2):287–94. doi:10.1067/mai.2001.111141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Esposito V, Lucariello A, Savarese L, Cinelli MP, Ferraraccio F, Bianco A, et al. Morphology changes in human lung epithelial cells after exposure to diesel exhaust micron sub particles (PM1.0) and pollen allergens. Environ Pollut (2012) 171:162–7. doi:10.1016/j.envpol.2012.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Mazzarella G, Lucariello A, Bianco A, Calabrese C, Thanassoulas T, Savarese L, et al. Exposure to submicron particles (PM1.0) from diesel exhaust and pollen allergens of human lung epithelial cells induces morphological changes of mitochondria tonifilaments and rough endoplasmic reticulum. In vivo (2014) 28(4):557–61.
159. Chou CH, Hsu JY, Fu LS, Chu JJ, Chi CS. The air pollutant sodium sulfite enhances mite crude extract-stimulated detachment of A549 airway epithelium cells. J Microbiol Immunol Infect (2008) 41(1):26–31.
160. Bellanger AP, Bosch-Cano F, Millon L, Ruffaldi P, Franchi M, Bernard N. Reactions of airway epithelial cells to birch pollen grains previously exposed to in situ atmospheric Pb concentrations: a preliminary assay of allergenicity. Biol Trace Elem Res (2012) 150(1–3):391–5. doi:10.1007/s12011-012-9485-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Mazzarella G, Esposito V, Bianco A, Ferraraccio F, Prati MV, Lucariello A, et al. Inflammatory effects on human lung epithelial cells after exposure to diesel exhaust micron sub particles (PM1.0) and pollen allergens. Environ Pollut (2012) 161:64–9. doi:10.1016/j.envpol.2011.09.046
162. Wallace LA, Mitchell H, O’Connor GT, Neas L, Lippmann M, Kattan M, et al. Particle concentrations in inner-city homes of children with asthma: the effect of smoking, cooking, and outdoor pollution. Environ Health Perspect (2003) 111(9):1265–72. doi:10.1289/ehp.6135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Weitzman M, Gortmaker S, Walker DK, Sobol A. Maternal smoking and childhood asthma. Pediatrics (1990) 85(4):505–11.
164. Seymour BW, Pinkerton KE, Friebertshauser KE, Coffman RL, Gershwin LJ. Second-hand smoke is an adjuvant for T helper-2 responses in a murine model of allergy. J Immunol (1997) 159(12):6169–75.
165. Min MG, Song DJ, Miller M, Cho JY, McElwain S, Ferguson P, et al. Coexposure to environmental tobacco smoke increases levels of allergen-induced airway remodeling in mice. J Immunol (2007) 178(8):5321–8. doi:10.4049/jimmunol.178.8.5321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev (2007) 87(3):1047–82. doi:10.1152/physrev.00048.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Brody JS, Steiling K. Interaction of cigarette exposure and airway epithelial cell gene expression. Annu Rev Physiol (2011) 73:437–56. doi:10.1146/annurev-physiol-012110-142219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: airway epithelium, proteinases, pollutants, airway inflammation, HDM allergens, cockroach allergens, fungal allergens
Citation: Gandhi VD and Vliagoftis H (2015) Airway epithelium interactions with aeroallergens: role of secreted cytokines and chemokines in innate immunity. Front. Immunol. 6:147. doi: 10.3389/fimmu.2015.00147
Received: 17 September 2014; Accepted: 18 March 2015;
Published online: 02 April 2015.
Edited by:
Charles Dudley Mills, BioMedical Consultants, USAReviewed by:
Masaaki Murakami, Hokkaido University, JapanYuri Sykulev, Thomas Jefferson University, USA
Jian Zhang, The Ohio State University, USA
Rakesh K. Kumar, University of New South Wales, Australia
Copyright: © 2015 Gandhi and Vliagoftis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harissios Vliagoftis, Pulmonary Research Group, Department of Medicine, University of Alberta, 550A Heritage Medical Research Centre, Edmonton, AB T6G 2S2, Canada e-mail:aGFyaUB1YWxiZXJ0YS5jYQ==