 Kai Fang
Kai Fang Matthew B. Grisham
Matthew B. Grisham Christopher G. Kevil
Christopher G. Kevil- 1Division of Digestive Diseases, Inflammatory Bowel Disease Center, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA
- 2Department of Immunology and Molecular Microbiology, Texas Tech University Health Sciences Center, Lubbock, TX, USA
- 3Department of Pathology, Louisiana State University Health Sciences Center, Shreveport, LA, USA
- 4Department of Molecular and Cellular Physiology, Louisiana State University Health Sciences Center, Shreveport, LA, USA
Experimental models of colitis in mice have been used extensively for analyzing the molecular events that occur during inflammatory bowel disease (IBD) development. However, it is uncertain to what extent the experimental models reproduce features of human IBD. This is largely due to the lack of precise methods for direct and comprehensive comparison of mouse and human inflamed colon tissue at the molecular level. Here, we use global gene expression patterns of two sets of pediatric IBD and two mouse models of colitis to obtain a direct comparison of the genome signatures of mouse and human IBD. By comparing the two sets of pediatric IBD microarray data, we found 83 genes were differentially expressed in a similar manner between pediatric Crohn’s disease and ulcerative colitis. Up-regulation of the chemokine (C–C motif) ligand 2 (CCL2) gene that maps to 17q12, a confirmed IBD susceptibility loci, indicates that our comparison study can reveal known genetic associations with IBD. In comparing pediatric IBD and experimental colitis microarray data, we found common signatures amongst them including: (1) up-regulation of CXCL9 and S100A8; (2) cytokine–cytokine receptor pathway dysregulation; and (3) over-represented IRF1 and IRF2 transcription binding sites in the promoter region of up-regulated genes, and HNF1A and Lhx3 binding sites were over-represented in the promoter region of the down-regulated genes. In summary, this study provides a comprehensive view of transcriptome changes between different pediatric IBD populations in comparison with different colitis models. These findings reveal several new molecular targets for further study in the regulation of colitis.
Introduction
Ulcerative colitis (UC) and Crohn’s disease (CD) are the two major forms of inflammatory bowel disease (IBD). The incidence rate of pediatric CD in the US is 43 per 100,000 and 28 per 100,000 for pediatric UC (1). As recently reported, the incidence and prevalence of pediatric IBD is rising in both developed and developing countries (2). Growth retardation poses a significant threat to the quality of life of 15–40% of children and adolescents with IBD (3). Although environmental factors, microbes in the gastroenterological tract, genetic susceptibility, and immune system dysfunction have been implicated, the etiology of pediatric IBD remains incompletely understood.
During the development of IBD, the colon tissue changes its genome transcription in response to pathological conditions, which is a result of dysregulated interaction between the immune system and enteric bacteria. The common feature of UC and CD inflamed tissue genome transcription provides new clues for pediatric IBD treatment. Although the microarray assay has been performed on pediatric IBD, there is no comprehensive genome transcription analysis for pediatric UC or CD. Here, we performed transcriptome analysis using two sets of pediatric IBD microarray data (4, 5), T-cell transfer colitis model microarray data (6), and dextran sodium sulfate (DSS)-induced colitis microarray data (7) generated from our laboratory and deposited in the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) database. Network and promoter analysis was performed to identify differentially expressed genes in the inflamed colon tissue from pediatric IBD patients versus experimental animal models. Comparison between pediatric IBD and experimental colitis microarray data revealed the similarly expressed genes and over-represented transcription factor binding sites (TFBS) in the promoter regions of the dysregulated genes.
Materials and Methods
Pediatric IBD Microarray Datasets
To get a comprehensive view of the pediatric IBD genome transcription profile, two sets of pediatric IBD microarray data were selected from NCBI. Those two sets of microarray data were obtained by using Affymetrix GeneChip Human Genome HG-U133 plus 2.0 arrays that provide the most comprehensive coverage of transcribed human genome and contain probes for approximately 22,634 genes. The microarray data were generated from pediatric colon in healthy controls, colon only CD, and colon only UC. The dataset GSE10616 contained data from 11 control samples, 14 CD samples, and 10 UC samples (4); the dataset GSE9686 contained data from 8 control samples, 11 CD samples, and 5 UC samples (5). Colon RNA was isolated from biopsies obtained from patients and healthy controls at diagnosis. The pediatric Crohn’s Disease Activity Index (PCDAI) and Pediatric Ulcerative Colitis Clinical Activity Index (PUCAI) were used to assess the clinical severity of the IBD sample.
GeneSifter Analysis
Two sets of pediatric IBD microarray data were uploaded to GeneSifter software1 and normalized for comparison by Robust Multichip Average (RMA) method. The gene expression difference threshold was set to 2 with no upper limit. Data were analyzed with a Student’s t-test followed by a Benjamini and Hochberg post test to limit false discovery rates, as we previously reported (7).
Ingenuity Pathway Analysis
To see the relationship between differentially expressed genes, the selected genes identified as dysregulated in pediatric CD and pediatric UC microarray data were then imported to IPA2 for network analysis. Genes that were related to each other in biological functions and/or diseases were organized into networks according to the Ingenuity Pathways Knowledge Base (IPKB). IPKB is a database derived from the data mining of the expression of and functional relationships between molecules; this information was extracted from published papers found in NCBI PubMed, Medline, and several other databases.
Cis-Regulatory Elements Analysis
To identify common properties of promoter regions of differentially expressed genes, the Affymetrix gene ID of identified genes in pediatric UC and CD were uploaded to the cREMaG system3 (8). The sequence upstream the transcription start site (TSS) is 5000 base pairs, and the downstream sequence of TSS is 1000 base pairs. Promoter sequences were scanned with TFBS matrices obtained from the JASPAR database and the public release of the TRANSFAC database using the TFBS BioPerl module (9, 10). The top 10 of the most over-represented binding sites were selected for comparison analysis.
Comparison between Pediatric IBD and Experimental Colitis Microarray Data
By directly comparing differential gene expression between human and mouse inflamed colon tissue, we assessed the similarity between human and mouse colitis. The dysregulated gene in DSS-colitis (GEO data base accession number GSE22307) and T-cell transfer colitis model (accession number GSE27302) were divided into eight classes according to the genes expression trends. In the DSS-colitis model, there were 1609 genes that were significantly altered during the colitis development, with 501 progressively up-regulated genes and 173 progressively down-regulated genes (7). In the T-cell transfer colitis model, there were 1775 gene expressions that were significantly changed, with 341 progressively up-regulated genes and 361 progressively down-regulated genes (6). The two sets of microarray data were obtained by using the same platform, Mouse Genome 430 2.0 Array (Affymetrix), which provided the most comprehensive annotated coverage of the mouse genome, composing of over 34,000 well-characterized mouse genes. The genes whose expression progressively changed were correlated with inflammation development and were selected for promoter binding sites analysis. The over-presented promoter binding sites were further compared with the over-presented binding sites obtained from pediatric IBD array data.
Results
GeneSifter Analysis Pediatric IBD Microarray Data
Analysis of GSE 9686 pediatric CD microarray data showed that 242 genes were differentially expressed, 173 genes had up-regulated expression, while 69 genes had down-regulated expression. Analysis of the GSE 10616 pediatric CD microarray data showed that there were 298 genes differentially expressed (the expression of 209 genes were up-regulated and 89 genes were down-regulated). After comparing two sets of pediatric CD microarray data, we found the expression of 167 genes was similarly changed. Among those 167 genes, 117 genes were up-regulated (Table S1 in Supplementary Material), and 50 gene expressions were down-regulated (Table S2 in Supplementary Material).
In GSE9686 pediatric UC microarray data, there were 3860 genes differentially expressed (1717 genes were up-regulated, and 2143 genes were down-regulated). While in GSE10616 pediatric UC, there were 1826 genes differentially expressed (1122 genes were up-regulated and 704 genes were down-regulated). After comparing the two sets of pediatric UC data, we found that 1071 genes were similarly up-regulated (Table S3 in Supplementary Material), and 736 genes were down-regulated (Table S4 in Supplementary Material).
After comparing the data in Tables S1 and S3 in Supplementary Material, we found that there were 65 genes up-regulated in pediatric CD and pediatric UC, as shown in Table 1 and Figure 1A. By comparing Tables S2 and S4 in Supplementary Material, we found that there were 18 genes down-regulated in pediatric CD and pediatric UC, as shown in Table 2 and Figure 1B.
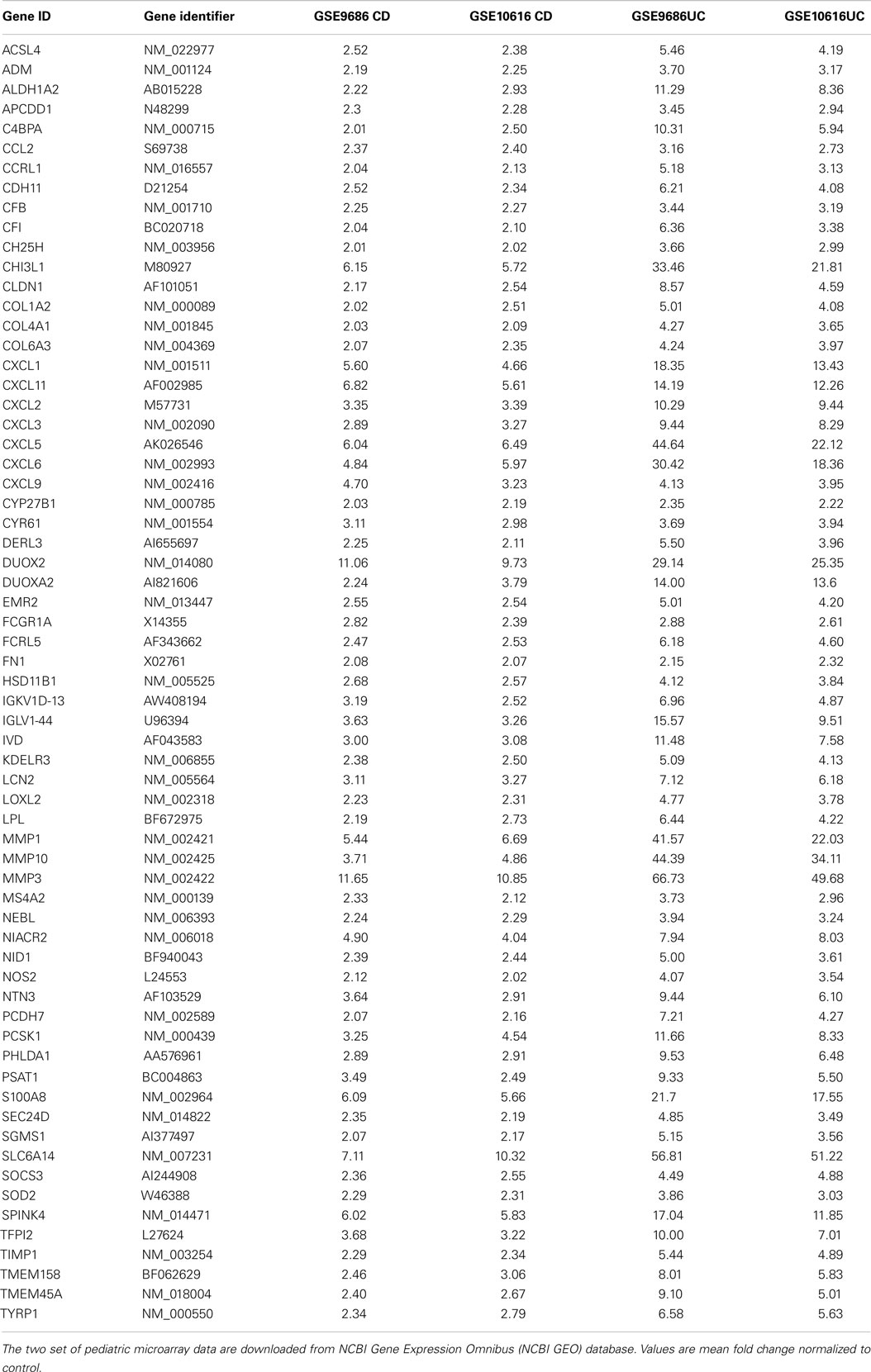
Table 1. Similarly up-regulated genes in pediatric IBD.
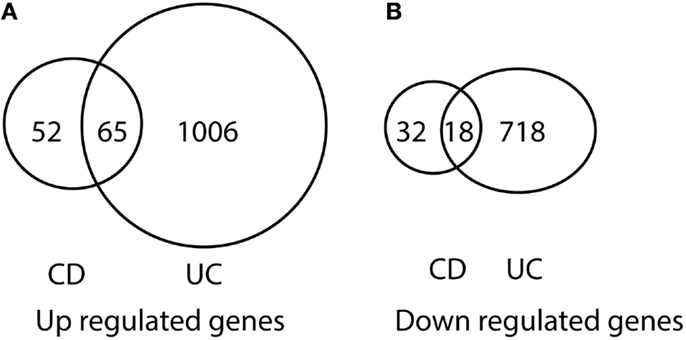
Figure 1. Venn diagram illustration of gene expression similarity between pediatric CD and UC patient sample microarray data. (A) One-hundred seventeen genes were up-regulated from pediatric CD compared with 1071 up-regulated genes from pediatric UC patients, with 65 genes being common between the two groups. (B) Fifty genes were down-regulated from pediatric CD compared from 736 down-regulated gene from pediatric UC patients, with 18 genes being common between the two groups.
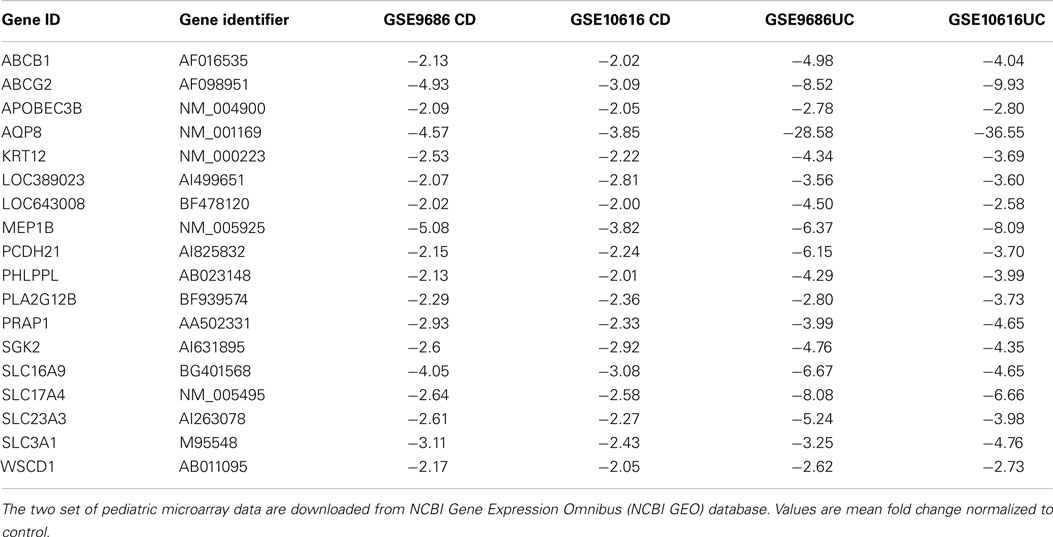
Table 2. Similarly down-regulated genes in pediatric IBD.
Of the up-regulated genes, seven were from the CXC chemokine family: CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL9, and CXCL11, which are the key components of the cytokine–cytokine receptor interaction pathway. CXCL1 is expressed by epithelial cells, macrophages, and neutrophils (11, 12) and has neutrophil chemoattractant activity (13). CXCL2 is secreted by macrophages and monocytes and is a chemoattractant for polymorphonuclear cells, leukocytes, and hematopoietic stem cells (11, 14, 15). CXCL5 is expressed in eosinophils and stimulates the chemotaxis of neutrophils (16). CXCL6 is a chemoattractant for neutrophils (17). CXCL9 is an interferon (IFN)-dependent CXC chemokine, which plays a pro-inflammatory role and has been found to be expressed at high levels in UC tissue (18). CXCL11 is a chemoattractant for activated T cells (19).
Of the down-regulated genes, four were solute carrier genes: SLC16A9, SLC17A4, SLC23A3, and SLC3A1. The functions of those down-regulated genes related to the transport of amino acids (20), monocarboxylate (21), glutamate (22), sodium-phosphate (23), and ascorbic acid (24).
Ingenuity Analysis of Pediatric IBD Microarray Data
Those genes differentially expressed in pediatric CD and UC were uploaded to Ingenuity software for network analysis. Those differentially expressed genes in pediatric CD inflamed colon tissue were organized into eight networks. The molecules in each network and their top functions are listed in Table S5 in Supplementary Material. Those differentially expressed genes in pediatric UC inflamed colon tissue were organized into 25 networks, and the molecules in each network and their top functions are listed in Table S6 in Supplementary Material.
Figure 2 shows the first network of pediatric CD inflamed colon tissue differentially expressed genes with their cell-to-cell signaling functions and their interactions, as they relate to gastrointestinal and hepatic system disease. As shown in Figure 2, the transcription of nine chemokine genes was up-regulated, and those genes indirectly react with the NF-κB complex. Figure 3 shows the network 2 differentially expressed genes in pediatric CD inflamed colon tissue, which is composed of 15 up-regulated genes with functions related to connective tissue and genetic disorders. MMP-1 and MMP-3 are located in the center of pediatric CD network 2. ChI3l1 is also implicated in this network through its indirect reaction with IGFBP5.
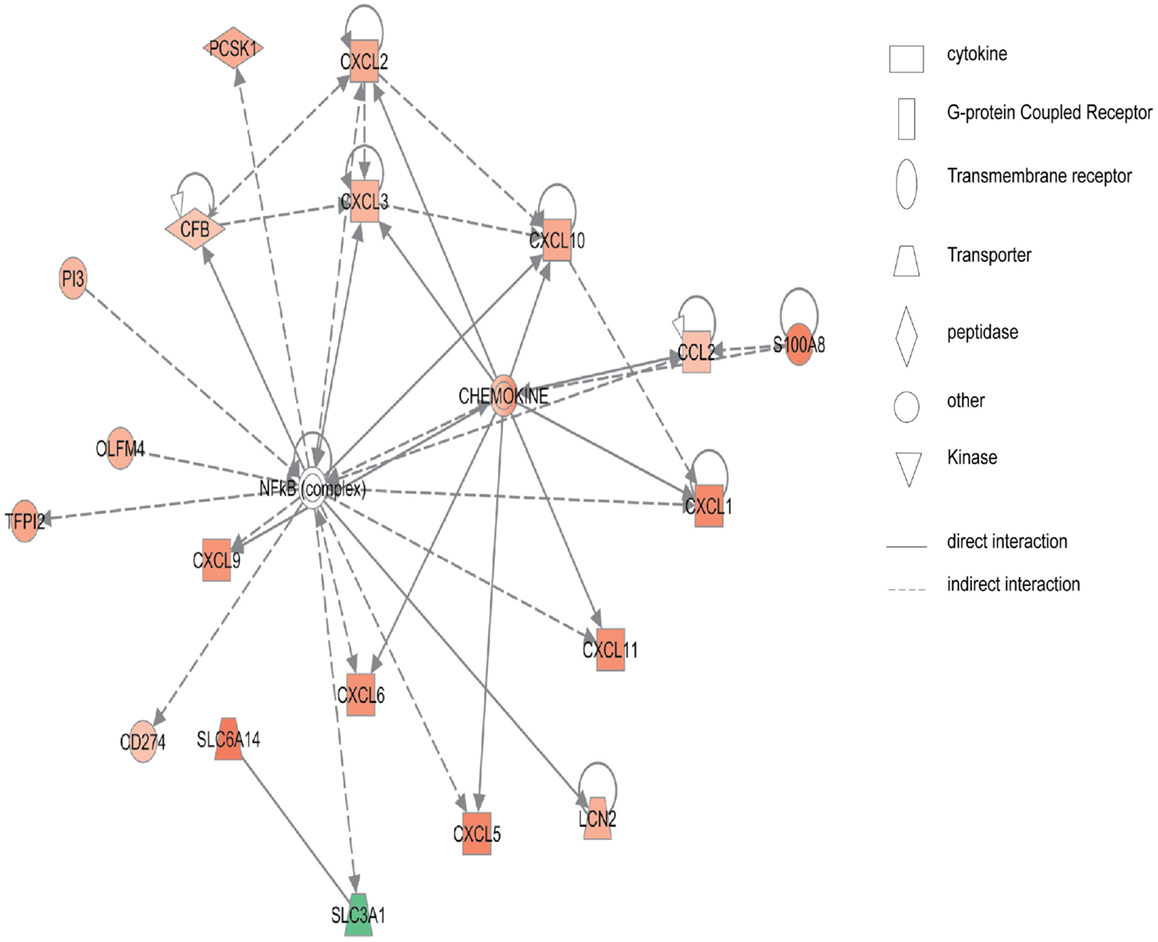
Figure 2. Network 1 of pediatric CD has biological functions associated with cell-to-cell signaling and interaction, gastrointestinal disease, hepatic system disease. Red shading indicates up-regulation, whereas green shading shows down-regulation.
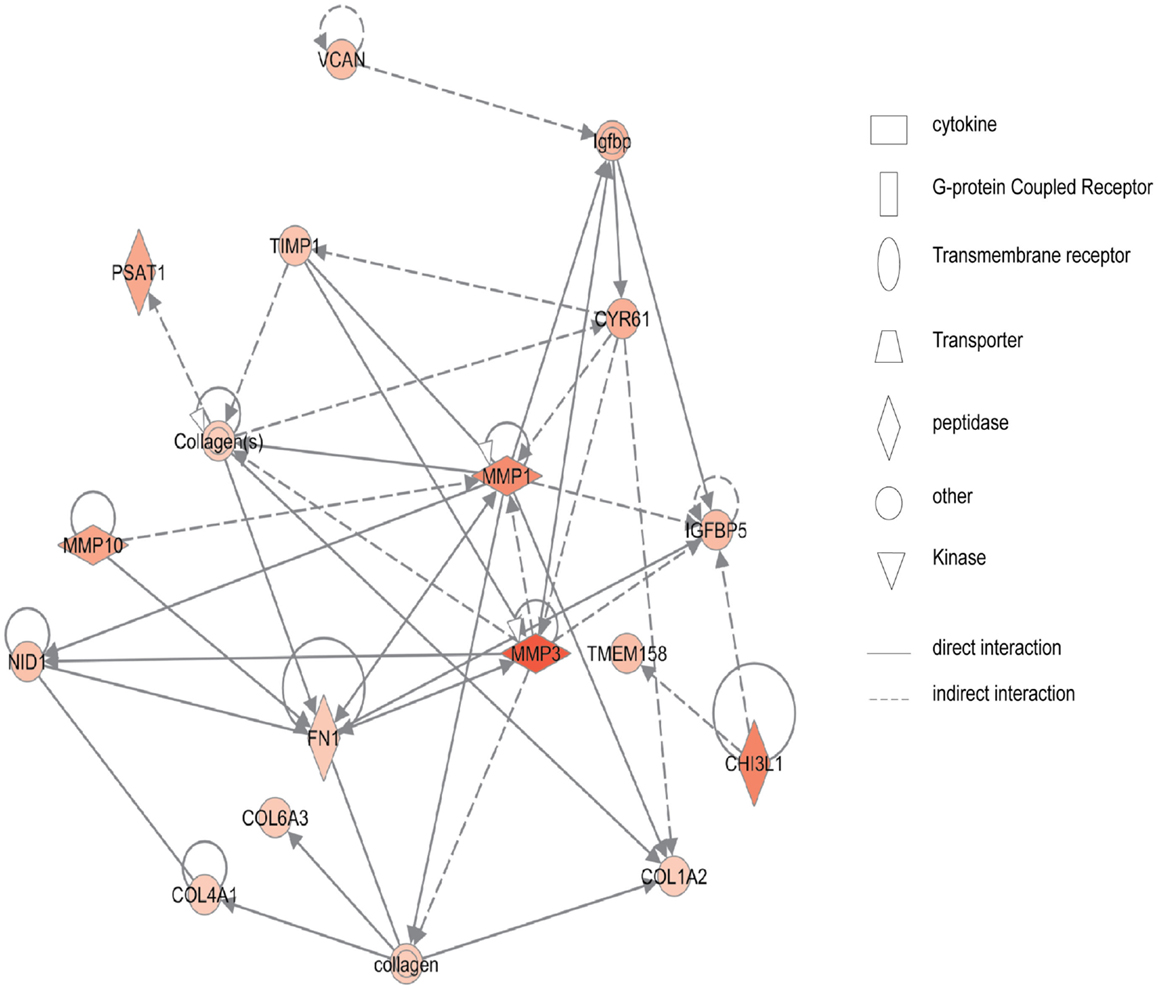
Figure 3. Network 2 of pediatric CD has biological functions associated with connective tissue disorders, genetic disorder, and cardiovascular disease. Red shading indicates up-regulation, whereas green shading shows down-regulation.
Figure 4 shows the first network of pediatric UC with functions related to cellular movement and signaling. Pediatric UC network 1 is mainly composed of eleven G-protein-coupled receptors, which were all up-regulated. Transcription of eight members of the collage family was up-regulated as shown in Figure 5, with functions related to connective tissue. Interestingly, ChI3L1 was in the center of the pediatric UC network 2 (Figure 5), and ChI3L1 indirectly interacts with COL16A2 and TNC.
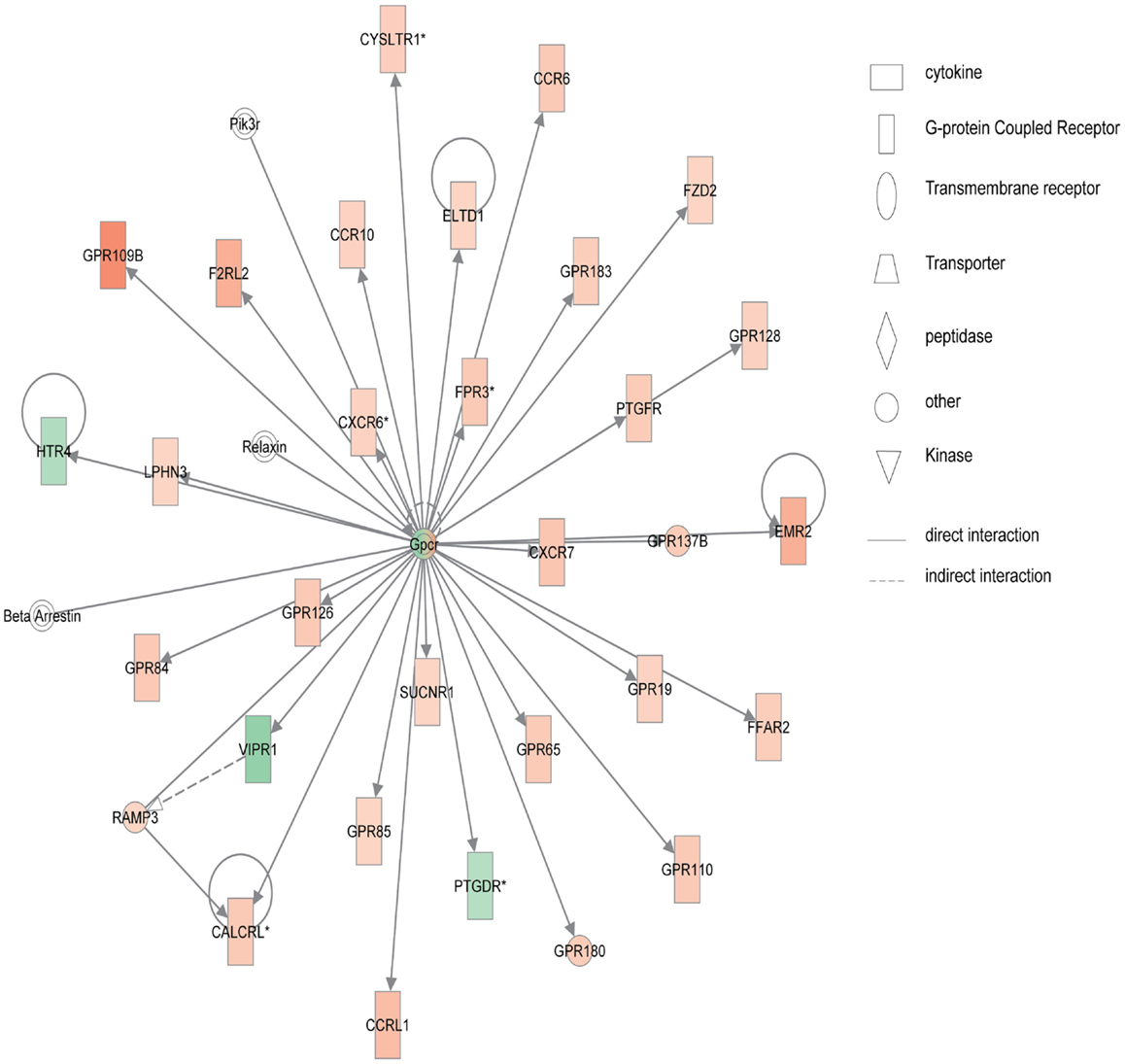
Figure 4. Network 1 of pediatric UC has biological functions associated with cellular movement, cell signaling, and nucleic acid metabolism. Red shading indicates up-regulation, whereas green shading shows down-regulation.
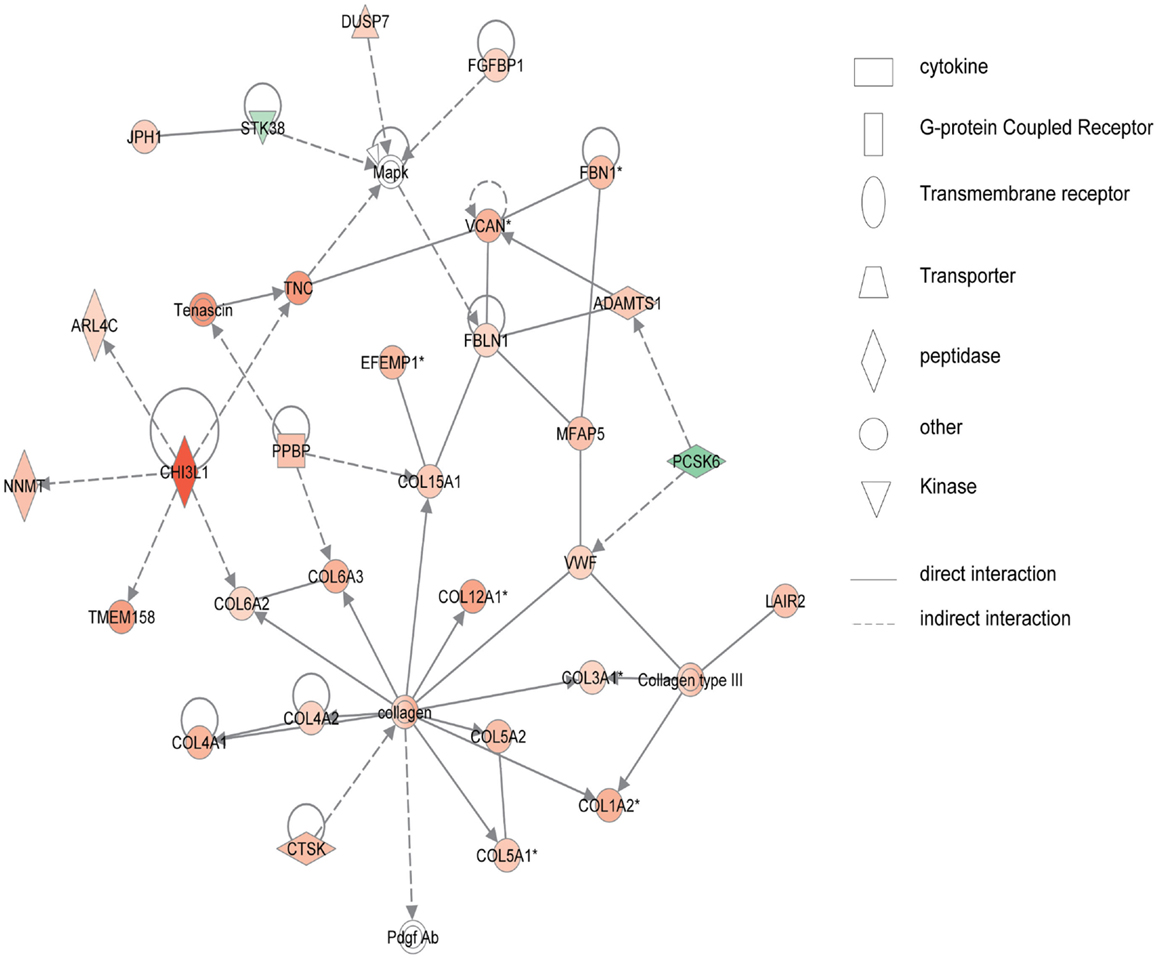
Figure 5. Network 2 of pediatric UC has biological functions associated with connective tissue disorders, genetic disorder, and dermatological diseases and conditions. Red shading indicates up-regulation, whereas green shading shows down-regulation.
Promoter Analysis of Pediatric IBD Microarray Data
Using the CREMaG system, we indentified over-presented TFBS in the differentially expressed genes. The over-presented TFBS of differentially regulated genes in pediatric CD is shown in Tables S7–S10 in Supplementary Material. TFBS over-presented in pediatric UC differentially expressed genes are shown in Table S11 in Supplementary Material (for up-regulated genes) and in Table S12 in Supplementary Material (for down-regulated genes). The fold-difference in TFBS frequency was computed by dividing the observed TFBS number by the background number.
By comparison, we found that there were six promoter sequences (RELA, NF-κB, IRF2, Evi1, and IRF1) that were over-presented in genes that were up-regulated in pediatric IBD. There were six TFBS (Lhx3, MEF2A, HNF1A, Nobox, NR2F1, and Foxa2) that were over-presented in the genes that were down-regulated in pediatric IBD-inflamed colon tissue.
In the pediatric CD microarray data analysis, the NF-κB binding sequence was over-presented in the inflammatory-related genes, such as CCL2, CXCL10, CXCL2, CXCL3, CXCL6, CXCL9, and in other up-regulated genes in the pediatric CD-inflamed colon tissue. The network analysis of pediatric CD (Figure 2) also showed that NF-κB regulates chemokine gene expression. Not surprisingly, in the pediatric UC microarray data promoter analysis, the NF-κB binding site was also shown to be over-represented in the promoter region of the up-regulated genes, as it is well known that NF-κB plays a pivotal role in the expression of inflammatory mediators. The promoter region of 89 up-regulated genes (ICAM-1, COL1A1, WNT5A, CXCL5, IL-1B, CXCL2, IL-6, IL-11, and others) in pediatric UC has the NF-κB binding site.
Comparison of Pediatric IBD and Experimental Colitis Microarray Data
The dysregulated KEGG pathway of GSE9686 pediatric UC and GSE10616 pediatric UC is shown in Tables S13 and S14 in Supplementary Material. The over-represented TFBS of progressively up-regulated or down-regulated genes in the T-cell transfer colitis model and the DSS-colitis model are shown in Tables S15–S18 in Supplementary Material.
Comparison of differentially expressed genes in pediatric IBD and progressively up-regulated or down-regulated genes in experimental colitis is shown in Table 3 and Figures 6 and 7. The comparison of the over-presented promoters in the differentially expressed genes is shown in Table 4. Among them, the cytokine–cytokine receptor pathway was dysregulated in all the four sets of microarray data. CXCL9 and S100A8 were up-regulated in all the four sets of microarray data. The expression of S100A8 was also found to be up-regulated in the trinitrobenzene sulfonic acid (TNBS)-induced colitis rat model and T-cell-mediated colitis in SCID mice (25, 26).

Table 3. Common dysregulated pathways and genes.
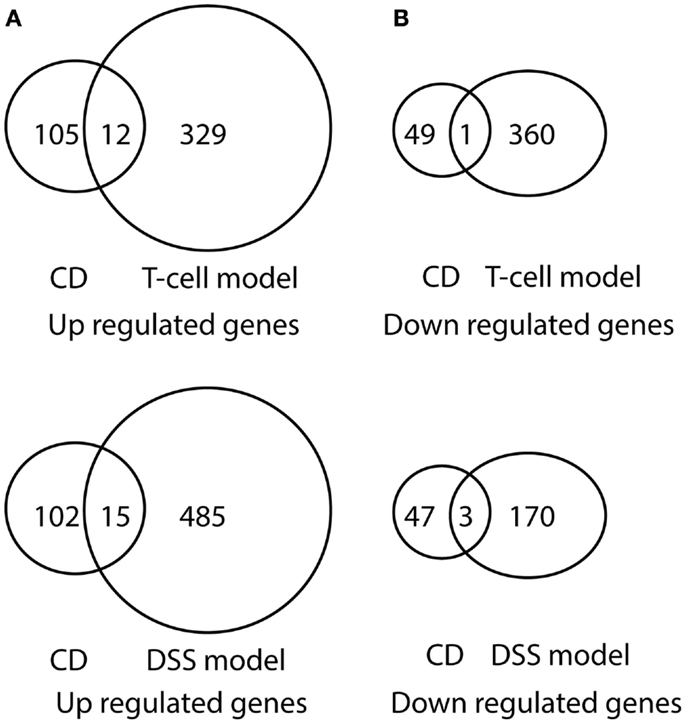
Figure 6. Venn diagram illustration of gene expression similarity between pediatric CD and experimental colitis model microarray data. (A) One-hundred seventeen genes were up-regulated from pediatric CD compared with 341 up-regulated genes from T-cell colitis model and 501 up-regulated genes from DSS-colitis model. (B) Fifty genes were down-regulated from pediatric CD compared from 361 down-regulated gene from T-cell colitis model and 173 down-regulated genes from DSS-colitis model.
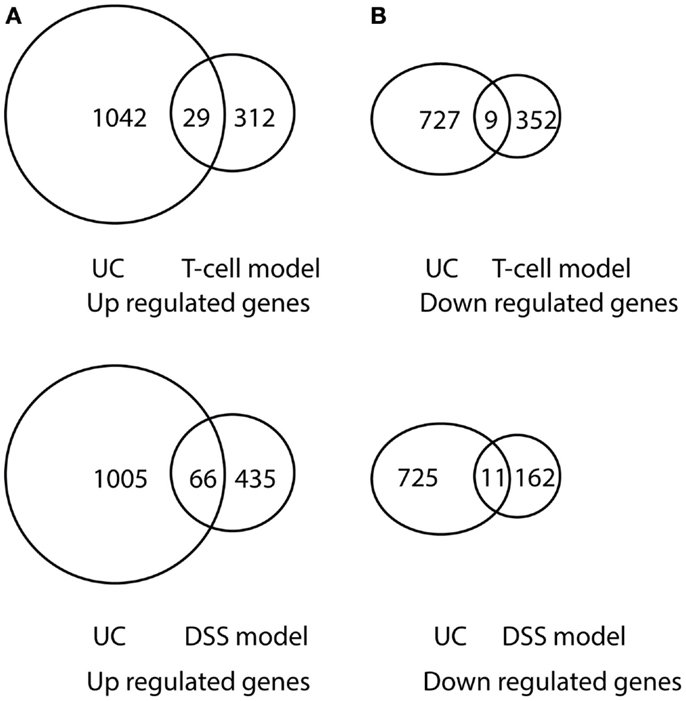
Figure 7. Venn diagram illustration of gene expression similarity between pediatric UC patient sample microarray data and experimental colitis model microarray data. (A) One-thousand seventy one genes were up-regulated from pediatric UC compared with 341 up-regulated genes from T-cell colitis model and 501 up-regulated genes from DSS-colitis model. (B) Seven-hundred thirty six genes were down-regulated from pediatric UC compared from 361 down-regulated gene from T-cell colitis model and 173 down-regulated genes from DSS-colitis model.
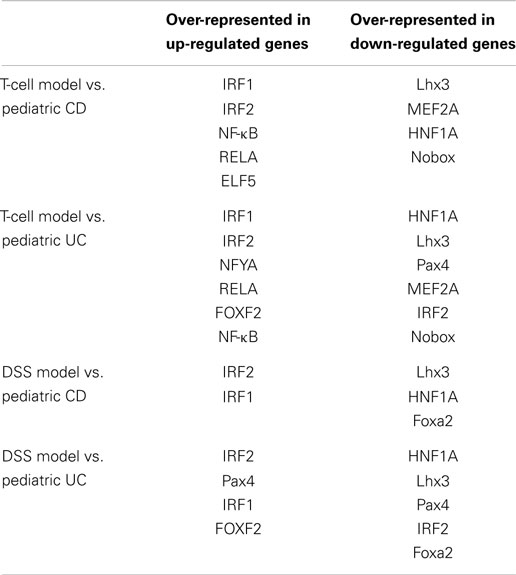
Table 4. Comparison promoter between animal model and pediatric IBD.
Promoter analysis provided the possible common regulatory mechanism of the expression of the dysregulated genes. As shown in Table 4, the IRF1 and IRF2 binding sites were over-represented in the up-regulated genes in pediatric IBD and experimental colitis. The HNF1A and Lhx3 binding sites were over-presented in the down-regulated genes in the four sets of microarray data. HNF1A is a transcription factor that regulates the expression of cytokine-driven C-reactive protein, which is a clinical marker of inflammation (27). Lhx3 is a transcription factor that is required for pituitary and motor neuron development (28).
As shown in Table 3, cytokine–cytokine receptor interaction pathway is dysregulated in pediatric UC, CD, T-cell transfer, and DSS-colitis model. While chemokine signaling pathway is dysregulated in pediatric UC, CD, and T-cell transfer colitis model, but not in DSS-colitis model. Additionally, NF-κB binding site is over-presented in the promoter region of up-regulated genes in pediatric CD, UC, and T-cell transfer colitis, but not in DSS-colitis model (Table 4). Thus, the comparison of dysregulated KEGG pathway (Table 3) and the over-represented TFBS (Table 4) showed that the T-cell transfer colitis model was better than the DSS-induced colitis model at simulating pediatric IBD; however, the DSS-colitis model was more similar to pediatric UC than pediatric CD, as the DSS model has more common dysregulated pathways and molecules (Table 3) and over-represented TFBS in the dysregulated genes with pediatric UC than CD (Table 4).
Discussion
Our microarray analysis revealed that chitinase 3-like 1 (cartilage glycoprotein-39, CHI3L1) was up-regulated in pediatric IBD samples. CHI3L1 has the ability to enhance the adhesion and internalization of bacteria in epithelial cells (29). In vivo, neutralizing CHI3L1 with an antibody suppresses DSS-induced colitis, and this neutralization dramatically decreases bacteria adhesion and invasion of epithelial cells. It has been demonstrated that CHI3L1 expression is up-regulated in epithelial cells under inflammatory conditions. CHI3L1 also activates Akt signaling in epithelial cells through its chitin binding motif, and increases secretion of IL-8 and TNF-α in a dose-dependent manner (30). Fecal CHI3L1 levels are positively correlated with pathology score (31). Serum concentration of CHI3L1 is also elevated in IBD patients (32). Thus, CHI3L1 might be selected as both a target and a marker of pediatric IBD.
Cysteine-rich, angiogenic inducer 61 (CYR61 or CCN1) was up-regulated in pediatric IBD. It has been demonstrated that CCN1 up-regulates pro-inflammatory gene transcription, such as TNF-α, IL-1α, IL-1β, IL-6, and IL-12b in mice macrophages (33). This induction results from CCN1 direct activation of NF-κB and increased TNF-α synthesis. CCN1 supports macrophage adhesion through integrin αMβ2 and syndecan-4. Because mice lacking CCN1 cannot develop in utero, involving vascular defects in the placenta, CCN1 is also related with vasculogenesis during embryogenesis (34). Moreover, CCN1 construct transfected mice showed increased angiogenesis in colon tissue (35). Together, these suggest that CCN1 might be a unique target to treat pediatric IBD through inhibition of pro-inflammatory gene expression and angiogenesis.
Chemokine (C–C motif) ligand 2 (CCL2), also known as monocyte chemotactic protein-1 or small inducible cytokine A2, was up-regulated in pediatric inflamed colon tissue. CCL2 attracts monocytes, memory T cells, and dendritic cells to sites of tissue injury, infection, and inflammation (36, 37). Interestingly, CCL2 is located in the confirmed CD and UC susceptibility loci 19q12 (38, 39). Increased expression of CCL2 (Table 1) in pediatric inflamed colon tissue supports the idea that CCL2 might be one of the causal genes of pediatric IBD. Interestingly, this has been demonstrated by nanomolar concentrations of CCL2 stimulating inflammatory responses of monocytes and effector T cells, whereas picomolar CCL2 exerts a global suppressive effect on T-cell trafficking into inflamed lymph nodes (40), as confirmed by picomolar levels of CCL2 ameliorating TNBS- and DSS-induced colitis (41). Thus, before targeting CCL2 for IBD therapy more information is needed regarding its dose effect on human colon tissue inflammation responses.
One major difference of genome transcription in adult IBD versus pediatric IBD colon tissue is that there is fewer common dysregulated genes found in adult IBD. Feng et al. found that 25 genes were up-regulated and 18 genes were down-regulated in adult IBD inflamed colon tissue (42). We found that 65 genes were up-regulated and 18 genes were down-regulated in pediatric IBD colon tissue. Compared with those two studies, CXCL2 and CXCL3 were both up-regulated in pediatric and adult IBD, and only ABCB1 was down-regulated in pediatric and adult IBD. This comparison suggests that there is a large difference between pediatric and adult IBD patients. Additionally, both studies show that there is a difference between CD and UC genome transcription patterns, which suggests that CD and UC have distinctive pathogenesis.
Promoter analysis provided potential targets at the transcription factor level. NF-κB transcription factors are comprised of five family members in mammalian cells: RelA (p65), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2). Those members form homo- or hetero-dimers of NF-κB complexes to regulate the expression of a variety of genes (43). Interestingly, Stronati et al. found that nuclear NF-κB and the binding activity of NF-κB to a consensus DNA sequence were significantly increased in the inflamed mucosa of patients, compared to controls (44). In IBD patients, the increased NF-κB expression in mucosal macrophages is accompanied with an increasing capacity of these cells to produce and secrete TNF-α, IL-1, and IL-6 (45). Many of the established immunosuppressive drugs for IBD, like corticosteroids, play anti-inflammatory roles, at least partly via the inhibition of the NF-κB activity (46). The fact that the NF-κB binding sites are over-represented in the T-cell transfer colitis model, combined with the fact that the NF-κB binding sites are over-represented in pediatric IBD inflamed colon tissue suggests that NF-κB likely plays an important role in pediatric IBD. In agreement with this idea, NF-κB has been extensively studied, and different ways to block NF-κB have been evaluated for IBD treatment. Unfortunately, due to significant side effects and liver toxicity, optimal ways to block NF-κB to treat IBD has not been realized (47).
Interferon regulatory factor-1 and -2 (IRF1 and IRF2) are transcription factors that regulate expression of inflammatory-related genes, but are primarily identified as transcription factors which regulate the human IFN-α/β gene (48). Interestingly, our promoter analysis showed that binding site of IRF1 and IFR2 are over-represented in the pediatric IBD up-regulated genes. Additionally, Clavell et al. found increased expression of IRF1 in lamina propria mononuclear cells from patients with CD (49). Compared with wild-type mice, production of TNF-α and IFN-γ in IRF1−/− mice is greatly impaired (50). Mice with a target mutation in IRF2 (IRF2−/−) exhibit significant inhibition of IL-12, IL-12R, IFN-γ, IL-1 β, and IL-6 expression (51). It has been demonstrated that IRF2 recruits the NF-κB transcription factor into the nucleus via physical interaction, which enhances TNF-α-induced NF-κB transcription (52). Thus, IRF1 and IRF2 have the potential to be selective and potentially effective targets for the treatment of both experimental colitis and pediatric IBD.
In conclusion, we performed pediatric IBD transcriptome analysis and its cross-species comparison with experimental colitis models. Identification of common dysregulated gene expression profiles, over-represented transcription binding sites, and related transcription factors controlling dysregulated gene expression changes reveal several molecular targets that serve as novel pathways for further study and potential therapy for pediatric IBD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by NIH grant DK 43875-18 projects 1 and 4, and Cores A, B, and C. Some work in this study was also supported by grants from the DOD (W81XWH-11-1-0666 to MBG) and NIH (R01-DK091269 to MBG).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2015.00165/abstract
Footnotes
References
1. Park KT, Bass D. Inflammatory bowel disease-attributable costs and cost-effective strategies in the United States: a review. Inflamm Bowel Dis (2011) 17:1603–9. doi:10.1002/ibd.21488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: a systematic review of international trends. Inflamm Bowel Dis (2011) 17:423–39. doi:10.1002/ibd.21349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Shamir R. Nutritional aspects in inflammatory bowel disease. J Pediatr Gastroenterol Nutr (2009) 48(Suppl 2):S86–8. doi:10.1097/MPG.0b013e3181a15ca0
4. Kugathasan S, Baldassano RN, Bradfield JP, Sleiman PM, Imielinski M, Guthery SL, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet (2008) 40:1211–5. doi:10.1038/ng.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Carey R, Jurickova I, Ballard E, Bonkowski E, Han X, Xu H, et al. Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm Bowel Dis (2008) 14:446–57. doi:10.1002/ibd.20342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Fang K, Zhang S, Glawe J, Grisham MB, Kevil CG. Temporal genome expression profile analysis during t-cell-mediated colitis: identification of novel targets and pathways. Inflamm Bowel Dis (2012) 18:1411–23. doi:10.1002/ibd.22842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Fang K, Bruce M, Pattillo CB, Zhang S, Stone R 2nd, Clifford J, et al. Temporal genomewide expression profiling of DSS colitis reveals novel inflammatory and angiogenesis genes similar to ulcerative colitis. Physiol Genomics (2011) 43:43–56. doi:10.1152/physiolgenomics.00138.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Piechota M, Korostynski M, Przewlocki R. Identification of cis-regulatory elements in the mammalian genome: the cREMaG database. PLoS One (2010) 5:e12465. doi:10.1371/journal.pone.0012465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, et al. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res (2008) 36:D102–6. doi:10.1093/nar/gkm955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Lenhard B, Wasserman WW. TFBS: computational framework for transcription factor binding site analysis. Bioinformatics (2002) 18:1135–6. doi:10.1093/bioinformatics/18.8.1135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Iida N, Grotendorst GR. Cloning and sequencing of a new gro transcript from activated human monocytes: expression in leukocytes and wound tissue. Mol Cell Biol (1990) 10:5596–9.
12. Becker S, Quay J, Koren HS, Haskill JS. Constitutive and stimulated MCP-1, GRO alpha, beta, and gamma expression in human airway epithelium and bronchoalveolar macrophages. Am J Physiol (1994) 266:L278–86.
13. Moser B, Clark-Lewis I, Zwahlen R, Baggiolini M. Neutrophil-activating properties of the melanoma growth-stimulatory activity. J Exp Med (1990) 171:1797–802. doi:10.1084/jem.171.5.1797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci U S A (1989) 86:612–6. doi:10.1073/pnas.86.2.612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. O’Donovan N, Galvin M, Morgan JG. Physical mapping of the CXC chemokine locus on human chromosome 4. Cytogenet Cell Genet (1999) 84:39–42. doi:10.1159/000015209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Persson T, Monsef N, Andersson P, Bjartell A, Malm J, Calafat J, et al. Expression of the neutrophil-activating CXC chemokine ENA-78/CXCL5 by human eosinophils. Clin Exp Allergy (2003) 33:531–7. doi:10.1046/j.1365-2222.2003.01609.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Proost P, Wuyts A, Conings R, Lenaerts JP, Billiau A, Opdenakker G, et al. Human and bovine granulocyte chemotactic protein-2: complete amino acid sequence and functional characterization as chemokines. Biochemistry (1993) 32:10170–7. doi:10.1021/bi00089a037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Egesten A, Eliasson M, Olin AI, Erjefält JS, Bjartell A, Sangfelt P, et al. The proinflammatory CXC-chemokines GRO-alpha/CXCL1 and MIG/CXCL9 are concomitantly expressed in ulcerative colitis and decrease during treatment with topical corticosteroids. Int J Colorectal Dis (2007) 22:1421–7. doi:10.1007/s00384-007-0370-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med (1998) 187:2009–21. doi:10.1084/jem.187.12.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Palacin M, Kanai Y. The ancillary proteins of HATs: SLC3 family of amino acid transporters. Pflugers Arch (2004) 447:490–4. doi:10.1007/s00424-003-1062-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch (2004) 447:619–28. doi:10.1007/s00424-003-1067-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Reimer RJ, Edwards RH. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch (2004) 447:629–35. doi:10.1007/s00424-003-1087-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Collins JF, Bai L, Ghishan FK. The SLC20 family of proteins: dual functions as sodium-phosphate cotransporters and viral receptors. Pflugers Arch (2004) 447:647–52. doi:10.1007/s00424-003-1088-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Takanaga H, Mackenzie B, Hediger MA. Sodium-dependent ascorbic acid transporter family SLC23. Pflugers Arch (2004) 447:677–82. doi:10.1007/s00424-003-1104-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Martínez-Augustin O, Merlos M, Zarzuelo A, Suárez MD, de Medina FS. Disturbances in metabolic, transport and structural genes in experimental colonic inflammation in the rat: a longitudinal genomic analysis. BMC Genomics (2008) 9:490. doi:10.1186/1471-2164-9-490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Brudzewsky D, Pedersen AE, Claesson MH, Gad M, Kristensen NN, Lage K, et al. Genome-wide gene expression profiling of SCID mice with T-cell-mediated colitis. Scand J Immunol (2009) 69:437–46. doi:10.1111/j.1365-3083.2009.02243.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Armendariz AD, Krauss RM. Hepatic nuclear factor 1-alpha: inflammation, genetics, and atherosclerosis. Curr Opin Lipidol (2009) 20:106–11. doi:10.1097/MOL.0b013e3283295ee9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Kriström B, Zdunek AM, Rydh A, Jonsson H, Sehlin P, Escher SA. A novel mutation in the LIM homeobox 3 gene is responsible for combined pituitary hormone deficiency, hearing impairment, and vertebral malformations. J Clin Endocrinol Metab (2009) 94:1154–61. doi:10.1210/jc.2008-0325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kawada M, Hachiya Y, Arihiro A, Mizoguchi E. Role of mammalian chitinases in inflammatory conditions. Keio J Med (2007) 56:21–7. doi:10.2302/kjm.56.21
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Chen CC, Llado V, Eurich K, Tran HT, Mizoguchi E. Carbohydrate-binding motif in chitinase 3-like 1 (CHI3L1/YKL-40) specifically activates Akt signaling pathway in colonic epithelial cells. Clin Immunol (2011) 140:268–75. doi:10.1016/j.clim.2011.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Aomatsu T, Imaeda H, Matsumoto K, Kimura E, Yoden A, Tamai H, et al. Faecal chitinase 3-like-1: a novel biomarker of disease activity in paediatric inflammatory bowel disease. Aliment Pharmacol Ther (2011) 34:941–8. doi:10.1111/j.1365-2036.2011.04805.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Vind I, Johansen JS, Price PA, Munkholm P. Serum YKL-40, a potential new marker of disease activity in patients with inflammatory bowel disease. Scand J Gastroenterol (2003) 38:599–605. doi:10.1080/00365520310000537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Bai T, Chen CC, Lau LF. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J Immunol (2010) 184:3223–32. doi:10.4049/jimmunol.0902792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol (2002) 22:8709–20. doi:10.1128/MCB.22.24.8709-8720.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Koon HW, Zhao D, Xu H, Bowe C, Moss A, Moyer MP, et al. Substance P-mediated expression of the pro-angiogenic factor CCN1 modulates the course of colitis. Am J Pathol (2008) 173:400–10. doi:10.2353/ajpath.2008.080222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A (1994) 91:3652–6. doi:10.1073/pnas.91.9.3652
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Xu LL, Warren MK, Rose WL, Gong W, Wang JM. Human recombinant monocyte chemotactic protein and other C-C chemokines bind and induce directional migration of dendritic cells in vitro. J Leukoc Biol (1996) 60:365–71.
38. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42:1118–25. doi:10.1038/ng.717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet (2011) 43:246–52. doi:10.1038/ng.764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Flaishon L, Hart G, Zelman E, Moussion C, Grabovsky V, Lapidot Tal G, et al. Anti-inflammatory effects of an inflammatory chemokine: CCL2 inhibits lymphocyte homing by modulation of CCL21-triggered integrin-mediated adhesions. Blood (2008) 112:5016–25. doi:10.1182/blood-2007-12-129122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Maharshak N, Hart G, Ron E, Zelman E, Sagiv A, Arber N, et al. CCL2 (pM levels) as a therapeutic agent in inflammatory bowel disease models in mice. Inflamm Bowel Dis (2010) 16:1496–504. doi:10.1002/ibd.21254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Wu F, Dassopoulos T, Cope L, Maitra A, Brant SR, Harris ML, et al. Genome-wide gene expression differences in Crohn’s disease and ulcerative colitis from endoscopic pinch biopsies: insights into distinctive pathogenesis. Inflamm Bowel Dis (2007) 13:807–21. doi:10.1002/ibd.20110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol (2007) 8:49–62. doi:10.1038/nrm2083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Stronati L, Negroni A, Merola P, Pannone V, Borrelli O, Cirulli M, et al. Mucosal NOD2 expression and NF-kappaB activation in pediatric Crohn’s disease. Inflamm Bowel Dis (2008) 14:295–302. doi:10.1002/ibd.20332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Neurath MF, Pettersson S, Meyer zum Büschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med (1996) 2:998–1004. doi:10.1038/nm0996-998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Atreya I, Atreya R, Neurath MF. NF-kappaB in inflammatory bowel disease. J Intern Med (2008) 263:591–6. doi:10.1111/j.1365-2796.2008.01953.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kucharzik T, Maaser C, Lügering A, Kagnoff M, Mayer L, Targan S, et al. Recent understanding of IBD pathogenesis: implications for future therapies. Inflamm Bowel Dis (2006) 12:1068–83. doi:10.1097/01.mib.0000235827.21778.d5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol (2001) 19:623–55. doi:10.1146/annurev.immunol.19.1.623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Clavell M, Correa-Gracian H, Liu Z, Craver R, Brown R, Schmidt-Sommerfeld E, et al. Detection of interferon regulatory factor-1 in lamina propria mononuclear cells in Crohn’s disease. J Pediatr Gastroenterol Nutr (2000) 30:43–7. doi:10.1097/00005176-200001000-00016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Senaldi G, Shaklee CL, Guo J, Martin L, Boone T, Mak TW, et al. Protection against the mortality associated with disease models mediated by TNF and IFN-gamma in mice lacking IFN regulatory factor-1. J Immunol (1999) 163:6820–6.
51. Cuesta N, Salkowski CA, Thomas KE, Vogel SN. Regulation of lipopolysaccharide sensitivity by IFN regulatory factor-2. J Immunol (2003) 170:5739–47. doi:10.4049/jimmunol.170.11.5739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Chae M, Kim K, Park SM, Jang IS, Seo T, Kim DM, et al. IRF-2 regulates NF-kappaB activity by modulating the subcellular localization of NF-kappaB. Biochem Biophys Res Commun (2008) 370:519–24. doi:10.1016/j.bbrc.2008.03.136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: interferon regulatory factor, chemokines, chitinase 3-like 1, transcription, bioinformatics
Citation: Fang K, Grisham MB and Kevil CG (2015) Application of comparative transcriptional genomics to identify molecular targets for pediatric IBD. Front. Immunol. 6:165. doi: 10.3389/fimmu.2015.00165
Received: 10 December 2014; Accepted: 26 March 2015;
Published online: 08 April 2015.
Edited by:
Pietro Ghezzi, Brighton and Sussex Medical School, UKReviewed by:
Manuela Mengozzi, Brighton and Sussex Medical School, UKWenzhong Xiao, Massachusetts General Hospital and Harvard Medical School, USA
Copyright: © 2015 Fang, Grisham and Kevil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Fang, Division of Digestive Diseases, Inflammatory Bowel Disease Center, David Geffen School of Medicine at UCLA, 675 Charles E Young Drive S, MRL1519, Los Angeles, CA 90095-7019, USA e-mail:a2ZhbmdAbWVkbmV0LnVjbGEuZWR1;
Christopher G. Kevil, Department of Pathology, Louisiana State University Health Science Center Shreveport, 1501 Kings Highway, Shreveport, LA 71103, USA e-mail:Y2tldmlsQGxzdWhzYy5lZHU=