 Christoph Eckert
Christoph Eckert Niklas Klein
Niklas Klein Miroslaw Kornek
Miroslaw Kornek Veronika Lukacs-Kornek
Veronika Lukacs-Kornek- Department of Medicine II, Saarland University Medical Center, Homburg, Germany
In recent years, it has been an explosion of information regarding the role of various myeloid cells in liver pathology. Macrophages and dendritic cell (DC) play crucial roles in multiple chronic liver diseases such as fibrosis and non-alcoholic fatty liver disease (NAFLD). The complexity of myeloid cell populations and the missing exclusive marker combination make the interpretation of the data often extremely difficult. The current review aims to summarize the multiple roles of macrophages and DCs in chronic liver diseases, especially pointing out how these cells influence liver immune and parenchymal cells thereby altering liver function and pathology. Moreover, the review outlines the currently known marker combinations for the identification of these cell populations for the study of their role in liver immunology.
Liver as an Immune Milieu
The liver functions as a metabolic center to ensure the proper processing of nutrients and the clearance of toxins; yet, plays multiple roles in systemic immune responses and in immune surveillance. The liver receives blood from both the systemic circulation and the intestine that mixes within the liver sinusoids (1). Approximately, two-third of the hepatic blood flow procures from the oxygen rich arteria hepatica and one-third is from the vena porta carrying microbial and food-derived antigens and molecules (1, 2). The mixed blood travels through the sinusoids that are specialized blood vessels lined by the liver sinusoidal endothelial cells (LSECs). LSECs assemble a discontinuous endothelium that is in contact with various passenger and organ-resident immune cells (3). Besides LSECs, the liver contains other parenchymal cells such as hepatocytes and hepatic stellate cells (HSCs). The activation status and extracellular matrix production of HSCs are critical for the progression of multiple liver diseases (4, 5). Importantly, these liver parenchymal cells interact with the variety of immune cells, influence memory T cells, respond to danger signals, and additionally take on the role of antigen presenting cells (APCs) within the liver (6, 7). As APCs, they present antigens in the context of immunosuppressive cytokines and inhibitory surface molecules resulting largely in tolerance (6, 7). The liver also encompasses large populations of hematopoetic cells such as innate lymphocytes (NK, NKT cells, and γδT cells) and myeloid cells [dendritic cells (DCs) and macrophages] (6). Multiple cross-talks exist between hematopoetic cells and liver parenchymal cells at steady state and during injury. This review focuses on the physiological and pathological roles of liver DCs and macrophages paying special attention to chronic liver diseases such as fibrosis and non-alcoholic fatty liver disease (NAFLD).
Liver Dendritic Cells
Dendritic cells are present in all tissues and represent the major APCS within the body (8). They constantly sense their environment and capable of recognizing pathogens and various danger signals. Activation of DCs results in their maturation toward several functionally distinct “effector DCs” (9) that drive T cell responses, such as T helper cell differentiation, induction of CTL, and T cell tolerance (9). Additionally, DCs communicate with innate lymphocytes (e.g., NK, NKT cells), therefore, can influence both innate and adaptive immune responses (8).
Murine liver DC population, similarly as in most non-lymphoid organs (except the lamina propria and dermis), consists of three types of DCs (Table 1): the cDC1s (classical type 1 DCs), the cDC2s (classical type 2 DCs), and pDCs (10, 11). Despite of this categorization, in most liver studies, DCs are evaluated as either CD11c+ or MHCII+ cells. Although neither of the molecules pinpoint exclusively DCs, using these markers liver DCs are primarily located within the portal area and rarely scattered within the parenchyma (6). The cDC1 cells resemble lymphoid tissue CD8+ DCs, show migratory capacity in various non-lymphoid organs, and can efficiently cross-present cell-associated antigens (10, 11). Although the role of DC migration in liver pathology has not been explored in details, antigen injected or targeted to the liver reaches the draining LN and induces T cell activation (12, 13). Additionally, migratory DCs could be identified within the portal lymphatic vessels in electronmicroscopy analyses (1, 14).
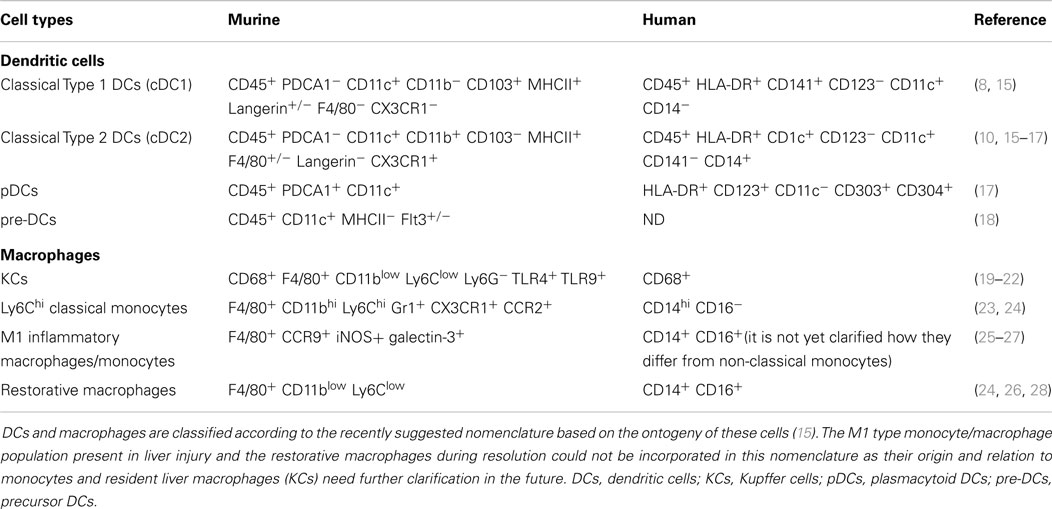
Table 1. Summary of DC and macrophage population in healthy and injured liver.
The cDC2s within non-lymphoid organs are heterogeneous and partially monocyte lineage derived (10, 11). Their specific role is less understood in non-lymphoid organs, involving the liver as well. While the development of this subset depends in most non-lymphoid organs on the presence of FLT3L and M-CSF, in the liver, these cells are not exclusively dependent on these growth factors but yet on an unidentified molecule (10). The liver, similarly to other non-lymphoid tissues, contains not only fully differentiated DCs but precursor DC population as well. From these pre-DCs, either FLT3L or GM-CSF can induce liver DC development bestowing DC homeostasis in situ (29, 30).
Functionally, CD11c+ cells isolated from healthy mouse liver are less mature, have lower capacity to endocytose antigen, and induce less efficient allogenic T cell activation as secondary lymphoid organ (SLO)-derived DCs (31, 32). The inhibitory/tolerogenic capacity of liver DCs could be attributed to the specific microenvironment provided by parenchymal cells of the liver. Fibroblastic and VCAM+ cells derived from the liver could induce hematopoetic progenitor cells to differentiate toward tolerogenic DCs in vitro that can inhibit experimental autoimmune hepatitis (33). It is assumed that circulatory DCs during their translocation within the liver sinusoids toward the lymphatics receive such tolerogenic education from liver parenchymal cells (14, 34). Yet, its in vivo relevance needs to be elucidated.
Freshly isolated murine liver CD11c+ cells promote Th2 rather than Th1 T cell differentiation and via interacting with NK cells induce regulatory T cell (Treg) development (35, 36). Moreover, liver DCs produce increased amount of IL-10, IL-27 but less IL-12 upon LPS stimuli (37, 38). This hyporesponsive behavior toward TLR stimuli, known as endotoxin tolerance, involves LPS/TLR4 but also extends toward other TLRs (6). This is especially important, as the liver is constantly exposed to gut derived microbial products. The breakdown in this tolerance could be observed in colitis where pro-inflammatory DC/macrophage population expands within the liver due to the increased amount of bacterial products present in the portal blood. This creates an inflammatory environment in the liver despite the absence of direct liver damage (39). The tolerant state toward TLRs is an active process and involves the action of various negative regulators of the TLR signaling pathway (6). Interestingly, under steady state, liver DCs rather respond to ECM stimuli (collagen-type I, laminin, fibronectin) that induces MHC-II upregulation and maturation of GM-CSF expanded liver DCs in vitro (40).
In humans, the cDC2 cells (CD11c+ BDCA1+) are the most abundant in the liver and they exhibit similar immature, tolerogenic capacity as their murine counterpart (16, 41) (Table 1). The cDC1 cell population that expresses CD141+ has been recently identified as a counterpart of murine CD8α+ cells (42). These cells induce pro-inflammatory allogeneic MLRs, resulting in IFN-γ and IL-17 production by activated T cells (17). Importantly, as opposite to cDC2s and pDCs, cDC1s (identified in the study as CD141+ cells) were markedly decreased during liver diseases but among the DC-subsets produced the highest level of IFN-λ (17). It is possible that functional differences are reflected among the DC subsets and each subset represents different aspects of liver immunity and tolerance. In line with this, a classification of murine liver DCs according to their lipid content distinguishes between immunogenic and tolerogenic liver DCs. Due to their acetyl-CoA carboxylase activity, HL-DCs (high lipid DCs) mount strong immunogenic CTLs while the LL-DCs (low lipid DCs) with low lipid content are tolerogenic (43). Notably, the marker combinations used for this study showed that both HL-DCs and LL-DCs include multiple DC-subsets distinguished by currently known surface markers and were not restricted to one specific subset. Novel surface molecules are needed to specifically explore their functional diversity.
pDCs are the major source of type-I IFN, regulate NK cell activity, and play important role in the induction of antiviral immunity (44, 45). The murine liver is especially rich in pDCs; yet, the human counterpart contains a smaller proportion of this population among all DCs (17) (Table 1). Under steady state condition, pDCs express low level of costimulatory molecules, are weak T cell stimulators, and induce apoptosis in activated T cells in a Treg dependent manner (46). Later could indicate a cellular interplay between pDCs and Tregs in the liver microenvironment in order to maintain the tolerogenic milieu. Accordingly, pDCs can induce efficient CD4 and CD8 T cell tolerance to orally administered antigens (47).
Microbial products, such as muramyl dipeptide present in the portal blood, upregulate PDL-1 in pDCs and reduce their response to TLR9 stimuli (48). This is another example for the TLR-mediated hyporesponsiveness (“endotoxin tolerance”) in the liver. Strikingly, upon FLT3L treatment, the expanded liver pDCs display strong immunostimulatory properties (49). It is unclear whether this could be due to the expansion of a specific subpopulation of pDCs, or their modified interaction with Tregs, or the result of the complete rearrangement in the myeloid cell compartment, and the consequent imbalance in the tolerogenic milieu.
Taken together, the multiple DC subsets within the liver participate in guarding the tolerogenic environment and primarily skewed toward suppressing T cell responses and toward induction of Tregs. While DCs are the main APCs and inducers of T cell immunity in SLOs, within the liver environment the question still remains: how immunity can be induced by DCs in such suppressive microenvironment? Induction of immunity might be attributed to special DC subpopultaions such as the CD141+ cDC1s in humans (17) and the CD103+ cDC1s in mice (50). Moreover, the appearance of novel DC population, such as monocyte-derived DCs present in iMATEs (intrahepatic myeloid-cell aggregates for T cell population expansion), participates in efficient CLT expansion within the liver (51). Alternatively, immunity is induced by migratory DCs reaching the draining LN, thus, outside of the liver suppressive environment. In line with this, antigen specifically expressed in draining LN results in hepatitis inducing CD8 T cell activation, while the same antigen within the liver induces tolerance (52). Additionally, the liver can provide newly formed structures for T cell activation resulting in immunity. Portal tract associated lymphatic structures (PALPs) during Propionibacterium acnes granuloma formation and tertiary lymphoid structures in biliary cirrhosis represent locations where possible T and B cell activation takes place, respectively (53, 54).
Kupffer Cells – Resident Macrophage Population of the Liver
Kupffer cells (KCs) are tissue resident macrophages and they represent the largest hematopoetic cell population within the liver. They arise from yolk sac during fetal development (55), adjust themselves to the local microenvironment (56, 57), and renew their population at steady state locally throughout adult life with no or minimal contribution of hematopoetic progenitors or blood monocytes (58–60). In mice, KCs can be distinguished from monocytes among the F4/80+ cells as Ly6Clow CD11blow cell population (20, 21) (Table 1) and possess functional specifications according to their positioning within the sinusoid (61). Recent study could distinguish two KC functional groups: the one with higher phagocytosis capacity and the one with preference toward cytokine production (61, 62). Additionally, macrophages are functionally grouped into two classes M1 and M2. While such plain classification is questionable and often overstated, still provide a simple but distinguishable concept for functional categorization of these cells. M1 (termed classically activated) macrophages are pro-inflammatory, while the M2 (termed alternatively activated) macrophages are suppressive and involved in cellular repair (63). According to this, KCs belong to the M2 type of cells and play fundamental role in homeostasis, immune surveillance, and tissue repair (63).
Their importance as tolerogenic APCs in the liver microenvironment is demonstrated in liver transplantation where they prolong allograft survival (31). At steady state, they inhibit DC mediated T cell activation within the sinusoids and presentation of high affinity peptide by KCs results in deletional CD8 T cell tolerance (6, 64). Furthermore, they promote the suppressive capacity of Tregs toward hepatic antigens (65, 66).
As all tissue resident macrophages, KCs express a wide repertoire of receptors for the recognition of pathogens and danger signals such as Toll-like receptors, members of the inflammasome, and scavenger receptors (31). In the presence of TLR ligands such as LPS and CpG, KCs become immunogenic, and can induce CD8 T cell activation, and the generation of efficient CLT response (67, 68). Thus, during liver infection, they support the development of antimicrobial T cell responses. Unfortunately, KCs induce efficient CTL against antigens from the systemic circulation such as the case in influenza infection (69). This CTL response results in bystander hepatitis, often accompanying systemic viral infections. Besides CD8 T cell responses, recent study describes naive CD4 T cell activation in the murine liver by antigens expressed in hepatocytes. This process is independent from lymphoid tissue but dependent on clodronate-sensitive liver APC population possibly involving KCs as well (70). Thus, KCs participate in the generation of both CD4 and CD8 T cell responses.
Using their scavenger receptor repertoire, KCs are involved in the clearance of apoptotic cell debris and central to iron homeostasis (71). KCs interact with multiple immune cells within the sinusoids, such as Tregs, DCs, DC precursors, and innate lymphocytes (7, 53, 72, 73). After recognizing any danger signals, KCs primarily drive the influx of inflammatory leukocytes such as neutrophils and monocytes (63). Thus, KCs function as sentinels and central orchestrators of cellular processes in healthy and injured liver. Additionally, while they support the tolerogenic milieu within the liver, their presence also ensures the protection of the liver during pathogen invasion.
Tools to Study Liver DCs and Macrophages
In order to characterize the specific physiological and pathological roles of DCs and macrophages, various animal models, tools have been developed (Table 2) Among these models, there are mouse lines deficient in transcription factors that are responsible for the development of one or multiple subsets of myeloid cells. Due to the multiple cell types affected in these models, the broader impact of each of these genes makes it difficult to unequivocally pinpoint subset specific functions. Nevertheless, these transgenic animals helped significantly to establish broader understanding of macrophage and DC development and their role under steady state and inflammation (63, 74). However, just few of these models have been evaluated so far in fibrosis and non-alcoholic steatohepatitis (NASH) models (Table 2) but also in liver immunology. This might extend in the future as genetic model lacking cDC1s have just recently demonstrated that cDC1s respond to hepatotropic viral infection and are keys in the induction of anti-viral CD8 T cell response in situ (50).

Table 2. Summary of the available models to study liver macrophages and DCs.
The most frequently used cell-depleting tools in liver immunology are the clodronate liposome mediated depletion of mononuclear cells and the CD11c-/CD11b-DTR (diphtheria toxin receptor) transgenic system (87, 89). Clodronate-encapsulated liposomes are taken up by mononuclear cells and the clodronate bisphosphonate within the cell induces apoptosis that results in depletion of the phagocytic cell population. Multiple phagocytic cell types are affected using this depletion method such as KCs, macrophages, and some members of the DC population as well. Since more than one cell types are affected, the effects can be extrapolated to a group of cells and not to individual subtypes (89). Additionally, the release of inflammatory mediators (such as TNF) has been associated with this type of cell depletion further complicating the interpretation of experimental results (90).
The other widely used tool for liver biology is the CD11c-DTR-based depletion system. Here, the human diphtheria toxin receptor is expressed under the CD11c promoter and administration of diphtheria toxin results in the depletion of CD11c+ cells. This model is used to dissect the role of conventional DCs. The major disadvantage in this system is that multiple cell types are affected such as marginal zone macrophages, monocytes, activated CD8 T cells, NK cells, and plasmacytoid DCs (89). Two different CD11c-DTR mouse lines have been generated: the one encompassing only a short piece of the CD11c promoter (3) and the one with the full-length promoter (91). Although, they differ in the list of affected cell types, they gave important insights in the role of CD11c+ cells in liver immunology (Table 2). Novel DTR tools have been developed in recent years that aim to restrict the expression of DTR more specifically to DCs. The zbtb-46-DTR model uses the transcription factor zbtb46 that is exclusively expressed by DCs and DC-committed precursors (92). Unfortunately, zbtb46 is upregulated in monocytes stimulated with GM-CSF and IL-4, suggesting some limitations to this promoter (93, 94). Another promising promoter is the DC NK lectin group receptor-1 (DNGR-1) that seems to be highly restricted to the DC linage (95). Not only for DCs but also for the study of macrophages, the perfect targeting tool still needs to be developed. The primary tool for analyzing macrophages is the CD11b-DTR system. However, CD11b is a widely expressed marker among multiple immune cell types causing caveat for the interpretation of cell types using this model for understanding liver diseases (Table 2) (87).
To follow myeloid cells in situ using in vivo imaging, flow cytometry or microscopy multiple reporter mouse models have been developed. The promoters from different molecules such as e.g., CD11c, Csfr1, CCR2, MHC-II, or CX3CR1 were used to generate these animal models (88) (Table 2). These models have their own limitations according to their expression profile that have been reviewed elsewhere (88) (Table 2). Some of the promoters are also utilized to express Cre recombinase (Table 2). Crossing these Cre expressing lines with animals carrying a flox-ed gene allows the analyses of the cell specific depletion of the gene of interest. Certainly, the specificity and limitation of the models are determined by the expression pattern of the promoter used for Cre expression (87). Despite the availability of these models, only limited have been exploited for understanding specifically liver fibrosis and NASH (Table 2).
Taken together, multiple models are available to answer liver immunological questions. While each of the available models has its own limitation, the combination of these models with each other can still pinpoint important contribution of DCs and macrophages in liver pathology.
The Role of Dendritic Cells and Macrophages in Chronic Liver Diseases
Liver Fibrosis Progression
Liver fibrosis is a common endpoint of many chronic liver diseases such as viral hepatitis, primary biliary cirrhosis, alcoholic and NASH, or autoimmune liver disorder (96, 97). To investigate liver fibrogenesis, several rodent models have been developed inducing toxic (CCL4), biliary (bile duct ligation), oxidative (TAA-induced), or metabolic (MCD/methionine choline deficient diet induced) liver damage (96, 97). The MCD diet contains high sucrose and fat (usually 40% sucrose and 10% fat) but lacks the amino acid methionine and the small molecule choline that are essential for hepatic β-oxidation and production/secretion of very low density lipoprotein (VLDL). As a result, lipids are deposited in the liver and steatosis, and NASH develops in these animals (98).
Remarkably, even though the molecular mechanisms leading to hepatic cell death are very different, the process of fibrogenesis and the cellular components involved share common hallmarks. Such common components, that have established their role in liver fibrosis, are the macrophages and the recruited inflammatory monocytes.
Major evidence for the involvement of macrophages in liver fibrosis is demonstrated in in vivo depletion studies using the CD11b-DTR system and the clodronate-liposome mediated cell depletion. In CCL4 induced liver injury, the progression of fibrosis was attenuated in the absence of CD11b+ cells and the number of HSC-derived myofibroblasts was greatly reduced (79). The administration of clodronate liposomes similarly suggested that macrophages are pro-fibrogenic and affect the survival of HSCs via TNF and IL-1 induced NF-kb signaling (84, 99). Liver macrophage populations change during liver injury. One of the major changes is the recruitment of inflammatory monocytes to the injured liver and their differentiation toward tissue macrophages (24, 26, 28, 100). Resident KCs in liver injury rapidly secrete pro-inflammatory cytokines such as IL-1β, TNF, CCL2, and CCL5 resulting in recruitment of multiple immune cells involving monocytes as well. The accumulation of circulating Ly6Chi monocytes within the liver is greatly dependent on CCR2/CCL2 and CCL1/CCR8 axis (100). The monocyte recruiting chemokines, however, not only originate from KCs but also from TLR-activated HSCs (101). Moreover, senescent hepatocytes and NF-kb-inducing kinase (NIK) activation in hepatocytes lead to the release of numerous chemokines (86, 102). These chemokines can influence the migration or activation state of macrophages that in turn induce hepatocyte apoptosis. Accordingly, hepatocyte-specific expression of the NIK in vivo triggers massive liver inflammation and hepatocyte apoptosis leading to liver fibrosis (86). Thus, the macrophage–hepatocyte cross-talk seems to greatly influence cell recruitment and the activation state of macrophages, thereby affecting the progression of liver injury. The fact that in the above study KC/macrophage depletion using clodronate reversed NIK-induced damage, also strongly suggests this.
Monocyte recruitment to the injured liver can be observed early within 24 h after the induction of CCL4 damage (25). These early recruited cells are CCR9+, colocalize and interact with CCR9+ HSCs (27). Furthermore, these monocyte-derived macrophages are characterized as CD11b+F4/80+iNOS+ cells that exhibit profibrogenic properties via promoting HSC activation, Th1 cell differentiation, and TGFβ release (25, 26). In addition to this, profibrogenic Ly6Chi macrophages express PDGF, IL-13 and IL-4 that directly act on HSC derived myofibroblasts and induce ECM production (25, 26). Macrophages produce various lectins among them galectin-3 is required for TGFβ mediated myofibroblast activation and matrix production that further underline their profibrogenic capacity (103).
Another chemokine that affects the infiltrating monocytes is the fractalkine receptor (CX3CR1). Fractalkine is released by hepatocytes and HSCs during liver injury. It ensures the survival of infiltrating monocytes and influences their differentiation toward tissue macrophages (25). In the absence of CX3CR1, infiltrating monocytes develop into highly inflammatory macrophages that die early via apoptosis. This perpetuates further inflammation and recruitment of Ly6Chi cells. Additionally, CX3CR1 on KCs increase their IL-10 expression and reduces their TNF and TGFβ (104). Thus, fractalkine represent a negative feedback on the extension of liver inflammation through affecting KCs and the presence and destiny of Ly6Chi cells at least in the murine system. It requires future research to clarify how the changes in monocyte and macrophage subsets observed in mice are reflected in humans.
Regarding the M1–M2 classification of macrophages, during the progression phase of liver fibrosis and during fibrosis resolution, both types of cells are present in the liver side by side (105). Interestingly, based on histological analyses these M1 and M2 macrophages localize near to the fibrotic septa and could indicate further undiscovered cross-talk among these cells in liver pathology. Of note, transcriptional analyses of macrophages that are present in the resolution phase display a profile that cannot be classified according to the M1/M2 nomenclature (28).
Accumulation of macrophages within the injured liver caused just partially by the recruited monocytes and their differentiation toward tissue macrophages. There is some evidence that local multiplication of resident and monocyte-derived macrophage population contribute to this process. Ki67 staining during CCL4 mediated liver injury demonstrated the presence of proliferating KCs and monocyte-derived macrophages (28, 62). In most recent study, Listeria infection of the liver resulted in monocyte-derived macrophage proliferation via IL-4 and IL-33 (106). Whether these cytokines are also involved in this process during other types of liver injury and in humans as well remain to be elucidated.
Multiple animal studies reported that the number of dendritic cells, pre-DCs, and pDCs increase during the progression phase of liver fibrosis (76, 107). This raised the assumption that DCs might contribute to fibrosis progression. Using the CD11c-DTR model, it has been demonstrated that CD11c+ cells provide a pro-inflammatory milieu by producing IL-1β and TNF during injury (76). Moreover, isolated cells contribute to HSC survival in vitro suggesting a clear profibrogenic capacity of these cells (37). This phenomenon, despite of the relatively broad CD11c expression among other myeloid cells as discussed above (Tables 1 and 2), was attributed to DCs.
Another study determined using the same CD11c-DTR system that DC depletion accelerates the development of fibrosis due to their influence on angiogenesis. DCs seem to be the source of the anti-angiogenic VEGF receptor 1 (also known as sFlt-1) and thus influence the bioavailability of VEGF during fibrogenesis (78). Notably, recent study has demonstrated that VEGF+ inflammatory monocytes/monocyte derived macrophages colocalize with newly formed vessels in injured liver and pharmacological inhibition of CCL2 mediated recruitment of inflammatory monocytes reduces fibrosis-induced angiogenesis without affecting fibrosis progression (108). Thus, recruited monocytes/macrophages seem to counterbalance the anti-angiogenic property of DCs during fibrosis progression. Whether classical DCs or pDCs truly contribute to fibrosis progression or play other role during liver injury still remain to be clarified in the future.
Liver Fibrosis Regression
During liver fibrosis, the increased production of ECM is accompanied by high expression of MMPs and the presence of collagenase activity, suggesting alterations and adjustments in the fibrotic ECM. In fact, the fibrotic ECM seems to be different biochemically than ECM produced during a steady state turnover (109). In lung fibrosis, the pathological ECM activates fibroblastic cells to build further matrix indicating a positive cross-talk between fibroblast and matrix components (109). Also during liver fibrosis, heavily cross-linked, modified ECM could be identified (110); however, it remains to be elucidated whether similar regulatory loop as in the lung operates in liver fibrosis as well.
Importantly, after removal of the noxious agents causing liver damage, fibrotic scars degrade and normal liver architecture can be restored. This process is called resolution. While this functions well in various animal models, in humans this seems to be a point of no return where fibrosis and cirrhosis progresses nonetheless (97).
In resolution, the role of macrophages has been demonstrated in multiple animal studies. Depletion of CD11b+ cells during fibrosis progression, as above discussed, reduced scarring while during fibrosis resolution led to a failure in matrix degradation (79, 80). This strongly suggests the dominant presence of two functionally different macrophage populations. According to this, Ramachandran et al. have identified a subset of Ly6Clow “restorative” macrophages during resolution (28). These cells originated from Ly6Chi recruited monocytes expressed MMPs including MMP9, MMP12, and phagocytosis related genes. Importantly, based on gene expression profiling, they could not be fit in the M1/M2 macrophage classification. Moreover, phagocytosis of liposomes or cellular debris by liver macrophages could recapitulate this type of restorative phenotype (28). In addition to this, recent study demonstrated that scar associated myeloid cells attract endothelial cells to the scar tissue via VEGF and that genetic ablation of VEGF in myeloid cells resulted in the increase of MMP2 and MMP13 and decrease of TIMP1 in the liver. While macrophages have not been unequivocally identified as myeloid cells in this study, the results indicate that the myeloid cells induced angiogenesis gears the balance toward fibrolysis (111). This is in line with recent findings that demonstrated that VEGF signaling plays key role during liver fibrosis resolution. Anti-VEGF antibody treatment during resolution led to impaired tissue repair. Mechanistically, VEGF regulated endothelial permeability, monocyte recruitment, and affected the CXCL9 and MMP13 expression of scar-associated macrophages. Importantly, depletion of Cfsr1+ cells (including macrophages, monocytes, and DCs Table 2) impaired fibrosis resolution (112).
Based on these findings, macrophages can be grouped in profibrogenic and restorative macrophage populations beyond the M1/M2 scheme, a classification that might be much more beneficial for finding new targets for fibrosis therapy. However, multiple open questions remain concerning the balance of the heterogeneous population in liver diseases and the relation to each other. One molecule could provide a better understanding to the problem, the chemokine CX3CL1. Ramachandran et al. showed a higher expression of CX3CR1 within the restorative macrophage population, then in the profibrotic subset (28). Consistent with these findings, HSC and hepatocyte-derived fractalkine led to the induction of Arginase 1 in a mixed Kupffer-cell/macrophage cell population, a marker that has been associated with the fibrolytic macrophage subset (28, 104). Thus, an intriguing possibility is the progressive class switch between macrophage populations during fibrosis progression and regression. This possibility is underlined by the fact that the overall number of profibrogenic Ly6Chi macrophages strongly decreases in resolution despite the presence of their strong proliferation activity at early time points of fibrosis regression. At the same time, the number of Ly6Clo macrophages increases (28). Along this line, blocking CCL2 dependent liver infiltration by Ly6Chi monocytes during fibrosis regression leads to a higher relative amount of Ly6Clo macrophages (113). Moreover, the Ly6Clo macrophages could be shown to be postphagocytic and seem to appear in the phase of reduced hepatocyte death, further supporting the switch concept (28). It remains to be clarified in the future how the macrophage populations interact and relate to each other. Similarly as the murine restorative macrophages in humans, this population is likely represented by the CD14+ CD16+ cells (24, 26). They display phagocytic activity but as opposite to the murine cells express a variety of pro-inflammatory and pro-fibrogenic molecules as well.
Besides macrophages, DCs have also been implicated in liver fibrosis resolution. Jiao et al. have demonstrated that depletion of CD11c+ cells leads to delay in fibrosis resolution and delayed clearance of activated HSCs. To more precisely pinpoint DCs in this process, adoptive transfer of purified DCs or expansion of endogenous DCs using FLT3L could accelerate regression. Moreover, DCs were the source of MMP9 and therefore seem to complement restorative macrophages in this process (75).
Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome that includes hypertension, hyperlipidemia, insulin resistance, and visceral adiposity, and shows a worldwide increasing tendency among chronic liver diseases (114, 115). In most cases, the liver steatosis is mild. However, up to one-fifth of the cases progresses toward NASH that is characterized by intrahepatic inflammation, increased steatosis with hepatocellular ballooning, and often accompanied by progressive fibrosis (114, 115). NASH is prone to the development of cirrhosis and liver cancer (115). While the precise cellular and molecular mechanisms of NASH are not yet fully understood, multiple studies have investigated macrophages and DCs in this disease.
Similarly as during liver fibrogenesis, in NASH, the two main components that show alterations are the response of macrophages/KCs and the recruited inflammatory monocytes. The key role of macrophages/KCs in NASH has been demonstrated in studies where these cells were specifically depleted using gadolinium chloride or clodronate liposomes (81–83, 85). In the absence of KCs, the steatohepatitis was markedly reduced. In addition to this, KCs display an M1 TNF expressing pro-inflammatory phenotype and increase triglyceride accumulation, decrease fatty acid oxidation and insulin responsiveness of hepatocytes (82, 83). KC-derived TNF production seems to be central in NASH development, as silencing liver TNF or using TNFR1/2 deficient animals attenuate liver steatosis compared with control wild-type animals (85, 116).
Multiple triggers have been identified for KC activation and for the induction of their pro-inflammatory cytokine production in NASH. TLR4 deficient animals showed reduced liver damage and KC depletion prevented the increase in TLR4 expression during MCD diet (81). Bacterial product induced KC activation is in accordance with clinical data that demonstrate bacterial translocation in NASH patients (117). Notably, TLR4 can be triggered not only by LPS but also by free fatty acids and high mobility group box-1 protein (HMGB1) that is increased in obesity and during hepatocyte injury. Not only LPS but also translocated nucleic acids have been implicated in the development of NASH via triggering TLR9 mediated KC activation and IL-1β release (118).
Lipidomics and mass spectrometry profiling revealed that KCs accumulate toxic lipids due to the dysregulation of lipid metabolism during high fat diet. Moreover, these lipid-loaded KCs produce pro-inflammatory cytokines and chemokines (119). The balance between the M1 and M2 type of KCs seem to be a key for NASH progression. Mice fed with high-fat diet displayed a predominant M2 KC polarization, the apoptosis of M1 KCs and resistance to hepatocyte steatosis. In vitro experiments demonstrated that M2 macrophages release IL-10 that in return increase the sensitivity of M1 macrophages to undergo apoptosis (120).
The other hallmark of NASH is the increased monocyte recruitment to the injured liver. Activated KCs upregulate their MCP-1 expression that is the major chemokine involved in the recruitment of Ly6Chi cells. These Ly6Chi cells are pro-inflammatory and further perturb hepatic inflammation (85). Consequently, CCR2 deficient animals show decreased steatosis (113, 121). On the other hand, CCR2 signaling, when MCD diet is replaced with control diet, counteracts tissue resolution by perpetuating inflammation (113). This is a similar phenomenon as observed in fibrosis resolution (113).
Non-alcoholic steatohepatitis is associated with increased number of hepatic DCs identified by MHCII+ CD11c+ cells (77). Depletion of these cells using CD11c-DTR mouse model exacerbates hepatic inflammation whereas during the resolution phase delays the reconstitution of normal tissue homeostasis. Importantly, these cells take up apoptotic cells, inhibit TLR expression, T cell expansion, and cytokine production by innate cells (77). This strongly suggests DCs as an important negative regulator of NASH inflammation. As opposite to this, another study has classified CD11c+ cells during MCD-diet based on their lipid content (43). It remains to be clarified whether the tolerogenic LL-DC (low lipid DC) population is equivalent with the same immunoregulatory DCs in NASH as described by Henning et al. Of note, based on the surface marker expression profile of HL-DCs and LL-DCs, they rather seemed to be a part of a heterogeneous population, despite that all cells expressed various level of CD11c (43).
Summary and Conclusion
Taken together, the liver represents a unique immunological niche within the body. Its parenchymal and non-parenchymal cells guard its tolerogenic and suppressive microenvironment while supporting its sentinel task of the portal and systemic circulation (Figure 1). Most liver injuries trigger the activation of resident KC/macrophage population that rapidly releases pro-inflammatory mediators such as TNF and IL-1β. This is followed by a chain of events that seem to be commonly shared by many injuries causing NASH and leading to liver fibrosis. The response involves the alterations within the myeloid cell composition primarily affecting macrophages. Importantly, other immune cells such as DCs, neutrophils, innate cells, and activated T cell are also recruited to the injured liver and play various roles in disease progression (6, 97). The exact role of liver DCs during chronic liver injury is yet to be determined. Nevertheless, they seem to be similarly pro-inflammatory as the Ly6Chi recruited monocytes. This feature is shared with infectious liver diseases; thus, it supposes to induce liver protecting immunity (2, 7). During chronic liver diseases, the overwhelming presence of pro-inflammatory immune cells together with liver damaging noxious agents eventually lead to extensive cell death and scar formation, a common outcome for chronic liver disorders. While KC activation alarms other immune cells to travel to the liver, it influences metabolic processes and survival of hepatocytes. During disease progression, Ly6Chi cells seem to develop into Ly6Clo restorative macrophages. These cells, if the harmful agent vanishes, lead to resolution and can restore normal tissue architecture (Figure 1). Especially, in this process, DCs are complementing the macrophage population. In infection, recent report demonstrated that necroptosis of KCs was necessary to induce the Th2 mediated tissue repair (106) that remains to be tested to affect fibrosis resolution in the future. Equally important is the more detailed understanding of the factors involved in the switch from the pro-inflammatory to the restorative macrophage population.
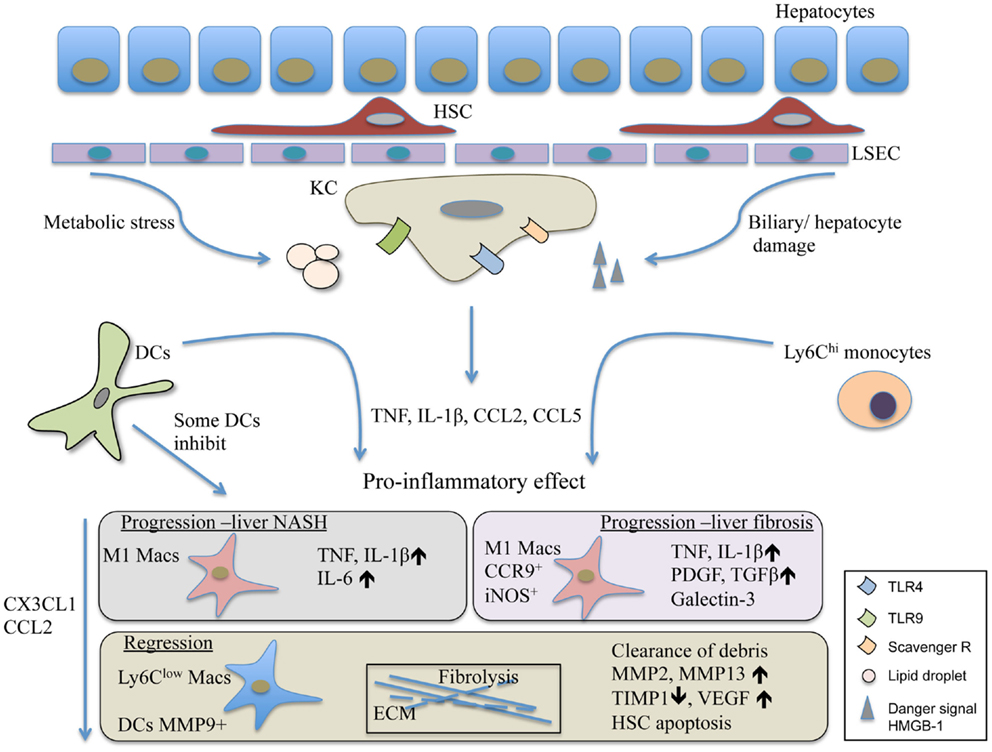
Figure 1. The contribution of DCs and macrophages to the pathomechanism of liver fibrosis and NASH. Liver injury triggers the activation of Kuppfer cells, the resident macrophage population of the liver. Their activation leads to the release of inflammatory mediators and chemokines such as TNF, IL-1β, and CCL2. This is followed by the recruitment of various immune cells involving inflammatory monocytes and DCs. The Ly6Chi monocytes differentiate into M1 CCR9+iNOS+ macrophages, and together with DCs in the progression phase of liver injury, act in a pro-inflammatory manner and perpetuate inflammation. Some DCs, possibly the LL-DCs, seem to inhibit liver steatohepatitis and protect liver damage. In resolution, the Ly6Clow restorative macrophages together with MMP9+ DCs promote fibrolysis and the restoration of normal tissue architecture. HMGB-1, high mobility group box-1 protein; HSC, hepatic stellate cells; KC, Kupffer cells; LL-DC, low lipid containing DCs; LSEC, liver sinusoidal endothelial cells.
Despite of the significant amount of data available in mice, we have just limited understanding about the course of events in human liver diseases. It will need future studies to analyze DC, monocyte, and macrophage populations within human liver samples not only phenotypically and functionally but also on genomic level in comparison with their murine counterparts. This can lead to better understanding of liver diseases but also for identifying novel therapeutic targets. A promising clinical perspective is to target chemokines in the early phase of the liver response to avoid inflammatory cell recruitment and further inflammation. One possibility is affecting the CCL2 axis. Currently, Cenicriviroc, an inhibitor of CCR2, is tested (Centaur study, phase 2 clinical trial, NCT:022117475) to attenuate fibrosis progression in NASH patients. Along this line, other chemokines that could affect the differentiation of monocytes to inflammatory macrophages could be a possible target in the future. Additionally, DCs and restorative macrophages could become novel objectives for inducing fibrolysis and reversing liver damage. Notably, autologous transfer of expanded mononuclear cells to chronic viral hepatitis-associated fibrotic patients showed improved outcome as indicated by reduced Child-Pugh score (122), suggesting a great potential of myeloid cell transfer-based therapeutic procedures in the future.
Author Contributions
CE, NK contributed to the writing of the manuscript, prepared the tables. MK supervised the students and critically read the manuscript. VL-K developed the concept of the manuscript, supervised, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Alexander von Humboldt Foundation, Sofja Kovalevskaja Award to VL-K by a Deutsche Krebshilfe grant #111184 to MK.
Abbreviations
APC, antigen presenting cell; BDL, bile duct ligation; BM, bone marrow; DCs, dendritic cells; Batf3, basic leucine zipper transcription factor, ATF-like 3; CCL4, carbon tetrachloride; CCR9, chemokine (C–C motif) receptor 9; CTL, cytotoxic T lymphocyte; DNGR-1, DC NK lectin group receptor-1; DT, diphtheria toxin; DTR, diphtheria toxin receptor; ECM, extracellular matrix; FA, fatty acid; Flt3L, FMS-like tyrosine kinase 3 ligand; GM-CSF, granulocyte macrophage colony-stimulating factor; HSC, hepatic stellate cells; HL-DC, high-lipid liver dendritic cell; Id2, inhibitor of DNA binding 2; IL, interleukin; KC, Kupffer cells; LL-DC, low-lipid liver dendritic cell; LSEC, liver sinusoidal endothelial cells; LPS, lipopolysaccharide; MCD, methionine choline deficient; MCP-1, monocyte chemotactic protein-1; MMP-9, matrix metalloproteinase-9; MMP13, matrix metalloproteinase-13; NASH, non-alcoholic steatohepatitis; pDC, plasmacytoid dendritic cell; PDL-1, programed cell death 1 ligand; TNFα, tumor necrosis factor-alpha; TAA, thioacetamide; TLR, toll like receptor; zbtb46, zinc finger and BTB domain containing.
References
1. Ohtani O, Ohtani Y. Lymph circulation in the liver. Anat Rec (Hoboken) (2008) 291:643–52. doi:10.1002/ar.20681
2. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol (2013) 14:996–1006. doi:10.1038/ni.2691
3. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology (2006) 43:S54–62. doi:10.1002/hep.21060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Winau F, Quack C, Darmoise A, Kaufmann SH. Starring stellate cells in liver immunology. Curr Opin Immunol (2008) 20:68–74. doi:10.1016/j.coi.2007.10.006
5. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol (2013) 3:1473–92. doi:10.1002/cphy.c120035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol (2010) 10:753–66. doi:10.1038/nri2858
7. Crispe IN. Immune tolerance in liver disease. Hepatology (2014) 60:2109–17. doi:10.1002/hep.27254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature (2007) 449:419–26. doi:10.1038/nature06175
9. Hopp AK, Rupp A, Lukacs-Kornek V. Self-antigen presentation by dendritic cells in autoimmunity. Front Immunol (2014) 5:55. doi:10.3389/fimmu.2014.00055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206:3115–30. doi:10.1084/jem.20091756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol (2012) 13:888–99. doi:10.1038/ni.2370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Yoneyama H, Narumi S, Zhang Y, Murai M, Baggiolini M, Lanzavecchia A, et al. Pivotal role of dendritic cell-derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J Exp Med (2002) 195:1257–66. doi:10.1084/jem.20011983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Barbier L, Tay SS, McGuffog C, Triccas JA, McCaughan GW, Bowen DG, et al. Two lymph nodes draining the mouse liver are the preferential site of DC migration and T cell activation. J Hepatol (2012) 57:352–8. doi:10.1016/j.jhep.2012.03.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Matsuno K, Kudo S, Ezaki T. The liver sinusoids as a specialized site for blood-lymph translocation of rat dendritic cells. Adv Exp Med Biol (1997) 417:77–81. doi:10.1007/978-1-4757-9966-8_13
15. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14:571–8. doi:10.1038/nri3712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Bosma BM, Metselaar HJ, Mancham S, Boor PP, Kusters JG, Kazemier G, et al. Characterization of human liver dendritic cells in liver grafts and perfusates. Liver Transpl (2006) 12:384–93. doi:10.1002/lt.20659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Kelly A, Fahey R, Fletcher JM, Keogh C, Carroll AG, Siddachari R, et al. CD141(+) myeloid dendritic cells are enriched in healthy human liver. J Hepatol (2014) 60:135–42. doi:10.1016/j.jhep.2013.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev (2010) 234:45–54. doi:10.1111/j.0105-2896.2009.00879.x
19. Guo S, Yang C, Mei F, Wu S, Luo N, Fei L, et al. Down-regulation of Z39Ig on macrophages by IFN-gamma in patients with chronic HBV infection. Clin Immunol (2010) 136:282–91. doi:10.1016/j.clim.2010.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Nascimento M, Huang SC, Smith A, Everts B, Lam W, Bassity E, et al. Ly6Chi monocyte recruitment is responsible for Th2 associated host-protective macrophage accumulation in liver inflammation due to schistosomiasis. PLoS Pathog (2014) 10:e1004282. doi:10.1371/journal.ppat.1004282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol (2014) 193:344–53. doi:10.4049/jimmunol.1400574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Strauss O, Dunbar PR, Bartlett A, Phillips A. The immunophenotype of antigen presenting cells of the mononuclear phagocyte system in normal human liver – a systematic review. J Hepatol (2015) 62:458–68. doi:10.1016/j.jhep.2014.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology (2009) 50:261–74. doi:10.1002/hep.22950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology (2013) 57:385–98. doi:10.1002/hep.26016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, et al. The fractalkine receptor CX(3)CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology (2010) 52:1769–82. doi:10.1002/hep.23894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Zimmermann HW, Seidler S, Nattermann J, Gassler N, Hellerbrand C, Zernecke A, et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS One (2010) 5:e11049. doi:10.1371/journal.pone.0011049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Chu PS, Nakamoto N, Ebinuma H, Usui S, Saeki K, Matsumoto A, et al. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology (2013) 58:337–50. doi:10.1002/hep.26351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A (2012) 109:E3186–95. doi:10.1073/pnas.1119964109
29. Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol (2007) 8:578–83. doi:10.1038/ni1462
30. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science (2009) 324:392–7. doi:10.1126/science.1170540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Thomson AW, Geller DA, Gandhi C, Murase N, Demetris AJ, Beer-Stolz D. Hepatic antigen-presenting cells and regulation of liver transplant outcome. Immunol Res (2011) 50:221–7. doi:10.1007/s12026-011-8223-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Lukacs-Kornek V, Schuppan D. Dendritic cells in liver injury and fibrosis: shortcomings and promises. J Hepatol (2013) 59:1124–6. doi:10.1016/j.jhep.2013.05.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Xia S, Guo Z, Xu X, Yi H, Wang Q, Cao X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood (2008) 112:3175–85. doi:10.1182/blood-2008-05-159921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Kudo S, Matsuno K, Ezaki T, Ogawa M. A novel migration pathway for rat dendritic cells from the blood: hepatic sinusoids-lymph translocation. J Exp Med (1997) 185:777–84. doi:10.1084/jem.185.4.777
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Khanna A, Morelli AE, Zhong C, Takayama T, Lu L, Thomson AW. Effects of liver-derived dendritic cell progenitors on Th1- and Th2-like cytokine responses in vitro and in vivo. J Immunol (2000) 164:1346–54. doi:10.4049/jimmunol.164.3.1346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Jinushi M, Takehara T, Tatsumi T, Yamaguchi S, Sakamori R, Hiramatsu N, et al. Natural killer cell and hepatic cell interaction via NKG2A leads to dendritic cell-mediated induction of CD4 CD25 T cells with PD-1-dependent regulatory activities. Immunology (2007) 120:73–82. doi:10.1111/j.1365-2567.2006.02479.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Abe M, Tokita D, Raimondi G, Thomson AW. Endotoxin modulates the capacity of CpG-activated liver myeloid DC to direct Th1-type responses. Eur J Immunol (2006) 36:2483–93. doi:10.1002/eji.200535767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Chen Y, Jiang G, Yang HR, Gu X, Wang L, Hsieh CC, et al. Distinct response of liver myeloid dendritic cells to endotoxin is mediated by IL-27. J Hepatol (2009) 51:510–9. doi:10.1016/j.jhep.2009.04.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Mikami Y, Mizuno S, Nakamoto N, Hayashi A, Sujino T, Sato T, et al. Macrophages and dendritic cells emerge in the liver during intestinal inflammation and predispose the liver to inflammation. PLoS One (2014) 9:e84619. doi:10.1371/journal.pone.0084619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Lu L, Woo J, Rao AS, Li Y, Watkins SC, Qian S, et al. Propagation of dendritic cell progenitors from normal mouse liver using granulocyte/macrophage colony-stimulating factor and their maturational development in the presence of type-1 collagen. J Exp Med (1994) 179:1823–34. doi:10.1084/jem.179.6.1823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol (2009) 182:1901–11. doi:10.4049/jimmunol.0803404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity (2012) 37:60–73. doi:10.1016/j.immuni.2012.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology (2012) 143:1061–72. doi:10.1053/j.gastro.2012.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity (2008) 29:352–61. doi:10.1016/j.immuni.2008.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity (2010) 33:955–66. doi:10.1016/j.immuni.2010.11.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Tokita D, Sumpter TL, Raimondi G, Zahorchak AF, Wang Z, Nakao A, et al. Poor allostimulatory function of liver plasmacytoid DC is associated with pro-apoptotic activity, dependent on regulatory T cells. J Hepatol (2008) 49:1008–18. doi:10.1016/j.jhep.2008.07.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity (2008) 29:464–75. doi:10.1016/j.immuni.2008.06.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Castellaneta A, Sumpter TL, Chen L, Tokita D, Thomson AW. NOD2 ligation subverts IFN-alpha production by liver plasmacytoid dendritic cells and inhibits their T cell allostimulatory activity via B7-H1 up-regulation. J Immunol (2009) 183:6922–32. doi:10.4049/jimmunol.0900582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Kingham TP, Chaudhry UI, Plitas G, Katz SC, Raab J, Dematteo RP. Murine liver plasmacytoid dendritic cells become potent immunostimulatory cells after Flt-3 ligand expansion. Hepatology (2007) 45:445–54. doi:10.1002/hep.21457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Krueger PD, Kim TS, Sung SS, Braciale TJ, Hahn YS. Liver-resident CD103+ dendritic cells prime antiviral CD8+ T cells in situ. J Immunol (2015) 194:3213–22. doi:10.4049/jimmunol.1402622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Huang LR, Wohlleber D, Reisinger F, Jenne CN, Cheng RL, Abdullah Z, et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat Immunol (2013) 14:574–83. doi:10.1038/ni.2573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Bowen DG, Zen M, Holz L, Davis T, McCaughan GW, Bertolino P. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J Clin Invest (2004) 114:701–12. doi:10.1172/JCI21593
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Yoneyama H, Matsuno K, Zhang Y, Murai M, Itakura M, Ishikawa S, et al. Regulation by chemokines of circulating dendritic cell precursors, and the formation of portal tract-associated lymphoid tissue, in a granulomatous liver disease. J Exp Med (2001) 193:35–49. doi:10.1084/jem.193.1.35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Link A, Hardie DL, Favre S, Britschgi MR, Adams DH, Sixt M, et al. Association of T-zone reticular networks and conduits with ectopic lymphoid tissues in mice and humans. Am J Pathol (2011) 178:1662–75. doi:10.1016/j.ajpath.2010.12.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336:86–90. doi:10.1126/science.1219179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell (2014) 159:1327–40. doi:10.1016/j.cell.2014.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159:1312–26. doi:10.1016/j.cell.2014.11.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38:792–804. doi:10.1016/j.immuni.2013.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science (2013) 342:1242974. doi:10.1126/science.1242974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38:79–91. doi:10.1016/j.immuni.2012.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol (2010) 53:903–10. doi:10.1016/j.jhep.2010.04.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Bouwens L, Baekeland M, De Zanger R, Wisse E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology (1986) 6:718–22. doi:10.1002/hep.1840060430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14:986–95. doi:10.1038/ni.2705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Kuniyasu Y, Marfani SM, Inayat IB, Sheikh SZ, Mehal WZ. Kupffer cells required for high affinity peptide-induced deletion, not retention, of activated CD8+ T cells by mouse liver. Hepatology (2004) 39:1017–27. doi:10.1002/hep.20153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Wiegard C, Frenzel C, Herkel J, Kallen KJ, Schmitt E, Lohse AW. Murine liver antigen presenting cells control suppressor activity of CD4+CD25+ regulatory T cells. Hepatology (2005) 42:193–9. doi:10.1002/hep.20756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology (2009) 50:612–21. doi:10.1002/hep.23043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, Van Rooijen N, et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol (2010) 11:295–302. doi:10.1038/ni.1855
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Beattie L, Phillips R, Brown N, Owens BM, Chauhan N, Dalton JE, et al. Interferon regulatory factor 7 contributes to the control of Leishmania donovani in the mouse liver. Infect Immun (2011) 79:1057–66. doi:10.1128/IAI.00633-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Polakos NK, Cornejo JC, Murray DA, Wright KO, Treanor JJ, Crispe IN, et al. Kupffer cell-dependent hepatitis occurs during influenza infection. Am J Pathol (2006) 168:1169–78. doi:10.2353/ajpath.2006.050875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Tay SS, Wong YC, Roediger B, Sierro F, Lu B, McDonald DM, et al. Intrahepatic activation of naive CD4+ T cells by liver-resident phagocytic cells. J Immunol (2014) 193:2087–95. doi:10.4049/jimmunol.1400037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Ganz T. Macrophages and systemic iron homeostasis. J Innate Immun (2012) 4:446–53. doi:10.1159/000336423
72. Uwatoku R, Suematsu M, Ezaki T, Saiki T, Tsuiji M, Irimura T, et al. Kupffer cell-mediated recruitment of rat dendritic cells to the liver: roles of N-acetylgalactosamine-specific sugar receptors. Gastroenterology (2001) 121:1460–72. doi:10.1053/gast.2001.29594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Schmieg J, Yang G, Franck RW, Van Rooijen N, Tsuji M. Glycolipid presentation to natural killer T cells differs in an organ-dependent fashion. Proc Natl Acad Sci U S A (2005) 102:1127–32. doi:10.1073/pnas.0408288102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity (2014) 41:21–35. doi:10.1016/j.immuni.2014.06.013
75. Jiao J, Sastre D, Fiel MI, Lee UE, Ghiassi-Nejad Z, Ginhoux F, et al. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology (2012) 55:244–55. doi:10.1002/hep.24621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Connolly MK, Bedrosian AS, Mallen-St Clair J, Mitchell AP, Ibrahim J, Stroud A, et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J Clin Invest (2009) 119:3213–25. doi:10.1172/JCI37581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology (2013) 58:589–602. doi:10.1002/hep.26267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Blois SM, Piccioni F, Freitag N, Tirado-Gonzalez I, Moschansky P, Lloyd R, et al. Dendritic cells regulate angiogenesis associated with liver fibrogenesis. Angiogenesis (2014) 17:119–28. doi:10.1007/s10456-013-9382-5
79. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115:56–65. doi:10.1172/JCI22675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol (2007) 178:5288–95. doi:10.4049/jimmunol.178.8.5288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Rivera CA, Adegboyega P, Van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol (2007) 47:571–9. doi:10.1016/j.jhep.2007.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes (2010) 59:347–57. doi:10.2337/db09-0016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Lanthier N, Molendi-Coste O, Cani PD, Van Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J (2011) 25:4301–11. doi:10.1096/fj.11-189472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Sunami Y, Leithauser F, Gul S, Fiedler K, Guldiken N, Espenlaub S, et al. Hepatic activation of IKK/NFkappaB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology (2012) 56:1117–28. doi:10.1002/hep.25711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem (2012) 287:40161–72. doi:10.1074/jbc.M112.417014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Shen H, Sheng L, Chen Z, Jiang L, Su H, Yin L, et al. Mouse hepatocyte overexpression of NF-kappaB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology (2014) 60:2065–76. doi:10.1002/hep.27348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Chow A, Brown BD, Merad M. Studying the mononuclear phagocyte system in the molecular age. Nat Rev Immunol (2011) 11:788–98. doi:10.1038/nri3087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol (2011) 89:525–38. doi:10.1189/jlb.0810472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Bar-On L, Jung S. Defining in vivo dendritic cell functions using CD11c-DTR transgenic mice. Methods Mol Biol (2010) 595:429–42. doi:10.1007/978-1-60761-421-0_28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Torres PF, Slegers TP, Peek R, Van Rooijen N, Van Der Gaag R, Kijlstra A, et al. Changes in cytokine mRNA levels in experimental corneal allografts after local clodronate-liposome treatment. Invest Ophthalmol Vis Sci (1999) 40:3194–201.
91. Hochweller K, Striegler J, Hammerling GJ, Garbi N. A novel CD11c.DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells. Eur J Immunol (2008) 38:2776–83. doi:10.1002/eji.200838659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med (2012) 209:1153–65. doi:10.1084/jem.20112675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Meredith MM, Liu K, Kamphorst AO, Idoyaga J, Yamane A, Guermonprez P, et al. Zinc finger transcription factor zDC is a negative regulator required to prevent activation of classical dendritic cells in the steady state. J Exp Med (2012) 209:1583–93. doi:10.1084/jem.20121003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med (2012) 209:1135–52. doi:10.1084/jem.20120030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Schraml BU, Van Blijswijk J, Zelenay S, Whitney PG, Filby A, Acton SE, et al. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell (2013) 154:843–58. doi:10.1016/j.cell.2013.07.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Liedtke C, Luedde T, Sauerbruch T, Scholten D, Streetz K, Tacke F, et al. Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair (2013) 6:19. doi:10.1186/1755-1536-6-19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol (2014) 14:181–94. doi:10.1038/nri3623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol (2012) 18:2300–8. doi:10.3748/wjg.v18.i19.2300
99. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (2013) 58:1461–73. doi:10.1002/hep.26429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol (2014) 60:1090–6. doi:10.1016/j.jhep.2013.12.025
101. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med (2007) 13:1324–32. doi:10.1038/nm1663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Irvine KM, Skoien R, Bokil NJ, Melino M, Thomas GP, Loo D, et al. Senescent human hepatocytes express a unique secretory phenotype and promote macrophage migration. World J Gastroenterol (2014) 20:17851–62. doi:10.3748/wjg.v20.i47.17851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A (2006) 103:5060–5. doi:10.1073/pnas.0511167103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology (2010) 52:1390–400. doi:10.1002/hep.23795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Beljaars L, Schippers M, Reker-Smit C, Martinez FO, Helming L, Poelstra K, et al. Hepatic localization of macrophage phenotypes during fibrogenesis and resolution of fibrosis in mice and humans. Front Immunol (2014) 5:430. doi:10.3389/fimmu.2014.00430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity (2015) 42:145–58. doi:10.1016/j.immuni.2014.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Bleier JI, Katz SC, Chaudhry UI, Pillarisetty VG, Kingham TP III, Shah AB, et al. Biliary obstruction selectively expands and activates liver myeloid dendritic cells. J Immunol (2006) 176:7189–95. doi:10.4049/jimmunol.176.12.7189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Mockel D, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut (2014) 63:1960–71. doi:10.1136/gutjnl-2013-306294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Parker MW, Rossi D, Peterson M, Smith K, Sikstrom K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest (2014) 124:1622–35. doi:10.1172/JCI71386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology (2004) 126:1795–808. doi:10.1053/j.gastro.2004.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Kantari-Mimoun C, Castells M, Klose R, Meinecke AK, Lemberger UJ, Rautou PE, et al. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology (2015). doi:10.1002/hep.27635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Yang L, Kwon J, Popov Y, Gajdos GB, Ordog T, Brekken RA, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology (2014) 146:1339–1350 e1331. doi:10.1053/j.gastro.2014.01.061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology (2014) 59:1060–72. doi:10.1002/hep.26783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol (2013) 10:686–90. doi:10.1038/nrgastro.2013.171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol (2013) 10:656–65. doi:10.1038/nrgastro.2013.183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut (2006) 55:415–24. doi:10.1136/gut.2005.071118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology (2009) 49:1877–87. doi:10.1002/hep.22848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology (2010) 139:323–334 e327. doi:10.1053/j.gastro.2010.03.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol (2012) 57:141–9. doi:10.1016/j.jhep.2012.02.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology (2014) 59:130–42. doi:10.1002/hep.26607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61:416–26. doi:10.1136/gutjnl-2011-300304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: dendritic cells, Kupffer cells, liver fibrosis, NASH, inflammatory monocytes
Citation: Eckert C, Klein N, Kornek M and Lukacs-Kornek V (2015) The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front. Immunol. 6:179. doi: 10.3389/fimmu.2015.00179
Received: 27 February 2015; Accepted: 30 March 2015;
Published online: 20 April 2015.
Edited by:
Florent Ginhoux, Singapore Immunology Network, SingaporeReviewed by:
Frank Tacke, University Hospital Aachen, GermanyCharles-Antoine Dutertre, Duke-NUS Graduate Medical School, Singapore
Copyright: © 2015 Eckert, Klein, Kornek and Lukacs-Kornek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veronika Lukacs-Kornek, Department of Medicine II, Saarland University Medical Center, Kirrbergerstr. 100, Homburg 66424, Germany e-mail:dmVyb25pa2EubHVrYWNzLWtvcm5la0B1bmlrbGluaWt1bS1zYWFybGFuZC5kZQ==