 Chulbul M. I. Ahmed
Chulbul M. I. Ahmed Joseph Larkin III
Joseph Larkin III Howard M. Johnson
Howard M. Johnson- Department of Microbiology and Cell Science, University of Florida, Gainesville, FL, USA
Suppressors of cytokine signaling (SOCS) are inducible intracellular proteins that play essential regulatory roles in both immune and non-immune function. Of the eight known members, SOCS1 and SOCS3 in conjunction with regulatory T cells play key roles in regulation of the immune system. Molecular tools such as gene transfections and siRNA have played a major role in our functional understanding of the SOCS proteins where a key functional domain of 12-amino acid residues called the kinase inhibitory region (KIR) has been identified on SOCS1 and SOCS3. KIR plays a key role in inhibition of the JAK2 tyrosine kinase, which in turn plays a key role in cytokine signaling. A peptide corresponding to KIR (SOCS1-KIR) bound to the activation loop of JAK2 and inhibited tyrosine phosphorylation of STAT1α transcription factor by JAK2. Cell internalized SOCS1-KIR is a potent therapeutic in the experimental allergic encephalomyelitis (EAE) mouse model of multiple sclerosis and showed promise in a psoriasis model and a model of diabetes-associated cardiovascular disease. By contrast, a peptide, pJAK2(1001–1013), that corresponds to the activation loop of JAK2 is a SOCS1 antagonist. The antagonist enhanced innate and adaptive immune response against a broad range of viruses including herpes simplex virus, vaccinia virus, and an EMC picornavirus. SOCS mimetics and antagonists are thus potential therapeutics for negative and positive regulation of the immune system.
Introduction
The immune system consists of both positive and negative regulators that act in harmony to maintain immune homeostasis. A family of intracellular proteins called suppressors of cytokine signaling (SOCS) negatively regulates both receptor-associated tyrosine kinases such as the JAKs and receptor tyrosine kinases such as EGF receptor (1–5). The JAKs play a key role in cytokine signaling by phosphorylation and activation of transcription factors called STATs (6). As these tyrosine kinases thus play key roles in cytokine, growth factor, and hormone signaling, various SOCS play an indispensable role in regulation of their activities (7, 8). The SOCS group has a membership of eight, SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, SOCS7, and SH2 cytokine-inducible protein. SOCS1 is important for its regulation of the JAK2 tyrosine kinase as well as the related kinases TYK2 and JAK1 (2, 8). Receptor tyrosine kinases such as EGF receptor are also regulated by SOCS1 (4). Deficiency in SOCS1 as per knockout of the gene (SOCS1−/−) results in a neonatal fatal inflammatory disease where the cytokine gamma interferon (IFNγ) is the chief cause of the resultant pathology (9).
Among other regulatory players in the immune response, regulatory T cells (Tregs), particularly so-called natural Tregs, also play a key role in immune homeostasis (10–13). We and others have shown that there is an obligatory cross-talk between SOCS1 and peripheral Tregs where SOCS1 plays a key role in maintaining Treg stability (12–14). Molecular genetics via transfections and siRNA approaches have generally been the only way to regulate the intracellular activities of SOCS such as SOCS1 (15). SOCS1 protein has not lent itself to a functional approach to immune regulation for various technical reasons; however, SOCS3 protein has been obtained and used in structure studies of SOCS3/JAK2 interaction (16). We have, by contrast, been able to develop small peptide mimetics and antagonists of SOCS1 (17–19). This has been achieved for the following reasons. First, we have identified one of the key regions of the SOCS1 molecule called the kinase inhibitory region (KIR) as a target for development of SOCS1 mimetics (17). Having done that, we focused on the JAK2 tyrosine kinase activation or autocatalytic region, which interacts with the SOCS1 mimetic as a target for development of SOCS1 (and SOCS3) antagonists (18). Second, it has been well established among the structural biology community that many key proteins, particularly those associated with signaling, lack stable tertiary structure yet carry out numerous biological functions (20). The KIRs of SOCS1 and SOCS3 are slightly homologous and solution structure of SOCS3 has shown that KIR is unstructured (21). The activation loop of JAK2 is similarly unstructured (20). We reasoned that unstructured sequences of proteins are candidates for intrinsic function independent of other regions of the proteins. With this understanding, we were not conceptually restricted in the development of small peptide mimetics and antagonists of SOCS1. We show here that the SOCS1 mimetics are effective in treatment of autoimmune and inflammatory disorders and that the SOCS1 antagonists enhance the immune response for effective handling of viral infections by tilting immune homeostasis toward increased immune activity. This review/perspective thus demonstrates the success of our approach.
Development of SOCS1 Mimetics
We focused on the activation loop of JAK2 to first develop inhibitors of its enzymatic activity independent of the physiological regulation of JAKs by SOCS. The autophosphorylation or activation loop region of human JAK2 became a focal point in our development of SOCS1 mimetics. The amino acid sequence of this region is 1001LPQDKEYYKVKEP for human JAK2 and is depicted symbolically as pJAK2(1001–1013) with the p of pJAK2 indicative of phosphorylation of tyrosine 1007, reflecting its activation state (22). Our first SOCS1 mimetic was developed using an algorithm based on hydropathic complementarity to the JAK2 activation loop (23, 24). We thus synthesized a 12-amino acid peptide, WLVFFVIFYFFR, and determined that it specifically bound the activation loop of JAK2, inhibited IFNγ activation of JAK2 and STAT1α as well as phosphorylation of the IFNγ receptor subunit IFNGR1 (4). Functionally, the tyrosine kinase inhibitory peptide (Tkip) inhibited antiviral activity of IFNγ as well as upregulation of MHC class I molecules on fibroblast cells (4). When compared to the KIR residues of SOCS1, Tkip showed homology with the KIR residues F56, F59, and R60 (22). F59 has been shown to be critical for binding of SOCS1 to activated JAK2 (22). The sequences of Tkip and SOCS1-KIR are shown aligned in Table 1 to illustrate the critical residues.
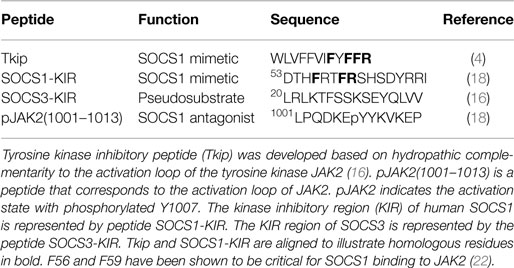
Table 1. SOCS1 mimetics and antagonists.
The KIR region of SOCS1 and SOCS3 are located in the N-terminus of the proteins and are adjacent to a similarly short extended SH2 sequence (ESS). C-terminus to ESS is the SRC homology 2 (SH2) domain, followed by the 40 amino acid SOCS box (3). The SOCS box is shared in all SOCS proteins, in contrast to the restriction of KIR to SOCS1 and SOCS3. The SOCS box is involved in proteasomal degradation of the associated tyrosine kinases. Thus, SOCS1-KIR only inhibits JAK2 kinase activity, while the intact SOCS1 protein inhibits JAK2 kinase activity and initiates its proteasomal degradation.
Peptides corresponding to the 12-amino acid KIR (SOCS1-KIR) and the adjacent extended SH2 sequence (ESS) regions of SOCS1 were compared to Tkip for binding to JAK2 activation loop, pJAK2(1001–1013). Both Tkip and SOCS1-KIR, but not the ESS peptide, bound to pJAK2(1001–1013). Tkip, thus, fortuitously led us to the KIR region of SOCS1 as a potential SOCS1 mimetic. Dose–response competitive binding suggested that Tkip and SOCS1-KIR similarly recognized the activation loop as per the pJAK2(1001–1013) peptide. Tkip inhibited JAK2 autophosphorylation; SOCS1-KIR did not. Both inhibited STAT1α activation, IFNγ activation of macrophages, and antigen-specific lymphocyte proliferation. The use of the SOCS1 mimetics in treatment of autoimmune diseases in mouse models is presented under a separate section below.
Remarkable progress has been made in structure studies of SOCS3, which also contains a KIR region in its N-terminus (3). Care should be exercised, however, in the extrapolation of the structure findings with SOCS3 to SOCS1, particularly concerning KIR. In crystallographic determination of the structure of SOCS3/JAK2 complex, it was shown that KIR was required for complex formation, but the SOCS3-KIR peptide alone did not block JAK2 kinase activity (16). In the same experiment, however, the authors did show that SOCS1-KIR blocked JAK2 activity, which confirms our original observation that SOCS1-KIR peptide mimics SOCS1 protein in inhibiting JAK2 (18). For SOCS3, KIR is thus described as a “pseudosubstrate” in that it is thought to block substrate access to the activation loop of JAK2 (16). Since SOCS3-KIR is required for SOCS3 binding to JAK2, it seems reasonable to suppose that it is binding to something on JAK2. Since the crystallographic studies showed that unstructured KIR and JAK2 activation loop were in close proximity, it is possible that SOCS3-KIR could bind to the activation loop in such a manner that would not block JAK2 kinase activity, but would block substrate access, possibly in concert with another region(s) of SOCS3.
Development of SOCS1 Antagonist
Both SOCS1 and SOCS3-KIR are required for the binding of those SOCS to JAK2 as well as TYK2 and JAK1 tyrosine kinases (3). As indicated, SOCS1-KIR can inhibit JAK2 kinase activity, and although SOCS3-KIR functions as a pseudosubstrate, it does not inhibit JAK2 in the absence of other domains of SOCS3. In any event, both the KIRs of SOCS1 and SOCS3 are intimately associated with the activation loop of JAK2 as reflected in the JAK2 peptide of Table 1. In the activated state, the JAK2 kinase domain is associated with the pseudokinase domain, suggesting that this association is responsible for JAK2 autoinhibition. Importantly, the activation loop is in the kinase domain and phosphorylation of Y1007 in the activation loop is associated with activation of JAK2 (16). Thus, identification of the SOCS1-KIR binding site on JAK2 leads conceptually to development of a SOCS1 antagonist for enhancement of the immune response. As indicated above, JAK2 activation loop peptide, pJAK2(1001–1013), binds SOCS1-KIR. Cell-penetrating lipo-pJAK2(1001–1013) increased IFNγ and IL-6 biological activity by blocking SOCS1 function in cells (18). Consistent with increasing IFNγ activity, JAK2 activation loop peptide enhanced IFN activity against herpes simplex virus-1 (HSV-1) in keratinocytes (25). The anti-HSV-1 effects of the SOCS1 antagonist is expanded upon in the antiviral section below with respect to other viruses in both cultures and animal models. The development of SOCS1 mimetic and antagonist is illustrated in Figure 1.
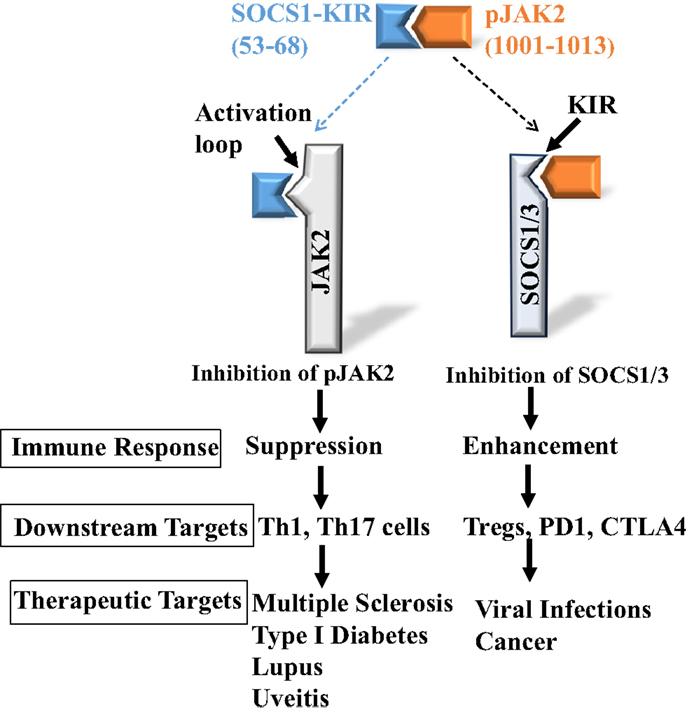
Figure 1. Development of SOCS1 mimetic and antagonist. Peptides corresponding to SOCS1-KIR, SOCS1-KIR (53–68), and pJAK2 activation loop, pJAK2(1001–1013), interact with each other, demonstrating complementarity. They also interact with the cognate SOCS1 and pJAK2 proteins, respectively, and inhibit pJAK2 and SOCS1 functions, resulting in either suppressed or enhanced immune response.
Cross-Talk between SOCS1 and Regulatory T Cells
Forkhead box P3 (FoxP3) natural or constitutive regulatory T cells (Tregs) play an essential role in homeostatic regulation as FoxP3 Treg-deficient mice (scurfy) die of inflammatory disease by 3 weeks after birth (13). The intracellular SOCS proteins, particularly SOCS1, similarly play an indispensible role in such disorders. In this regard, SOCS1 knockout (SOCS1−/−) mice uniformly suffer neonatal death by 3 weeks after birth, similar to the case for FoxP3 Treg deficiency, with unregulated IFNγ playing the central role in the malignant inflammation that is responsible for the fatal outcome (9). Thus, despite their absolute requirement for normal development, there has been little interest concerning the possibility of cross-talk between Tregs and SOCS1. Intuitively, it would seem that such communication is necessary; otherwise, one should expect difficulties in terms of maintaining lymphocyte homeostasis.
We and others have recently shown that SOCS1 and FoxP3 Tregs do indeed interact in such a way that is particularly beneficial to Tregs. Specifically, SOCS1−/− mice were deficient in peripheral Tregs despite enhanced thymic development (10–12). Adoptive transfer of CD4+ T lymphocytes that express the SOCS1 gene or parental administration of the SOCS1 mimetic SOCS1-KIR each induced significant but short-term survival of SOCS1−/− mice. However, adoptive transfer of the CD4+ T cells combined with administration of SOCS1-KIR to SOCS1−/− neonatal mice resulted in increased survival long term (12). In contrast to the periphery, FoxP3 expression in thymic lymphocytes was similar in wild-type and SOCS1−/− mice. Consistent with increased survival, combined CD4+/SOCS1-KIR treatment resulted in decreased leukocyte organ infiltration, reduction in serum IFNγ, and enhanced accumulation of FoxP3 Tregs in SOCS1−/− mice. These data suggest that SOCS1 is required for normal peripheral FoxP3 Treg function, and in fact may play a hierarchical role in SOCS1/Treg cross-talk as stable expression of FoxP3 in Tregs is dependent on SOCS1 in these cells (14).
Consistent with the above, it has been shown that SOCS1−/− Tregs produce high levels of IFNγ and rapidly lose FoxP3 when transferred into immunodeficient Rag2−/− mice or when cultured in vitro (14). The SOCS1−/− Tregs showed hyperactivation of transcription factors STAT1 and STAT3, and it has been proposed that such activation is responsible for Treg instability and loss of suppressive functions (14). How STAT activation is mechanistically linked to loss of FoxP3 and Treg instability is, however, not known.
There is evidence that a subset of Treg cells can convert to a T helper 1 (Th1) or T helper 17 (Th17) phenotype under inflammatory and autoimmune conditions (26). Th1 and/or Th17 cells are the effectors in such diseases as type I diabetes and multiple sclerosis (MS) (17, 27). Thus, Treg cells may initially respond to control an inflammatory or autoimmune state but then undergo conversion and actually exacerbate the condition. Focus on the E2 ubiquitin-conjugating enzyme Ubc13 has provided some insight into Treg plasticity (26). Ubc13 is involved in the formation and conjugation of lysine 63-linked polyubiquitin chains to phosphorylated inhibitor of NF-κB (IκB) where phosphorylation is mediated by IκB kinase (IKK) (26). IκB is then separated from NF-κB, freeing NF-κB to carry out specific transcription. Mice that had Ubc13 specifically ablated or knocked out in Treg cells suffered from systemic autoimmunity with reduction in weight and inflammatory lymphocyte infiltration of the heart, kidney, liver, and lung. Ubc13-deficient Treg cells were shown to be capable of causing the autoimmune condition. Related to this, Ubc13-deficient Treg cells were defective in SOCS1 and IL-10 induction. Reporter gene assays showed that active NF-κB was required for SOCS1 induction but Ubc13 ablated cells lacked active NF-κB because of lack of an effect on IκB. Treatment of cells with the SOCS1 mimetic SOCS1-KIR suppressed IL-17 production in cells from Ubc13-deficient mice. Further, loss of weight and a normal T cell profile were partially restored in SOCS1-KIR treated mice. This study thus showed that Ubc13 plays a critical role in preventing Treg cells from undergoing harmful phenotype changes and that Ubc13 regulated downstream signaling via SOCS1 is key to maintaining Treg cell homeostasis. Translationally, it suggests a role for SOCS1 mimetics in treating inflammatory and autoimmune diseases where Ubc13-like dysregulation may be involved.
SOCS1, regulatory T cells, the programed death-1 (PD-1), and T-lymphocyte-associated protein 4 (CTLA-4) immune mediators are all involved in negative modulation of the immune response. As was shown above with SOCS1 and Tregs, it appears that all of these regulatory players including SOCS1 are interconnected and interdependent, probably in complex ways. It was recently shown, for example, that there is cross-talk between SOCS1 and PD-1, where siRNA silencing of SOCS1 expression resulted in inhibition of PD-1 upregulation (28). Similarly, CTLA-4 has been shown to be a key effector molecule in Treg function (29, 30). The modulatory effect of SOCS1 mimetic and antagonist on Tregs, thus, probably extends to an effect on these other players in positive and negative regulation of immune function (see Figure 1 for example). In principle, this suggests a global approach to positive and negative regulation of immune functions via the SOCS1 mimetic and antagonist. Currently, specific reagents are used to attack various players such as PD-1 and CTLA-4 for enhancement of immune system against cancer (31). Theoretically, the SOCS1 antagonist should affect these molecules along with its effects on Tregs.
Effect of SOCS1-KIR in Autoimmunity: The Experimental Allergic Encephalomyelitis Model as Well as Other Autoinflammatory Disease Models
In the study of the possible role of SOCS1 and/or SOCS3 in the therapeutic efficacy of IFNβ in the treatment of relapsing–remitting MS, astrocytes treated with IFNβ showed upregulation of SOCS1 and SOCS3 (32). This upregulation was due to the corresponding activation of STAT1α and STAT3 by SOCS1 and SOCS3, respectively. The chain of events affected chemokine production and lymphocyte infiltration of the central nervous system (CNS). Given that IFNβ is an effective therapeutic for relapsing–remitting MS (33, 34), it is possible that SOCS1 and SOCS3 play an important role in the effectiveness. In a model of Th17-mediated experimental allergic encephalomyelitis (EAE), the loss of SOCS1 in T cells resulted in increased IFNγ activity and a shift of the T cell population from Th17 to Th1, thus alleviating the EAE (35). In our mouse model of EAE where both Th1 and Th17 cells were involved, loss of SOCS1 from T cells did not protect from EAE (17). Specifically, quantitative reverse transcription polymerase chain reaction (qRT-PCR) results for SOCS1 and SOCS3 mRNA profiles in CNS infiltrating T cells showed a complex pattern. For example, SOCS1 mRNA levels were absent, while SOCS3 levels were modestly increased. By comparison, splenic CD4+ T cell profiles of mRNA were modest for both SOCS1 and SOCS3. Significantly, non-CD4+ monocytic cells showed a high level of SOCS3 in the CNS relative to that of SOCS1. Thus, endogenous SOCS may help control cells like macrophage, microglia, or dendritic cells in the CNS, while the infiltrating effector T cells, not dependent on the accessory cells (36), may be little affected due to the absence of endogenous SOCS1 and/or SOCS3. SOCS mimetics could potentially augment or replace the reduced endogenous SOCS activity of CNS infiltrating lymphocytes and thus have a therapeutic effect in EAE.
We have shown that the Tkip SOCS1 mimetic is therapeutic against relapsing–remitting EAE in mice, dampening both the cellular and humoral immune responses against myelin basic protein (MBP) (37). In a more comprehensive study of treating EAE mice with SOCS1-KIR, we similarly showed SOCS1 mimetic therapeutic protection (17). Mice had lymphocyte infiltration of the CNS at the beginning of treatment, but such infiltration was cleared after 3 weeks of treatment. Both CD4+ Th1 and Th17 cells were suppressed, as well as the IFNγ and IL-17A cytokines are, respectively, associated with them. We further showed for the case of Th17 cells that the polarizing cytokine IL-23 was inhibited by SOCS-KIR. There is evidence that Th1 and Th17 cells function cooperatively in the pathology of EAE where Th1 cells compromise the blood–brain barrier (BBB) and facilitate infiltration by Th17 cells (38, 39). The restoration of a pathologic brain to a normal state by SOCS1-KIR in our EAE model is consistent with its inhibitory effects on both the Th1 and Th17 arms of EAE and possibly MS.
SOCS1-KIR or its analogs also show promise in other autoimmune and/or inflammatory disorders. Psoriasis is an autoinflammatory disorder of the skin that involves interaction between cells of the immune system with keratinocytes of the skin [reviewed in Ref. (40)]. Both the innate and adaptive immune systems play a role in the pathogenesis of psoriasis. Under homeostatic conditions, microbial attack of the skin results in activation of innate immune cells such as dendritic cells, which produce the interleukins 12 (IL-12) and 23 (IL-23), which are involved in the activation of Th1 and Th17 cells, respectively. Keratinocytes in their capacity as innate immune cells play a role in dendritic cell activation as per their production of IL-1, IL-6, and tumor necrosis factor α (TNFα). Th1 and Th17 cells in turn act on keratinocytes via IFNγ (Th1) and IL-17A and F (Th17). This further activates the keratinocytes to produce chemokines, interleukins, and other proteins that have both direct and indirect effect on the microbial insult. Psoriasis is thought to develop because of a dysregulated feedback loop between the innate and adaptive immune systems. Psoriasis can be controlled but the dysregulation cannot be converted back to the homeostatic state.
An understanding of the role of SOCS1 and/or SOCS3 in the pathogenesis of psoriasis is in its infancy. It has been reported that keratinocytes with deleted SOCS3, but not SOCS1 in mice resulted in psoriasis-like skin pathology (41). Others have reported that SOCS1 plays a key role in Th1 and IFNγ-induced form of psoriasis-like skin disorder in experiments where human keratinocytes were transiently transfected with SOCS1 genes (42).
Treatment of human keratinocytes or human skin explants with IFNγ in an experimental setting produced an inflammatory pattern similar to that of psoriasis with JAK2 involvement in activation of STAT1α (43). This in turn induced the epidermal expression of the integrin ICAM-1, HLA-DR MHC, and chemokines CXCL10 and CCL2. Immunohistochemically, the IFNγ-treated explants were similar to those from psoriasis patients. The SOCS1-KIR mimetic analog significantly blocked STAT1α activation as well as expression of the integrin, MHC, and chemokines. It was proposed that the SOCS mimetic was a potential therapeutic for psoriasis (43).
Obesity is a major problem in the United States as well as the world at large (44). It is associated with a condition called metabolic syndrome, which is characterized by insulin resistance reviewed in Ref. (45). The adipose tissue associated with the metabolic syndrome fuels the activation of macrophages, which in turn play a major role in the systemic inflammation that affects key tissues and organs such as the liver. The inflammatory cytokines that result from the systemic inflammation cause insulin resistance by inhibiting insulin receptor substrates (IRSs) activity, which are the mediators of insulin signaling via interaction with the insulin receptor tyrosine kinase.
SOCS1 and SOCS3 have been shown to play a paradoxical role in insulin activity in type 2 diabetes. Evidence suggests that these SOCS dampen the inflammatory response associated with metabolic syndrome and thus play a role in increasing insulin sensitivity (45). The flip side of this is that SOCS1 (as well as SOCS3) is also associated with insulin resistance via its competition with IRS 1 and 2 for binding sites on the insulin receptor, through its targeting of IRS1/2 and insulin receptor for proteasomal degradation, and through its inhibitory effect on insulin receptor and JAK kinase activities (46–49).
A recent quite interesting use of SOCS1-KIR was in the treatment of streptozotocin-induced diabetes in a mouse model (27). SOCS1 mimetic was particularly effective in reducing vascular plaque accumulation of lipid, macrophages, and T cells. The atheroprotective effect was accompanied by systemic reduction in proinflammatory Ly6Chigh monocytes as well as local reduction in aorta expression of chemokines and cytokines. Thus, SOCS1-KIR and other related SOCS mimetics have therapeutic potential to retard the vascular problems associated with diabetes (27). Unlike SOCS1, SOCS1-KIR lacks the ability to target IRS for proteasomal degradation, since it lacks the required SOCS box (3). This could possibly play a role in its effectiveness in reducing the vascular inflammation in the mouse model of diabetes (27).
Broad Antiviral Activity of SOCS1 Antagonist
There is a constitutive presence of IFNβ (50, 51) and SOCS1 (19) in cells. The constitutive IFNβ plays a key role in optimizing the activity of induced types I and II IFNs (19, 50, 51). At the same time, the constitutive IFNβ as well as induced and added IFNs are regulated by SOCS1 (19). For example, if we treat cells with lipo-pJAK2(1001–1013), we decrease constitutive and induced SOCS1, which in turn increases the levels and activities of the constitutive, induced, and added IFNs. Our demonstration that keratinocytes treated with lipo-pJAK2(1001–1013) possessed enhanced antiviral activity against HSV-1 (25) supported our suppositions about antiviral activity, particularly in vivo, against disparate viruses such as vaccinia virus (19) and encephalomyocarditis virus (EMCV) (19) described below in a stringent test of broad, effective antiviral activity.
Vaccinia Virus
Poxviruses are large, complex, double-stranded DNA viruses that have wrecked havoc on human existence over the ages, because of their innate ability to neutralize the IFN system (52, 53). The smallpox virus is historically responsible for some of the most devastating pandemics in the history of humankind and has been estimated to cause approximately 500 million deaths globally in the past century alone (53). Human monkeypox virus is a zoonotic poxvirus with a clinical presentation similar to that of smallpox. The majority of human monkeypox infections occur in Central Africa and if a more infectious, virulent variant should arise, it could represent a health concern and hysteria similar to that involving the recent Ebola virus epidemic in Africa (54).
The testing of the SOCS1 antagonist in a vaccinia virus poxvirus model could thus serve two purposes. First, it tests the antiviral effect of the antagonist. Second, it provides a potential much needed drug therapeutic to poxvirus infections. We reasoned that independent of all other therapeutics that the SOCS1 antagonist, lipo-pJAK2(1001–1013), would tilt the immune balance in mice infected with a lethal dose of vaccinia virus toward a more vigorous IFN and innate protective immune response against the virus (19). Antagonist protected mice as shown in Figure 2 in a dose-dependent fashion with complete protection at 200 μg lipo-pJAK2(1001–1013), while controls succumbed to virus by day 9.
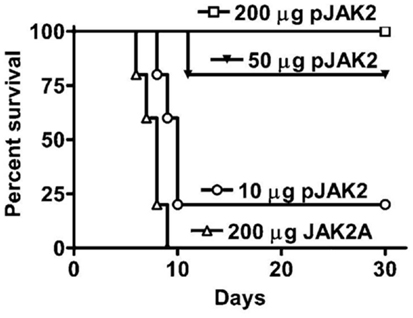
Figure 2. SOCS1 antagonist is an effective therapeutic for lethal intranasal vaccinia virus infection of C57BL/6 mice. Lipo-pJAK2(1001–1013), pJAK2 at 10 μg (○), 50 μg (▼), or 200 μg (□) was injected intraperitoneally on days −2, −1, and 0, relative to virus challenge. Lipo-JAK2(1001–1013)2A (JAK2A) has alanine substituted for tyrosines at 1007 and 1008, and is an inactive control. For details, see Ref. (19).
The question arises as to whether the SOCS antagonist inhibits virus replication or virus spread. Accordingly, vaccinia virus susceptible cells were treated with lipo-pJAK2(1001–1013) at a final concentration of 50 μM and then challenged with a dose of virus that ensured that all of the cells were infected at the same time, resulting in a one-step growth curve. Lipo-pJAK2(1001–1013) inhibited virus replication by approximately 92% as determined by intracellular virus yield when compared to the control variant lipo-pJAK2(1001–1013)2A. Inhibition was approximately 83% as determined by extracellular virus yield. Thus, SOCS1 antagonist inhibited vaccinia virus replication and not simply its release from the cells. This observation is in stark contrast to inhibition of vaccinia virus release but not replication from cells by two different tyrosine kinase inhibitors (55, 56). Thus, the SOCS1 antagonist inhibited vaccinia virus replication and not simply its release, the latter being less reliable in protecting infected mice.
Encephalomyocarditis Virus
Encephalomyocarditis virus is a plus strand rodent picornavirus but is capable of infecting other species including humans (57). Treatment of L929 fibroblasts with the SOCS1 antagonist at 24 μM prior to infection with EMCV (200 PFU) inhibited EMCV growth by approximately 50% (19). The antagonist variant, by contrast, was only 7% protective. In vivo studies involved treatment of C57BL/6 mice with 50, 100, or 200 μg lipo-JAK2(1001–1013) every other day beginning at day 2 and resulted in 60–80% protection at the higher doses. Variant lipo-pJAK2(1001–1013)2A by contrast, was not protective at 200 μg with the death of all the mice by day 5. The extent of protection by the antagonist is remarkable, given the aggressive virulence of EMCV in these experiments. Thus, the SOCS1 antagonist is protective against EMCV as well as against vaccinia virus.
Conclusion
As key as SOCS proteins such as SOCS1 are to normal immune and non-immune functions, they do not lend themselves to controlled and practical manipulation to restoration of homeostasis in autoimmune and inflammatory disorders. Intrinsically disordered proteins (IDPs) and IDP regions lack stable tertiary structure but are responsible for numerous biological functions, particularly those associated with signaling, transcription, DNA interactions, and cellular division and differentiation (58, 59). We took advantage of this important aspect of protein function in our development of small peptide SOCS1 mimetics based on the KIR region of SOCS1. We similarly developed a SOCS1 antagonist based on the activation loop of JAK2, which is the target of the KIR of SOCS1. We view these peptide mimetics and antagonists as templates of a larger and more general approach to development of mimetics and antagonists of other molecules involved in signaling.
Author Contributions
CA, performed research; HJ and JL designed research; HJ wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grant R01 AI 056152 (HJ), a grant to the University of Florida by HJ, and a grant from Lupus Research Institute (JL). We thank Ms. Antia Cain for help with Figure 1. Permission from Journal of Immunology has been granted for the use of Figure 2 and narrative as per Ref. (4, 18, 19, 37). Permission from Journal of Neuroimmunology has been granted for use of narrative from Ref. (17). All of the materials for use have been granted under copyright of the journals.
References
1. Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol (2004) 22:503–29. doi:10.1146/annurev.immunol.22.091003.090312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol (2008) 19(4):414–22. doi:10.1016/j.semcdb.2008.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol (2007) 7(6):454–65. doi:10.1038/nri2093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Flowers LO, Johnson HM, Mujtaba MG, Ellis MR, Haider SM, Subramaniam PS. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J Immunol (2004) 172(12):7510–8. doi:10.4049/jimmunol.172.12.7510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Ahmed CM, Johnson HM. The role of a non-canonical JAK-STAT pathway in IFN therapy of poxvirus infection and multiple sclerosis: an example of Occam’s broom? JAKSTAT (2013) 2(4):e26227. doi:10.4161/jkst.26227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Brivanlou AH, Darnell JE. Signal transduction and the control of gene expression. Science (2002) 295(5556):813–8. doi:10.1126/science.1066355
7. Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol (2002) 2(6):410–6. doi:10.1038/nri818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Kazi JU, Kabir NN, Flores-Morales A, Rönnstrand L. SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell Mol Life Sci (2014) 71(17):3297–310. doi:10.1007/s00018-014-1619-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell (1999) 98(5):609–16. doi:10.1016/S0092-8674(00)80048-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Takahashi R, Yoshimura A. SOCS1 and regulation of regulatory T cells plasticity. J Immunol Res (2014) 2014:943149. doi:10.1155/2014/943149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol (2014) 14(3):154–65. doi:10.1038/nri3605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Collins EL, Jager LD, Dabelic R, Benitez P, Holdstein K, Lau K, et al. Inhibition of SOCS1−/− lethal autoinflammatory disease correlated to enhanced peripheral Foxp3+ regulatory T cell homeostasis. J Immunol (2011) 187(5):2666–76. doi:10.4049/jimmunol.1003819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Ramsdell F, Ziegler SF. FOXP3 and scurfy: how it all began. Nat Rev Immunol (2014) 14(5):343–9. doi:10.1038/nri3650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Takahashi R, Nishimoto S, Muto G, Sekiya T, Tamiya T, Kimura A, et al. SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-{gamma} and IL-17A production. J Exp Med (2011) 208(10):2055–67. doi:10.1084/jem.20110428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Evel-Kabler K, Song XT, Aldrich M, Huang XF, Chen SY. SOCS1 restricts dendritic cells’ ability to break self tolerance and induce antitumor immunity by regulating IL-12 production and signaling. J Clin Invest (2006) 116(1):90–100. doi:10.1172/JCI26169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol (2013) 20(4):469–76. doi:10.1038/nsmb.2519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Jager LD, Dabelic R, Waiboci LW, Lau K, Haider MS, Ahmed CM, et al. The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J Neuroimmunol (2011) 232(1–2):108–18. doi:10.1016/j.jneuroim.2010.10.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol (2007) 178(8):5058–68. doi:10.4049/jimmunol.178.8.5058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Ahmed CM, Dabelic R, Martin JP, Jager LD, Haider SM, Johnson HM. Enhancement of antiviral immunity by small molecule antagonist of suppressor of cytokine signaling. J Immunol (2010) 185(2):1103–13. doi:10.4049/jimmunol.0902895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, et al. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell (2006) 22(2):205–16. doi:10.1016/j.molcel.2006.03.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Babon JJ, Yao S, DeSouza DP, Harrison CF, Fabri LJ, Liepinsh E, et al. Secondary structure assignment of mouse SOCS3 by NMR defines the domain boundaries and identifies an unstructured insertion in the SH2 domain. FEBS J (2005) 272(23):6120–30. doi:10.1111/j.1742-4658.2005.05010.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J (1999) 18(5):1309–20. doi:10.1093/emboj/18.5.1309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Villain M, Jackson PL, Manion MK, Dong WJ, Su Z, Fassina G, et al. De novo design of peptides targeted to the EF hands of calmodulin. J Biol Chem (2000) 275(4):2676–85. doi:10.1074/jbc.275.4.2676
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Blalock JE, Bost KL. Binding of peptides that are specified by complementary RNAs. Biochem J (1986) 234(3):679–83.
25. Frey KG, Ahmed CM, Dabelic R, Jager LD, Noon-Song EN, Haider SM, et al. HSV-1-induced SOCS-1 expression in keratinocytes: use of a SOCS-1 antagonist to block a novel mechanism of viral immune evasion. J Immunol (2009) 183(2):1253–62. doi:10.4049/jimmunol.0900570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Chang JH, Xiao Y, Hu H, Jin J, Yu J, Zhou X, et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat Immunol (2012) 13(5):481–90. doi:10.1038/ni.2267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Recio C, Oguiza A, Lazaro I, Mallavia B, Egido J, Gomez-Guerrero C. Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler Thromb Vasc Biol (2014) 34(9):1953–60. doi:10.1161/ATVBAHA.114.304144
28. Zhang Y, Ma CJ, Ni L, Zhang CL, Wu XY, Kumaraguru U, et al. Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. J Immunol (2011) 186(5):3093–103. doi:10.4049/jimmunol.1002006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity (2014) 41(6):1013–25. doi:10.1016/j.immuni.2014.12.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor ctla-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity (2014) 41(6):1026–39. doi:10.1016/j.immuni.2014.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol (2008) 181(5):3167–76. doi:10.4049/jimmunol.181.5.3167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Javed A, Reder AT. Therapeutic role of beta-interferons in multiple sclerosis. Pharmacol Ther (2006) 110(1):35–56. doi:10.1016/j.pharmthera.2005.08.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Stüve O. Knowns and unknowns in the future of multiple sclerosis treatment. J Neurol Sci (2009) 287(Suppl 1):S30–6. doi:10.1016/S0022-510X(09)71298-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol (2008) 180(6):3746–56. doi:10.4049/jimmunol.180.6.3746
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Oukka M. Interplay between pathogenic Th17 and regulatory T cells. Ann Rheum Dis (2007) 66(Suppl 3):iii87–90. doi:10.1136/ard.2007.078527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Mujtaba MG, Flowers LO, Patel CB, Patel RA, Haider MI, Johnson HM. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J Immunol (2005) 175(8):5077–86. doi:10.4049/jimmunol.175.8.5077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol (2009) 66(3):390–402. doi:10.1002/ana.21748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol (2008) 9(2):137–45. doi:10.1038/ni1551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med (2009) 361(5):496–509. doi:10.1056/NEJMra0804595
41. Eriksen KW, Woetmann A, Skov L, Krejsgaard T, Bovin LF, Hansen ML, et al. Deficient SOCS3 and SHP-1 expression in psoriatic T cells. J Invest Dermatol (2010) 130(6):1590–7. doi:10.1038/jid.2010.6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Federici M, Giustizieri ML, Scarponi C, Girolomoni G, Albanesi C. Impaired IFN-gamma-dependent inflammatory responses in human keratinocytes overexpressing the suppressor of cytokine signaling 1. J Immunol (2002) 169(1):434–42. doi:10.4049/jimmunol.169.1.434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Madonna S, Scarponi C, Doti N, Carbone T, Cavani A, Scognamiglio PL, et al. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur J Immunol (2013) 43(7):1883–95. doi:10.1002/eji.201343370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. World Health Organization Fact Sheet [Internet] (2015). Available from: http://www.who.int/mediacentre/factsheets/fs311/en/
45. Galic S, Sachithanandan N, Kay TW, Steinberg GR. Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem J (2014) 461(2):177–88. doi:10.1042/BJ20140143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol (2004) 24(12):5434–46. doi:10.1128/MCB.24.12.5434-5446.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, et al. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J Biol Chem (2001) 276(51):47944–9. doi:10.1074/jbc.M104602200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Mooney RA, Senn J, Cameron S, Inamdar N, Boivin LM, Shang Y, et al. Suppressors of cytokine signaling-1 and -6 associate with and inhibit the insulin receptor. A potential mechanism for cytokine-mediated insulin resistance. J Biol Chem (2001) 276(28):25889–93. doi:10.1074/jbc.M010579200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Dey BR, Furlanetto RW, Nissley P. Suppressor of cytokine signaling (SOCS)-3 protein interacts with the insulin-like growth factor-I receptor. Biochem Biophys Res Commun (2000) 278(1):38–43. doi:10.1006/bbrc.2000.3762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol (2001) 2(5):378–86. doi:10.1038/35073080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, et al. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science (2000) 288(5475):2357–60. doi:10.1126/science.288.5475.2357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Moss B. Fields Virology. In: Knipe DM, Howley PM, editors. 3rd edn. Poxviridae. Philadelphia, PA: Lippincott, Williams, and Wilkins (2007). p. 2905–45.
53. Moss B, Shisler JL. Immunology 101 at poxvirus U: immune evasion genes. Semin Immunol (2001) 13(1):59–66. doi:10.1006/smim.2000.0296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. McCollum AM, Damon IK. Human monkeypox. Clin Infect Dis (2014) 58(2):260–7. doi:10.1093/cid/cit703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Reeves PM, Bommarius B, Lebeis S, McNulty S, Christensen J, Swimm A, et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med (2005) 11(7):731–9. doi:10.1038/nm1265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Yang H, Kim SK, Kim M, Reche PA, Morehead TJ, Damon IK, et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest (2005) 115(2):379–87. doi:10.1172/JCI200523220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Racaniello VR. Fields Virology. In: Knipe DM, Howley PM, editors. 3rd edn. Picornaviridae: The Viruses and Their Replication. Philadelphia, PA: Lippincott, Williams, and Wilkins (2007). p. 795–838.
58. Dunker AK, Bondos SE, Huang F, Oldfield CJ. Intrinsically disordered proteins and multicellular organisms. Semin Cell Dev Biol (2014) 37C:44–55. doi:10.1016/j.semcdb.2014.09.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: immune therapy, SOCS mimetics, SOCS antagonist, antivirals, poxvirus
Citation: Ahmed CMI, Larkin J III and Johnson HM (2015) SOCS1 mimetics and antagonists: a complementary approach to positive and negative regulation of immune function. Front. Immunol. 6:183. doi: 10.3389/fimmu.2015.00183
Received: 05 February 2015; Accepted: 02 April 2015;
Published: 21 April 2015
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Akihiko Yoshimura, Keio University, JapanAlexander Dalpke, University of Heidelberg, Germany
Copyright: © 2015 Ahmed, Larkin and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Howard M. Johnson, Department of Microbiology and Cell Science, University of Florida, P. O. Box 110700, Gainesville, FL 32611-0700, USAam9obnNvbmhAdWZsLmVkdQ==