 Magda Carneiro-Sampaio1*
Magda Carneiro-Sampaio1* Antonio Coutinho2
Antonio Coutinho2
- 1Department of Pediatrics, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
- 2Instituto Gulbenkian de Ciência, Oeiras, Portugal
Autoimmune disorders (AID) have been increasingly observed in association with primary immunodeficiencies (PIDs). Here, we discuss the interface between PID and AID, focusing on autoimmune manifestations early in life, which can be diagnostic clues for underlying PIDs. Inflammatory bowel disease in infants and children has been associated with IL-10 and IL-10R deficiencies, chronic granulomatous disease, immunedysregulation-polyendocrinopathy-enteropathy-X-linked syndrome (IPEX), autoinflammatory disorders, and others. Some PIDs have been identified as underlying defects in juvenile systemic lupus erythematosus: C1q-, IgA-, IgM deficiencies, alterations of the IFN-α pathway (in Aicardi–Goutières syndrome due to TREX1 mutation). IPEX (due to FOXP3 mutation leading to Treg cell deficiency), usually appearing in the first months of life, was recently observed in miscarried fetuses with hydrops who presented with CD3+ infiltrating lymphocytes in the pancreas. Hemophagocytic lymphohistiocytosis due to perforin deficiency was also identified as a cause of fetal hydrops. In conclusion, PID should be suspected in any infant with signs of autoimmunity after excluding transferred maternal effects, or in children with multiple and/or severe AID.
Introduction
An increasing number of reports have shown that a substantial proportion of patients with primary immunodeficiencies (PID) develop autoimmune disorders (AID), in addition to increased susceptibility to infections (1, 2). These findings are paradoxical if natural tolerance is thought to result from negative selection, but they are compatible with the notion that physiological autoimmunity is necessary for self-tolerance. In this case, immunodeficiency is the cause of autoimmunity; in fact, they are the two sides of the same coin. Monogenic PIDs may provide unique opportunities to uncover the genetic bases and mechanisms of complex AID, the study of which has already helped to unravel the pathways that drive autoimmunity. For example, studies of immunedysregulation-polyendocrinopathy-enteropathy-X-linked syndrome (IPEX), which is caused by FOXP3 gene mutations, have critically established the fundamental role of regulatory T cells (Tregs) in natural tolerance, and PID-AID associations have consolidated the notion of dominant tolerance. Thus, contrary to classical notions of “recessive” tolerance (lymphocyte deletion or anergy) that interpret autoimmunity as resulting from excess (auto) reactivity, the PID-AID associations would rather indicate deficiency in mechanisms that physiologically regulate autoreactivities that are always present.
Autoimmune phenomena, however, have also been observed in other types of PIDs, notably in innate immunity defects (1, 2). Although the association between complement deficiencies and systemic lupus erythematosus (SLE) has long been known, it is now recognized that defects in phagocytes and other innate immunity mechanisms also present autoimmunity. In our view, the concept of AID should actually be expanded to include innate disorders that are classified as “autoinflammatory diseases” (3); thus, there is no a priori reason, evolutionary or otherwise, to restrict AID to adaptive immunity. On the contrary, all “ridding” functions are potentially aggressive if they escape regulation, particularly in the innate system. This view predicts that AID should eventually be found in invertebrates.
Here, we discuss the interface between primary immunodeficiency and AID, focusing on early-onset (including intrauterine) manifestations. Our aim is to draw attention to AID in early life as being clinical manifestations of underlying PIDs.
Heterogeneity of PID-AID Associations
As previously noted (1), a handful of PIDs are systematically associated with AID, such that we consider them to be monogenic autoimmune diseases. However, these PIDs are heterogeneous in terms of the age of onset and clinical manifestations (Table 1). IPEX is a life-threatening condition, with some patients developing AID already in fetal life and rarely surviving infancy in the absence of hematopoietic stem cell transplantation (HSCT); type I diabetes, autoimmune enteropathy leading to chronic diarrhea, inflammatory bowel disease (IBD), and cytopenias are the most common autoimmune manifestations; affected infants also present severe allergy and high IgE levels (4, 5). Omenn syndrome (OS), which is also a life-threatening condition, usually appears in the first months after birth through a combination of severe immunodeficiency with allergic hyperinflammation and autoimmunity, with predominant skin involvement and high levels of IgE. OS is also a consequence of loss of regulation because of the highly restricted, oligoclonal CD4 TCR (T-cell receptor) repertoire, resulting from hypomorphic (severe but not null) mutations in critical genes for V-region rearrangement or lymphocyte development (RAG1, RAG2, and other SCID-related genes, including IL7RA, IL2RG, ADA, DCLRE1C, Ligase IV gene, CHD7, RMRP, AK2) (6). Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED), which is caused by AIRE gene defects, is the prototypic disorder of central (thymic) tolerance and is much less severe, with mucocutaneous candidiasis and autoimmune endocrinopathies usually appearing in childhood (7). Mucocutaneous candidiasis is likely of autoimmune origin, caused by auto-antibodies to cytokines involved in anti-fungal immunity (anti-IL-17) (8). Autoimmune lymphoproliferative syndrome (ALPS) represents a group of lymphocyte apoptosis disorders (due to Fas mutations in most cases) with AID targets predominantly in blood cells (2). ALPS usually appears in childhood and may improve with age. Complete C1q deficiency is intimately associated with juvenile SLE development (9). Note that different autoimmune phenotypes are associated with different genetic defects (1), and a major question concerning the mechanisms of disease continues to be the restricted tissue “targeting” of these conditions.
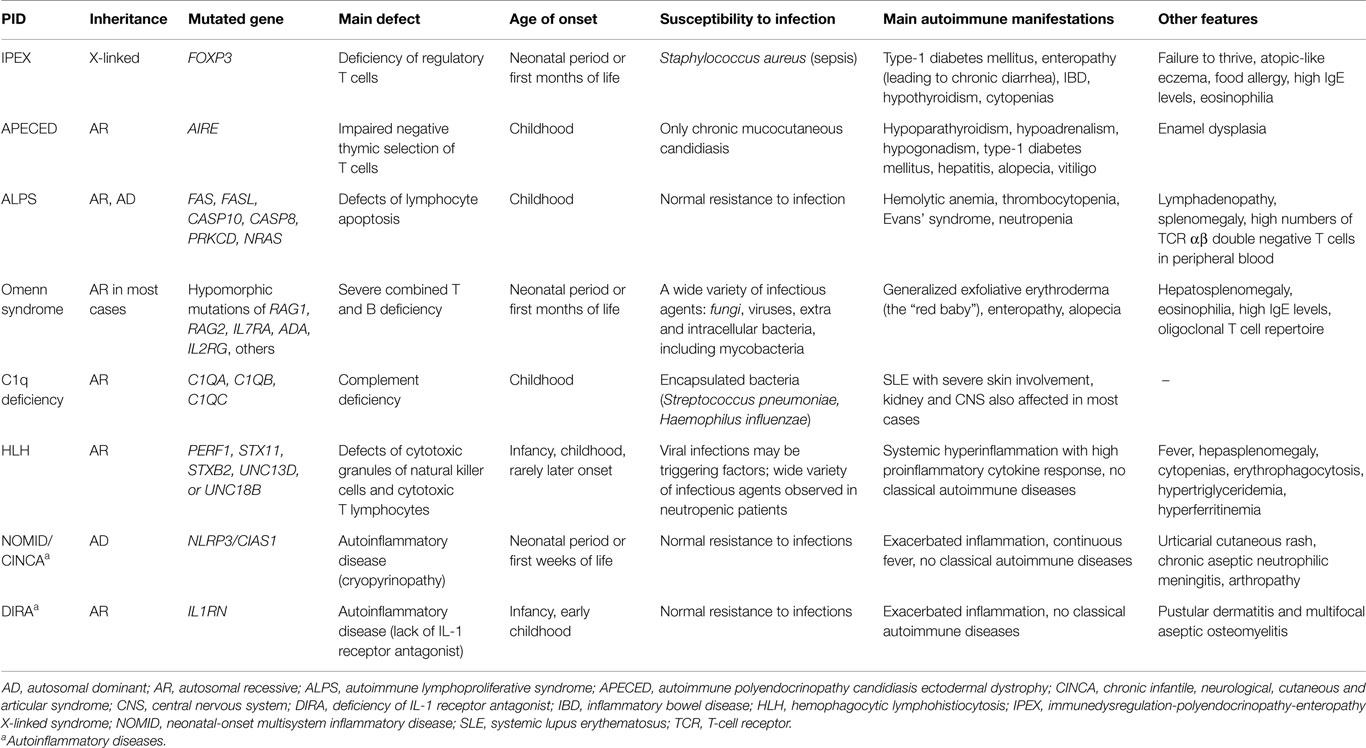
Table 1. Main characteristics of some monogenic PIDs systematically associated to autoimmune manifestations.
There is another group of PIDs that are strongly associated with AID (more than 20% of affected patients demonstrate autoimmune manifestations), in which two of the most frequent PIDs are included: IgA deficiency and common variable immunodeficiency (CVID) (Table 2) (1, 2); the mechanisms underlying pathogenic autoreactivity are not entirely clear yet.
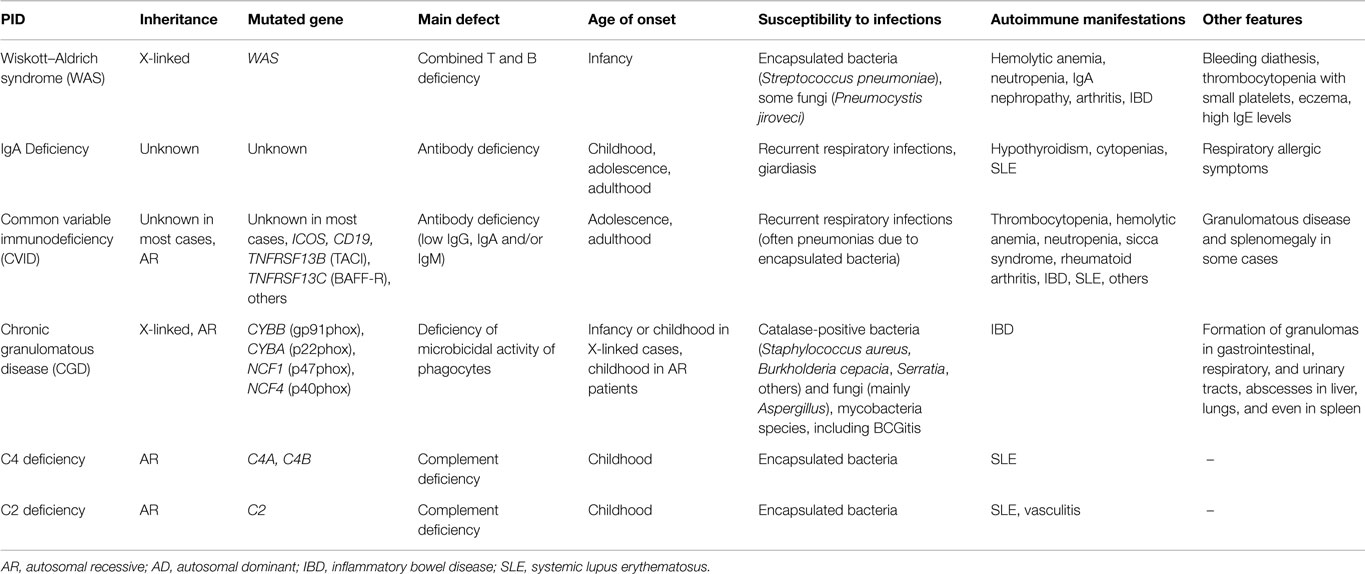
Table 2. Main characteristics of some PIDs strongly associated to autoimmune manifestations (present in more than 20% of the PID cases).
There are a few PIDs in which AID is rarely or never observed, even in patients with chronic or recurrent infections (1). Examples of such PIDs include IL-12/IL-23 – IFN-γ axis deficiencies, IRAK-4 deficiency, autosomal dominant Hyper-IgE syndrome (due to STAT3 mutations leading to deficiency of Th17 cells), and congenital asplenia.
Fetal Autoimmune Diseases Associated with PIDs
IPEX typically manifests early in life: 45% of patients in the literature presented symptoms in the neonatal period, and there were 11 cases at birth (4). These observations clearly suggest fetal onset of AID, which we have recently demonstrated in two unrelated families (5). In one family, a newborn with intrauterine growth restriction presented at birth with type-1 diabetes, diarrhea, thrombocytopenia, eczematous dermatitis, eosinophilia, high IgE levels, and autoantibodies to pancreatic islet antigens at 4 days of age, with negative maternal serology. In a second family, a different FOXP3 mutation was identified in two miscarried monochorionic twin male fetuses, who died at 21 weeks of gestation due to hydrops; the fetuses demonstrated CD3+ infiltrating lymphocytes in their pancreases. Both families had a history of repeated miscarriages of males in consecutive generations, as well as premature babies who developed severe fatal diarrhea immediately after birth. Additionally, at the Children’s Hospital of Philadelphia, in the same family, two miscarried fetuses who died due to hydrops were identified as carrying another FOXP3 mutation (Sara L. Reichert, personal communication, 2014).
We are aware of only limited additional observations of fetal AID: a case of OS at birth due to an IL7Rα deficiency (10) and a few reports on fetal hydrops associated with HLH (hemophagocytic lymphohistiocytosis), including two perforin-deficient twins (11); these cases were described as non-immune hydrops but, as discussed above, we would classify them as AID due to innate immunity mechanisms. Otherwise, in IPEX, autoimmune hemolytic anemia may be the cause of fetal hydrops (5).
In terms of classical views of tolerance, it is surprising that “break of tolerance” can occur when immune system is still immature. Rather than being “broken”, however, we suggest that establishment of tolerance itself was severely compromised in these cases. Thus, given the current knowledge of the age-dependent development of the various lymphocyte classes and effector functions, as well as on the ability of fetuses to produce immune responses (12), it is not surprising that pathological fetal autoimmunity exists and that there is enough time before birth to produce autoantibodies and to develop autoreactive T lymphocytes, which cause destruction of pancreatic beta-cells or other lesions.
Autoimmune diseases in fetal life are, indeed, extremely rare and have always been associated with severe PIDs; hence, all newborns with suspected AID should be investigated for PID.
PIDs Associated with Juvenile Systemic Lupus Erythematosus
Deficiencies of the early components of the classical complement pathway have long been recognized as strong risk factors for SLE development (13). Complete C1q deficiency may be considered to be a monogenic SLE form, because more than 90% of affected patients develop severe disease, usually beginning early in life. IgA deficiency was identified in 1–6% of SLE patients and is 10–50 times more frequent in SLE than in the general population. We have recently investigated complement and immunoglobulin deficiencies in patients with both juvenile- and adult-onset SLE (9, 14). Among 72 children and adolescents, 16 (22%) were identified with complement [C1q (n = 2), C2 (n = 2), C4 (n = 2)] or immunoglobulin [IgA (n = 3), IgM (n = 3), IgG2 (n = 4)] deficiencies. The only two cases identified before 2 years of age were associated to PIDs: one C1q deficiency of fatal course and a persistent IgM deficiency. In 5 of the 6 cases with complement deficiencies, SLE manifestations began before 10 years of age. In contrast, no complement deficiency was identified among 300 adult-onset SLE, whereas 3 patients had selective IgA deficiency, and 24 showed isolated IgM deficiency (<30 mg/dL), which was repeatedly observed in the follow-up of 5 patients. Although IgM deficiency has not been consistently reported as being a risk factor for SLE development, note the following three details: (a) observations in murine models indicate that IgM autoantibodies may be protective as neutralizers of IgG autoantibodies; (b) deficiencies in serum and/or secretory IgM resulted in elevated levels of lupus-type IgG antibodies; and (c) these clinical cases all support the idea that a lack of IgM production may be relevant to SLE pathophysiology [reviewed in Ref. (9)]. Severe hypogammaglobulinemia was not observed in either juvenile or adult patients in our cohort, but it has been reported that SLE complicates the clinical course of CVID (13). Analyzing gene copy numbers for C4A and C4B in the same groups, juvenile SLE patients presented lower values than healthy controls and adult-onset SLE patients (Kaline Pereira and Luis Andrade, personal communication).
Aicardi–Goutières syndrome (AGS), another form of early-onset SLE, is seen to be a monogenic form of lupus that brought light to the role of type I interferons and TREX1 gene mutations in SLE pathophysiology (15). AGS is a genetically determined encephalopathy that phenotypically overlaps with SLE (erythematous skin lesions, oral ulcers, arthritis, thrombocytopenia, leukopenia, positive antinuclear antibodies), and its autosomal-recessive (AR) form is associated with high IFN-α production. Similar to AGS, SLE is associated with alterations in type I interferon metabolism, with upregulation of type I IFN-inducible genes in peripheral blood cells. TREX1, which encodes an exonuclease (a DNAse type III), is the most frequently mutated gene in AGS (15), resulting in intracellular accumulation of abnormal single-stranded DNA, which may explain the high levels of IFN-α observed. TREX1 mutations were observed more frequently (0.5–3%) in a large cohort of SLE patients than in healthy controls, thus supporting the hypothesis of TREX1 involvement in lupus pathogenesis.
Systemic lupus erythematosus has also been described in some patients with AR Hyper-IgE syndrome caused by DOCK8 mutations, as well as in some patients with prolidase deficiency, a rare metabolic disease (15). In both monogenic diseases, the patients also presented with atopic-like skin manifestations and high serum IgE levels.
A homozygous missence mutation in the PRKCD gene (encoding protein-kinase delta) was recently identified in three SLE children from a consanguineous family (16). It has been observed that Prkcd participates in the deletion of autoreactive B cells, and deficient mice have demonstrated systemic autoimmunity and B cell proliferation.
Interestingly, other PIDs are systematically associated with autoimmune diseases but not with SLE (Table 1). APECED (due to AIRE mutations) is of particular interest because hundreds of patients reportedly reached adulthood with several organ-specific AID, but did not develop SLE, suggesting that defective thymic selection of lymphocytes should not represent a relevant pathogenic mechanism (7, 13). Reinforcing this idea, SLE has rarely been described among Down syndrome patients, who demonstrate lower thymic expression of AIRE and are at high risk of developing organ-specific autoimmune diseases, but not lupus (17).
The evidence above that an underlying defect is more common in pediatric SLE than in adult forms suggests that SLE appearance in the first years of life should lead to the suspicion of a PID.
PIDs in Early-Onset Inflammatory Bowel Disease
Inflammatory bowel disease in infants and young children has been increasingly recognized as being associated with monogenic defects, particularly PIDs such as IL-10 and IL-10R deficiencies, chronic granulomatous disease, Wiskott-Aldrich syndrome, IPEX, IPEX-like syndromes (CD25 deficiency and gain of function in STAT1 signaling), atypical SCID (severe combined immunodeficiency), OS, NEMO (nuclear factor kB essential modulator) deficiency, XIAP (X-linked inhibitor of apoptosis) deficiency, and autoinflammatory disorders (Tables 1 and 2) (4, 18, 19). PIDs associated with “early“ or “very early-onset” IBD (identified before 10 or 6 years of age, respectively) thus represent various defects of adaptive immunity (including regulatory T cells and anti-inflammatory cytokines), innate immunity, as well as neutrophil dysfunctions, but not complement deficiencies (19).
In a multicentric study with 66 IBD patients younger than 5 years of age and different ethnic origins, 16 (24%) cases demonstrated IL-10 or IL-10R deficiencies (18), with precocious symptoms in the first months of life (infantile IBD forms), and perianal disease in most cases. Although age of onset, location, and other characteristics of the intestinal disease, together with clinical and laboratorial features, may indicate an underlying deficiency, phenotypes usually overlap, and genetic testing should be performed not only to identify monogenic defects but also to select appropriate treatment (18, 19). In some entities, including IL-10 signaling defects, HSCT represents a promising therapeutic option and sometimes the only option to ensure patient survival. As a general rule, abnormal susceptibility to infections and the presence of other autoimmune and/or inflammatory phenomena suggest an underlying PID in infants and young children with an IBD picture.
Most Autoinflammatory Diseases Manifest Early in Life
Monogenic autoinflammatory diseases (MAID) are classified as PIDs (2) and comprise a growing number of recently identified defects, most of which manifest in childhood, and some of which typically manifest in infancy (3, 20). The study of MAID patients has unraveled key regulatory and effector innate immunity pathways and demonstrated that uncontrolled pro-inflammatory cytokine and chemokine responses can be life threatening and cause significant organ damage as do classical AID mediated by autoantibodies and autoreactive T lymphocytes. Familial Mediterranean fever (FMF) and tumor necrosis factor) Receptor Associated Periodic Syndrome (TRAPS) have been identified as the two most frequent entities in various population groups, including a Brazilian one, FMF presenting significant prevalence in eastern Mediterranean populations (3).
Monogenic autoinflammatory diseases characteristically present with non-infectious fever (continuous in some diseases or periodic in others) and systemic and/or disease-specific organ inflammation. The skin, bones, joints, eyes, and central nervous system are the most common affected sites in MAIDs (3). Extensive skin lesions in infants should be a warning sign for MAIDs: generalized urticarial, pustular, erythematonodular or purpuric rash have been described in different diseases. Abdominal pain is also a frequent feature in FMF and TRAPS.
Some MAIDs begin in the first months of life (Table 1). Neonatal-onset multisystem inflammatory disease (NOMID), also called chronic infantile, neurological, cutaneous and articular (CINCA) syndrome, is the most severe form of cryopyrin (a component of il-1β activating complex) associated periodic syndromes (CAPS). CAPS are caused by autosomal dominant gain-of-function mutation in the NLRP3/CIAS1 gene, and approximately 40% of patients with clinical NOMID/CINCA are negative for germ line mutation by Sanger sequencing. This defect can be diagnosed clinically and it is characterized by continuous low-grade fever, urticarial cutaneous rash, chronic aseptic neutrophilic meningitis, and arthropathy, starting in the first weeks of life (3). If untreated, patients develop permanent organ damage as a consequence of persistent inflammation in the affected organs. IL-1 blocking agents currently represent the most effective treatment for NOMID/CINCA and the other cryopyrinopathies.
Another early-onset MAID is the deficiency of interleukin 1 receptor antagonist (DIRA) (Table 1). Affected patients present with pustular (sparse, in crops or generalized) dermatitis, and multifocal aseptic osteomyelitis, and periostitis; highly elevated levels of acute phase reactants are also observed (3, 21). Fever is often low grade or even absent in DIRA. A recombinant IL-1R antagonist (anakinra) has been successfully used in DIRA patients with rapid resolution of symptoms and normalization of acute phase reactant levels.
Thus, MAID should be suspected in patients with manifestations of excessive inflammatory responses in infancy and childhood.
Autoimmune Diseases as Warning Signs for PIDs in Infants and Children
After excluding transferred maternal effects, any AID in infants should raise a PID suspicion. Persistent neonatal diabetes mellitus, autoimmune hypothyroidism, and autoimmune cytopenias (primarily thrombocytopenia and hemolytic anemia) may be precocious features of IPEX. Autoimmune cytopenias may also complicate the course of DiGeorge syndrome, combined immunodeficiencies due to RAG1 gene mutations, and even SCID in babies (10, 22, 23). Regarding AID in young children, an underlying PID (or other monogenic disorder) should be investigated in patients with multiple and/or severe AID, primarily if recurrent or serious infections are also present. In addition to fever of unknown origin (episodic or continuous), extensive skin lesions of various types, as well as persistent inflammation in the joints, bones, eyes, and central nervous system should represent warning signs for MAIDs in early life.
PIDs should also be considered as the underlying defect in cases of fetal hydrops and recurrent miscarriages of unknown cause (5, 11).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs. Adriana Almeida de Jesus, Maria de Lourdes Brizot, Mariana Nutti de Almeida Cordon, and Ricardo Toma, for their helpful suggestions for this manuscript, as well the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), for supporting MC-S’s research activities. Funding: Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP).
References
1. Carneiro-Sampaio M, Coutinho A. Tolerance and autoimmunity: lessons at the bedside of primary immunodeficiencies. Adv Immunol (2007) 95:51–82. doi: 10.1016/S0065-2776(07)95002-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol (2013) 147:155–74. doi:10.1016/j.clim.2013.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol (2012) 3:211. doi:10.3389/fimmu.2012.00211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Xavier-da-Silva MM, Moreira-Filho CA, Suzuki EK, Patricio F, Coutinho A, Carneiro-Sampaio M. Fetal-onset IPEX: report of two families and review of literature. Clin Immunol (2015) 156:131–40. doi:10.1016/j.clim.2014.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Milner JD, Fasth A, Etzioni A. Autoimmunity in severe combined immunodeficiency (SCID): lessons from patients and experimental models. J Clin Immunol (2008) 28(Suppl 1):S29–33. doi:10.1007/s10875-007-9159-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Arstila TP, Jarva H. Human APECED; a sick thymus syndrome? Front Immunol (2013) 4:313. doi:10.3389/fimmu.2013.00313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med (2010) 207(2):299–308. doi:10.1084/jem.20091669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Jesus AA, Liphaus BL, Silva CA, Andrade LEC, Bando SY, Coutinho A, et al. Complement and antibody primary immunodeficiencies in juvenile systemic lupus erythematosus. Lupus (2011) 20:1275–84. doi:10.1177/0961203311411598
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Zago CA, Jacob CM, de Albuquerque Diniz EM, Lovisolo SM, Zerbini MC, Dorna M, et al. Autoimmune manifestations in SCID due to IL7R mutations: omenn syndrome and cytopenias. Hum Immunol (2014) 75:662–6. doi:10.1016/j.humimm.2014.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Balta G, Topcuoglu S, Gursoy T, Gurgey A, Ovali F. Association of nonimmune hydrops fetalis with familial hemophagocytic lymphohistiocytosis in identical twin neonates with perforin His222Arg (c665A >G) mutation. J Pediatr Hematol Oncol (2013) 35:e332–4. doi:10.1097/MPH.0b013e31827078c6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Mold JE, McCune JM. Immunological tolerance during fetal development: from mouse to man. Adv Immunol (2012) 115:73–111. doi:10.1016/B978-0-12-394299-9.00003-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Carneiro-Sampaio M, Liphaus BL, Jesus AA, Silva CA, Oliveira JB, Kiss MH. Understanding systemic lupus erythematosus physiopathology in the light of primary immunodeficiencies. J Clin Immunol (2008) 28(Suppl 1):S34–41. doi:10.1007/s10875-008-9187-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Perazzio SF, Silva NP, Salomão R, Andrade LEC. Comprehensive screening for primary immunodeficiencies shows unexpectedly high frequency of selective IgM deficiency in systemic lupus erythematosus. Arthritis Rheum (2011) 63:S257.
15. Malattia C, Martini A. Paediatric-onset systemic lupus erythematosus. Best Pract Res Clin Rheumatol (2013) 27:351–62. doi:10.1016/j.berh.2013.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, et al. Protein kinase C d deficiency causes mendelian systemic lupus erythematosus with B-cell defective apoptosis and hyperproliferation. Arthritis Rheum (2013) 65:2161–71. doi:10.1002/art.38008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Lima FA, Moreira-Filho CA, Ramos PL, Brentani H, Lima Lde A, Arrais M, et al. Decreased AIRE expression and global thymic hypofunction in down syndrome. J Immunol (2011) 187:3422–30. doi:10.4049/jimmunol.1003053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143:347–55. doi:10.1053/j.gastro.2012.04.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology (2014) 147:990.e–1007.e. doi:10.1053/j.gastro.2014.07.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol (2015) 33:823–74. doi:10.1146/annurev-immunol-032414-112227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Jesus AA, Osman M, Silva CA, Kim PW, Pham TH, Gadina M, et al. A novel mutation of IL1RN in the deficiency of interleukin-1 receptor antagonist syndrome: description of two unrelated cases from Brazil. Arthritis Rheum (2011) 63:4007–17. doi:10.1002/art.30588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Henderson LA, Frugoni F, Hopkins G, de Boer H, Pai SY, Lee YN, et al. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity. J Allergy Clin Immunol (2013) 132:.e1–2. doi:10.1016/j.jaci.2013.06.032
Keywords: primary immunodeficiency, fetal autoimmunity, IPEX, juvenile systemic lupus erythematosus, inflammatory bowel disease
Citation: Carneiro-Sampaio M and Coutinho A (2015) Early-onset autoimmune disease as a manifestation of primary immunodeficiency. Front. Immunol. 6:185. doi: 10.3389/fimmu.2015.00185
Received: 30 January 2015; Accepted: 03 April 2015;
Published: 24 April 2015
Edited by:
Edward K. L. Chan, University of Florida, USAReviewed by:
Rosa Bacchetta, Fondazione Centro San Raffaele Del Monte Tabor, ItalyJolan Eszter Walter, Massachusetts General Hospital, USA
Sandro Félix Perazzio, Universidade Federal De São Paulo, Brazil
Copyright: © 2015 Carneiro-Sampaio and Coutinho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magda Carneiro-Sampaio, Instituto da Criança, Hospital das Clínicas da FMUSP, Avenue Dr. Enéas de Carvalho Aguiar, 647, São Paulo 05403-900, Brazil,bWFnZGFzY3NAdXNwLmJy