 Lucillia Bezu1,2,3,4†Ligia C. Gomes-da-Silva1,2,3,5†Heleen Dewitte6,7Karine Breckpot7Jitka Fucikova8,9Radek Spisek8,9
Lucillia Bezu1,2,3,4†Ligia C. Gomes-da-Silva1,2,3,5†Heleen Dewitte6,7Karine Breckpot7Jitka Fucikova8,9Radek Spisek8,9 Lorenzo Galluzzi1,2,10,11,12‡Oliver Kepp1,2,3‡*Guido Kroemer1,2,3,9,11,12,13‡*
Lorenzo Galluzzi1,2,10,11,12‡Oliver Kepp1,2,3‡*Guido Kroemer1,2,3,9,11,12,13‡*- 1Equipe 11 labellisée par la Ligue Nationale contre le Cancer, Centre de Recherche des Cordeliers, Paris, France
- 2U1138, INSERM, Paris, France
- 3Metabolomics and Cell Biology Platforms, Gustave Roussy Campus Cancer, Villejuif, France
- 4Faculté de Medecine, Université Paris-Sud, Le Kremlin-Bicêtre, France
- 5Department of Chemistry, University of Coimbra, Coimbra, Portugal
- 6Laboratory for General Biochemistry and Physical Pharmacy, Faculty of Pharmacy, Ghent University, Ghent, Belgium
- 7Laboratory of Molecular and Cellular Therapy, Vrije Universiteit Brussel, Jette, Belgium
- 8Sotio a.c., Prague, Czech Republic
- 9Department of Immunology, 2nd Faculty of Medicine, University Hospital Motol, Charles University, Prague, Czech Republic
- 10Gustave Roussy Campus Cancer, Villejuif, France
- 11Université Paris Descartes, Paris, France
- 12Université Pierre et Marie Curie, Paris, France
- 13Pôle de Biologie, Hopitâl Européen George Pompidou, AP-HP, Paris, France
The term “immunogenic cell death” (ICD) is commonly employed to indicate a peculiar instance of regulated cell death (RCD) that engages the adaptive arm of the immune system. The inoculation of cancer cells undergoing ICD into immunocompetent animals elicits a specific immune response associated with the establishment of immunological memory. Only a few agents are intrinsically endowed with the ability to trigger ICD. These include a few chemotherapeutics that are routinely employed in the clinic, like doxorubicin, mitoxantrone, oxaliplatin, and cyclophosphamide, as well as some agents that have not yet been approved for use in humans. Accumulating clinical data indicate that the activation of adaptive immune responses against dying cancer cells is associated with improved disease outcome in patients affected by various neoplasms. Thus, novel therapeutic regimens that trigger ICD are urgently awaited. Here, we discuss current combinatorial approaches to convert otherwise non-immunogenic instances of RCD into bona fide ICD.
Introduction
The expression “immunogenic cell death” (ICD) generally refers to a functionally peculiar case of regulated cell death (RCD) that – in immunocompetent hosts – is capable of activating an adaptive immune response against dead cell-associated antigens (1–5). Of note, ICD generally (but not obligatorily) manifests with apoptotic morphological features, and at least some of its manifestations depend on components of the apoptotic apparatus (6–8). Irrespective of these morphological and biochemical considerations, immunocompetent mice injected s.c. with cancer cells succumbing to bona fide ICD (in the absence of any adjuvant) develop a cellular immune response associated with the establishment of immunological memory that protects them from a subsequent challenge with living cells of the same type (1–3). Importantly, vaccination experiments of this type, involving murine cells and syngeneic mice, remain the gold-standard method to identify bona fide ICD, though several tests have been developed to detect some of its cellular manifestations (see below) (2, 3, 9, 10).
Only a few lethal stimuli are intrinsically endowed with the ability to trigger ICD (9, 11–14). These include some chemotherapeutic agents that are employed in the clinic, including (1) various anthracyclines (i.e., doxorubicin, epirubicin, and idarubicin), which are commonly used against a wide panel of malignant conditions (15–17); (2) mitoxantrone, an anthracenedione generally used for the treatment of acute myeloid leukemia, breast carcinoma, non-Hodgkin’s lymphoma, and prostate carcinoma (15, 16); (3) oxaliplatin, a platinum derivative approved for use in combination with 5-fluorouracil to treat advanced colorectal carcinoma (18, 19); (4) cyclophosphamide, an alkylating agent that is employed against various neoplastic and autoimmune conditions (20–23); and (5) bortezomib, a proteasomal inhibitor approved for the therapy of multiple myeloma and mantle cell lymphoma (24–26). Specific forms of irradiation as well as photodynamic therapy, both of which are habitually employed for the treatment of various neoplasms, have also been shown to trigger bona fide ICD (27–34). Finally, a bunch of hitherto experimental agents is intrinsically endowed with the capacity to initiate ICD, including (but not limited to) some oncolytic viruses (35–39), the microtubular inhibitor patupilone (40–42), and elevated hydrostatic pressures (43).
According to accepted models, ICD relies on the establishment of adaptive stress responses that promote the spatiotemporally coordinated emission of endogenous danger signals from dying cells (44, 45). The endogenous molecules that dispatch danger signals in response to stress are cumulatively known as “damage-associated molecular patterns” (DAMPs) and operate upon binding to receptors expressed by bystander cells, including cellular components of both the innate and adaptive immune system (2, 46–49). As it stands, four DAMPs have been shown to be required for RCD as induced by anthracyclines to be perceived as immunogenic, namely, (1) the exposure of the endoplasmic reticulum (ER) chaperone calreticulin (CALR) on the outer surface of the plasma membrane (16); (2) the secretion of ATP (50); (3) the production of type I interferon (IFN) (51); and (4) the release of the non-histone chromatin-binding protein high-mobility group box 1 (HMGB1) into the extracellular space (52). This said, it cannot be formally excluded that other hitherto undiscovered DAMPs are required for anthracycline-elicited RCD to promote an adaptive immune response. Along similar lines, not all these DAMPs may be required for RCD as induced by agents other than anthracyclines to be perceived as immunogenic (53–55).
In this context, i.e., anthracycline-induced ICD, CALR exposure obligatorily relies on the establishment of a pre-mortem ER stress response centered around the phosphorylation of eukaryotic translation initiation factor 2A, 65 kDa (EIF2A) (7, 56), ATP secretion requires the induction of autophagy (57), and type I IFN production stems from toll-like receptor 3 (TLR3) signaling (51). The molecular mechanisms underlying the ability of anthracyclines and other ICD inducers to promote HMGB1 release remain obscure (2, 3). Cumulatively, these DAMPs recruit antigen-presenting cells (APCs) to sites of active ICD and stimulate the uptake, processing, and presentation of dead cell-associated antigens, eventually resulting in the priming of an adaptive immune response (2, 3). In particular, CALR promotes antigen uptake by APCs by binding to low density lipoprotein receptor-related protein 1 (LRP1, best known as CD91) (58); ATP stimulates the recruitment of APCs and their activation upon binding to purinergic receptor P2Y, G-protein coupled, 2 (P2RY2) and purinergic receptor P2X, ligand-gated ion channel, 7 (P2RX7), respectively (50, 59, 60); type I IFNs exert immunostimulatory effects via IFN (alpha, beta, and omega) receptors (IFNARs) (51); and HMGB1 does so through TLR4 and advanced glycosylation end product-specific receptor (AGER, best known as RAGE) (52, 61).
A detailed discussion of the molecular and cellular mechanisms involved in the detection of ICD-associated DAMPs goes beyond the scope of this review and can be found in Ref. (2, 3). However, it is important to note that the failure of cancer cells to emit one (or more) of these DAMPs completely compromises the immunogenicity of RCD (2, 3). Thus, at odds with their wild-type counterparts, Calr−/− murine CT26 colorectal cells exposed to anthracyclines are unable to vaccinate mice against a subsequent inoculation with malignant cells of the same type (16). The same holds true in several other situations in which adaptive responses cannot proceed normally, including the genetic inhibition of autophagy (e.g., upon the expression of short-hairpin RNAs targeting the essential autophagy proteins Atg5 or Atg7) or the unfolded protein response (e.g., upon the expression of a non-phosphorylatable variant of EIF2A) (7, 57, 62, 63).
Accumulating clinical evidence indicates that the (re-)activation of a proficient immune response against malignant cells is associated with improved disease outcome in patients affected by a wide panel of neoplasms (64–68), in particular when malignant lesions are highly infiltrated by immune effector cells prior to therapy (69). Considerable efforts are therefore being devoted to the development of clinically implementable strategies that (re-)instate anticancer immunosurveillance (70, 71). So far, the most successful of these approaches involves the administration of monoclonal antibodies (mAbs) that block immunosuppressive receptors expressed by activated T cells, such as cytotoxic T lymphocyte-associated protein 4 (CTLA4) and programed cell death 1 (PDCD1, best known as PD-1) (72, 73). Three distinct checkpoint blockers of this type, namely, the CTLA4-targeting mAb ipilimumab and the PD-1-targeting mAbs nivolumab and pembrolizumab, are approved by the US Food and Drug Administration and other regulatory agencies worldwide for use as standalone immunotherapeutic interventions in melanoma patients (74–77). In addition, the administration of checkpoint blockers has been shown to improve the clinical profile of various chemotherapeutic and immunotherapeutic agents (78). Along similar lines, various combinatorial immuno(chemo)therapeutic regimens are being investigated in clinical trials for their ability to mediate superior antineoplastic effects as compared to monotherapies based on their constituents (79, 80). In this framework, various attempts are being made to render immunogenic otherwise non-immunogenic instances of therapy-induced RCD, thereby converting them into bona fide ICD (79, 81–84). This can be due to molecular defects that prevent cancer cells from emitting DAMPs appropriately, as mentioned above, as well as to the intrinsic features of the therapeutic agent under consideration (Table 1). For instance, at odds with its derivative oxaliplatin, cisplatin is intrinsically unable to trigger ICD since it does not stimulate the exposure of CALR on the outer surface of the plasma membrane (18, 19, 85).
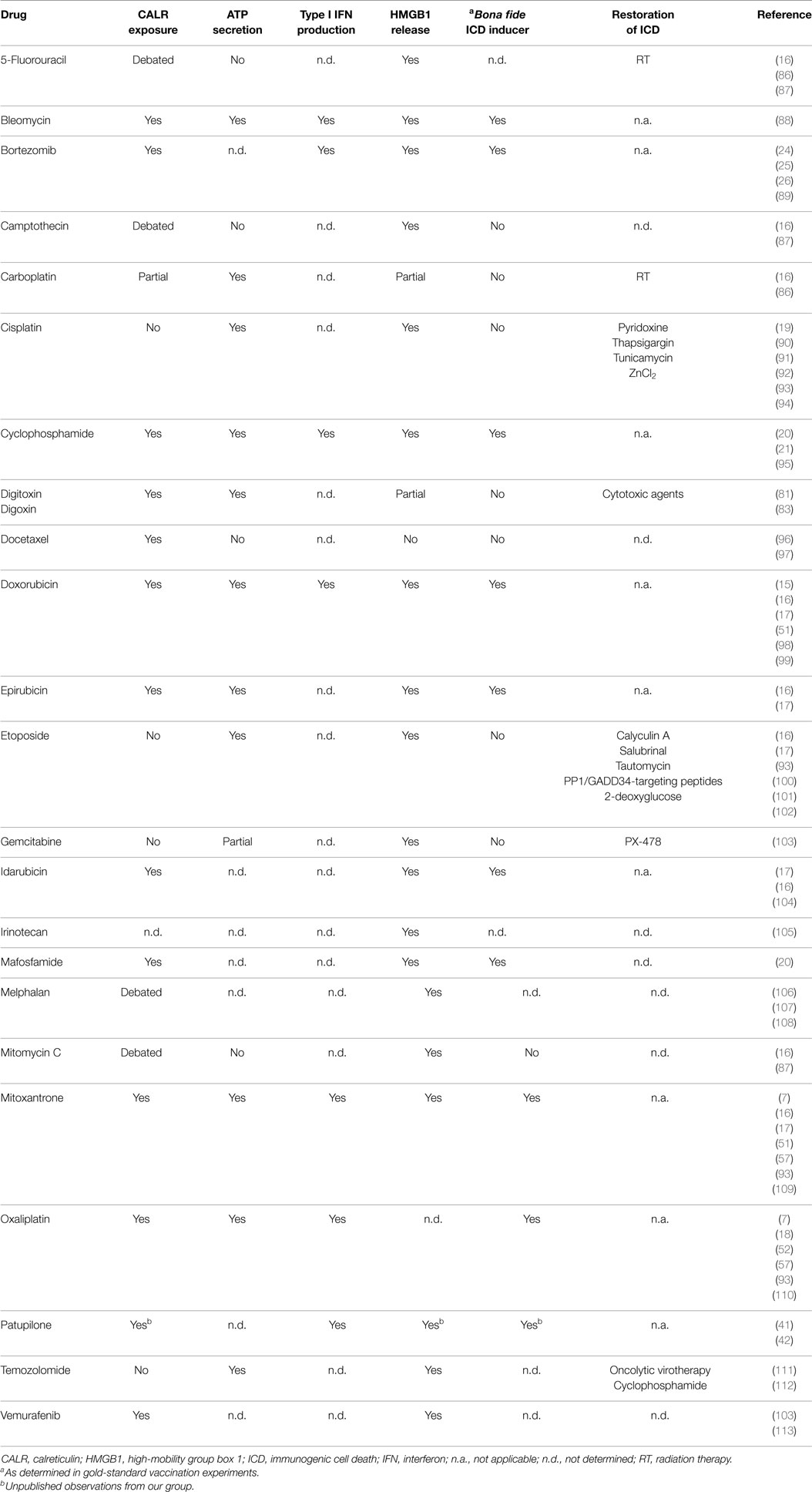
Table 1. Immunogenicity of chemotherapy-induced regulated cell death (examples).
Here, we discuss strategies to convert non-immunogenic instances of RCD into bona fide ICD. In particular, we will review approaches for (1) correcting the incapacity of some therapeutic agents to kill cancer cells while provoking the emission of one or more DAMP(s); or (2) complementing the missing DAMP(s) with exogenous interventions. On the contrary, we will not dwell on strategies that boost the immunogenicity of RCD by operating downstream of DAMP-sensing receptors.
Combinatorial Strategies to Restore CALR Exposure
Some anticancer therapeutics efficiently kill cancer cells (hence promoting the release of HMGB1) and stimulate the secretion of both ATP and type I IFNs, but selectively fail to promote CALR exposure. Most often, such a defect originates from the inability of these agents to trigger an ER stress response resulting in EIF2A phosphorylation (56, 114), and hence can be corrected by the co-administration of an ER stressors. As mentioned above, cisplatin is one of the antineoplastic agents that fail to trigger bona fide ICD as it does not drive a robust ER stress response (18, 19, 85). The ER-stressing agents that have been shown to correct this defect, hence rendering cisplatin-induced RCD immunogenic, include thapsigargin, an inhibitor of various members of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) (19, 114); tunicamycin, an inhibitor of N-glycosylation (19, 94, 114); pyridoxine, a cell-permeant precursor of bioactive vitamin B6 (90, 91, 115); and ZnCl2 (92). Similar results have been obtained by establishing an ER stress response through the enforced overexpression of reticulon 1 (RTN1), an ER protein involved in vesicular trafficking and secretion (116, 117). The latter approach is obviously incompatible with clinical applications. Nonetheless, these data reinforce the notion that the immunogenicity of cisplatin-induced RCD can be restored by various interventions that induce an ER stress (94).
Another strategy that successfully restores CALR exposure in cells succumbing to chemicals that per se do not enable this phenomenon consists in the co-administration of inhibitors of the EIF2A phosphatase composed of protein phosphatase 1, regulatory subunit 15A (PPP1R15A, best known as GADD34), and pyrophosphatase (inorganic) 1 (PPA1, best known as PP1), resulting in accrued EIF2A phosphorylation even in the absence of overt ER stress (16). Thus, whereas CT26 cells treated with etoposide (a topoisomerase II inhibitor currently approved for the treatment of various malignancies) (118, 119) do not expose CALR as they die, and hence fail to vaccinate mice against a subsequent challenge with neoplastic cells of the same type, they efficiently do so in the presence of tautomycin, calyculin A, and salubrinal (three distinct GADD34/PP1 inhibitors) (16). Similar results have been obtained with the small-interfering RNA (siRNA)-mediated downregulation of PP1 or GADD34 (16), as well as with short cell-permeant peptides that disrupt the physical interaction between these two proteins (102). Although siRNA- and peptide-based strategies may not be easily implemented in clinical settings, these results corroborate the specificity of tautomycin, calyculin A, and salubrinal, and lend further support to the notion that interventions that stimulate EIF2A phosphorylation efficiently promote CALR exposure even in the absence of overt ER stress (120).
At least theoretically, the co-administration of ER stressors or molecules that promote EIF2A phosphorylation can be harnessed to reconstitute the immunogenicity of RCD induced by all anticancer agents that per se do not stimulate CALR exposure on the cell surface but provoke ATP secretion, type I IFN production, and HMGB1 release. In addition, the inability of some anticancer agents to cause the translocation of CALR to the outer leaflet of the plasma membrane can be corrected, at least in some settings, by the co-administration of exogenous, recombinant CALR (7, 16, 106). CALR is indeed relatively “sticky” and its absorption on malignant cells succumbing to non-immunogenic RCD in vitro has been shown to fully restore the ability of these cells to vaccinate syngeneic mice against a subsequent neoplastic challenge (16). To the best of our knowledge, however, whether the systemic or intratumoral administration of recombinant CALR to tumor-bearing mice treated with non-immunogenic therapeutics is able to convert them into bona fide ICD inducers has not been tested yet. As compared to administration of small molecules that establish an ER stress response or promote EIF2A phosphorylation, the use of recombinant CALR appears advantageous in that (at least theoretically) it would complement the lack of CALR exposure in all scenarios, irrespective of the underlying molecular defects (including the downregulation or loss of CALR itself). However, such an approach may not be implementable in the clinic, owing to pharmacodynamic and pharmacokinetic issues (e.g., distribution of the recombinant protein, serum half-life, etc…) as well as economic considerations. Current efforts are therefore being focused on the identification of novel (and the refinement of existing) small molecule-based strategies to stimulate CALR exposure upon the establishment of an ER stress or the induction of EIF2A phosphorylation.
Combinatorial Strategies to Boost ATP Secretion
In some settings, anticancer agents kill malignant cells in an efficient fashion (which corresponds to a consistent release of HMGB1), while stimulating the exposure of CALR and the production of type I IFN, but this is not accompanied by the accumulation of extracellular ATP (57, 121), a defect that can stem from at least three different causes. First, some therapeutic agents are unable to stimulate (or even inhibit) autophagic responses, which are required for dying cells to secrete ATP in sufficient amount for signaling via P2RY2 and P2RX7 receptors (57). Second, some malignant cells bear genetic or epigenetic defects that affect the molecular machinery for autophagy (122, 123). These cells are intrinsically unable to preserve the intracellular ATP pool in the course of stress responses, resulting in limited ATP secretion during death (124). Third, some neoplastic cells express high levels of either ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, best known as CD39) or 5′-nucleotidase, ecto (NT5E, best known as CD73), two membrane-bound nucleotidases that degrade extracellular ATP (125).
So far, one general strategy has been shown to restore extracellular ATP concentrations to levels that are compatible with the efficient recruitment and activation of APCs, namely, the pharmacological inhibition of CD39. Thus, CT26 cells lacking essential components of the autophagic machinery, such as Atg5, Atg7, or Beclin 1 (Becn1), secrete limited amounts of ATP as they succumb to anthracyclines, and hence are incapable of vaccinating syngeneic mice against a subsequent challenge with malignant cells of the same type (57). Such a functional defect can be corrected by the co-administration of ARL67156, a broad spectrum inhibitor of extracellular nucleotidases (57). Further confirming these findings, CT26 engineered to overexpress CD39 and exposed to anthracyclines are unable to protect syngeneic mice against a subsequent injection with neoplastic cells of the same type (57, 125). This defect can be corrected by the co-administration of ARL67156, along with the restoration of RCD-associated ATP secretion (57, 125). Taken together, these results indicate that inhibitors of extracellular nucleotidases may constitute a convenient manner to boost the immunogenicity of RCD instances that are normally not associated with ATP secretion.
Importantly, the pharmacological activation of autophagy does not suffice for cancer cells to become immunogenic (16, 57). Nonetheless, combining anticancer agents that per se are unable to trigger ATP secretion with molecules that upregulate the autophagic flux, such as inhibitors of mechanistic target of rapamycin (MTOR) complex I (MTORCI), may efficiently convert non-immunogenic RCD instances into bona fide ICD. This hypothesis awaits formal experimental confirmation. Indeed, while other inducers of autophagy such as the glycolytic inhibitor 2-deoxyglucose (126) have been shown to reinstate the immunogenicity of etoposide-elicited RCD, such an effect was ascribed to the restoration of CALR exposure (indeed, etoposide kills malignant cells while promoting ATP secretion) (100). Finally, it should be noted that the establishment of an ATP gradient around dying cells may not constitute a general requirement for the perception of RCD as immunogenic (127). Moreover, at least in some settings, autophagy may actually inhibit ICD by limiting the production of reactive oxygen species in the course of adaptive stress responses, hence counteracting the establishment of ER stress and consequent CALR exposure (54, 55). Thus, further work is required to precisely identify malignancies in which autophagy supports ICD. Only in these scenarios, the co-administration of autophagy inducers may constitute a proper approach to reinstate the immunogenicity of RCD.
Combinatorial Strategies to Promote Type I IFN Production
Whereas the role of type I IFN in the regulation of innate and adaptive immune responses is well known (128, 129), type I IFN signaling in malignant cells has been identified as a requirement for (anthracycline-induced) ICD only recently (51). Thus, cancer cells respond to various anthracyclines by activating a TLR3-elicited signal transduction cascade resulting in type I IFN release, autocrine/paracrine type I IFN signaling, and chemokine (C–X–C motif) ligand 10 (CXCL10) secretion, two phenomena that underlie their vaccinating potential. At odds with their wild-type counterparts, Tlr3−/− and Ifnar1−/− murine cancer cells exposed to anthracyclines fail to vaccinate syngeneic mice against a subsequent injection of living cells of the same type (51). It has already been demonstrated that the inability of Tlr3−/− cells to undergo ICD can be corrected by the co-administration of recombinant type I IFNs or recombinant CXCL10. Similarly, Ifnar1−/− cells succumbing to anthracyclines turn immunogenic in the presence of recombinant CXCL10 (but not type I IFNs) (51).
Various synthetic TLR3 agonists are available and some of them, including polyinosinic:polycytidylic acid (polyI:C) and its clinical grade analog polyI:polyC12U (also known as rintatolimod and Ampligen™), have been extensively tested as immunostimulants in cancer patients (130, 131). It is therefore tempting to speculate that the co-administration of TLR3 agonists may restore the ability of anticancer agents that per se do not promote type I IFN release to trigger bona fide ICD. This hypothesis awaits urgent experimental confirmation. For the considerations presented above, small molecules that trigger TLR3 signaling would indeed be more convenient as clinical tools to restore type I IFN signaling than recombinant type I IFN or CXCL10 themselves.
Combinatorial Strategies to Substitute for HMGB1 Release
HMGB1 release occurs upon (nuclear and) plasma membrane permeabilization, i.e., it constitutes a post-mortem event (5, 132). Thus, all antineoplastic agents that efficiently kill malignant cells (as opposed to molecules that exert cytostatic effects or induce cell senescence) (133) promote HMGB1 release, perhaps with different kinetics (5, 132). However, the expression levels of HMGB1 vary in different tumor types and evolve along with tumor progression, implying that some malignant cells may express HMGB1 to levels that are not compatible with the activation of TLR4 and RAGE in immune cells upon release (134, 135). Importantly, the immunogenicity of anthracycline-induced RCD is compromised in these cells, as well in cells artificially depleted of HMGB1 by means of specific siRNAs (135). Recent results indicate that this defect can be efficiently corrected by the exogenous supply of a synthetic TLR4 agonist, i.e., dendrophilin, at least in experimental models (135). Since dendrophilin has not yet entered clinical development (130, 131), it will be interesting to see whether TLR4 agonists that are already licensed by regulatory agencies for use in humans, such as the Bacillus Calmette–Guérin (BCG) (80) and monophosphoryl lipid A (MPL) (136), are also able to restore the immunogenicity of HMGB1-deficient cells succumbing to ICD.
In this context, it is worth noting that cancer cells exposing CALR, secreting ATP, producing type I IFNs but releasing limited amounts of HMGB1 as they respond to a lethal stimulus in a suboptimal manner fail to elicit adaptive immune responses (137). Upon inoculation into immunocompetent mice, these cells actually form tumors at the vaccination site (as a significant fraction of them is not dying) and the animals are unable to control a subsequent challenge with cell of the same type (3). We have observed this to occur in murine cancer cells treated with digoxin or digitoxin, two glycosides approved in many countries for the treatment of cardiac conditions (81). These molecules efficiently inhibit the human Na+/K+ ATPase, which explains their pharmacological properties and their ability to kill some neoplastic cells of human origin, but not its murine counterpart (83). Thus, cardiac glycosides per se are unable to trigger ICD, at least in the murine system. However, clinical data indicate that they may convert non-immunogenic RCD as elicited by a very large panel of chemotherapeutics into bona fide ICD (83). From another standpoint, any anticancer agent that efficiently kills malignant cells could be considered as a means to restore the immunogenicity of cells responding to cardiac glycosides. We have recently initiated a clinical trial to prospectively test this hypothesis in head and neck squamous carcinoma patients.
Concluding Remarks
In spite of old beliefs, cancer cells continuously interact with the immune system: first, as they are generated by healthy cells upon malignant transformation; second, as they evolve and acquire additional neoplastic features; and third, when they are challenged with therapeutic interventions. During the last decade, such a conceptual revolution, i.e., considering tumors as entities that can be detected and destroyed by the immune system, has paved the way toward the development of novel therapeutic agents conceived to re(instate) anticancer immunity, and some of these interventions have already been licensed for use in humans by international regulatory agencies. In addition, it has become clear that many therapeutics that had been used for decades in the clinic are efficient (for the most part) because they engage the host immune system against malignant cells. ICD is one of the several mechanisms through which cytotoxic chemotherapeutics, targeted anticancer agents as well as some forms of radiotherapy can elicit tumor-targeting immune responses. Identifying novel ICD inducers as well as measures that convert non-immunogenic RCD into bona fide ICD is of primordial importance. Promising preclinical results and preliminary clinical findings suggest, indeed, that agents that promote CALR exposure, ATP secretion, type I IFN production, HMGB1 release or stimulate the downstream signal transduction pathway may considerably improve the clinical profile of conventional therapeutic regimens (Figure 1). A systematic investigation of the ability of currently available anticancer agents to elicit the abovementioned ICD-associated processes in human cancer cells of distinct histological origin is urgently awaited. These data may pave the way to the clinical implementation of combinatorial immuno(chemo)regimens that efficiently promote ICD and hence mediate complete tumor regression in a high proportion of patients.
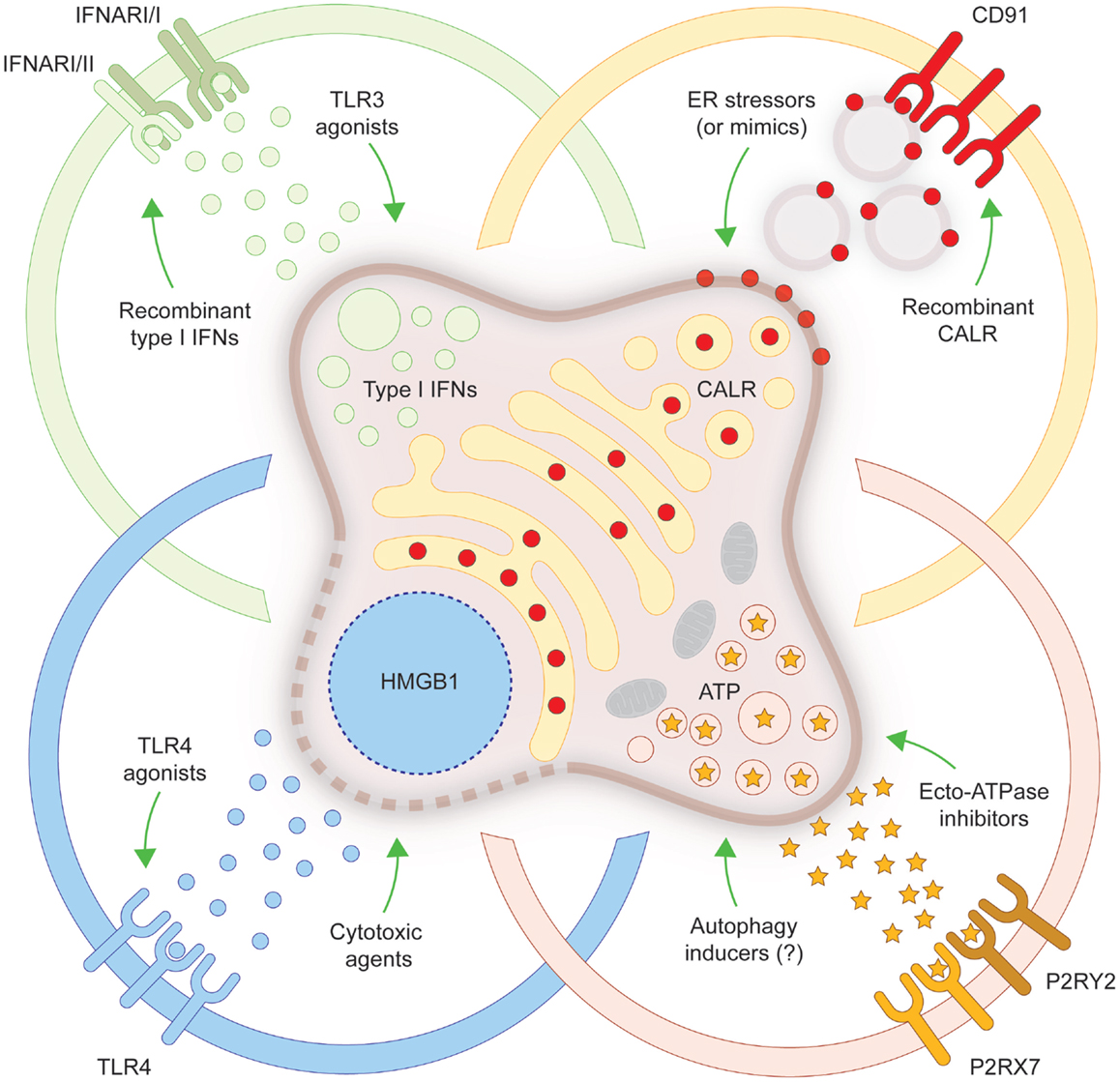
Figure 1. Strategies to convert non-immunogenic RCD into bona fide ICD. Upon inoculation into immunocompetent syngeneic hosts, cancer cells responding to a panel of lethal stimuli trigger an adaptive immune response against dead cell-associated antigens. Such an immunogenic variant of regulated cell death (RCD), commonly known as immunogenic cell death (ICD), relies on the exposure of calreticulin (CALR) on the cell surface, on the secretion of ATP, on the production of type I interferons (IFNs) and on the release of high-mobility group box 1 (HMGB1, which accompanies cell death). When any of these damage-associated molecular patterns cannot be emitted (in the appropriate spatiotemporal order), dying cancer cells cannot be perceived anymore as immunogenic by the host immune system. Several strategies have been conceived to correct these defects, hence converting non-immunogenic RCD into bona fide ICD. ER, endoplasmic reticulum; IFNAR, interferon (alpha, beta, and omega) receptor; P2RX7, purinergic receptor P2X, ligand gated ion channel, 7; P2RY2, purinergic receptor P2Y, G-protein coupled, 2; TLR, toll-like receptor.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Guest Associate Editor, Patrizia Agostinis, declares that despite having co-authored a few manuscripts with the authors Lorenzo Galluzzi, Oliver Kepp and Guido Kroemer in the past 2 years, there has been no conflict of interest during the review and handling of this manuscript.
Acknowledgments
GK is supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Abbreviations
APC, antigen-presenting cell; CALR, calreticulin; CTLA4, cytotoxic T lymphocyte-associated protein 4; CXCL10, chemokine (C–X–C motif) ligand 10; DAMP, damage-associated molecular pattern; EIF2A, eukaryotic translation initiation factor 2A, 65 kDa; ER, endoplasmic reticulum; HMGB1, high-mobility group box 1; ICD, immunogenic cell death; IFN, interferon; IFNAR, interferon (alpha, beta, and omega) receptor; mAb, monoclonal antibody; P2RX7, purinergic receptor P2X, ligand gated ion channel, 7; P2RY2, purinergic receptor P2Y, G-protein coupled, 2; RCD, regulated cell death; siRNA, small-interfering RNA; TLR, toll-like receptor.
References
1. Cirone M, Di Renzo L, Lotti LV, Conte V, Trivedi P, Santarelli R, et al. Activation of dendritic cells by tumor cell death. Oncoimmunology (2012) 1:1218–9. doi: 10.4161/onci.20428
2. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12:860–75. doi:10.1038/nrc3380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
4. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol (2014) 35:24–32. doi:10.1016/j.semcdb.2014.02.006
5. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ (2015) 22:58–73. doi:10.1038/cdd.2014.137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ (2009) 16:3–11. doi:10.1038/cdd.2008.150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J (2009) 28:578–90. doi:10.1038/emboj.2009.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ (2012) 19:107–20. doi:10.1038/cdd.2011.96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology (2014) 3:e955691. doi:10.4161/21624011.2014.955691
10. Sukkurwala AQ, Adjemian S, Senovilla L, Michaud M, Spaggiari S, Vacchelli E, et al. Screening of novel immunogenic cell death inducers within the NCI mechanistic diversity set. Oncoimmunology (2014) 3:e28473. doi:10.4161/onci.28473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev (2013) 24:319–33. doi:10.1016/j.cytogfr.2013.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Vacchelli E, Senovilla L, Eggermont A, Fridman WH, Galon J, Zitvogel L, et al. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology (2013) 2:e23510. doi:10.4161/onci.23510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Kepp O, Senovilla L, Kroemer G. Immunogenic cell death inducers as anticancer agents. Oncotarget (2014) 5:5190–1.
14. Vacchelli E, Aranda F, Eggermont A, Galon J, Sautes-Fridman C, Cremer I, et al. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology (2014) 3:e27878. doi:10.4161/onci.27878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202:1691–701. doi:10.1084/jem.20050915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13:54–61. doi:10.1038/nm1523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res (2011) 71:4821–33. doi:10.1158/0008-5472.CAN-11-0950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene (2010) 29:482–91. doi:10.1038/onc.2009.356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene (2011) 30:1147–58. doi:10.1038/onc.2010.500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res (2011) 71:768–78. doi:10.1158/0008-5472.CAN-10-2788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol (2011) 33:369–83. doi:10.1007/s00281-011-0245-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Stoetzer OJ, Fersching DM, Salat C, Steinkohl O, Gabka CJ, Hamann U, et al. Circulating immunogenic cell death biomarkers HMGB1 and RAGE in breast cancer patients during neoadjuvant chemotherapy. Tumour Biol (2013) 34:81–90. doi:10.1007/s13277-012-0513-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Ziccheddu G, Proietti E, Moschella F. The Janus face of cyclophosphamide: a sterile inflammatory response that potentiates cancer immunotherapy. Oncoimmunology (2013) 2:e25789. doi:10.4161/onci.25789
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Demaria S, Santori FR, Ng B, Liebes L, Formenti SC, Vukmanovic S. Select forms of tumor cell apoptosis induce dendritic cell maturation. J Leukoc Biol (2005) 77:361–8. doi:10.1189/jlb.0804478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood (2007) 109:4839–45. doi:10.1182/blood-2006-10-054221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Cirone M, Di Renzo L, Lotti LV, Conte V, Trivedi P, Santarelli R, et al. Primary effusion lymphoma cell death induced by bortezomib and AG 490 activates dendritic cells through CD91. PLoS One (2012) 7:e31732. doi:10.1371/journal.pone.0031732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Korbelik M, Sun J, Cecic I. Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res (2005) 65:1018–26.
28. Korbelik M, Zhang W, Merchant S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol Immunother (2011) 60:1431–7. doi:10.1007/s00262-011-1047-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother (2012) 61:215–21. doi:10.1007/s00262-011-1184-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Galluzzi L, Kepp O, Kroemer G. Immunogenic cell death in radiation therapy. Oncoimmunology (2013) 2:e26536. doi:10.4161/onci.26536
31. Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautes-Fridman C, Galon J, et al. Trial watch: anticancer radioimmunotherapy. Oncoimmunology (2013) 2:e25595. doi:10.4161/onci.25595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Bloy N, Pol J, Manic G, Vitale I, Eggermont A, Galon J, et al. Trial watch: radioimmunotherapy for oncological indications. Oncoimmunology (2014) 3:e954929. doi:10.4161/21624011.2014.954929
33. Garg AD, Agostinis P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem Photobiol Sci (2014) 13:474–87. doi:10.1039/c3pp50333j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol (2015) 25:11–7. doi:10.1016/j.semradonc.2014.07.005
35. Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther (2013) 20:7–15. doi:10.1038/gt.2011.205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Vacchelli E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, et al. Trial watch: oncolytic viruses for cancer therapy. Oncoimmunology (2013) 2:e24612. doi:10.4161/onci.25238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Workenhe ST, Mossman KL. Rewiring cancer cell death to enhance oncolytic viro-immunotherapy. Oncoimmunology (2013) 2:e27138. doi:10.4161/onci.27138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Workenhe ST, Pol JG, Lichty BD, Cummings DT, Mossman KL. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol Res (2013) 1:309–19. doi:10.1158/2326-6066.CIR-13-0059-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Pol J, Bloy N, Obrist F, Eggermont A, Galon J, Cremer I, et al. Trial watch: oncolytic viruses for cancer therapy. Oncoimmunology (2014) 3:e28694. doi:10.4161/onci.28185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Hoffmann J, Vitale I, Buchmann B, Galluzzi L, Schwede W, Senovilla L, et al. Improved cellular pharmacokinetics and pharmacodynamics underlie the wide anticancer activity of sagopilone. Cancer Res (2008) 68:5301–8. doi:10.1158/0008-5472.CAN-08-0237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Pellicciotta I, Yang CP, Goldberg GL, Shahabi S. Epothilone B enhances Class I HLA and HLA-A2 surface molecule expression in ovarian cancer cells. Gynecol Oncol (2011) 122:625–31. doi:10.1016/j.ygyno.2011.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science (2012) 337:1678–84. doi:10.1126/science.1224922
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Fucikova J, Moserova I, Truxova I, Hermanova I, Vancurova I, Partlova S, et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer (2014) 135:1165–77. doi:10.1002/ijc.28766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell (2010) 140:798–804. doi:10.1016/j.cell.2010.02.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Galluzzi L, Bravo-San Pedro JM, Kroemer G. Organelle-specific initiation of cell death. Nat Cell Biol (2014) 16:728–36. doi:10.1038/ncb3005
46. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta (2010) 1805:53–71. doi:10.1016/j.bbcan.2009.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol (2012) 13:780–8. doi:10.1038/nrm3479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Garg AD, Dudek AM, Agostinis P. Cancer immunogenicity, danger signals, and DAMPs: what, when, and how? Biofactors (2013) 39:355–67. doi:10.1002/biof.1125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Krysko O, Love Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis (2013) 4:e631. doi:10.1038/cddis.2013.156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi:10.1038/nm.2028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med (2014) 20:1301–9. doi:10.1038/nm.3708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi:10.1038/nm1622
53. Galluzzi L, Kepp O, Kroemer G. Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J (2012) 31:1055–7. doi:10.1038/emboj.2012.2
54. Garg AD, Dudek AM, Agostinis P. Autophagy-dependent suppression of cancer immunogenicity and effector mechanisms of innate and adaptive immunity. Oncoimmunology (2013) 2:e26260. doi:10.4161/onci.26260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy (2013) 9:1292–307. doi:10.4161/auto.25399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Kepp O, Menger L, Vacchelli E, Locher C, Adjemian S, Yamazaki T, et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev (2013) 24:311–8. doi:10.1016/j.cytogfr.2013.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334:1573–7. doi:10.1126/science.1208347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J (2012) 31:1062–79. doi:10.1038/emboj.2011.497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461:282–6. doi:10.1038/nature08296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38:729–41. doi:10.1016/j.immuni.2013.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418:191–5. doi:10.1038/nature00858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Ma Y, Adjemian S, Yang H, Catani JP, Hannani D, Martins I, et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology (2013) 2:e24568. doi:10.4161/onci.24568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Michaud M, Xie X, Bravo-San Pedro JM, Zitvogel L, White E, Kroemer G. An autophagy-dependent anticancer immune response determines the efficacy of melanoma chemotherapy. Oncoimmunology (2014) 3:e944047. doi:10.4161/21624011.2014.944047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Fridman WH, Galon J, Pages F, Tartour E, Sautes-Fridman C, Kroemer G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res (2011) 71:5601–5. doi:10.1158/0008-5472.CAN-11-1316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12:298–306. doi:10.1038/nrc3245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, et al. Trial watch: prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology (2012) 1:1323–43. doi:10.4161/onci.22009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity (2013) 39:782–95. doi:10.1016/j.immuni.2013.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Anitei MG, Zeitoun G, Mlecnik B, Marliot F, Haicheur N, Todosi AM, et al. Prognostic and predictive values of the immunoscore in patients with rectal cancer. Clin Cancer Res (2014) 20:1891–9. doi:10.1158/1078-0432.CCR-13-2830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi:10.1038/nature13954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov (2012) 11:215–33. doi:10.1038/nrd3626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39:74–88. doi:10.1016/j.immuni.2013.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Vacchelli E, Eggermont A, Galon J, Sautes-Fridman C, Zitvogel L, Kroemer G, et al. Trial watch: monoclonal antibodies in cancer therapy. Oncoimmunology (2013) 2:e22789. doi:10.4161/onci.25238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Aranda F, Vacchelli E, Eggermont A, Galon J, Fridman WH, Zitvogel L, et al. Trial watch: immunostimulatory monoclonal antibodies in cancer therapy. Oncoimmunology (2014) 3:e27297. doi:10.4161/onci.27297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Robert C, Thomas L, Bondarenko I, O’day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364:2517–26. doi:10.1056/NEJMoa1104621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Eggermont AM, Robert C. Melanoma: smart therapeutic strategies in immuno-oncology. Nat Rev Clin Oncol (2014) 11:181–2. doi:10.1038/nrclinonc.2014.36
76. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet (2014) 384:1109–17. doi:10.1016/S0140-6736(14)60958-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med (2015) 372:320–30. doi:10.1056/NEJMoa1412082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Galluzzi L, Kroemer G, Eggermont A. Novel immune checkpoint blocker approved for the treatment of advanced melanoma. Oncoimmunology (2014) 3:e967147. doi:10.1016/j.clindermatol.2012.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Vacchelli E, Prada N, Kepp O, Galluzzi L. Current trends of anticancer immunochemotherapy. Oncoimmunology (2013) 2:e25396. doi:10.4161/onci.25396
80. Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buque A, Senovilla L, Baracco EE, et al. Classification of current anticancer immunotherapies. Oncotarget (2014) 5:12472–508.
81. Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med (2012) 4:143ra199. doi:10.1126/scitranslmed.3003807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kono K, Mimura K. Immunogenic tumor cell death induced by chemoradiotherapy in a clinical setting. Oncoimmunology (2013) 2:e22197. doi:10.4161/onci.22197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Menger L, Vacchelli E, Kepp O, Eggermont A, Tartour E, Zitvogel L, et al. Trial watch: cardiac glycosides and cancer therapy. Oncoimmunology (2013) 2:e23082. doi:10.4161/onci.23082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Palombo F, Focaccetti C, Barnaba V. Therapeutic implications of immunogenic cell death in human cancer. Front Immunol (2014) 4:503. doi:10.3389/fimmu.2013.00503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene (2012) 31:1869–83. doi:10.1038/onc.2011.384
86. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology (2014) 3:e28518. doi:10.4161/onci.28518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Yamamura Y, Tsuchikawa T, Miyauchi K, Takeuchi S, Wada M, Kuwatani T, et al. The key role of calreticulin in immunomodulation induced by chemotherapeutic agents. Int J Clin Oncol (2015) 20:386–94. doi:10.1007/s10147-014-0719-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Bugaut H, Bruchard M, Berger H, Derangere V, Odoul L, Euvrard R, et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells. PLoS One (2013) 8:e65181. doi:10.1371/journal.pone.0065181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu TC, et al. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol (2012) 189:3209–20. doi:10.4049/jimmunol.1103826
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Aranda F, Bloy N, Galluzzi L, Kroemer G, Senovilla L. Vitamin B6 improves the immunogenicity of cisplatin-induced cell death. Oncoimmunology (2014) 3:e955685. doi:10.4161/21624011.2014.955685
91. Aranda F, Bloy N, Pesquet J, Petit B, Chaba K, Sauvat A, et al. Immune-dependent antineoplastic effects of cisplatin plus pyridoxine in non-small-cell lung cancer. Oncogene (2014). doi:10.1038/onc.2014.234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Cirone M, Garufi A, Di Renzo L, Granato M, Faggioni A, D’orazi G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology (2013) 2:e26198. doi:10.4161/onci.26198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle (2009) 8:3723–8. doi:10.4161/cc.8.22.10026
94. Mihailidou C, Chatzistamou I, Papavassiliou A, Kiaris H. Improvement of chemotherapeutic drug efficacy by endoplasmic reticulum stress. Endocr Relat Cancer (2015) 22(2):229–38. doi:10.1530/ERC-15-0019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Chen X, Yang Y, Zhou Q, Weiss JM, Howard OZ, McPherson JM, et al. Effective chemoimmunotherapy with anti-TGFbeta antibody and cyclophosphamide in a mouse model of breast cancer. PLoS One (2014) 9:e85398. doi:10.1371/journal.pone.0085398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang KY, Ferrone S, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer (2013) 133:624–36. doi:10.1002/ijc.28070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Wang W, Qin S, Zhao L. Docetaxel enhances CD3+ CD56+ cytokine-induced killer cells-mediated killing through inducing tumor cells phenotype modulation. Biomed Pharmacother (2015) 69:18–23. doi:10.1016/j.biopha.2014.10.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med (2011) 208:491–503. doi:10.1084/jem.20100269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD, et al. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res (2010) 70:9062–72. doi:10.1158/0008-5472.CAN-10-1825
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Beneteau M, Zunino B, Jacquin MA, Meynet O, Chiche J, Pradelli LA, et al. Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response. Proc Natl Acad Sci U S A (2012) 109:20071–6. doi:10.1073/pnas.1206360109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Boyd-Tressler A, Penuela S, Laird DW, Dubyak GR. Chemotherapeutic drugs induce ATP release via caspase-gated pannexin-1 channels and a caspase/pannexin-1-independent mechanism. J Biol Chem (2014) 289:27246–63. doi:10.1074/jbc.M114.590240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Kepp O, Galluzzi L, Giordanetto F, Tesniere A, Vitale I, Martins I, et al. Disruption of the PP1/GADD34 complex induces calreticulin exposure. Cell Cycle (2009) 8:3971–7. doi:10.4161/cc.8.23.10191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Zhao T, Ren H, Jia L, Chen J, Xin W, Yan F, et al. Inhibition of HIF-1alpha by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget (2015) 6:2250–62.
104. Wemeau M, Kepp O, Tesniere A, Panaretakis T, Flament C, De Botton S, et al. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis (2010) 1:e104. doi:10.1038/cddis.2010.82
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Frey B, Stache C, Rubner Y, Werthmoller N, Schulz K, Sieber R, et al. Combined treatment of human colorectal tumor cell lines with chemotherapeutic agents and ionizing irradiation can in vitro induce tumor cell death forms with immunogenic potential. J Immunotoxicol (2012) 9:301–13. doi:10.3109/1547691X.2012.693547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Dudek-Peric AM, Ferreira GB, Muchowicz A, Wouters J, Prada N, Martin S, et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res (2015). doi:10.1158/0008-5472.CAN-14-2089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Lu X, Ding ZC, Cao Y, Liu C, Habtetsion T, Yu M, et al. Alkylating agent melphalan augments the efficacy of adoptive immunotherapy using tumor-specific CD4+ T cells. J Immunol (2015) 194:2011–21. doi:10.4049/jimmunol.1401894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Zhou P, Teruya-Feldstein J, Lu P, Fleisher M, Olshen A, Comenzo RL. Calreticulin expression in the clonal plasma cells of patients with systemic light-chain (AL-) amyloidosis is associated with response to high-dose melphalan. Blood (2008) 111:549–57. doi:10.1182/blood-2007-11-125526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ (2008) 15:1499–509. doi:10.1038/cdd.2008.67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Gou HF, Huang J, Shi HS, Chen XC, Wang YS. Chemo-immunotherapy with oxaliplatin and interleukin-7 inhibits colon cancer metastasis in mice. PLoS One (2014) 9:e85789. doi:10.1371/journal.pone.0085789
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther (2013) 21:1212–23. doi:10.1038/mt.2013.51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Rubner Y, Muth C, Strnad A, Derer A, Sieber R, Buslei R, et al. Fractionated radiotherapy is the main stimulus for the induction of cell death and of Hsp70 release of p53 mutated glioblastoma cell lines. Radiat Oncol (2014) 9:89. doi:10.1186/1748-717X-9-89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Martin S, Dudek-Peric AM, Maes H, Garg AD, Gabrysiak M, Demirsoy S, et al. Concurrent MEK and autophagy inhibition is required to restore cell death associated danger-signalling in vemurafenib-resistant melanoma cells. Biochem Pharmacol (2015) 93:290–304. doi:10.1016/j.bcp.2014.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol (2012) 13:89–102. doi:10.1038/nrm3270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, et al. Prognostic impact of vitamin B6 metabolism in lung cancer. Cell Rep (2012) 2:257–69. doi:10.1016/j.celrep.2012.06.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Steiner P, Kulangara K, Sarria JC, Glauser L, Regazzi R, Hirling H. Reticulon 1-C/neuroendocrine-specific protein-C interacts with SNARE proteins. J Neurochem (2004) 89:569–80. doi:10.1111/j.1471-4159.2004.02345.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Michaud M, Sukkurwala AQ, Di Sano F, Zitvogel L, Kepp O, Kroemer G. Synthetic induction of immunogenic cell death by genetic stimulation of endoplasmic reticulum stress. Oncoimmunology (2014) 3:e28276. doi:10.4161/onci.28276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Dubois SG, Grier HE. Chemotherapy: the role of ifosfamide and etoposide in Ewing sarcoma. Nat Rev Clin Oncol (2009) 6:251–3. doi:10.1038/nrclinonc.2009.25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Johnson FM, Glisson BS. Chemotherapy: irinotecan or etoposide as front-line therapy for SCLC? Nat Rev Clin Oncol (2009) 6:562–3. doi:10.1038/nrclinonc.2009.141
120. Kepp O, Semeraro M, Pedro JM, Bloy N, Buque A, Huang X, et al. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol (2015). doi:10.1016/j.semcancer.2015.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity (2013) 39:211–27. doi:10.1016/j.immuni.2013.07.017
122. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (2011) 333:1109–12. doi:10.1126/science.1201940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34(7):856–80. doi:10.15252/embj.201490784
124. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ (2014) 21:79–91. doi:10.1038/cdd.2013.75
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Michaud M, Sukkurwala AQ, Martins I, Shen S, Zitvogel L, Kroemer G. Subversion of the chemotherapy-induced anticancer immune response by the ecto-ATPase CD39. Oncoimmunology (2012) 1:393–5. doi:10.4161/onci.19070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov (2013) 12:829–46. doi:10.1038/nrd4191
127. Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, et al. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer (2015) 136:E313–25. doi:10.1002/ijc.29202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi:10.1038/nri3581
129. McNab F, Mayer-Barber K, Sher A, Wack A, O’garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi:10.1038/nri3787
130. Vacchelli E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, et al. Trial watch: toll-like receptor agonists for cancer therapy. Oncoimmunology (2013) 2:e25238. doi:10.4161/onci.25238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Aranda F, Vacchelli E, Obrist F, Eggermont A, Galon J, Sautes-Fridman C, et al. Trial watch: toll-like receptor agonists in oncological indications. Oncoimmunology (2014) 3:e29179. doi:10.4161/onci.29179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol (2005) 5:331–42. doi:10.1038/nri1594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi:10.1016/j.cell.2013.05.039
134. Pang X, Zhang Y, Wei H, Zhang J, Luo Q, Huang C, et al. Expression and effects of high-mobility group box 1 in cervical cancer. Int J Mol Sci (2014) 15:8699–712. doi:10.3390/ijms15058699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ (2014) 21:69–78. doi:10.1038/cdd.2013.72
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Srivastava AK, Dinc G, Sharma RK, Yolcu ES, Zhao H, Shirwan H. SA-4-1BBL and monophosphoryl lipid A constitute an efficacious combination adjuvant for cancer vaccines. Cancer Res (2014) 74:6441–51. doi:10.1158/0008-5472.CAN-14-1768-A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, et al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev (2011) 30:61–9. doi:10.1007/s10555-011-9273-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ATP, autophagy, calreticulin, endoplasmic reticulum stress, HMGB1, type I interferon
Citation: Bezu L, Gomes-da-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O and Kroemer G (2015) Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 6:187. doi: 10.3389/fimmu.2015.00187
Received: 12 March 2015; Accepted: 06 April 2015;
Published: 24 April 2015
Edited by:
Patrizia Agostinis, University of Leuven, BelgiumReviewed by:
Stephan Gasser, National University of Singapore, SingaporeLuis De La Cruz-Merino, Hospital Universitario Virgen Macarena, Spain
Copyright: © 2015 Bezu, Gomes-da-Silva, Dewitte, Breckpot, Fucikova, Spisek, Galluzzi, Kepp and Kroemer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oliver Kepp and Guido Kroemer, INSERM U1138, Centre des Recherche des Cordeliers, Equipe 11, 15 rue de l’Ecole de Médecine, Paris 75005, Franceb2xpdmVyLmtlcHBAZ3VzdGF2ZXJvdXNzeS5mcg==;a3JvZW1lckBvcmFuZ2UuZnI=
†Equally contributed to this work.
‡Shared senior co-authorship.