 Mateusz Pawel Poltorak
Mateusz Pawel Poltorak Barbara Ursula Schraml
Barbara Ursula Schraml- Institute for Medical Microbiology, Immunology and Hygiene, Technische Universität München, Munich, Germany
Dendritic cells (DCs) are a heterogeneous group of mononuclear phagocytes with versatile roles in immunity. They are classified predominantly based on phenotypic and functional properties, namely their stellate morphology, expression of the integrin CD11c, and major histocompatibility class II molecules, as well as their superior capacity to migrate to secondary lymphoid organs and stimulate naïve T cells. However, these attributes are not exclusive to DCs and often change within inflammatory or infectious environments. This led to debates over cell identification and questioned even the mere existence of DCs as distinct leukocyte lineage. Here, we review experimental approaches taken to fate map DCs and discuss how these have shaped our understanding of DC ontogeny and lineage affiliation. Considering the ontogenetic properties of DCs will help to overcome the inherent shortcomings of purely phenotypic- and function-based approaches to cell definition and will yield a more robust way of DC classification.
Introduction
Dendritic cells (DCs) were originally identified in mouse spleen for their unique stellate morphology, their ability to adhere to certain glass surfaces and their superior capacity to activate naïve T lymphocytes that distinguished them from macrophages (MØs) (1–3). Mostly for historical reasons, DCs are considered part of the mononuclear phagocyte (MP) system, which groups all highly phagocytic cells derived from monocytes or their precursors based on the premise that tissue MØs arise from monocytes (4–9). This presumed relatedness of DCs, monocytes, and MØs coupled to the lack of reliable ways to distinguish MP subtypes has caused continuous debates over accurate cell-type identification and has led some to question whether DCs in fact constitute an independent cell lineage (6, 7, 10–14). However, today we have conclusive evidence demonstrating that DCs, monocytes, and MØs have distinct cellular origin and we further distinguish plasmacytoid DCs (pDCs) from two subsets of so-called conventional or classical DCs (cDCs) based on unique developmental requirements (7, 15–19). Nonetheless, DCs remain defined based on phenotypic and functional properties that often overlap with those of monocytes or MØs (19), although some have suggested a shift in paradigm toward a nomenclature that takes cell ontogeny into account (6, 7, 10).
Dendritic cells are generally identified by their high expression of major histocompatibility complex class II molecules (MHCII) and of the integrin CD11c, as well as their superior capacity to migrate from non-lymphoid to lymphoid organs and stimulate naïve T cells (3, 20–22). However, these characteristics are not absolute and can change in situations of inflammation or infection, thus complicating cell identification (6, 7, 23, 24). For instance, CD11c, considered the hallmark surface marker of DCs, is also found on B, T, and NK cells as well as some monocytes, MØs, and eosinophils (25–32). Dendritic protrusions have also been observed in some MØs and T cells (33–35). Further, surface markers, such as F4/80, CD14, or CD64 (Fc-gamma receptor 1), generally associated with monocytes or MØs can be found on DCs (36–38). One might argue that the most defining feature of DCs is their ability to activate T cells, however such definition discounts the fact that DCs potently regulate innate immune responses independent of their ability to migrate to lymphoid organs or stimulate T cells (39–44). Conversely, non-DCs can carry antigen to lymph nodes and activate naïve T cells in some instances (45–47).
Therefore, morphological and functional properties, as well as the expression of surface markers are insufficient to clearly distinguish DCs from monocytes and MØs, raising the necessity to find a more robust way of cell identification. Recent studies in mouse and human indicate that DCs, MØs, and monocytes have unique ontogenetic properties and thus can be considered distinct cell lineages (36, 48–54). Here, we review approaches that have been employed to track and define the progeny of DC precursors in vivo and discuss how such “fate mapping” approaches have improved our understanding of DC heterogeneity and ontogeny. These studies lay the foundation for moving toward cell ontogeny as a major lineage-determining criterion, which will allow for a more reliable and precise classification of DCs and DC subsets.
DC Development
Dendritic cells are short-lived and their maintenance relies on constant replenishment from bone marrow progenitors that originate from hematopoietic stem cells (HSCs) (19, 55). In the classic model of DC development monocytes and DCs arise from bi-potent progenitors, so-called MØ and DC progenitors (MDPs) (Figure 1) (56). MDPs further give rise to common DC progenitors (CDPs) restricted to the generation of pDCs and cDCs (Figure 1) (57, 58). pDCs terminally differentiate in the bone marrow, thus exit the bone marrow as fully developed cells and reach peripheral organs via the blood stream (Figure 1) (15, 59). In contrast, cDCs arise from another developmental intermediate termed pre-DC, which exits the bone marrow and migrates through the blood to seed lymphoid and non-lymphoid tissues (60, 61). There, pre-DCs terminally differentiate into cDCs, including the main CD11b− and CD11b+ subtypes (Figure 1) (60–63). In lymphoid tissues these are CD8α+CD11b− and CD11b+ resident cDCs, whereas in non-lymphoid tissues they comprise CD103+CD11b− and CD11b+ migratory cDCs (3, 60–63). Like pDCs, monocytes complete their development in the bone marrow but in tissues they differentiate into cells with DC- or MØ-like features (Figure 1) (23, 24, 64, 65). This plasticity is remarkably prominent in inflammatory or infectious environments, when monocyte-derived cells with qualities of DCs have been referred to as TNF-α/iNOS-producing DCs (Tip-DCs), monocyte-derived DCs (mo-DCs), and/or inflammatory DCs (23, 24, 64, 65).
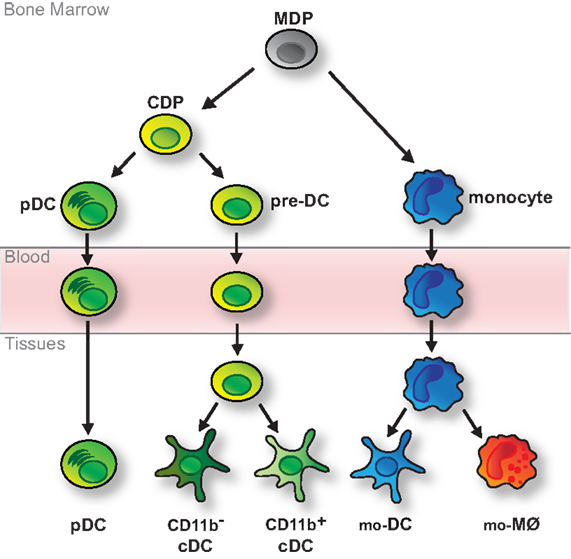
Figure 1. Classic model of DC development. DCs and monocytes are ancestrally related and arise from bi-potential MDPs residing in the bone marrow. MDPs further differentiate into monocytes and CDPs, which are restricted to the generation of various types of DCs. CDPs give rise to pDCs, which fully develop in the bone marrow, and pre-DCs, which migrate through the blood to tissues, where they fully differentiate into CD11b− (including CD8α+ cDCs in lymphoid tissue and migratory CD103+ cDCs in non-lymphoid tissue) and CD11b+ cDCs. Monocytes complete their development in the bone marrow and reach peripheral tissues via the bloodstream. There they further differentiate into monocyte-derived DCs (mo-DCs) or MØs (mo-MØs) in response to environmental cues.
Although most of our knowledge concerning DC development is derived from mouse studies, developmental parallels have been observed in other species (66–73). Especially the identification of putative equivalent DC progenitor populations in human holds promise for future research (72, 73). Yet, some uncertainties remain. Common lymphoid progenitors (CLPs) can give rise to DC descendants upon adoptive transfer (74), although it is now thought that DCs originate predominantly from myeloid progenitors (75, 76). Nonetheless, some pDCs, but not cDCs, show evidence of VDJ gene rearrangements, potentially indicating lymphoid lineage heritage (15, 59, 77). However, it remains unclear whether evidence of Rag gene expression history necessarily means that pDCs have dual lymphoid and myeloid origin. Contrary to the dogma that monocytes and DCs share a common immediate ancestor, recent data suggest that lineage divergence of HSC-derived myeloid cells occurs much earlier than previously predicted and that monocytes and DCs might arise independent of a bi-potential developmental intermediate (49, 78, 79). Elucidating such unresolved aspects pertaining to DC ontogeny may solve uncertainties in determining lineage affiliation, which, in turn, will aid to further decipher the unique functions of DCs in immunity.
Fate Mapping
Understanding cell development requires models with which the relationship of a precursor cell and its progeny can be defined in vivo. Such “fate mapping” can be achieved in various ways and relies on the selective labeling of the cell(s) of interest so that consequently the development of the marked cell can be followed in its natural environment (80). Tracing progenitors in vivo also offers the possibility to determine the fate of populations when lineage affiliation is most heavily debated, namely following experimental manipulation to generate conditions of inflammation or infection. While most fate mapping strategies follow the progeny of bulk cell populations, recently developed techniques have enabled the tracing of single cells, thus providing valuable information regarding their developmental potential at the clonal level (80, 81). In all fate mapping experiments, it is important to consider that their interpretation is dependent on the use of select, faithful and stable markers (82).
Precursor Transfers
The transfer of purified and pre-marked precursor cells into congenic recipients is the most accessible form of fate mapping as a variety of labeling options can be used to distinguish between donor and host cells (Figure 2A) (80). As a result, precursor transfers are commonly used to study cell development and lineage relationships and remain a standard protocol for defining the stemness of progenitor cells (80). Such experiments rely on the ability to purify sufficient precursors that, after cell isolation, retain the capacity to home to the appropriate anatomical niche and expand sufficiently into detectable progeny. To circumvent such limitations transfer studies are often combined with protocols to induce leukopenia, such as irradiation, in order to increase the niche available for cell engraftment (Figure 2A) (80). However, these manipulations can alter developmental signals, which, in turn, might impact on the interpretation of results (18, 54, 83). To best mimic the endogenous cellular environment, progenitors have been returned directly to their organs of origin, for instance by intra-bone injection (84).
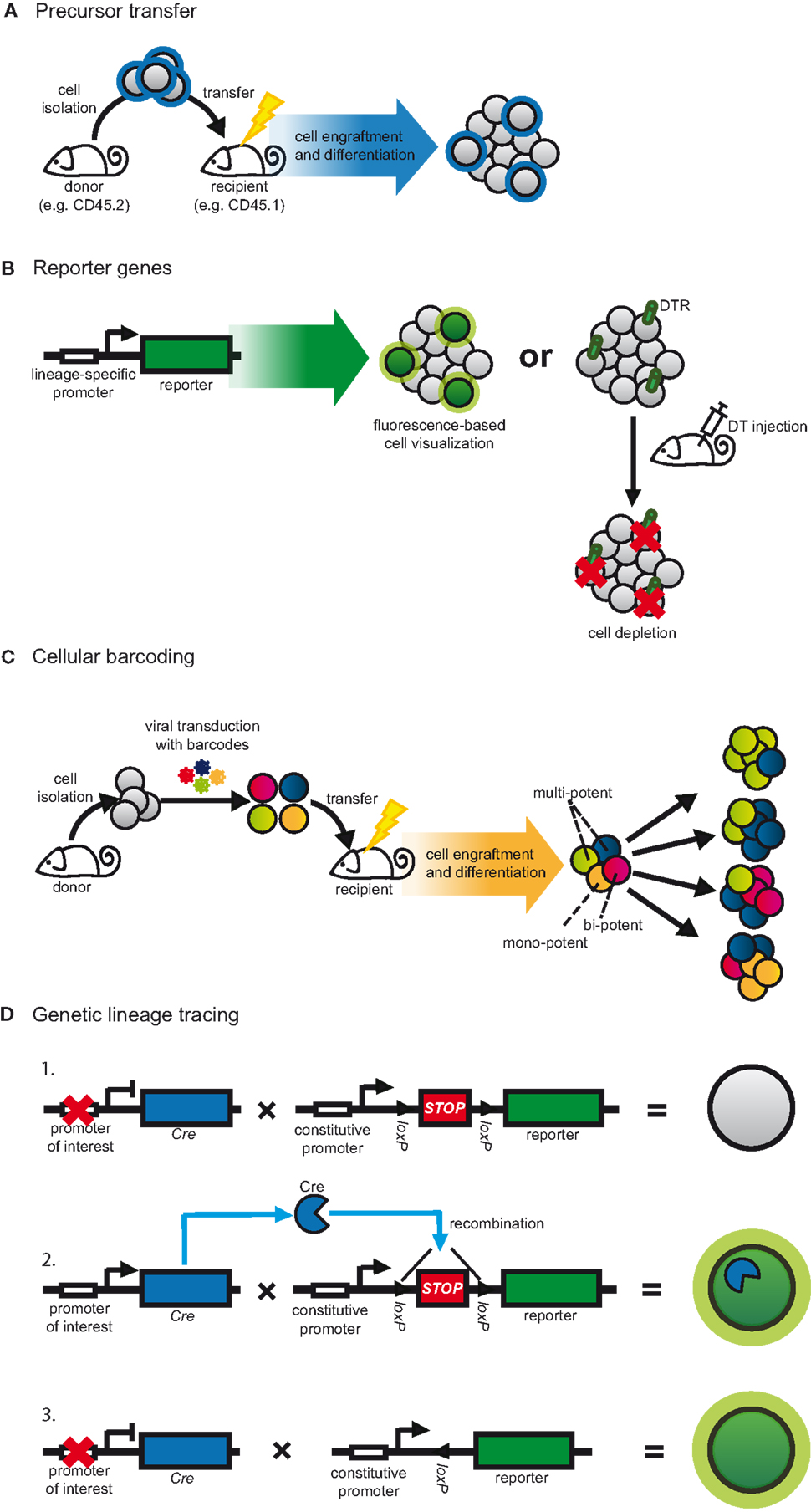
Figure 2. Strategies to fate map DCs. (A) Progenitors are adoptively transferred to assess their differentiation in the physiological context. Graft-derived cells are distinguished from host cells based on pre-defined labels, for instance congenic markers. This method is often combined with strategies to increase the niche available for cell engraftment, such as irradiation. (B) In transgenic approaches, lineage-restricted promoters can be used to drive a reporter gene. Target cell populations can be visualized by the expression of fluorescent proteins or can be depleted. In the latter case, cell-restricted expression of DTR allows for conditional cell ablation following DT injection. (C) Progenitors are transduced in vitro with semi-random DNA sequences (barcodes) by retro- or lentiviral vectors and subsequently transferred into irradiated congenic recipients. After differentiation, cell progeny are analyzed for their barcode repertoire using deep sequencing or microarray. The representation of a given barcode in multiple cell populations indicates multi-potency of the transferred cell. (D) Expression of Cre recombinase is driven by a lineage or cell-specific promoter. Additionally, a reporter gene, usually a fluorescent protein, is placed under control of a constitutive promoter. Expression of the reporter is blocked by inserting a loxP-flanked STOP cassette (1). Cre expression leads to excision of the STOP cassette resulting in expression of the reporter gene (2). Since the promoter-driving reporter gene expression is constitutively active, the target cell is irreversibly labeled irrespective of continuous Cre expression (3).
The DC progenitors MDP, CDP, and pre-DC were in part defined by assessing their developmental potential after adoptive transfer into mice (56–58, 60, 61, 84–86). In such experiments, MDPs give rise to DCs and monocytes, whereas CDPs and pre-DCs are restricted to the generation of DCs but do not generate monocytes or other leukocyte lineages (56–58, 61, 84–86). In combination with experiments assessing the differentiation potential of single progenitors in vitro (56–58), these studies have significantly shaped our view of DC development (Figure 1). Surprisingly, the existence of MDP as a bi-potential intermediate for DCs and monocytes has recently been questioned when single CX3CR1+ MDPs were unable to generate both DCs and monocytes upon differentiation in vitro (78). The authors further found that adoptively transferred CX3CR1+ MDPs, not only gave rise to DCs and monocytes but also neutrophils (78). However, such multi-potency of MDPs was not observed in earlier studies (52, 56, 61, 85, 86) and is not evident in genetic CX3CR1 fate mapping experiments (50). It is possible that these discrepancies may be explained by experimental variation such as differences in cell isolation, the timing of analysis or variances in the niche available for cell engraftment following irradiation (18, 54, 83). In light of these results it is noteworthy, however, that upon adoptive transfer MDPs exhibit pDC potential only in some studies (52, 86) but not others (56, 85), whereas the presumed downstream CDPs produce both pDCs and cDCs (57, 58). Taken together these experiments raise some doubt about the existence of a MDP as a key developmental intermediate for monocytes, cDCs, and pDCs. However, resolving this matter will require the use of better models to trace single cells in vivo as experiments relying on the isolation and analysis of bulk progenitor populations are inherently prone to disparities in gating strategy or cell purity.
In DC ontogeny, these issues are augmented because MDP and CDP exhibit substantial phenotypic overlap: both lack lineage-defining markers, are characterized by expression of CX3CR1, CD115 (M-CSFR, Csf1R) as well as CD135 (FMS-like tyrosine kinase 3, FLT3) and, until recently, CDP could only be distinguished from MDP by lower expression of the receptor tyrosine kinase CD117 (c-kit) (56–58, 61, 86). We have recently found that the C-type lectin receptor DNGR-1 (Clec9a) marks cells resembling CDPs (36). Surprisingly, upon adoptive transfer, DNGR-1+CD115+ progenitors exhibit cDC-restricted differentiation potential and do not generate pDCs (36), suggesting that DNGR-1 marks cDC-restricted progenitors. These data are in line with a recent study demonstrating a strong bias for CD115+ CDPs to generate cDCs, whereas pDCs arise predominantly from CD115 negative cells (79). Therefore, cDCs and pDCs appear to have distinct developmental intermediates that can be distinguished by expression of CD115 (79) and DNGR-1 (36). Since CD115+ CDPs presumably express DNGR-1 (36), it is unclear why some CD115+ CDPs show combined cDC and pDC potential in clonal assays (57, 58, 79). It is possible that antibody-mediated triggering of DNGR-1 or growth factor receptors, such as CD115, during cell isolation skews DC differentiation toward a particular DC sub-lineage in an unforeseeable manner. The developmental potential of progenitors may also be influenced by the specific culture conditions used (78) or DCs could exhibit a degree of developmental plasticity (87). Nonetheless, the existence of a putative intermediate monocyte-restricted progenitor downstream of MDP (common monocyte progenitor, cMoP) (52) alongside the aforementioned pDC- and cDC-restricted progenitors supports a model in which monocytes, cDCs, and pDCs develop independently. The genuine point of lineage divergence, however, remains to be determined.
Questions regarding the lineage affiliation of DCs have been muddled significantly by the developmental plasticity of monocytes (6, 24). The phenotypic transformation of monocytes into DC-like cells is most prominent in inflamed environments (8, 19, 23, 24). It can also be mimicked in vitro by culturing monocytes in the presence of GM-CSF (granulocyte-macrophage colony-stimulating factor) ± IL-4 (Interleukin-4) (88, 89). However, in vivo the inflammation-induced differentiation of monocytes into cells with attributes of DCs appears GM-CSF-independent (90), highlighting that the developmental requirements underlying this phenotypic conversion in vitro might differ from those involved in vivo. In the absence of experimentally induced infection or inflammation, adoptively transferred monocytes readily acquire CD11c and MHCII expression as well as functional features of DCs in non-lymphoid tissues (91–95). This phenotypic conversion is also observed after adoptive transfer into unirradiated hosts, which most closely mimics steady-state conditions (63). In contrast, transferred monocytes do not generate DCs in lymphoid organs, even if the niche for engraftment is opened by depletion of CD11c+ cells (84). Importantly, in non-lymphoid tissues monocytes exclusively generate CD11b+, but not CD103+CD11b− cells, which is in contrast to CDPs and pre-DCs that generate CD11b+ as well as CD103+CD11b− cDCs (63, 91–95). Therefore, CD11c+MHCII+CD11b+ cells in non-lymphoid tissues appear to constitute a population of mixed cellular origin that can arise from monocytic progenitors as well as pre-DCs. Adoptive transfer experiments do not allow to determine the relative contribution of each progenitor to this population, although surrogate markers such as CD64 or Mar-1 can serve to distinguish monocyte-derived cells from bona fide pre-DC-derived cDCs (46, 93, 94).
Notably, in irradiated hosts transferred monocytes can also generate CD11c+MHCII+ cells of the epidermis, which resemble Langerhans cells (LCs) (96–99). LCs exhibit many phenotypic and functional features of DCs, such as the capacity to migrate to lymphoid organs and stimulate naïve T cells, and have long been considered a prototypical DC population (96–99). However, we now realize that the majority of LCs is established before birth and maintained under steady-state conditions by self-renewal from local progenitors (96, 97, 99–102). These properties thus ontogenetically separate LCs from bone marrow-derived DCs or monocyte-derived cells. Moreover, monocytes may not necessarily adopt features of DCs or MØs upon entry into tissues, as a recent study indicates that monocyte can also exist in tissues without further differentiation (45). When considering this immense plasticity it will be crucial to elucidate the environmental cues that shape the diverse fates of monocytes to further dissect the full functional spectrum of monocytes and monocyte-derived cells.
Lineage Restricted Reporters
When the availability of isolatable progenitor cells is limiting and when populations are ontogenetically heterogeneous or might be influenced by alterations in their surroundings, determining lineage affiliation requires models to trace cells directly in their natural environment. One way to achieve this is by engineering models in which lineage-restricted promoters or genetic elements drive the expression of reporter genes (Figure 2B) (80, 82, 103). It is important to bear in mind that such experiments assume that the expression of the selected marker is restricted to the cell lineage in question and therefore, the choice of stable and specific markers is essential (80, 82, 103). Additionally, the genetic elements used to drive expression of the reporter must faithfully mimic endogenous gene expression (80, 82, 103).
Genetic elements of the Itgax gene, which encodes CD11c, have extensively been used to generate reporters to study DCs (82, 103). As such, transgenic mice in which the CD11c promoter drives the expression of fluorescent proteins (Figure 2B) have been key to visualizing the distribution and cellular interactions of DCs in a variety of tissues, including lymphoid organs, heart, lung, and skin (103–107). But fate mapping can also be achieved by cell deletion. Transgenic expression of primate diphtheria toxin receptor (DTR) renders murine cells susceptible to diphtheria toxin (DT)-induced cell death and, thus, enables inducible target cell depletion (Figure 2B) (82, 108). In this sense mice in which DTR expression is controlled by the elements of the CD11c promoter have been widely used to characterize the in vivo functions of DCs (28, 109–112). In part through analyzing such reporter mice, however, it has become evident that CD11c expression is not entirely restricted to DCs. It is also expressed on alveolar MØs, Ly6Clow as well as activated monocytes, plasmablasts, NK cells, and some T cells (25–29, 113). In addition, CD11c-driven fate reporter expression varies depending on the specific promoter elements used for transgenesis. CD11c.DTR mice, which were generated by conventional transgenesis using a 5.5-kb promoter element of the Itgax gene (109, 114), efficiently deplete most CD11c-expressing cDCs, LCs, alveolar, splenic marginal zone, and metallophilic MØs, as well as plasmablasts and T cells (27, 109, 115). However, DT-induced cell depletion in these mice is incomplete and spares certain cell types that transcribe their endogenous Itgax allele, including pDCs and NK cells (82, 115). Additionally, prolonged cell depletion using CD11c.DTR mice requires the use of bone marrow chimeras, possibly because of aberrant DTR expression on non-immune cells (82, 108, 112). Notably, this is not the case in CD11c.DOG and CD11c.LuciDTR mice, which were generated using bacterial artificial chromosome (BAC) transgenesis to place DTR under control of the extended regulatory region of the Itgax gene and in which DTR expression seems to more faithfully represent endogenous CD11c expression (28, 112, 115, 116). In all models, the occurrence of systemic neutrophilia and monocytosis following CD11c+ cell depletion (28, 115, 117) adds another layer of complexity to deciphering the cellular function and lineage affiliation of DCs.
The realization that CD11c is not restricted to DCs in all instances nurtured the search for more specific lineage-defining markers. Two groups simultaneously identified the transcription factor Zbtb46 (zDC, Btbd4) as ideal candidate to distinguish cDCs, as it is expressed in pre-DCs and cDCs but not in pDCs or their precursors (37, 38). Consistently, CD8α+ and CD11b+ cDCs in lymphoid organs as well as CD103+ cDCs in non-lymphoid organs uniformly express Zbtb46 as assessed in Zbtb46-GFP (37) and Zbtb46-DTR (38) reporter mice generated by site-directed mutagenesis. In contrast, CD11c+MHCII+CD11b+ cells in non-lymphoid organs, including lung, small intestine, and kidney, exhibit partial Zbtb46 expression (37, 38) indicating that they represent a heterogeneous population. This is consistent with reports demonstrating that these cells are of mixed monocyte and pre-DC origin (63, 91, 92, 95). Subsequently, Zbtb46 reporter mice have been used to help establish lineage relationships in a variety of tissues including heart, pancreas, tumors, and thymus (118–121). The fact that Zbtb46 expression is also found in human DCs suggests that it may also help to identify DCs across species (48, 122).
Nevertheless, the use of Zbtb46 as lineage-defining marker requires a note of caution. Zbtb46 expression is downregulated after DC stimulation and it is found in some non-immune cells (37, 123). Despite its prominent expression in the cDC lineage, Zbtb46 appears largely dispensable for cDC development (37, 123). Instead, it may reinforce DC-specific transcriptional programs (37) and/or suppress DC activation (123). Interestingly, monocytes activated in the presence of GM-CSF ± IL-4 uniformly induce Zbtb46 expression, whereas monocyte-derived Tip-DCs that are generated following infection with Listeria monocytogenes do not (37). This raises the possibility that Zbtb46 may control DC-like features of monocyte-derived cells in some inflammatory situations and it will be interesting to determine if Zbtb46 controls transcriptional programs in monocytes. These data also highlight that despite its selective expression on cDC progenitors and their descendants, Zbtb46 is not necessarily an indicator of cell ontogeny.
Identifying Common Developmental Requirements
Establishing that the development and/or delineation of a cell type depends on a certain transcription or growth factor constitutes a powerful way of fate mapping that has extensively been applied to MPs (42, 51, 63, 124–141). We can now clearly delineate DCs into distinct subpopulations based on the transcriptional programs that govern their development. pDCs are distinguished from two subsets of cDCs by their dependence on E2-2 (67, 142). The differentiation of pre-DCs into CD8α+ cDCs in lymphoid organs and CD103+CD11b− cDCs in non-lymphoid tissues is controlled by a set of transcription factors, including Irf8, Nfil-3, Id2, and Batf3 (124–128). Therefore, CD8α+ cDCs and CD103+ cDCs represent a developmentally related lineage of cDCs (6, 7). Notably, these cells also exhibit a degree of functional relatedness that is, for instance, exemplified by their superior capacity to activate CD8+ T cells (124, 143–145). In contrast, the development of CD11b+ cDCs from pre-DCs is controlled by distinct transcription factors, including RelB, RbpJ, PU.1, and Irf4 (42, 129–136). Notably, expression of CD24 separates pre-DCs into cells that preferentially generate either CD8α+ or CD11b+ cDCs in spleen (60) suggesting a stepwise differentiation of pre-DCs into cDCs. It will be interesting to determine whether such heterogeneity of pre-DCs also exists in the bone marrow. Notably, the extent of transcription factor dependence is linked to the genetic background of the particular mouse strain analyzed (146–148), indicating that transcriptional requirements are not always absolute or redundant factors exist (148). Consistently, CD8α+ DCs can develop in the absence of Batf3, Id2, and Nfil-3 (149). The local microenvironment may also contribute to shaping the diversity of the DC compartment, as in some tissues, such as the spleen and intestinal system, CD11b+ cDCs can be divided into ontogenetically and functionally distinct subpopulations (36, 42, 91, 95, 131). Importantly, some of the transcription factors controlling DC differentiation in mice have also been implicated in the development of human DCs (67, 69, 71) and putative equivalent DC subpopulations exist in rat, chicken, sheep, and pig (150–153), highlighting that DC populations are conserved across species.
While several growth factors have been linked to DC differentiation, the development of all DC subsets is strongly dependent on FLT3 ligand (FLT3L) and downstream signaling events (7, 18, 154). FLT3L administration potently expands pDCs and cDCs in mice and humans (72, 73, 85, 155–157). In vitro, FLT3L promotes the differentiation of bone marrow progenitors from mice, humans, and pigs into functional subsets of DCs (66, 158, 159). Mice lacking FLT3L display a severe deficiency in DCs, which is also apparent, although to a lesser extent, in mice lacking its receptor CD135 or mice treated with CD135 inhibitors (63, 137, 160, 161). In contrast, FLT3L appears largely dispensable for monocyte and MØ development (137) and, therefore, FLT3L dependency is often used delineate DCs in vivo (18, 65, 162). The interpretation of fate mapping using mice deficient in CD135 or its ligand is however complicated by the fact that these animals also exhibit abnormalities in other hematopoietic lineages, including B, T, and NK cells (137, 163) and show evidence of systemic neutrophilia and monocytosis, as has been reported in other DC-deficient models (112, 117).
Despite the prominent expression of CD135 on DC progenitors it remains to be clarified exactly at what stage of cellular differentiation FLT3L impacts on DC development. Consistent with a role for FLT3L early in development, a reduction of bone marrow CDPs in FLT3L deficient animals has been reported but ranges from a mere twofold decrease (164) to near complete absence (78). In contrast, the numbers of MDPs and splenic pre-DCs appear largely unaffected by CD135 deficiency (85). The observation that pre-DC frequencies in non-lymphoid organs of FLT3L-deficient mice are reduced (63) and that transfer of DCs into a FLT3L-deficient environment decreases their homeostatic proliferation (85) indicates a role for FLT3L in the peripheral expansion of DCs rather than their differentiation. This interpretation would equally be consistent with the observation that DCs that develop in the absence of FLT3L are functional (137). In light of this finding it will be interesting to determine, to what extent FLT3L impacts on the development and functional regulation of other MPs. Addition of FLT3L to purified human monocytes cultured with GM-CSF ± IL-4 increases their T cell stimulatory capacity (165), although it is not clear whether this is also the case for murine monocytes. Culture of murine bone marrow with GM-CSF and IL-4 presumably mimics monocyte differentiation under the same conditions (166). When FLT3 signaling is inhibited in such bulk cultures the T cell stimulatory capacity of the output cells is reduced (161). Therefore, these data raise the possibility that FLT3L might influence monocyte differentiation into cells with functional properties of DCs also in the murine system, although a direct causality remains to be demonstrated. Further, comparative gene expression profiling revealed that upon migration to lymph nodes LCs induce CD135 expression (167), indicating that they might be capable of responding to FLT3L. Therefore, it is conceivable that FLT3L may control certain functional aspects generally associated with DCs, such as antigen presentation, in ontogenetically distinct MP subtypes, which will be interesting to formally address in the context of FLT3L or CD135 deficiency.
Dendritic cell progenitors also express CD115, the receptor for MØ colony-stimulating factor (M-CSF) (56–58, 61, 86). However, compared to the dominant role of FLT3L in DC differentiation, M-CSF-deficiency only mildly impacts on DC development (168). M-CSF deficient osteopetrotic (op/op) mice exhibit a two- to threefold reduction in splenic cDCs and pDCs, respectively, but the remaining DCs are capable of stimulating a mixed lymphocyte reaction and induce costimulatory molecules upon activation, thus appear functional (168). In contrast, M-CSF is strongly required for monocyte and MØ development (141, 169). Therefore, the observation that mice lacking CD115 exhibit reduced frequencies of CD11c+MHCII+CD11b+ cells in non-lymphoid organs (63, 91) likely reflects the ontogenetic heterogeneity of this population (63, 91–95). Consistently, M-CSF is also required for the generation of monocyte-derived cells with features of DCs during inflammation (90). Nonetheless, M-CSF may play a role in DC development. It can promote DC differentiation in vitro and in vivo even in the absence of FLT3L, although DCs generated by M-CSF alone phenotypically and functionally differ from those induced by FLT3L (170). M-CSF-induced DC poeisis is also more efficient in FLT3L-sufficient conditions (170). In vivo, antibody-mediated blockade of M-CSF in pregnant mice reduces pre-DC extravasation, translating into a reduction of CD11b+ DCs in the pregnant uterus (171). Whether M-CSF affects pre-DC migration also in other tissues and whether it acts in a cell intrinsic manner or by promoting the production of chemotactic factors by other cells remains to be determined (171).
In purified monocytes, GM-CSF induces phenotypic and functional attributes of DCs (88, 89, 172). Similarly, purified CD115+ MDPs respond to GM-CSF by differentiating into CD11c+MHCII+ DCs (85) and GM-CSF deficiency leads to a slight reduction of bone marrow MDPs and CDPs (164). However, GM-CSF is dispensable for the differentiation of lymphoid tissue DCs (85, 173) and, therefore, it seemed likely that GM-CSF would selectively regulate the differentiation of monocytes into cells resembling DCs (23). This speculation also lead to the hypothesis that monocytes cultured in the presence of GM-CSF represent the counterpart of mo-DCs generated under conditions of inflammation/infection in vivo (23). Surprisingly, GM-CSF does not appear to control monocyte differentiation in vivo (90) and thus, GM-CSF elicited monocyte-derived cells are unlikely to be fully equivalent to inflammatory monocyte-derived cells. Rather, GM-CSF influences the homeostasis of cDCs in a variety, but not all, non-lymphoid tissues, most likely by promoting cell survival (90). Importantly, GM-CSF deficiency leads to a greater reduction of CD103+ cDCs than of CD11b+ cDCs (90). However, the extent of cDC reduction in the absence of GM-CSF apparently relates to the markers used for cell identification (90, 147, 164). This is most likely because GM-CSF regulates certain phenotypic as well as functional features of DCs, such as CD103 expression (174) or their ability to cross-present antigen (90, 174, 175). Therefore, the above-mentioned growth factors not only influence lineage decisions but also impact on the functional regulation of DCs, monocytes, and MØs. Elucidating the exact roles of FLT3L, GM-CSF, and M-CSF in each cell type will help to decipher the functional heterogeneity of MPs.
Cellular Barcoding
The biggest challenge for fate mapping is to trace the developmental plasticity of individual cells. This can now be achieved using “cellular barcoding,” in which progenitors are tagged in vitro with semi-random, non-coding DNA sequences by transduction using retro- or lentiviral vectors (Figure 2C) (81). Therefore, the barcodes are heritable and by choosing conditions of low transduction efficiency one can ensure that each cell receives only a single barcode. Subsequently, barcode-labeled progenitors are adoptively transferred in numbers low enough to minimize the chance that two identically barcoded cells are transferred into the same recipient (Figure 2C). After differentiation in vivo, cell progeny are analyzed for their barcode repertoire using deep sequencing or custom microarray. Since each barcode represents an individual progenitor, the presence of the same barcode in more than one cell type indicates that they were generated from a single precursor (multi-potent or bi-potent, Figure 2C). On the other hand, if a barcode is only found in one cell type, the progenitor generated only a single cell lineage (mono-potent, Figure 2C) (81).
During maturation, HSCs are thought to progressively lose their self-renewal ability and become increasingly limited in their differentiation potential, ultimately giving rise to lineage-restricted progenitors (55, 176). Lymphoid primed multi-potent progenitors (LMPPs) are developmental intermediates downstream of HSCs that can give rise to various, but not all, cell lineages and are thus considered multi-potent (55, 176). Surprisingly, in barcoding experiments only a minority (3%) of single LMPPs exhibits true multi-potency, defined as the ability to generate all of the following cell lineages: B cells, DCs, and myeloid cells (monocytes and neutrophils) (49). Rather, single LMPPs differ drastically in terms of their cellular output: 10% of the progenitors contribute primarily to B cells, 10% primarily to myeloid cells but about 50% of transferred LMPPs produce predominantly DCs (49). The remaining fraction of progenitors exhibits bi-potentiality to generate combinations of the examined cell lineages (49). Therefore, LMPPs are multi-potent when analyzed as a population, however single cells exhibit unexpected lineage bias that is imprinted early in development. Why the majority of LMPPs is DC-committed (49), even though DCs constitute a minority lineage compared to B cells, remains to be clarified, although it is possible that some progenitors proliferate better than others or have certain competitive advantages. A major lineage divergence toward DCs seems to occur before or at the LMPP stage, as most HSCs analyzed by the same method are multi-potent, although even HSCs exhibit a degree of lineage bias (49, 177). Since CDPs might arise directly from LMPPs without additional developmental intermediates (79), these data infer that DCs diverge as a developmental lineage distinct from other myeloid cells early on (49).
This, again, questions the existence of a bi-potential MDP as central intermediate in the development of DCs and monocytes. Yet, it is noteworthy that even though DC-biased LMPPs are fivefold more frequent than bi-potent myeloid/DC LMPPs, mono-potent and bi-potent progenitors contribute equally to the final DC pool (49). Therefore, bi-potent progenitors seem to play a significant part in generating DCs, potentially because they have a proliferative advantage. Resolving these issues will require further refinement of the technique at hand. The differentiation potential of progenitors may be influenced by cell isolation, processing or in vitro manipulation (80) and virus-mediated transformation might skew cell fate in an unforeseeable manner, as evidenced by the fact that barcoded LMPPs cannot generate T cells (49, 81). This also means that barcoding does not yet uncover the full potential of single progenitors. The early lineage bias of HSCs and LMPPs suggests that cell development may follow a model of graded commitment rather than proceeding in a truly stepwise manner (178). It will be interesting to determine, to what extent this process is regulated by epigenetic modification and how inflammatory processes might impact on lineage divergence. Future studies will benefit from the development of models allowing for in vivo barcoding of single cells but the labor-intensive quantification and analysis of barcoding experiments makes it difficult to follow populations in real time.
Genetic Lineage Tracing
Dynamic mapping of populations of distinct origin in vivo can be achieved using genetic lineage tracing based on Cre-loxP technology (Figure 2D) (80, 179). It relies on inducible reporter genes that are placed under the control of constitutively active promoters, such as the Rosa26 locus. The reporter is most commonly a fluorescent protein that is preceded by a loxP-flanked STOP cassette and, therefore, its expression is induced only after Cre recombinase (Cre) mediated excision of the stop codon (Figure 2D). Since this form of labeling is genetic it is also heritable, meaning that any cell expressing Cre will pass on the label to all progeny, irrespective of continuous recombinase expression (Figure 2D). Since the promoter driving the reporter gene is constitutively active, labeling is irreversible and not affected by fluctuations in gene expression (Figure 2D) (80).
By crossing mice expressing Cre under the control of the Clec9a locus to Rosa26-STOP-flox-enhanced-yellow fluorescent protein (YFP) reporter mice (180), we have recently generated the first genetic model to trace the progeny of DNGR-1+ CDPs and pre-DCs (36). In these mice, YFP expression is restricted to DCs but is not found in monocytes or MØs even in inflammatory conditions, as tested after intestinal inflammation or infection with L. monocytogenes (36). Nonetheless, certain limitations need to be taken into account. DNGR-1 is also expressed on CD8α+/CD103+ cDCs and to a lower extent on pDCs (36, 71, 181, 182) and, therefore, in these populations labeling is not a strict indicator of cell ontogeny. Further, labeling of CDP and pre-DC progeny in mice heterozygous for Cre is incomplete, possibly due to a delay in Cre protein synthesis and DNA recombination in rapidly cycling progenitors (36). Consistently, penetrance of the YFP label is increased in mice homozygous for Cre (36). The efficiency of lineage tracing experiments in such cases or when Cre expression is low may be improved by using alternate reporter constructs in which the loxP sites are positioned closer together, thus facilitating recombination (183).
Genetic lineage tracing does not require prior knowledge of which markers are expressed by the output cells and, thus, enables unbiased monitoring of cell ontogeny. Therefore, we were able to identify CDP-derived cells in cell populations previously thought to constitute monocytes/MØs based on the expression of surface markers, such as CD64 (36). CD64+ CDP-derived cells do not express Clec9a message and are especially frequent in kidneys, although the presence of few YFP+ cells in the CD64+ component of lung and small intestine indicates that atypical CDP-derived cells also exist in other tissues (36). CD64+ kidney DCs resemble yolk sac-derived F4/80hi tissue-resident MØs, appear to lack Zbtb46 expression (37) and their affiliation as DCs or MØs has been debated (184). We, therefore, used adoptive transfer as additional method to confirm cell ontogeny. Surprisingly, neither purified DNGR-1+ CDPs nor total bone marrow generated F4/80hiCD64+ CDP progeny in kidneys 1 week after adoptive transfer into irradiated recipients (36). Since kidney DCs reportedly have a slow turnover (185), it is possible that CDPs had insufficient opportunity to reach their renal niche and expand during short-term transfer experiments. Consistent with this notion, F4/80hiCD64+ kidney leukocytes were efficiently generated from bone marrow progenitors in long-term reconstitution experiments (36). Therefore, our data strongly support a CDP origin of CD64+ kidney leukocytes, despite their phenotypic resemblance to monocytes or MØs (36). These data exemplify the power of lineage tracing in following cell ontogeny in an unbiased way, although it is possible that DNGR-1 is expressed on yet unidentified developmental intermediates.
Addressing this possibility might require tamoxifen-inducible Cre constructs that can be used to pulse label progenitor populations (80). In the future, combinatorial approaches, such as “split-Cre” fragments controlled by two different promoters (186) or an intersection where Cre and the inducible reporter are driven by two cell-specific promoters (187, 188) may be of benefit to generate improved models to lineage trace DCs. The identification of CDP-derived cells with attributes of monocytes/MØs exemplifies the insufficiency of phenotypic properties, such as surface markers, as means of accurate cell identification of MPs. It also raises the question why cells of distinct ontogeny but overlapping phenotype exist in the same tissue. Further elucidation of the specific functions of MPs in immunity will benefit from lineage tracing approaches that result in target cell deletion through the use of inducible DTR or DT subunit modules (82, 112, 189, 190).
Conclusion
The studies discussed above have significantly advanced our understanding of DC ontogeny but have also uncovered some uncertainties (Figure 3). While the bone marrow origin of DCs and monocytes is undisputed, the exact developmental intermediates and branching points between HSCs and DC progenitors remain to be clarified. Current data indicate that lineage imprinting toward DCs and monocytes may occur as early as LMPPs, potentially through epigenetic modification (Figure 3). This realization constitutes a major conceptual shift as it puts in question the existence of a bi-potential MDP and the resulting relatedness of DCs and monocytes. A definitive resolution of this question requires increasingly refined methods to genetically trace single progenitors or select DC and monocyte lineages. Nonetheless, it is clear that cDCs, pDCs, and monocytes can be separated based on their descendance from committed developmental intermediates (Figure 3). Their differentiation is further driven by unique factors indicating that their developmental paths are distinct (Figure 3). In stark contrast to pDCs, cDCs, and monocytes, most tissue MØs arise from embryonic progenitors and are predominantly maintained by self-renewal into adulthood (Figure 3).
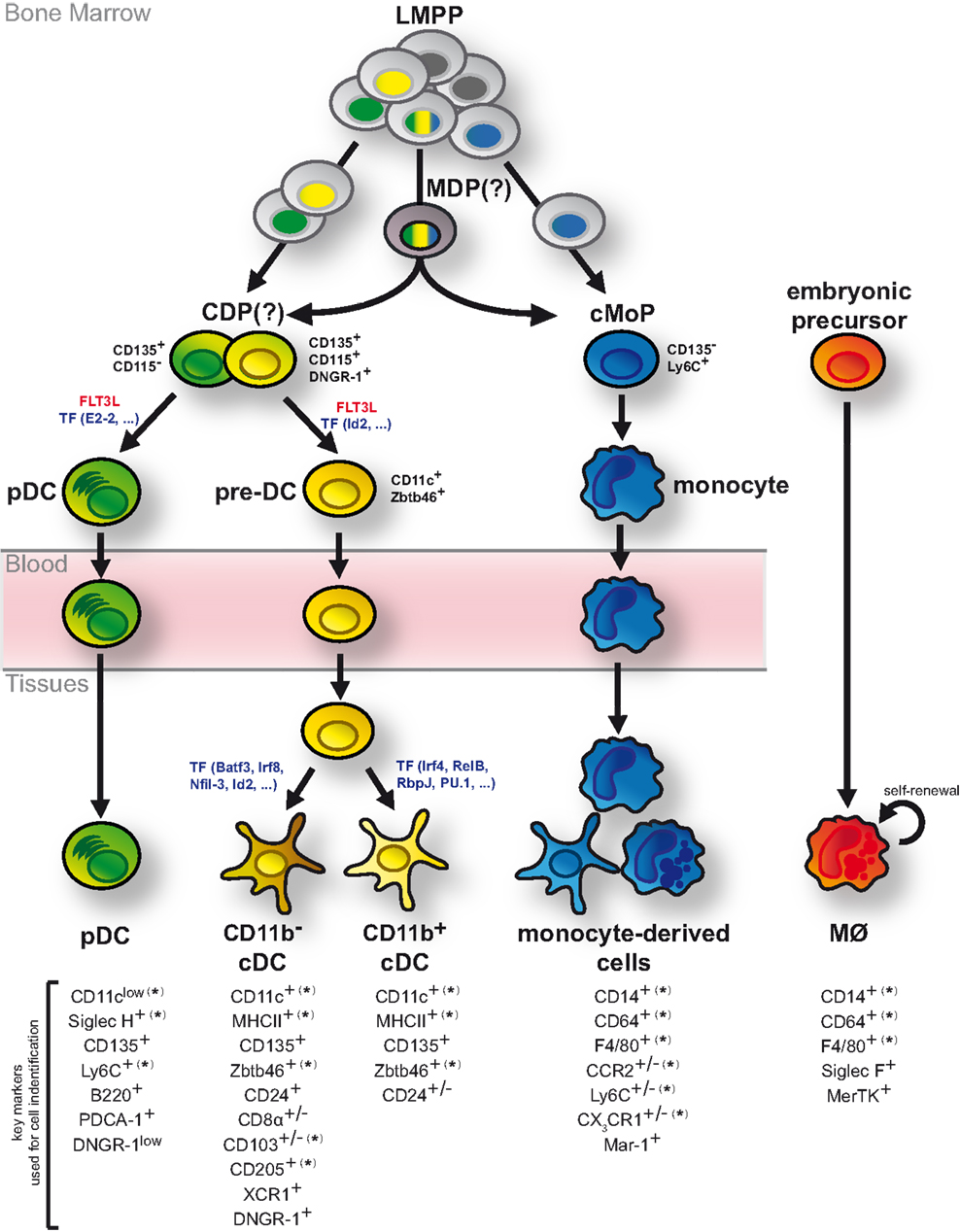
Figure 3. DCs develop as independent cell lineage. Although bone marrow LMPPs are generally considered multi-potent, single LMPPs exhibit a degree of lineage bias toward generating exclusively DCs or monocytes. Such mono-potent LMPPs (single colored nuclei) may generate DC progenitors and monocyte progenitors (cMoPs) directly without additional developmental intermediates. In contrast, multi-potent LMPPs (tri-colored nuclei) presumably give rise to DCs and monocytes via bi-potent MDPs and CDPs. CDPs separate into pDC- and cDC-biased DC progenitors and can be delineated from cMoPs based on the expression of surface markers, including CD135, DNGR-1, Ly6C, and CD115 (as indicated in black). While pDCs fully develop in the bone marrow, cDCs arise via pre-DCs, which migrate through the blood stream to lymphoid and non-lymphoid tissues, where they terminally differentiate into distinct cDC subsets. These developmental processes are strongly driven by FLT3L and controlled by several transcription factors (TF; indicated in blue). Monocytes also fully differentiate in the bone marrow but upon entry into lymphoid and non-lymphoid tissues and directed by environmental cues they can acquire features of DCs or MØs. In contrast to DCs and monocytes, most tissue-resident MØs arise from embryonic progenitors and are maintained by self-renewal. Markers commonly used to distinguish pDCs, cDCs, monocyte-derived cells, and MØs in mice are shown. (*) indicates that the specified marker can be expressed on ontogenetically distinct MP subtypes in some instances. (+/−) indicates markers with heterogeneous or tissue-dependent expression on the specified MP sublineage.
Taken together, these data unequivocally establish that DCs, monocytes, and MØs develop as unique cellular entities and although one could argue that most of this knowledge is derived from mouse studies, developmental parallels have been observed in other species (66–73). Despite these advances, we are at a loss for a universal definition of DCs that is readily accessible to experts within and outside the field of MP biology. In light of this recognition, it has been suggested to revise the current nomenclature of MPs into a system that takes cell ontogeny into account when defining subpopulations (6). Such system would greatly aid our understanding of phagocyte biology as it remains uncertain to what extent the cellular origin of DCs, monocytes, and MØs determines the unique functionality of these cells in immunity and/or tissue homeostasis. While global profiling has revealed a role for the local tissue microenvironment in shaping the transcriptional landscape of DCs, monocytes, and MØs from different organs, certain gene signatures and transcriptional features are set by ontogeny (167, 191–193). Therefore, the full functional diversity of DCs, monocytes, and MØs is likely shaped by both nature (ontogeny) and nurture (the environment). Since ontogeny is immutable it provides a more robust common denominator for cell definition that enables deciphering cellular functions without assuming preconceived functional or phenotypic relationships. DC classification based on cell ancestry is a work in progress but its implementation will ultimately yield a more robust and transparent way of cell definition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
BS and MP are funded by the German Research Foundation (Emmy Noether Grant: Schr 1444/1-1). We thank members of the Schraml lab and Clarissa Prazeres da Costa for critical reading of the manuscript.
References
1. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med (1973) 137(5):1142–62. doi: 10.1084/jem.137.5.1142
2. Steinman RM, Witmer MD. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc Natl Acad Sci U S A (1978) 75(10):5132–6. doi:10.1073/pnas.75.10.5132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Steinman RM, Idoyaga J. Features of the dendritic cell lineage. Immunol Rev (2010) 234(1):5–17. doi:10.1111/j.0105-2896.2009.00888.x
4. van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med (1968) 128(3):415–35. doi:10.1084/jem.128.3.415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46(6):845–52.
6. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi:10.1038/nri3712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Hashimoto D, Miller J, Merad M. Dendritic cell and macrophage heterogeneity in vivo. Immunity (2011) 35(3):323–35. doi:10.1016/j.immuni.2011.09.007
8. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5(12):953–64. doi:10.1038/nri1733
9. van Furth R. Identification of mononuclear phagocytes: overview and definitions. In: Adams DO, Edelson PJ, Koren H, editors. Methods for Studying Mononuclear Phagocytes. New York: Academic Press (1980). p. 243–52.
10. Schraml BU, Reis e Sousa C. Defining dendritic cells. Curr Opin Immunol (2015) 32:13–20. doi:10.1016/j.coi.2014.11.001
11. Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol (2010) 10(6):453–60. doi:10.1038/nri2784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Hume DA. Macrophages as APC and the dendritic cell myth. J Immunol (2008) 181(9):5829–35. doi:10.4049/jimmunol.181.9.5829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Hume DA. The mononuclear phagocyte system. Curr Opin Immunol (2006) 18(1):49–53. doi:10.1016/j.coi.2005.11.008
14. Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi T. The mononuclear phagocyte system revisited. J Leukoc Biol (2002) 72(4):621–7.
15. Reizis B. Regulation of plasmacytoid dendritic cell development. Curr Opin Immunol (2010) 22(2):206–11. doi:10.1016/j.coi.2010.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol (2009) 10(12):1237–44. doi:10.1038/ni.1822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Briseño CG, Murphy TL, Murphy KM. Complementary diversification of dendritic cells and innate lymphoid cells. Curr Opin Immunol (2014) 29C:69–78. doi:10.1016/j.coi.2014.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327(5966):656–61. doi:10.1126/science.1178331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Nussenzweig MC, Steinman RM, Witmer MD, Gutchinov B. A monoclonal antibody specific for mouse dendritic cells. Proc Natl Acad Sci U S A (1982) 79(1):161–5. doi:10.1073/pnas.79.1.161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Steinman RM, Kaplan G, Witmer MD, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. V. Purification of spleen dendritic cells, new surface markers, and maintenance in vitro. J Exp Med (1979) 149(1):1–16. doi:10.1084/jem.149.1.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med (1990) 171(5):1753–71. doi:10.1084/jem.171.5.1753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol (2007) 7(1):19–30. doi:10.1038/nri1996
24. Mildner A, Yona S, Jung S. A close encounter of the third kind: monocyte-derived cells. Adv Immunol (2013) 120:69–103. doi:10.1016/B978-0-12-417028-5.00003-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Carlens J, Wahl B, Ballmaier M, Bulfone-Paus S, Forster R, Pabst O. Common gamma-chain-dependent signals confer selective survival of eosinophils in the murine small intestine. J Immunol (2009) 183(9):5600–7. doi:10.4049/jimmunol.0801581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A (2004) 61(2):170–7. doi:10.1002/cyto.a.20064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Probst HC, Tschannen K, Odermatt B, Schwendener R, Zinkernagel RM, Van Den Broek M. Histological analysis of CD11c-DTR/GFP mice after in vivo depletion of dendritic cells. Clin Exp Immunol (2005) 141(3):398–404. doi:10.1111/j.1365-2249.2005.02868.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Hochweller K, Striegler J, Hämmerling GJ, Garbi N. A novel CD11c.DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells. Eur J Immunol (2008) 38(10):2776–83. doi:10.1002/eji.200838659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Huleatt JW, Lefrançois L. Antigen-driven induction of CD11c on intestinal intraepithelial lymphocytes and CD8+ T cells in vivo. J Immunol (1995) 154(11):5684–93.
30. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood (2011) 118(5):1305–15. doi:10.1182/blood-2011-01-331462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Drutman SB, Kendall JC, Trombetta ES. Inflammatory spleen monocytes can upregulate CD11c expression without converting into dendritic cells. J Immunol (2012) 188(8):3603–10. doi:10.4049/jimmunol.1102741
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hebel K, Griewank K, Inamine A, Chang H-D, Müller-Hilke B, Fillatreau S, et al. Plasma cell differentiation in T-independent type 2 immune responses is independent of CD11c(high) dendritic cells. Eur J Immunol (2006) 36(11):2912–9. doi:10.1002/eji.200636356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Kim K-W, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G, et al. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood (2011) 118(22):e156–67. doi:10.1182/blood-2011-04-348946
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Farache J, Koren I, Milo I, Gurevich I, Kim KW, Zigmond E, et al. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity (2013) 38(3):581–95. doi:10.1016/j.immuni.2013.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Bergstresser PR, Sullivan S, Streilein JW, Tigelaar RE. Origin and function of Thy-1+ dendritic epidermal cells in mice. J Invest Dermatol (1985) 85(1 Suppl):85s–90s. doi:10.1111/1523-1747.ep12275516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Schraml BU, van Blijswijk J, Zelenay S, Whitney PG, Filby A, Acton SE, et al. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell (2013) 154(4):843–58. doi:10.1016/j.cell.2013.07.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med (2012) 209(6):1135–52. doi:10.1084/jem.20120030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Meredith MM, Liu K, Darrasse-Jèze G, Kamphorst AO, Schreiber HA, Guermonprez P, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med (2012) 209(6):1153–65. doi:10.1084/jem.20112675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, et al. CD8α(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity (2011) 35(2):249–59. doi:10.1016/j.immuni.2011.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med (1997) 186(11):1819–29. doi:10.1084/jem.186.11.1819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Whitney PG, Bar E, Osorio F, Rogers NC, Schraml BU, Deddouche S, et al. Syk signaling in dendritic cells orchestrates innate resistance to systemic fungal infection. PLoS Pathog (2014) 10(7):e1004276. doi:10.1371/journal.ppat.1004276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Satpathy AT, Briseño CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol (2013) 14(9):937–48. doi:10.1038/ni.2679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity (2012) 36(2):276–87. doi:10.1016/j.immuni.2011.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Arora P, Baena A, Yu KOA, Saini NK, Kharkwal SS, Goldberg MF, et al. A single subset of dendritic cells controls the cytokine bias of natural killer T cell responses to diverse glycolipid antigens. Immunity (2014) 40(1):105–16. doi:10.1016/j.immuni.2013.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity (2013) 39(3):599–610. doi:10.1016/j.immuni.2013.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity (2013) 38(2):322–35. doi:10.1016/j.immuni.2012.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Pozzi LA, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol (2005) 175(4):2071–81. doi:10.4049/jimmunol.175.4.2071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. McGovern N, Schlitzer A, Gunawan M, Jardine L, Shin A, Poyner E, et al. Human dermal CD14+ cells are a transient population of monocyte-derived macrophages. Immunity (2014) 41(3):465–77. doi:10.1016/j.immuni.2014.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Naik SH, Perié L, Swart E, Gerlach C, van Rooij N, de Boer RJ, et al. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature (2013) 496(7444):229–32. doi:10.1038/nature12013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38(1):79–91. doi:10.1016/j.immuni.2012.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336(6077):86–90. doi:10.1126/science.1219179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko A-C, Krijgsveld J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol (2013) 14(8):821–30. doi:10.1038/ni.2638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Haniffa M, Ginhoux F, Wang X-N, Bigley V, Abel M, Dimmick I, et al. Differential rates of replacement of human dermal dendritic cells and macrophages during hematopoietic stem cell transplantation. J Exp Med (2009) 206(2):371–85. doi:10.1084/jem.20081633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38(4):792–804. doi:10.1016/j.immuni.2013.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Iwasaki H, Akashi K. Myeloid lineage commitment from the hematopoietic stem cell. Immunity (2007) 26(6):726–40. doi:10.1016/j.immuni.2007.06.004
56. Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science (2006) 311(5757):83–7. doi:10.1126/science.1117729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Naik SH, Sathe P, Park H-Y, Metcalf D, Proietto AI, Dakic A, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol (2007) 8(11):1217–26. doi:10.1038/ni1522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol (2007) 8(11):1207–16. doi:10.1038/ni1518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Shortman K, Sathe P, Vremec D, Naik S, O’Keeffe M. Plasmacytoid dendritic cell development. Adv Immunol (2013) 120:105–26. doi:10.1016/B978-0-12-417028-5.00004-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Naik SH, Metcalf D, Van Nieuwenhuijze A, Wicks I, Wu L, O’Keeffe M, et al. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol (2006) 7(6):663–71. doi:10.1038/ni1340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science (2009) 324(5925):392–7. doi:10.1126/science.1170540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol (2007) 8(6):578–83. doi:10.1038/ni1462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206(13):3115–30. doi:10.1084/jem.20091756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity (2003) 19(1):59–70. doi:10.1016/S1074-7613(03)00171-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Haniffa M, Collin M, Ginhoux F. Ontogeny and functional specialization of dendritic cells in human and mouse. Adv Immunol (2013) 120:1–49. doi:10.1016/B978-0-12-417028-5.00001-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Guzylack-Piriou L, Alves MP, McCullough KC, Summerfield A. Porcine Flt3 ligand and its receptor: generation of dendritic cells and identification of a new marker for porcine dendritic cells. Dev Comp Immunol (2010) 34(4):455–64. doi:10.1016/j.dci.2009.12.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell (2008) 135(1):37–48. doi:10.1016/j.cell.2008.09.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, Hoek J, et al. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood (2000) 96(3):878–84.
69. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med (2011) 365(2):127–38. doi:10.1056/NEJMoa1100066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Harada S, Kimura T, Fujiki H, Nakagawa H, Ueda Y, Itoh T, et al. Flt3 ligand promotes myeloid dendritic cell differentiation of human hematopoietic progenitor cells: possible application for cancer immunotherapy. Int J Oncol (2007) 30(6):1461–8. doi:10.3892/ijo.30.6.1461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood (2012) 119(25):6052–62. doi:10.1182/blood-2012-01-406967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Breton G, Lee J, Zhou YJ, Schreiber JJ, Keler T, Puhr S, et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J Exp Med (2015) 212(3):401–13. doi:10.1084/jem.20141441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Lee J, Breton G, Oliveira TYK, Zhou YJ, Aljoufi A, Puhr S, et al. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J Exp Med (2015) 212(3):385–99. doi:10.1084/jem.20141442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Traver D, Akashi K, Manz M, Merad M, Miyamoto T, Engleman EG, et al. Development of CD8alpha-positive dendritic cells from a common myeloid progenitor. Science (2000) 290(5499):2152–4. doi:10.1126/science.290.5499.2152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Schlenner SM, Madan V, Busch K, Tietz A, Läufle C, Costa C, et al. Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity (2010) 32(3):426–36. doi:10.1016/j.immuni.2010.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Luche H, Ardouin L, Teo P, See P, Henri S, Merad M, et al. The earliest intrathymic precursors of CD8α(+) thymic dendritic cells correspond to myeloid-type double-negative 1c cells. Eur J Immunol (2011) 41(8):2165–75. doi:10.1002/eji.201141728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Sathe P, Vremec D, Wu L, Corcoran L, Shortman K. Convergent differentiation: myeloid and lymphoid pathways to murine plasmacytoid dendritic cells. Blood (2013) 121(1):11–9. doi:10.1182/blood-2012-02-413336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Sathe P, Metcalf D, Vremec D, Naik SH, Langdon WY, Huntington ND, et al. Lymphoid tissue and plasmacytoid dendritic cells and macrophages do not share a common macrophage-dendritic cell-restricted progenitor. Immunity (2014) 41(1):104–15. doi:10.1016/j.immuni.2014.05.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Onai N, Kurabayashi K, Hosoi-Amaike M, Toyama-Sorimachi N, Matsushima K, Inaba K, et al. A clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity (2013) 38(5):943–57. doi:10.1016/j.immuni.2013.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Kretzschmar K, Watt FM. Lineage tracing. Cell (2012) 148(1–2):33–45. doi:10.1016/j.cell.2012.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Naik SH, Schumacher TN, Perié L. Cellular barcoding: a technical appraisal. Exp Hematol (2014) 42(8):598–608. doi:10.1016/j.exphem.2014.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Bar-On L, Jung S. Defining dendritic cells by conditional and constitutive cell ablation. Immunol Rev (2010) 234(1):76–89. doi:10.1111/j.0105-2896.2009.00875.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Plett PA, Frankovitz SM, Orschell-Traycoff CM. In vivo trafficking, cell cycle activity, and engraftment potential of phenotypically defined primitive hematopoietic cells after transplantation into irradiated or nonirradiated recipients. Blood (2002) 100(10):3545–52. doi:10.1182/blood.V100.10.3545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, et al. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J Exp Med (2007) 204(1):171–80. doi:10.1084/jem.20061011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Waskow C, Liu K, Darrasse-Jèze G, Guermonprez P, Ginhoux F, Merad M, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol (2008) 9(6):676–83. doi:10.1038/ni.1615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Auffray C, Fogg DK, Narni-Mancinelli E, Senechal B, Trouillet C, Saederup N, et al. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J Exp Med (2009) 206(3):595–606. doi:10.1084/jem.20081385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Schlitzer A, Loschko J, Mair K, Vogelmann R, Henkel L, Einwächter H, et al. Identification of CCR9- murine plasmacytoid DC precursors with plasticity to differentiate into conventional DCs. Blood (2011) 117(24):6562–70. doi:10.1182/blood-2010-12-326678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med (1994) 179(4):1109–18. doi:10.1084/jem.179.4.1109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Romani N, Gruner S, Brang D, Kämpgen E, Lenz A, Trockenbacher B, et al. Proliferating dendritic cell progenitors in human blood. J Exp Med (1994) 180(1):83–93. doi:10.1084/jem.180.1.83
90. Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity (2012) 36(6):1031–46. doi:10.1016/j.immuni.2012.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propria dendritic cell network. Immunity (2009) 31(3):513–25. doi:10.1016/j.immuni.2009.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity (2009) 31(3):502–12. doi:10.1016/j.immuni.2009.06.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van der Woude CJ, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol (2012) 42(12):3150–66. doi:10.1002/eji.201242847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Langlet C, Tamoutounour S, Henri S, Luche H, Ardouin L, Grégoire C, et al. CD64 expression distinguishes monocyte-derived and conventional dendritic cells and reveals their distinct role during intramuscular immunization. J Immunol (2012) 188(4):1751–60. doi:10.4049/jimmunol.1102744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Scott CL, Bain CC, Wright PB, Sichien D, Kotarsky K, Persson EK, et al. CCR2(+)CD103(-) intestinal dendritic cells develop from DC-committed precursors and induce interleukin-17 production by T cells. Mucosal Immunol (2014) 8(2):327–39. doi:10.1038/mi.2014.70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol (2002) 3(12):1135–41. doi:10.1038/ni852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Ginhoux F, Tacke F, Angeli V, Bogunovic M, Loubeau M, Dai X-M, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol (2006) 7(3):265–73. doi:10.1038/ni1307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol (2008) 8(12):935–47. doi:10.1038/nri2455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunol Rev (2010) 234(1):120–41. doi:10.1111/j.0105-2896.2009.00886.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Ginhoux F, Merad M. Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol (2010) 88(4):387–92. doi:10.1038/icb.2010.38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med (2012) 209(6):1167–81. doi:10.1084/jem.20120340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Chorro L, Sarde A, Li M, Woollard KJ, Chambon P, Malissen B, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med (2009) 206(13):3089–100. doi:10.1084/jem.20091586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol (2011) 89(4):525–38. doi:10.1189/jlb.0810472
104. Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, et al. Visualizing dendritic cell networks in vivo. Nat Immunol (2004) 5(12):1243–50. doi:10.1038/ni1139
105. Thornton EE, Looney MR, Bose O, Sen D, Sheppard D, Locksley R, et al. Spatiotemporally separated antigen uptake by alveolar dendritic cells and airway presentation to T cells in the lung. J Exp Med (2012) 209(6):1183–99. doi:10.1084/jem.20112667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Choi J-H, Do Y, Cheong C, Koh H, Boscardin SB, Oh Y-S, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med (2009) 206(3):497–505. doi:10.1084/jem.20082129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Khanna KM, Blair DA, Vella AT, McSorley SJ, Datta SK, Lefrançois L. T cell and APC dynamics in situ control the outcome of vaccination. J Immunol (2010) 185(1):239–52. doi:10.4049/jimmunol.0901047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Bennett CL, Clausen BE. DC ablation in mice: promises, pitfalls, and challenges. Trends Immunol (2007) 28(12):525–31. doi:10.1016/j.it.2007.08.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Jung S, Unutmaz D, Wong P, Sano G-I, de los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity (2002) 17(2):211–20. doi:10.1016/S1074-7613(02)00365-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Tittel AP, Heuser C, Ohliger C, Knolle PA, Engel DR, Kurts C. Kidney dendritic cells induce innate immunity against bacterial pyelonephritis. J Am Soc Nephrol (2011) 22(8):1435–41. doi:10.1681/ASN.2010101072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Bar-On L, Birnberg T, Kim K-W, Jung S. Dendritic cell-restricted CD80/86 deficiency results in peripheral regulatory T-cell reduction but is not associated with lymphocyte hyperactivation. Eur J Immunol (2011) 41(2):291–8. doi:10.1002/eji.201041169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. van Blijswijk J, Schraml BU, Reis e Sousa C. Advantages and limitations of mouse models to deplete dendritic cells. Eur J Immunol (2013) 43(1):22–6. doi:10.1002/eji.201243022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Racine R, Chatterjee M, Winslow GM. CD11c expression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J Immunol (2008) 181(2):1375–85. doi:10.4049/jimmunol.181.2.1375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Brocker T, Riedinger M, Karjalainen K. Driving gene expression specifically in dendritic cells. Adv Exp Med Biol (1997) 417:55–7. doi:10.1007/978-1-4757-9966-8_9
115. Tittel AP, Heuser C, Ohliger C, Llanto C, Yona S, Hämmerling GJ, et al. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nat Methods (2012) 9(4):385–90. doi:10.1038/nmeth.1905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Bar-On L, Jung S. Defining in vivo dendritic cell functions using CD11c-DTR transgenic mice. Methods Mol Biol (2010) 595:429–42. doi:10.1007/978-1-60761-421-0_28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Jiao J, Dragomir A-C, Kocabayoglu P, Rahman AH, Chow A, Hashimoto D, et al. Central role of conventional dendritic cells in regulation of bone marrow release and survival of neutrophils. J Immunol (2014) 192(7):3374–82. doi:10.4049/jimmunol.1300237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Gardner JM, Metzger TC, McMahon EJ, Au-Yeung BB, Krawisz AK, Lu W, et al. Extrathymic aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4+ T Cells. Immunity (2013) 39(3):560–72. doi:10.1016/j.immuni.2013.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40(1):91–104. doi:10.1016/j.immuni.2013.11.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell (2014) 26(5):638–52. doi:10.1016/j.ccell.2014.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, Unanue ER. A minor subset of Batf3-dependent antigen-presenting cells in islets of langerhans is essential for the development of autoimmune diabetes. Immunity (2014) 41(4):657–69. doi:10.1016/j.immuni.2014.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity (2013) 38(2):336–48. doi:10.1016/j.immuni.2012.10.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Meredith MM, Liu K, Kamphorst AO, Idoyaga J, Yamane A, Guermonprez P, et al. Zinc finger transcription factor zDC is a negative regulator required to prevent activation of classical dendritic cells in the steady state. J Exp Med (2012) 209(9):1583–93. doi:10.1084/jem.20121003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322(5904):1097–100. doi:10.1126/science.1164206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med (2010) 207(4):823–36. doi:10.1084/jem.20091627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med (2002) 196(11):1415–25. doi:10.1084/jem.20021263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Hacker C, Kirsch RD, Ju X-S, Hieronymus T, Gust TC, Kuhl C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol (2003) 4(4):380–6. doi:10.1038/ni903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Kashiwada M, Pham NL, Pewe LL, Harty JT, Rothman PB. NFIL3/E4BP4 is a key transcription factor for CD8alpha(+) dendritic cell development. Blood (2011) 117(23):6193–7. doi:10.1182/blood-2010-07-295873
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc Natl Acad Sci U S A (2004) 101(24):8981–6. doi:10.1073/pnas.0402139101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med (2007) 204(7):1653–64. doi:10.1084/jem.20062648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity (2011) 35(5):780–91. doi:10.1016/j.immuni.2011.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity (2013) 38(5):970–83. doi:10.1016/j.immuni.2013.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity (2013) 38(5):958–69. doi:10.1016/j.immuni.2013.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’shea JJ, et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol (2005) 174(5):2573–81. doi:10.4049/jimmunol.174.5.2573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Wu L, D’Amico A, Winkel KD, Suter M, Lo D, Shortman K. RelB is essential for the development of myeloid-related CD8alpha- dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity (1998) 9(6):839–47. doi:10.1016/S1074-7613(00)80649-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Guerriero A, Langmuir PB, Spain LM, Scott EW. PU.1 is required for myeloid-derived but not lymphoid-derived dendritic cells. Blood (2000) 95(3):879–85.
137. McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood (2000) 95(11):3489–97.
138. Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature (2009) 457(7227):318–21. doi:10.1038/nature07472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol (2012) 13(8):753–60. doi:10.1038/ni.2360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity (2012) 37(6):1050–60. doi:10.1016/j.immuni.2012.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Wiktor-Jedrzejczak WW, Ahmed A, Szczylik C, Skelly RR. Hematological characterization of congenital osteopetrosis in op/op mouse. Possible mechanism for abnormal macrophage differentiation. J Exp Med (1982) 156(5):1516–27. doi:10.1084/jem.156.5.1516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous expression of the transcription factor e2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity (2010) 33(6):905–16. doi:10.1016/j.immuni.2010.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest (2012) 122(11):4037–47. doi:10.1172/JCI60659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med (2011) 208(9):1789–97. doi:10.1084/jem.20110538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol (2009) 10(5):488–95. doi:10.1038/ni.1724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Murphy KM. Transcriptional control of dendritic cell development. Adv Immunol (2013) 120:239–67. doi:10.1016/B978-0-12-417028-5.00009-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Edelson BT, Bradstreet TR, Kc W, Hildner K, Herzog JW, Sim J, et al. Batf3-dependent CD11b(low/-) peripheral dendritic cells are GM-CSF-independent and are not required for Th cell priming after subcutaneous immunization. PLoS One (2011) 6(10):e25660. doi:10.1371/journal.pone.0025660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Tussiwand R, Lee W-L, Murphy TL, Mashayekhi M, Wumesh KC, Albring JC, et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature (2012) 490(7421):502–7. doi:10.1038/nature11531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Seillet C, Jackson JT, Markey KA, Brady HJM, Hill GR, MacDonald KPA, et al. CD8α+ DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood (2013) 121(9):1574–83. doi:10.1182/blood-2012-07-445650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Contreras V, Urien C, Guiton R, Alexandre Y, Vu Manh T-P, Andrieu T, et al. Existence of CD8α-like dendritic cells with a conserved functional specialization and a common molecular signature in distant mammalian species. J Immunol (2010) 185(6):3313–25. doi:10.4049/jimmunol.1000824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Marquet F, Bonneau M, Pascale F, Urien C, Kang C, Schwartz-Cornil I, et al. Characterization of dendritic cells subpopulations in skin and afferent lymph in the swine model. PLoS One (2011) 6(1):e16320. doi:10.1371/journal.pone.0016320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Vu Manh TP, Marty H, Sibille P, Le Vern Y, Kaspers B, Dalod M, et al. Existence of conventional dendritic cells in Gallus gallus revealed by comparative gene expression profiling. J Immunol (2014) 192(10):4510–7. doi:10.4049/jimmunol.1303405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Hubert FX, Voisine C, Louvet C, Heslan M, Josien R. Rat plasmacytoid dendritic cells are an abundant subset of MHC class II+ CD4+CD11b-OX62- and type I IFN-producing cells that exhibit selective expression of toll-like receptors 7 and 9 and strong responsiveness to CpG. J Immunol (2004) 172(12):7485–94. doi:10.4049/jimmunol.172.12.7485
154. Sathaliyawala T, O’Gorman WE, Greter M, Bogunovic M, Konjufca V, Hou ZE, et al. Mammalian target of rapamycin controls dendritic cell development downstream of Flt3 ligand signaling. Immunity (2010) 33(4):597–606. doi:10.1016/j.immuni.2010.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Brasel K, McKenna HJ, Morrissey PJ, Charrier K, Morris AE, Lee CC, et al. Hematologic effects of flt3 ligand in vivo in mice. Blood (1996) 88(6):2004–12.
156. Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med (1996) 184(5):1953–62. doi:10.1084/jem.184.5.1953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Pulendran B, Banchereau J, Burkeholder S, Kraus E, Guinet E, Chalouni C, et al. Flt3-ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J Immunol (2000) 165(1):566–72. doi:10.4049/jimmunol.165.1.566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol (2005) 174(11):6592–7. doi:10.4049/jimmunol.174.11.6592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen J-L, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207(6):1261–71. doi:10.1084/jem.20092618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Tussiwand R, Onai N, Mazzucchelli L, Manz MG. Inhibition of natural type I IFN-producing and dendritic cell development by a small molecule receptor tyrosine kinase inhibitor with Flt3 affinity. J Immunol (2005) 175(6):3674–80. doi:10.4049/jimmunol.175.6.3674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang T, et al. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci U S A (2005) 102(46):16741–6. doi:10.1073/pnas.0506088102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Jenkins SJ, Hume DA. Homeostasis in the mononuclear phagocyte system. Trends Immunol (2014) 35(8):358–67. doi:10.1016/j.it.2014.06.006
163. Mackarehtschian K, Hardin JD, Moore KA, Boast S, Goff SP, Lemischka IR. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity (1995) 3(1):147–61. doi:10.1016/1074-7613(95)90167-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Kingston D, Schmid MA, Onai N, Obata-Onai A, Baumjohann D, Manz MG. The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood (2009) 114(4):835–43. doi:10.1182/blood-2009-02-206318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Kim S-W, Choi S-M, Choo YS, Kim I-K, Song B-W, Kim H-S. Flt3 ligand induces monocyte proliferation and enhances the function of monocyte-derived dendritic cells in vitro. J Cell Physiol (2014). doi:10.1002/jcp.24824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol (2007) 179(11):7577–84. doi:10.4049/jimmunol.179.11.7577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol (2012) 13(9):888–99. doi:10.1038/ni.2370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. MacDonald KPA, Rowe V, Bofinger HM, Thomas R, Sasmono T, Hume DA, et al. The colony-stimulating factor 1 receptor is expressed on dendritic cells during differentiation and regulates their expansion. J Immunol (2005) 175(3):1399–405. doi:10.4049/jimmunol.175.3.1399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood (2002) 99(1):111–20. doi:10.1182/blood.V99.1.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Fancke B, Suter M, Hochrein H, O’Keeffe M. M-CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood (2008) 111(1):150–9. doi:10.1182/blood-2007-05-089292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Tagliani E, Shi C, Nancy P, Tay C-S, Pamer EG, Erlebacher A. Coordinate regulation of tissue macrophage and dendritic cell population dynamics by CSF-1. J Exp Med (2011) 208(9):1901–16. doi:10.1084/jem.20110866
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Schreurs MW, Eggert AA, de Boer AJ, Figdor CG, Adema GJ. Generation and functional characterization of mouse monocyte-derived dendritic cells. Eur J Immunol (1999) 29(9):2835–41. doi:10.1002/(SICI)1521-4141(199909)29:09<2835::AID-IMMU2835>3.3.CO;2-H
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Vremec D, Lieschke GJ, Dunn AR, Robb L, Metcalf D, Shortman K. The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur J Immunol (1997) 27(1):40–4. doi:10.1002/eji.1830270107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Zhan Y, Carrington EM, Van Nieuwenhuijze A, Bedoui S, Seah S, Xu Y, et al. GM-CSF increases cross-presentation and CD103 expression by mouse CD8+ spleen dendritic cells. Eur J Immunol (2011) 41(9):2585–95. doi:10.1002/eji.201141540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Sathe P, Pooley J, Vremec D, Mintern J, Jin J-O, Wu L, et al. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J Immunol (2011) 186(9):5184–92. doi:10.4049/jimmunol.1002683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Luc S, Buza-Vidas N, Jacobsen SE. Biological and molecular evidence for existence of lymphoid-primed multipotent progenitors. Ann N Y Acad Sci (2007) 1106:89–94. doi:10.1196/annals.1392.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Verovskaya E, Broekhuis MJ, Zwart E, Ritsema M, van Os R, de Haan G, et al. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood (2013) 122(4):523–32. doi:10.1182/blood-2013-01-481135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Paul F, Amit I. Plasticity in the transcriptional and epigenetic circuits regulating dendritic cell lineage specification and function. Curr Opin Immunol (2014) 30C:1–8. doi:10.1016/j.coi.2014.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Vorhagen S, Jackow J, Mohor SG, Tanghe G, Tanrikulu L, Skazik-Vogt C, et al. Lineage tracing mediated by cre-recombinase activity. J Invest Dermatol (2015) 135(1):e28. doi:10.1038/jid.2014.472
180. Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol (2001) 1:4. doi:10.1186/1471-213X-1-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Caminschi I, Proietto AI, Ahmet F, Kitsoulis S, Shin Teh J, Lo JCY, et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood (2008) 112(8):3264–73. doi:10.1182/blood-2008-05-155176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Sancho D, Mourão-Sá D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest (2008) 118(6):2098–110. doi:10.1172/JCI34584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Liu J, Willet SG, Bankaitis ED, Xu Y, Wright CVE, Gu G. Non-parallel recombination limits cre-loxP-based reporters as precise indicators of conditional genetic manipulation. Genesis (2013) 51(6):436–42. doi:10.1002/dvg.22384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol (2012) 23(2):194–203. doi:10.1681/ASN.2011070680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int (2005) 68(3):1096–108. doi:10.1111/j.1523-1755.2005.00502.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Hirrlinger J, Requardt RP, Winkler U, Wilhelm F, Schulze C, Hirrlinger PG. Split-CreERT2: temporal control of DNA recombination mediated by split-Cre protein fragment complementation. PLoS One (2009) 4(12):e8354. doi:10.1371/journal.pone.0008354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, et al. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J Exp Med (2013) 210(10):2025–39. doi:10.1084/jem.20130903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Diehl GE, Longman RS, Zhang J-X, Breart B, Galan C, Cuesta A, et al. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature (2013) 494(7435):116–20. doi:10.1038/nature11809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Ohnmacht C, Pullner A, King SBS, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med (2009) 206(3):549–59. doi:10.1084/jem.20082394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Voehringer D, Liang H-E, Locksley RM. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J Immunol (2008) 180(7):4742–53. doi:10.4049/jimmunol.180.7.4742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13(11):1118–28. doi:10.1038/ni.2419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159(6):1312–26. doi:10.1016/j.cell.2014.11.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell (2014) 159(6):1327–40. doi:10.1016/j.cell.2014.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: dendritic cell, ontogeny, fate mapping, lineage tracing, mononuclear phagocyte
Citation: Poltorak MP and Schraml BU (2015) Fate mapping of dendritic cells. Front. Immunol. 6:199. doi: 10.3389/fimmu.2015.00199
Received: 31 January 2015; Accepted: 13 April 2015;
Published: 04 May 2015
Edited by:
Shalin Naik, Walter & Eliza Hall Institute, AustraliaCopyright: © 2015 Poltorak and Schraml. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Ursula Schraml, Institute for Medical Microbiology, Immunology and Hygiene, Technische Universität München, Trogerstraße 30, Munich 81675, Germany,YmFyYmFyYS5zY2hyYW1sQHR1bS5kZQ==