 Karin de Punder
Karin de Punder Leo Pruimboom
Leo Pruimboom- 1Institute of Medical Psychology, Charité University Medicine, Berlin, Germany
- 2Natura Foundation, Numansdorp, Netherlands
Chronic non-communicable diseases (NCDs) are the leading causes of work absence, disability, and mortality worldwide. Most of these diseases are associated with low-grade inflammation. Here, we hypothesize that stresses (defined as homeostatic disturbances) can induce low-grade inflammation by increasing the availability of water, sodium, and energy-rich substances to meet the increased metabolic demand induced by the stressor. One way of triggering low-grade inflammation is by increasing intestinal barrier permeability through activation of various components of the stress system. Although beneficial to meet the demands necessary during stress, increased intestinal barrier permeability also raises the possibility of the translocation of bacteria and their toxins across the intestinal lumen into the blood circulation. In combination with modern life-style factors, the increase in bacteria/bacterial toxin translocation arising from a more permeable intestinal wall causes a low-grade inflammatory state. We support this hypothesis with numerous studies finding associations with NCDs and markers of endotoxemia, suggesting that this process plays a pivotal and perhaps even a causal role in the development of low-grade inflammation and its related diseases.
Introduction
Inflammation is the response of the innate immune system triggered by stimuli like microbial pathogens and injury. Acute systemic inflammation such as in sepsis, trauma, burns, and surgery is characterized by a quick increase in plasma levels (up to 100-fold) of pro-inflammatory cytokines and acute phase proteins, while in low-grade inflammation, there is a sustained but only two to threefold increase in circulation pro-inflammatory mediators (1). Chronic low-grade inflammation is characteristic for many non-communicable diseases (NCDs) including diabetes type II, cardiovascular disorders, autoimmune diseases, chronic fatigue syndrome, depression, and neurodegenerative pathologies, but until now the exact mechanism behind the elevated levels of inflammatory mediators found in these conditions is not well understood (2–5).
Inflammation can be induced by the binding of pathogen-associated molecular patterns (PAMPs) to toll-like receptors (TLRs), which are expressed on different cells types including immune cells, adipocytes, and endothelial cells. The most extensively studied PAMP is lipopolysaccharide (LPS) or endotoxin (the terms LPS and endotoxin will be used interchangeably throughout the rest of the article), a major cell wall component of Gram-negative bacteria, which is normally present in the human circulation in very low concentrations. It has been hypothesized that most of this circulating LPS is derived from the gut, since the gut-microbiota is the biggest source of Gram-negative bacteria-derived LPS. However, LPS found in the circulation could also be derived from Gram-negative bacteria residing in the oral cavity, respiratory, and genitourinary tracts, or can be food-derived (6–8). Under certain circumstances, there can be an increase of endotoxin translocation across the intestinal barrier, leading to mildly increased concentrations in the blood circulation. This process has been associated with several NCDs, like depression (9), chronic fatigue syndrome (10), chronic heart failure (11), type 2 diabetes (12), autism (13), non-alcoholic fatty liver disease (NAFLD) (14), and inflammatory bowel disease (IBD) (15), diseases that are all linked to chronic systemic low-grade inflammation, indicating that endotoxemia could be an important contributor in the development of these conditions.
Here, we hypothesize that stress-induction leads to a more permeable intestinal wall intended to facilitate an increase in the availability of water, sodium, and energy-rich substances necessary to meet the increased metabolic demand induced by the stressor. Modern life-style factors, such as long-term psychosocial stress and components of our “Western” diet constantly challenge the stress-axis and further compromise intestinal barrier function, resulting in endotoxemia, low-grade inflammation, and its related diseases. We support our hypothesis by describing literature surrounding stress- and immune system-activation processes and their relation to gut barrier function and explain how life-style choices impact all these systems. In addition, we present a vast amount of literature describing associations with NCDs and markers of endotoxemia. Overall, we conclude that stress-induced disrupted barrier function in parallel with elevated circulating endotoxin levels may underlie disease onset and progression and should be considered much more than just a risk factor for chronic disease; it could be a cause.
Bacterial Toxins Activate the Immune System via TLRs
Lipopolysaccharide, the major cell wall component of Gram-negative bacteria, is characterized by its capacity to induce inflammation, fever, shock, and death (1). Additionally in recent years, other cell wall components of Gram-negative and -positive bacteria have been recognized to have endotoxic properties (16), but these will not be further addressed in the rest of the paper. Endotoxins are released from bacteria during infection or as a consequence of bacterial lysis. Although both whole bacteria and bacterial toxins can translocate transcellular or paracellular into the lymph, blood, and mesenteric lymph nodes, it is still not precisely clear if the presence of endotoxin in the blood circulation (endotoxemia) also presents whole bacteria translocation across the intestinal wall (17).
Inflammation can be induced by the binding of LPS to TLR4. The lipid-A moiety of LPS interacts with the LPS-sensing machinery composed of TLR4, myeloid differential protein 2, CD14, and LPS-binding protein (LBP). LBP transports and delivers circulating aggregates of LPS to lipoproteins, resulting in hepatic clearance, or delivers LPS to CD14 (the membrane-bound or secreted, soluble form of this molecule), leading to TLR4 activation. TLR4 activation activates two transcription factors, activator protein (AP)-1 and nuclear factor κB (NF-κB) (18, 19), and stimulates the production of pro-inflammatory mediators such as prostaglandin 2 (PGE2) (20), tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, interferon (IFN)-γ, and the acute phase protein, C-reactive protein (CRP) (19). Simultaneously, an uncontrolled pro-inflammatory reaction is prevented by the induction of TLR4, NF-κB, and AP-1 signaling inhibitors, which are probably involved in creating endotoxin tolerance (21). LPS tolerance is defined as a reduced responsiveness to a LPS challenge following a first encounter of endotoxin (22). It has been suggested that the dose of LPS exposure is important for determining the switch between LPS tolerance and priming. For example, in macrophages, high LPS concentrations induced a robust pro-inflammatory response in parallel with the activation of inhibitory feedback mechanisms. Lower concentrations of LPS, like those observed in NCDs, removed transcriptional suppressors on the promoters of pro-inflammatory genes and induced a mild but persistent expression of pro-inflammatory mediators (21, 23).
Intestinal Barrier Function
The Paracellular Pathway is Important for Water, Mineral, and Nutrient Uptake
The intestinal barrier allows for the regulated uptake of water, minerals, and nutrients and protects the gut lumen from damage due to harmful substances. Components can cross the epithelial barrier by active transport and endocytosis (transcellular) or via the paracellular route. Because hydrophilic solutes are limited to cross lipid membranes of epithelial cells, the paracellular route is an important and major route for the transport of water, solutes, and minerals across the intestinal barrier (24, 25). Active glucose, sodium, and water uptake is mediated by the activity of sodium-dependent glucose co-transporters (SGLTs) (26). The transcellular absorption of glucose and sodium and the resulting basolateral disposition of glucose and sodium by these transporters opens up the paracellular pathway structure and allows the passive flow of water and small nutrients by creating an osmotic gradient (27).
Intestinal permeability is a measure of the barrier function of the gut and relates to the paracellular space surrounding the brush border surface of the enterocytes and the junctional complexes (28). The junctional complex, containing tight junctions, adherens junctions, and desmosomes is an important regulator of the paracellular pathway and allows the passage of water, solutes, and ions, but under normal conditions provides a barrier to larger molecules (28, 29). The claudin family of junctional transmembrane proteins has a substantial effect on paracellular permeability. While one group of sealing claudins makes the paracellular barrier less permeable, the other group of claudins is known to increase paracellular permeability by the formation of pores that increase permeability for small solutes (30, 31). The expression of claudin proteins varies between tissues, explaining the variances in permeability of tight junctions among tissues (27). The paracellular pathway can be divided into the pore and non-pore pathway. The pore pathway is mainly controlled by the expression of claudins, while the non-pore pathway is more sensitive to cytoskeletal disruptions (30). Cytoskeletal rearrangements can be induced by phosphorylation of the regulatory myosin light chain (MLC), induced by MLC-kinase (MLCK). Phosphorylation of the MLC facilitates myosin binding to actin and therefore aids in cytoskeletal contractility. MLCK can be activated by cytokines such as TNF-α, causing increases in tight junction permeability by actomyosin contraction and reorganization of the tight junction (32, 33). In addition, SGLT1 activation and associated increases in tight junction permeability are also paralleled with phosphorylation of MLC, indicating that MLCK is an important mediator in tight junction and paracellular permeability regulation (25, 34) (Figure 1).
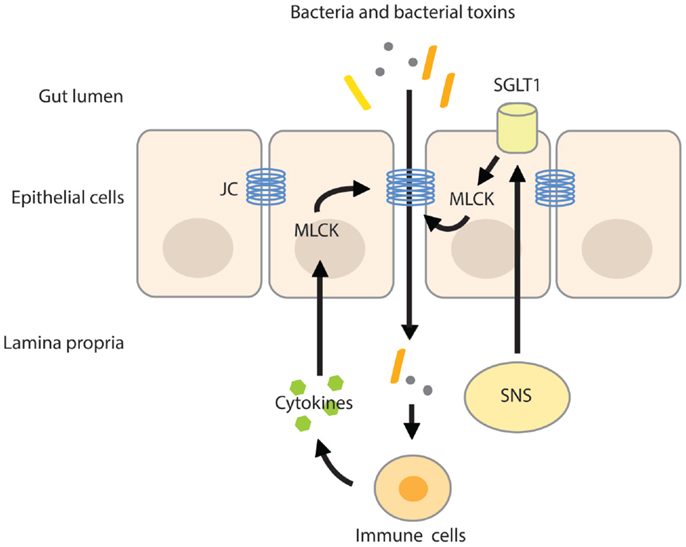
Figure 1. MLC phosphorylation increases intestinal permeability. Activation of the SNS increases intestinal permeability by stimulating the activity of SGLT1 on epithelial cells. Activation of SGLT1 is paralleled by MLC phosphorylation by MLCK, inducing actomyosin contraction and reorganization of the tight junction. The resulting increase in paracellular permeability raises the possibility of translocation of bacteria and/or their toxins across the more permeable gut barrier. Pro-inflammatory cytokines produced by activated immune cells residing in the lamina propria further increase intestinal permeability by activating MLCK. JC, junctional complex.
Increased intestinal permeability has been associated with autoimmune diseases, such as type 1 diabetes (35), rheumatoid arthritis, multiple sclerosis (36), and diseases related to chronic inflammation, like IBD (36, 37), asthma (38), chronic fatigue syndrome, and depression (10, 39). It has been hypothesized that chronic intestinal hyper-permeability results in a pro-inflammatory phenotype induced by the enhanced paracellular translocation of microbial (and dietary) antigens across the gut barrier (40).
Stress Increases Permeability of the Intestinal Barrier
Stressful stimuli activate the sympathetic nervous system (SNS) and hypothalamic–pituitary–adrenal (HPA)-axis. Activation of both systems increases the availability of water, minerals, and energy-rich substances in order to meet with the body’s metabolic demand (41, 42). The SNS responds instantly to physical and psychological stress by reallocating energy into different organs by neuronal regulation of heart rate, blood flow, release of catecholamines (adrenalin and noradrenalin) from the adrenal medulla (43), and stimulation of the renin–angiotensin–aldosterone system (44), involved in retention of water and sodium from the kidneys. In addition to the kidneys, water and sodium reabsorption can also be achieved at the level of the intestine. The intestinal wall is innervated by adrenergic sympathetic nerve fibers that upon stimulation increase water and sodium absorption (45, 46), which is paralleled by increases in intestinal permeability. The SNS-induced increase in permeability is likely mediated by β2-adrenergic receptors expressed on epithelial cells (47). Activation of the β2-adrenergic-receptors stimulated SGLT1-mediated glucose absorption from the gut (48, 49) and the resulting basolateral disposition of glucose and sodium by these transporters opens up the paracellular pathway (27) (Figure 1). Not surprisingly, blockage of the SNS by means of thoracic epidural anesthesia resulted in the blockage of the endotoxin-induced increase in intestinal permeability in rats (50).
Activation of the HPA-axis leads to the release of glucocorticoids that potentiate some of the actions of catecholamines. Essential to this response are the neurons in the paraventricular nucleus of the hypothalamus expressing corticotropin-releasing hormone (CRH) and other co-secretagogues, such as arginine vasopressin (AVP) and oxytocin, both involved in the regulation of water homeostasis. AVP and CRH trigger the immediate release of adrenocorticotropic hormone (ACTH) from the anterior pituitary, which in turn induces the release of glucocorticoids and to some extend mineralocorticoids from the adrenal cortex, stimulating gluconeogenesis and increasing sodium and water retention, respectively (51, 52). Intestinal permeability is regulated by several components of the HPA-axis.
In epithelial HT-29 monolayers, exposure to CRH resulted in an increased response to LPS as reflected by a decrease in transepithelial resistance and a significant increase in the expression of the pore forming protein, claudin-2. Interestingly enough, these effects were mediated by an increase in TLR4 expression, an observation that could be repeated in mice treated with the water-avoid stressor (53). TLR4 activation resulted in the activation of the transcription factor NF-κB, which has specific binding sites in the claudin-2 gene promoter (54), indicating that in epithelial cells CRH affects both intestinal permeability and inflammatory pathways.
In rats, exposure to restricted stress or swimming stress increased intestinal permeability throughout the whole intestinal tract as measured by the fractional secretion of the urinary recovery of sucrose (reflecting gastric permeability), the lactulose–mannitol ratio (as a marker for small intestinal permeability), and sucralose (reflecting both small intestinal and colonic permeability) (55). Other experimental animal stress models such as thermal injury or early maternal deprivation induced the development of gastric ulcers, altered gastrointestinal motility and ion secretion, and increased intestinal permeability [reviewed by Caso et al. (56)]. SGLT1 expression was markedly increased in the rat jejunum and ileum after 8 weeks of restraint stress. These findings were paralleled with an increase in intestinal lymphocytic infiltration and adrenal gland weight gain (26). The up-regulation of the SGLT1 is probably necessary to meet with the increased water, sodium, and nutrient demand, induced by chronic stress (42).
The effect of acute stress on intestinal permeability was also investigated in humans (57). In healthy volunteers subjected to a public speech test, high cortisol-responders displayed increased intestinal permeability as measured by the lactulose–mannitol ratio. Exogenous CRH administration also increased intestinal permeability, yet the CRH-induced hyper-permeability could be suppressed by the mast cell stabilizer disodium cromoglycate. Mast cell stabilization before the public speech test also did not alter intestinal permeability, however, it should be noted that in this experiment, a control group was not included. Nevertheless, these results identify CRH as an important factor in the stress-induced alterations of the intestinal barrier function. These alterations seemed to be mediated by intestinal mast cells that upon activation secrete pro-inflammatory mediators like IFN-γ and TNF-α. A variety of pro-inflammatory cytokines increases epithelial and endothelial paracellular permeability by modulating the structure of the tight junction and by inducing cytoskeletal disruptions via activation of MLCK (32, 34, 58) (Figure 1). For example, IFN-γ increased epithelial permeability of T84 monolayers to large molecules (10 kDa). Interestingly, the IFN-γ-induced increase in permeability also up-regulated the passage of FITC-labeled-endotoxin by 10-fold (59).
Neuroendocrine–Immune Interactions
The complex neuroendocrine–immune interactions are evidenced by the fact that emotional stressors influence the immune response and that pure immunological stimuli impact on cognitive performance (60). Inflammatory mediators activate the HPA-axis with the purpose to provoke disease behavior and redirect energy-rich nutrients toward the immune system (61). Cytokines have been shown to increase nutrient availability to meet with the inflammation-dependent increased metabolic demand. For example, the cytokine IL-1α increased whole body glucose metabolism on a central level (62) and cytokines like IL-6, TNF-α, IL-1, and IFN independently evoke a HPA-axis response (63–65). Immune mediators can communicate with the brain via several pathways. By stimulating afferent sensory nerve fibers, by entering the brain via the circumventricular organs or by binding to cerebral blood vessel endothelium, immune mediators effectively redirect energy-rich substrates toward the immune system (41, 42).
Besides inflammatory cytokines, prostaglandins synthesized via the cyclooxygenase system play a central role in inflammation and HPA-axis activation. Zimomra et al. (65) demonstrated that in rats the initial activation of the HPA-axis by LPS is mediated by prostaglandins, like PGE2, while inflammatory cytokines maintain corticosterone levels at later time-points. In this study, it was suggested that prostaglandins stimulated corticosterone release in a direct manner, since the peak in circulating corticosterone levels was observed long before the peak in circulating ACTH. This idea was confirmed by a study in rodents, showing that PGE2 directly stimulated the release of glucocorticoids from the adrenal gland (66). In human adrenal cells expressing TLR2 and TLR4, LPS stimulation resulted in the release of cortisol. This effect was mediated by PGE2, since inhibition of cyclooxygenase-2 attenuated cortisol release (67).
As indicated, TLR4 activation stimulates the release of PGE2 by immune cells, adipocytes, endothelial, epithelial, and probably also adrenal cells (68), inducing the peripheral release of glucocorticoids from the adrenal gland (66). PGE2 also activates glucocorticoid production through activation of the HPA-axis at the level of the hypothalamus and the pituitary (69). Macrophages, homing in blood vessels in the cranium, are directly activated by danger signals such as LPS. Activation of these special macrophages induces the production of PGE2 which directly stimulates the paraventricular nucleus of the hypothalamus, leading to higher production of glucocorticoids, which should probably protect against possible inflammation of the brain (69).
Acute Stress Stimulates Pro-Inflammatory Pathways by Increasing Intestinal Permeability
Acute stress modulates the immune response and changes immune cell distribution. These neuroendocrine effects on the immune system are mediated by stress-hormones released from the adrenal gland, by direct innervation of sympathetic nerve fibers into lymphoid organs and by stress hormone receptors expressed on immune cells, like glucocorticoid receptors (GRs) and α- and β-adrenergic receptors (70–72). It has been suggested that by mobilizing immune cells, the stress response, also known as the “fight–flight reaction,” prepares the immune system for oncoming challenges (70).
In addition, acute stress increases circulating pro-inflammatory mediators (73–75). In subjects exposed to acute stress, NF-κB was up-regulated in peripheral blood mononuclear cells in parallel with elevated levels of circulating catecholamines and glucocorticoids (76). Until now, it is not completely understood what causes this pro-inflammatory response. Glucocorticoids mostly have an inhibitory effect on inflammatory pathways and catecholamines a rather modulating than activating influence on the immune system (71, 72, 77), however, it has been shown that activation of the β-adrenergic receptor by noradrenalin (but not adrenalin) increased NF-κB binding to DNA in monocytes in vitro (76). A recent study in rodents showed that acute stress-induced neuro-inflammation could be prevented by a pre-stress treatment with antibiotics or an inhibitor of MLCK. In addition, these treatments prevented stress-induced hyper-permeability and endotoxemia, indicating that it is not the stress-factor itself producing a pro-inflammatory response of the immune system, but the fact that stress increases barrier permeability and the translocation of endotoxin. Pre-stress probiotic treatment with Lactobacillus farciminis had similar effects, which could be explained by its ability to enhance intestinal barrier function (78). In agreement with these results, it could be hypothesized that the (short-lasting) pro-inflammatory activity in humans observed during acute stress is initiated by a stress-induced increase in intestinal permeability, mediated by the SNS and components of the HPA-axis, and resulting in higher levels of translocating endotoxin interacting with TLRs on immune cells, adipocytes, and epithelial cells. A schematic overview of the complex neuroendocrine–immune interactions and their relation to gut barrier function are displayed in Figure 2.
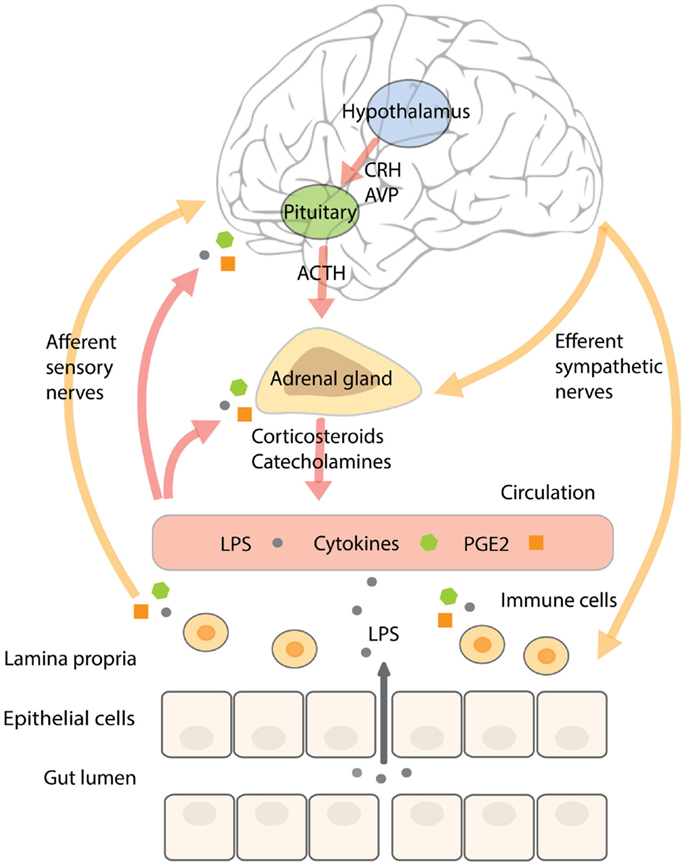
Figure 2. The complex neuroendocrine–immune interactions and their relation to gut barrier function. Stressors, including inflammatory mediators, activate the SNS and HPA-axis. Activation of the HPA-axis stimulates neurons in the paraventricular nucleus of the hypothalamus to secrete CRH and AVP that trigger the release of ACTH from the anterior pituitary, resulting in the secretion of corticosteroids from the adrenal cortex. CRH has been shown to affect intestinal permeability. SNS activation results in the release of catecholamines from the adrenal medulla. The intestinal wall is innervated by adrenergic sympathetic nerve fibers that upon stimulation increase water, sodium, and glucose absorption, paralleled by increased intestinal permeability. The resulting increase in translocation of endotoxin across the intestinal barrier can stimulate immune cells in the underlying lamina propria to secrete pro-inflammatory cytokines and prostaglandins like PGE2. Inflammatory mediators communicate with the brain by stimulating afferent sensory nerve fibers, by entering the brain via the circumventricular organs or by binding to cerebral blood vessel endothelium. Continuous stress-induced impairment of the intestinal barrier creates a vicious circle whereby inflammatory cytokines will persistently activate the SNS and HPA-axis resulting in barrier disruption, increased endotoxin translocation, and a pro-inflammatory state.
Chronic Stress Dysregulates the HPA-Axis and Changes Immune Function
Chronic psychological stress is known to dysregulate the immune system. These alterations are accompanied by low-grade inflammation, delayed wound healing, and increased susceptibility to infectious diseases (79). Chronic stress leads to hypercortisolemia (77), long-term permeability of barriers, endotoxemia, and low-grade inflammation (our hypothesis and theory). Normally, the release of glucocorticoids puts a limit on the maximum activity of the immune system; however, chronic HPA-axis stimulation can result in glucocorticoid resistance at the level of the immune system, making it insensitive to its inhibitory and modulatory actions (2). This process is observed in several conditions (including conditions related to psychosocial stress), whereby immune cells from patients are less responsive to the inhibitory actions of glucocorticoids on cytokine release and cell proliferation after stimulation in vitro (80–83). In addition, chronic stress induces a shift in the production of type 1 cytokines toward type 2 cytokine production. It can be deducted that by this mechanism, the part of the immune system involved in the clearance of extracellular bacteria and bacterial toxins (the type 2 response) is prevented from being suppressed and protection against ongoing microbial infiltration (endotoxemia) is guaranteed, while the type 1 response, involved in clearance of intracellular pathogens (like viruses) is inhibited (71, 84).
Life-Style-Related Factors Induce Endotoxemia
The fact that stress increases barrier permeability and thereby enhances the availability of water, sodium, and nutrients, makes sense from an evolutionary perspective. However, the question arises if the accompanied translocation of bacteria and their toxins should also be considered beneficial for the host. We speculate that when the composition of the microbiota is physiological, and barrier opening is short-lasting, acute stress will not produce low-grade inflammation. However, modern people suffer from new multi-factorial stressors, such as chronic psychosocial stress and the consumption of a “Western diet,” which constantly challenge the stress-axis, alter microbiota composition, and thereby compromise intestinal barrier function. This next section discusses how modern life-style factors impact the gut–brain–immune-axis and promote endotoxemia, low-grade inflammation, and its related diseases.
Gut-Microbiota Modulate Stress-Axis and Influence Gut Barrier Function
Large differences in the composition of the gut-microbiota and an overall reduction in microbial diversity are observed in Western populations when compared to traditional Hunter-gatherers or people from rural Africa (85, 86). These environment and diet-induced changes in gut-microbiota have been connected to an increased susceptibility to chronic diseases, like IBD, obesity, and type 1 and type 2 diabetes (87). The gut-microbiota influences inflammatory (88) and metabolic processes (89) and has been shown to influence the development of the HPA-axis and immune system (90, 91). For example, exposure to LPS during developmental periods can exaggerate the HPA-axis and immune response to stress (92, 93), but also the absence of bacteria can induce these effects. Animals raised in germ-free environments showed an exaggerated HPA-axis response, which was normalized by colonization with fecal matter from specifically germ-free animals or by the administration of the Gram-positive Bifidobacterium infantis (94). Vice versa, exposure to social stress changed the composition of the gut-microbiota in mice (95, 96) and prenatal stress altered the microbiome in rhesus monkeys by reducing the overall numbers of the Gram-positive Bifidobacteria and Lactobacilli (97), indicating that chronic stress affected the composition of the gut microbiome. Stress influences gut motility, secretions, and mucin production, thereby altering the habitat of resident bacteria, promoting changes in the composition of the gut microbiome (98), and allowing the growth of pathogenic bacteria (99).
Increasing evidence supports an important role for microbiota on the homeostasis of the intestinal barrier. Certain strains of the Gram-positive Lactobacilli decreased intestinal permeability in several animal and human disease models (78, 100). B. infantis reduced intestinal permeability (as assessed by 70-kDa fluorescein isothiocyanate–dextran transmucosal flux) and ameliorated symptoms in a neonatal necrotizing enterocolitis mouse model (101). Further evidence indicating the influence of the gut-microbiota on intestinal permeability was presented in detoxifying alcoholic-dependent subjects: lower levels of Ruminococcaceae and higher abundance of Lachnospiraceae (Dorea) and Blautia were associated with increased intestinal permeability (102). In addition, higher levels of certain pathogenic bacteria can increase intestinal permeability by disrupting the epithelial barrier and triggering cell death and inflammation. These bacteria have the ability to bind and/or translocate through endothelial and microfold cells and have been shown to secrete toxins or other effector molecules via specialized secretion systems. Although the exact mechanisms are not well described, most pathogenic gut bacteria including Escherichia coli, Helicobacter pylori, Staphylococcus aureus, Cholera Pseudomonas fluorescens, Pseudomonas aeruginosa, Yersinia enterocolitica, Campylobacter jejuni, and Salmonella typhimurium alter paracellular permeability by disassembling tight junctions and generating cytoskeleton changes by increasing inflammation [reviewed by Barreau et al. (103)]. As an example, a strain of E. coli, normally present in the human gut, induced focal leaks in colonic epithelial monolayers and in rat distal colon by using α-hemolysin, allowing for its paracellular translocation across the epithelial layer (104).
High-Caloric and High-Fat Diets Induce Inflammation and Increase Circulating Endotoxin Levels
Compared to healthy individuals, patients suffering from obesity have higher circulating endotoxin levels together with greater levels of circulating pro-inflammatory cytokines and insulin resistance (105). Food intake can produce post-prandial immune activation and elevate endotoxin levels when a meal is high in calories (106) or has a high fat content (6, 107–109).
Rodents fed a 4-week high-fat diet (72% fat) showed a constant elevation in circulating endotoxin levels, while in control animals, endotoxin levels only increased during feeding hours. Furthermore, a high-fat diet produced fasting glycemia, insulin resistance, general weight gain, and weight gain of the liver and visceral and subcutaneous adipose tissue. In addition, adipose tissue F4/80-positive cells (indicating the infiltration of macrophages), markers of inflammation, and liver triglyceride content were increased. Interestingly, almost similar effects were observed in mice subcutaneously infused with LPS (resulting in similar circulating LPS levels as observed in the high-fat fed mice). These effects were mediated by TLR4, since mice lacking CD14, which is important for the recognition of LPS to this receptor, showed a delayed response to a high-fat diet or LPS injections (107).
In healthy humans, a 910 calories high-fat and high-carbohydrate meal resulted in increased circulating endotoxin levels and elevated levels of LBP in parallel with higher inflammatory markers and increased protein expression of TLR2 and TLR4 in isolated leukocytes. A meal high in fruits and fiber did not induce these effects (108). Plasma endotoxin levels, pro-inflammatory markers, and leukocyte TLR4 expression increased after the intake of cream (300 calories), while the intake of 300 calories of glucose resulted only in a pro-inflammatory response and the intake of orange juice and water showed none of these effects (110). In healthy individuals, plasma endotoxin levels increased about 50% after the intake of a high-fat meal (900 calories) (6) and 4 weeks consumption of a Western-style diet raised plasma endotoxin activity levels by 71% (111).
How exactly the intake of a high-caloric meal increases circulating endotoxin levels is still unclear but has been explained by several mechanisms [reviewed by Kelly et al. (112)]. One of these suggested mechanisms is that the introduction of a high-fat diet modulates the expression of genes involved in the barrier function in epithelial cells, thereby directly compromising the integrity of the tight junction (113). Another explanation could be that a high-caloric/high-fat meal induces high levels of insulin and leptin, hormones that directly activate the SNS (114, 115). Moreover, insulin enhances SGLT1-mediated glucose absorption (116). Activation of the SGLT1 and the SNS leads to increased permeability of the gut barrier, which may induce the observed post-prandial endotoxemia (our hypothesis and theory).
Gliadin Compromises the Integrity of Tight Junctions
The intake of wheat and other cereal grains has been implicated in the development of inflammation-related diseases, by inducing inflammation and increasing intestinal permeability (40). Gliadin, a component of gluten, has been demonstrated to increase permeability in human Caco-2 intestinal epithelial cells by reorganizing actin filaments and altering expression of junctional complex proteins (117). Several studies by the group of Fasano et al. showed that the binding of gliadin to the chemokine receptor CXCR3 on epithelial IEC-6 and Caco-2 cells releases zonulin, a protein that directly compromises the integrity of the junctional complex (118, 119).
Alcohol Consumption Increases Intestinal Permeability
Alcohol consumption is an important risk factor for disease and is one of the major causes of chronic liver disease. Increased intestinal permeability has been observed during chronic alcohol consumption. In an animal model of chronic alcoholic liver disease, alcohol feeding for 8 weeks increased intestinal permeability (120). In humans, alcohol-dependence induced changes in the gut-microbiota composition that were associated with increased intestinal permeability (102). Furthermore, increased intestinal permeability and higher circulating endotoxin levels were observed in patients with chronic alcohol abuse (121–123).
Exercise-Induced Heat-Stress Increases Intestinal Permeability
Exercise increases body temperature, reduces intestinal blood flow (reallocated to the muscles and cardiac system), and increases intestinal permeability by activating the SNS and HPA-axis. Already in 1992, Oktedalen et al. (124) showed that marathon runners displayed a significant increase in intestinal permeability. In addition, studies have indicated that strenuous exercise induced higher circulating endotoxin levels and activated the immune system (125–128). Further evidence of exercise- and heat-induced increased intestinal permeability, leading to gastrointestinal complaints in people engaging in physical activity, has been recently reviewed (129).
Endotoxemia is Associated with Diseases Related to Chronic Inflammation
Multiple human studies have emerged that find associations with NCDs and markers of endotoxemia. Even aging, associated with higher sympathetic nerve activity (130) and higher circulating inflammatory mediators like IL-6, has been linked to higher plasma concentrations of LPS and LBP (131). In further support of our theory, in this section, an overview is given of human studies finding changes in levels of endotoxin or endotoxin-related markers in NCDs (Table 1).
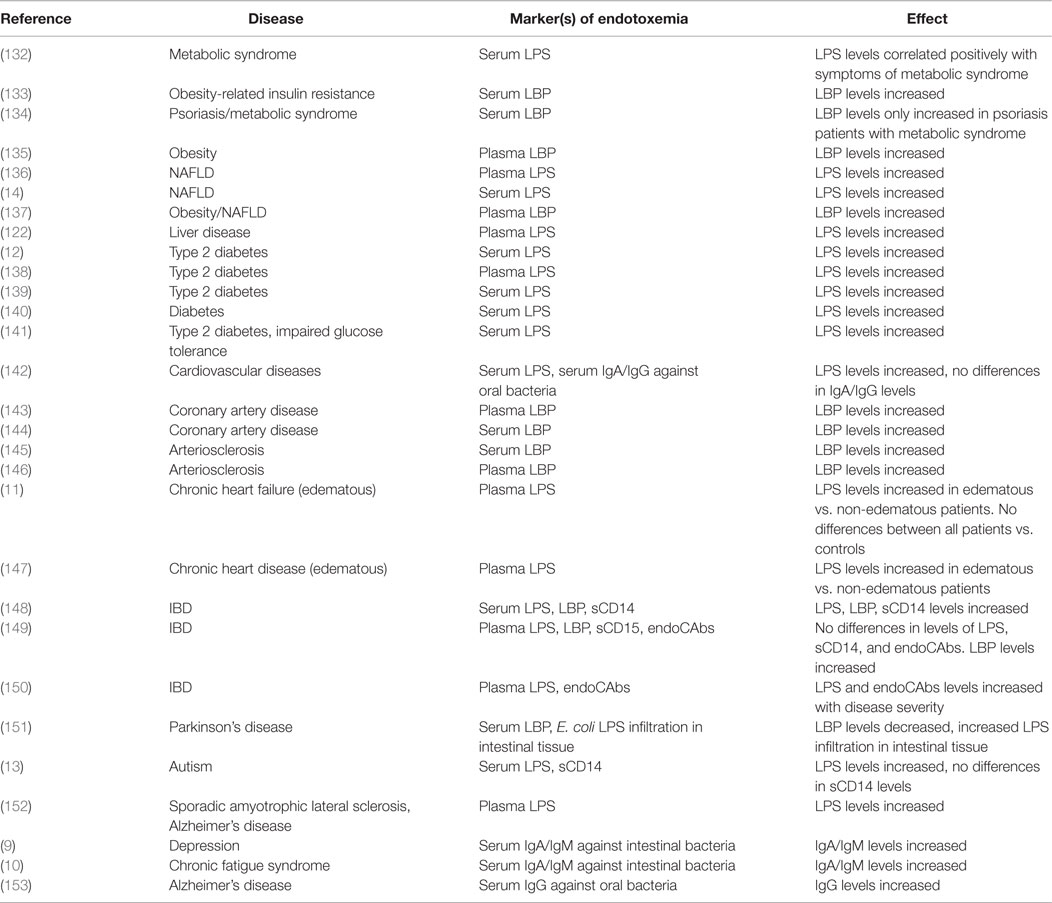
Table 1. Associations found between markers of endotoxemia and disease.
Metabolic Syndrome
Metabolic syndrome is accompanied by an increased risk for NAFLD, obesity, type 2 diabetes, and cardiovascular diseases. All of these conditions are related to and even predicted by increased sympathetic nerve activity (154) and a dysregulated HPA-axis (155). Higher circulating endotoxin and LBP levels are associated with risk factors of the metabolic syndrome, like insulin resistance, obesity, dyslipidemia, and chronic inflammation (132–135). Patients suffering from NAFLD exhibited significantly higher serum endotoxin levels in contrast to healthy controls (14). Farhardi et al. (136) indicated that elevated plasma endotoxin levels in these patients were related to an impaired intestinal barrier function, because, only in the patient group, the intake of a permeability stressor (aspirin) increased the 0–24 h urinary excretion of sucralose (a marker of whole-gut permeability). Furthermore, augmented plasma LBP levels in concert with increased plasma levels of TNF-α were observed in obese NAFLD patients compared to healthy controls (137).
Elevated circulating levels of endotoxin and LBP were detected in type 2 diabetics (12, 133, 138–140). Compared to healthy controls, obese individuals and type 2 diabetics showed higher endotoxin levels after the intake of a high-fat meal. Increased endotoxin levels were observed in all challenged individuals, yet higher endotoxin levels were seen in individuals suffering from metabolic illnesses, suggesting an increased intestinal permeability in these patients (141). This was further indicated by a recent study showing that increased serum levels of endotoxin, IL-6, and TNF-α were found in type 2 diabetic patients compared to healthy individuals. The level of endotoxin was positively related to zonulin, a marker for intestinal permeability (12).
A large cohort of patients with coronary artery disease identified increased serum LBP levels to be associated with total and cardiovascular mortality (144). Moreover, circulating LBP levels were associated with carotid intima media thickness (a marker of atherosclerosis), obesity, insulin resistance, and high-sensitive CRP (145).
Patients suffering from chronic heart failure with aggravated renal function displayed increased circulating endotoxin levels and an impairment of the intestinal barrier (11). Wiedermann et al. (146) showed that subjects with the highest levels of circulating endotoxin (90th percentile) had a threefold increased risk of incident atherosclerosis. Higher serum endotoxin and pro-inflammatory cytokine concentrations were seen in patients with edematous chronic heart disease compared to stable patients and healthy controls. Intriguingly, after short-term diuretic treatment, circulating endotoxin concentrations decreased in edematous patients (147). Diuretic treatment [like angiotensin-converting enzyme (ACE) inhibitors] ameliorated intestinal inflammation, perhaps by impacting on intestinal permeability through interference with the renin–angiotensin–aldosterone system. Several components of this system (renin, ACE, and angiotensin II) have been shown to stimulate pro-inflammatory pathways (44, 156).
Inflammatory Bowel Disease
Ulcerative colitis and Crohn’s disease are intestinal inflammatory disorders, also known as IBD, which have been causally linked to chronic psychological stress (157), altered immune function, changes in the gut-microbiota, increased intestinal permeability, and endotoxemia (158). For example, increased plasma endotoxin and LBP levels were measured in both patient groups, but were more pronounced in patients with active disease compared to inactive disease and were associated with disease severity (148). In addition, detectable plasma endotoxin levels and higher plasma levels of LBP were more frequently observed in IBD patients compared to controls (15, 149) and were correlated with disease severity and circulating TNF-α levels (150).
Psychiatric Diseases
Over the last decade, the role of the gut–brain axis has emerged as an important mediator in the development of psychiatric and mood disorders (159). Moreover, higher endotoxin levels and intestinal barrier dysfunction are observed in several of these conditions. For example, Parkinson’s patients exhibited increased total intestinal permeability and a more intense staining for E. coli LPS and oxidative stress markers in intestinal sigmoid mucosa samples. However, in these patients, endotoxin levels resembled control samples and serum LBP concentrations were lower compared to healthy individuals (151). Higher serum endotoxin levels are associated with severe autism, sporadic amyotrophic lateral sclerosis, and Alzheimer’s disease (13, 152). Furthermore, increased IgA and IgM responses against LPS of commensal bacteria were seen in chronic fatigue syndrome (10) and depression (9). Intriguingly, chronic oral infection of periodontitis was associated with Alzheimer’s disease where higher antibody levels against oral pathogens were observed years before the onset of symptoms in people suffering from Alzheimer’s disease (153), suggesting there was an increased translocation of bacteria and/or bacterial toxins from the mouth into the bloodstream.
Conclusion
Chronic low-grade inflammation is an eminent feature of NCDs. In addition, many studies report increased circulating endotoxin levels and increased gut permeability in patients suffering from these conditions. As reviewed in this paper, stress-induced increases in intestinal permeability, in combination with modern life-style factors, raise the possibility of translocation of bacteria and/or their toxins across the more permeable gut barrier. The resulting, long lasting, endotoxemia should be considered much more than just a risk factor for chronic disease; it could be a cause. Notwithstanding the fact that the exact origin and sequence of events involved in development of NCDs remain to be unsolved, evidence indicates that a disrupted barrier function in parallel with elevated circulating endotoxin levels may underlie disease onset and progression. For this reason, therapies aimed at restoring intestinal barrier function, life-style changes, and stress management should be considered important strategies in preventing and attenuating the pro-inflammatory state observed in NCDs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Alicia Lammerts van Bueren for the editorial work on the manuscript.
References
1. Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem (2008) 15(17):1697–705. doi: 10.2174/092986708784872393
2. Bosma-den Boer MM, van Wetten ML, Pruimboom L. Chronic inflammatory diseases are stimulated by current lifestyle: how diet, stress levels and medication prevent our body from recovering. Nutr Metab (2012) 9(1):32. doi:10.1186/1743-7075-9-32
3. Eckel RH, Alberti KG, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet (2010) 375(9710):181–3. doi:10.1016/S0140-6736(09)61794-3
4. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol (2006) 27(1):24–31. doi:10.1016/j.it.2005.11.006
5. Ruiz-Nunez B, Pruimboom L, Dijck-Brouwer DA, Muskiet FA. Lifestyle and nutritional imbalances associated with Western diseases: causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J Nutr Biochem (2013) 24(7):1183–201. doi:10.1016/j.jnutbio.2013.02.009
6. Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr (2007) 86(5):1286–92.
7. Moreira AP, Texeira TF, Ferreira AB, Peluzio Mdo C, Alfenas Rde C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxemia. Br J Nutr (2012) 108(5):801–9. doi:10.1017/S0007114512001213
8. Vaishnavi C. Translocation of gut flora and its role in sepsis. Indian J Med Microbiol (2013) 31(4):334–42. doi:10.4103/0255-0857.118870
9. Maes M, Mihaylova I, Leunis JC. Increased serum IgM antibodies directed against phosphatidyl inositol (Pi) in chronic fatigue syndrome (CFS) and major depression: evidence that an IgM-mediated immune response against Pi is one factor underpinning the comorbidity between both CFS and depression. Neuro Endocrinol Lett (2007) 28(6):861–7.
10. Maes M, Mihaylova I, Leunis JC. Increased serum IgA and IgM against LPS of enterobacteria in chronic fatigue syndrome (CFS): indication for the involvement of Gram-negative enterobacteria in the etiology of CFS and for the presence of an increased gut-intestinal permeability. J Affect Disord (2007) 99(1–3):237–40. doi:10.1016/j.jad.2006.08.021
11. Sandek A, Bjarnason I, Volk HD, Crane R, Meddings JB, Niebauer J, et al. Studies on bacterial endotoxin and intestinal absorption function in patients with chronic heart failure. Int J Cardiol (2012) 157(1):80–5. doi:10.1016/j.ijcard.2010.12.016
12. Jayashree B, Bibin YS, Prabhu D, Shanthirani CS, Gokulakrishnan K, Lakshmi BS, et al. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol Cell Biochem (2014) 388(1–2):203–10. doi:10.1007/s11010-013-1911-4
13. Emanuele E, Orsi P, Boso M, Broglia D, Brondino N, Barale F, et al. Low-grade endotoxemia in patients with severe autism. Neurosci Lett (2010) 471(3):162–5. doi:10.1016/j.neulet.2010.01.033
14. Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (2010) 7:15. doi:10.1186/1476-9255-7-15
15. Caradonna L, Amati L, Lella P, Jirillo E, Caccavo D. Phagocytosis, killing, lymphocyte-mediated antibacterial activity, serum autoantibodies, and plasma endotoxins in inflammatory bowel disease. Am J Gastroenterol (2000) 95(6):1495–502. doi:10.1111/j.1572-0241.2000.02085.x
16. Wang JE, Dahle MK, McDonald M, Foster SJ, Aasen AO, Thiemermann C. Peptidoglycan and lipoteichoic acid in gram-positive bacterial sepsis: receptors, signal transduction, biological effects, and synergism. Shock (2003) 20(5):402–14. doi:10.1097/01.shk.0000092268.01859.0d
17. Giannelli V, Di Gregorio V, Iebba V, Giusto M, Schippa S, Merli M, et al. Microbiota and the gut-liver axis: bacterial translocation, inflammation and infection in cirrhosis. World J Gastroenterol (2014) 20(45):16795–810. doi:10.3748/wjg.v20.i45.16795
18. Bohannon JK, Hernandez A, Enkhbaatar P, Adams WL, Sherwood ER. The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock (2013) 40(6):451–62. doi:10.1097/SHK.0000000000000042
19. Neves AL, Coelho J, Couto L, Leite-Moreira A, Roncon-Albuquerque R Jr. Metabolic endotoxemia: a molecular link between obesity and cardiovascular risk. J Mol Endocrinol (2013) 51(2):R51–64. doi:10.1530/JME-13-0079
20. Weinlich R, Bortoluci KR, Chehab CF, Serezani CH, Ulbrich AG, Peters-Golden M, et al. TLR4/MYD88-dependent, LPS-induced synthesis of PGE2 by macrophages or dendritic cells prevents anti-CD3-mediated CD95L upregulation in T cells. Cell Death Differ (2008) 15(12):1901–9. doi:10.1038/cdd.2008.128
21. Maitra U, Gan L, Chang S, Li L. Low-dose endotoxin induces inflammation by selectively removing nuclear receptors and activating CCAAT/enhancer-binding protein delta. J Immunol (2011) 186(7):4467–73. doi:10.4049/jimmunol.1003300
22. Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care (2006) 10(5):233. doi:10.1186/cc5055
23. Deng H, Maitra U, Morris M, Li L. Molecular mechanism responsible for the priming of macrophage activation. J Biol Chem (2013) 288(6):3897–906. doi:10.1074/jbc.M112.424390
24. Kapus A, Szaszi K. Coupling between apical and paracellular transport processes. Biochem Cell Biol (2006) 84(6):870–80. doi:10.1139/o06-202
25. Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol (1997) 273(4 Pt 1):C1378–85.
26. Lee CY. Chronic restraint stress induces intestinal inflammation and alters the expression of hexose and lipid transporters. Clin Exp Pharmacol Physiol (2013) 40(6):385–91. doi:10.1111/1440-1681.12096
27. Turner JR. Show me the pathway! Regulation of paracellular permeability by Na(+)-glucose cotransport. Adv Drug Deliv Rev (2000) 41(3):265–81. doi:10.1016/S0169-409X(00)00046-6
28. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol (2009) 9(11):799–809. doi:10.1038/nri2653
29. Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol (2004) 286(6):C1213–28. doi:10.1152/ajpcell.00558.2003
30. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol (2009) 1(2):a002584. doi:10.1101/cshperspect.a002584
31. Overgaard CE, Daugherty BL, Mitchell LA, Koval M. Claudins: control of barrier function and regulation in response to oxidant stress. Antioxid Redox Signal (2011) 15(5):1179–93. doi:10.1089/ars.2011.3893
32. Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, et al. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem (2006) 281(36):26205–15. doi:10.1074/jbc.M602164200
33. Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, et al. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci (2006) 119(Pt 10):2095–106. doi:10.1242/jcs.02915
34. Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci (2012) 1258:34–42. doi:10.1111/j.1749-6632.2012.06526.x
35. Secondulfo M, Iafusco D, Carratu R, deMagistris L, Sapone A, Generoso M, et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig Liver Dis (2004) 36(1):35–45. doi:10.1016/j.dld.2003.09.016
36. Fasano A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol (2012) 42(1):71–8. doi:10.1007/s12016-011-8291-x
37. Keita AV, Soderholm JD. The intestinal barrier and its regulation by neuroimmune factors. Neurogastroenterol Motil (2010) 22(7):718–33. doi:10.1111/j.1365-2982.2010.01498.x
38. Hijazi Z, Molla AM, Al-Habashi H, Muawad WM, Molla AM, Sharma PN. Intestinal permeability is increased in bronchial asthma. Arch Dis Child (2004) 89(3):227–9. doi:10.1136/adc.2003.027680
39. Maes M, Kubera M, Leunis JC, Berk M, Geffard M, Bosmans E. In depression, bacterial translocation may drive inflammatory responses, oxidative and nitrosative stress (O&NS), and autoimmune responses directed against O&NS-damaged neoepitopes. Acta Psychiatr Scand (2013) 127(5):344–54. doi:10.1111/j.1600-0447.2012.01908.x
40. de Punder K, Pruimboom L. The dietary intake of wheat and other cereal grains and their role in inflammation. Nutrients (2013) 5(3):771–87. doi:10.3390/nu5030771
41. Straub RH. Evolutionary medicine and chronic inflammatory state – known and new concepts in pathophysiology. J Mol Med (2012) 90(5):523–34. doi:10.1007/s00109-012-0861-8
42. Straub RH. TRPV1, TRPA1, and TRPM8 channels in inflammation, energy redirection, and water retention: role in chronic inflammatory diseases with an evolutionary perspective. J Mol Med (2014) 92(9):925–37. doi:10.1007/s00109-014-1175-9
43. Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol (2009) 54(19):1747–62. doi:10.1016/j.jacc.2009.05.015
44. Groeschel M, Braam B. Connecting chronic and recurrent stress to vascular dysfunction: no relaxed role for the renin-angiotensin system. Am J Physiol Renal Physiol (2011) 300(1):F1–10. doi:10.1152/ajprenal.00208.2010
45. Brunsson I, Eklund S, Jodal M, Lundgren O, Sjovall H. The effect of vasodilatation and sympathetic nerve activation on net water absorption in the cat’s small intestine. Acta Physiol Scand (1979) 106(1):61–8. doi:10.1111/j.1748-1716.1979.tb06370.x
46. Levens NR, Peach MJ, Carey RM. Interactions between angiotensin peptides and the sympathetic nervous system mediating intestinal sodium and water absorption in the rat. J Clin Invest (1981) 67(4):1197–207. doi:10.1172/JCI110135
47. Lange S, Delbro DS. Adrenoceptor-mediated modulation of Evans blue dye permeation of rat small intestine. Dig Dis Sci (1995) 40(12):2623–9. doi:10.1007/BF02220451
48. Aschenbach JR, Borau T, Gabel G. Glucose uptake via SGLT-1 is stimulated by beta(2)-adrenoceptors in the ruminal epithelium of sheep. J Nutr (2002) 132(6):1254–7.
49. Ishikawa Y, Eguchi T, Ishida H. Mechanism of beta-adrenergic agonist-induced transmural transport of glucose in rat small intestine. Regulation of phosphorylation of SGLT1 controls the function. Biochim Biophys Acta (1997) 1357(3):306–18. doi:10.1016/S0167-4889(97)00043-8
50. Schaper J, Wagner A, Enigk F, Brell B, Mousa SA, Habazettl H, et al. Regional sympathetic blockade attenuates activation of intestinal macrophages and reduces gut barrier failure. Anesthesiology (2013) 118(1):134–42. doi:10.1097/ALN.0b013e3182784c93
51. de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci (2005) 6(6):463–75. doi:10.1038/nrn1683
52. Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol (2012) 350(2):151–62. doi:10.1016/j.mce.2011.07.034
53. Yu Y, Liu ZQ, Liu XY, Yang L, Geng XR, Yang G, et al. Stress-derived corticotropin releasing factor breaches epithelial endotoxin tolerance. PLoS One (2013) 8(6):e65760. doi:10.1371/journal.pone.0065760
54. Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem (2011) 286(36):31263–71. doi:10.1074/jbc.M111.238147
55. Meddings JB, Swain MG. Environmental stress-induced gastrointestinal permeability is mediated by endogenous glucocorticoids in the rat. Gastroenterology (2000) 119(4):1019–28. doi:10.1053/gast.2000.18152
56. Caso JR, Leza JC, Menchen L. The effects of physical and psychological stress on the gastro-intestinal tract: lessons from animal models. Curr Mol Med (2008) 8(4):299–312. doi:10.2174/156652408784533751
57. Vanuytsel T, van Wanrooy S, Vanheel H, Vanormelingen C, Verschueren S, Houben E, et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut (2013) 63(8):1293–9. doi:10.1136/gutjnl-2013-305690
58. Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta (2009) 1788(4):864–71. doi:10.1016/j.bbamem.2008.08.027
59. Watson CJ, Hoare CJ, Garrod DR, Carlson GL, Warhurst G. Interferon-gamma selectively increases epithelial permeability to large molecules by activating different populations of paracellular pores. J Cell Sci (2005) 118(Pt 22):5221–30. doi:10.1242/jcs.02630
60. Wersching H, Duning T, Lohmann H, Mohammadi S, Stehling C, Fobker M, et al. Serum C-reactive protein is linked to cerebral microstructural integrity and cognitive function. Neurology (2010) 74(13):1022–9. doi:10.1212/WNL.0b013e3181d7b45b
61. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9(1):46–56. doi:10.1038/nrn2297
62. Lang CH, Molina PE, Yousef KA, Tepper PG, Abumrad NN. Role of IL-1 alpha in central nervous system immunomodulation of glucoregulation. Brain Res (1993) 624(1–2):53–60. doi:10.1016/0006-8993(93)90059-V
63. Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci (2000) 917:608–17. doi:10.1111/j.1749-6632.2000.tb05426.x
64. Goebel MU, Baase J, Pithan V, Exton M, Saller B, Schedlowski M, et al. Acute interferon beta-1b administration alters hypothalamic-pituitary-adrenal axis activity, plasma cytokines and leukocyte distribution in healthy subjects. Psychoneuroendocrinology (2002) 27(8):881–92. doi:10.1016/S0306-4530(01)00099-3
65. Zimomra ZR, Porterfield VM, Camp RM, Johnson JD. Time-dependent mediators of HPA axis activation following live Escherichia coli. Am J Physiol Regul Integr Comp Physiol (2011) 301(6):R1648–57. doi:10.1152/ajpregu.00301.2011
66. Mohn CE, Fernandez-Solari J, De Laurentiis A, Prestifilippo JP, de la Cal C, Funk R, et al. The rapid release of corticosterone from the adrenal induced by ACTH is mediated by nitric oxide acting by prostaglandin E2. Proc Natl Acad Sci U S A (2005) 102(17):6213–8. doi:10.1073/pnas.0502136102
67. Vakharia K, Hinson JP. Lipopolysaccharide directly stimulates cortisol secretion by human adrenal cells by a cyclooxygenase-dependent mechanism. Endocrinology (2005) 146(3):1398–402. doi:10.1210/en.2004-0882
68. Zacharowski K, Zacharowski PA, Koch A, Baban A, Tran N, Berkels R, et al. Toll-like receptor 4 plays a crucial role in the immune-adrenal response to systemic inflammatory response syndrome. Proc Natl Acad Sci U S A (2006) 103(16):6392–7. doi:10.1073/pnas.0601527103
69. Serrats J, Schiltz JC, Garcia-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron (2010) 65(1):94–106. doi:10.1016/j.neuron.2009.11.032
70. Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune cells – from barracks to boulevards to battlefields: a tale of three hormones – Curt Richter award winner. Psychoneuroendocrinology (2012) 37(9):1345–68. doi:10.1016/j.psyneuen.2012.05.008
71. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev (2000) 52(4):595–638.
72. Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev (2001) 53(4):487–525.
73. Prather AA, Carroll JE, Fury JM, McDade KK, Ross D, Marsland AL. Gender differences in stimulated cytokine production following acute psychological stress. Brain Behav Immun (2009) 23(5):622–8. doi:10.1016/j.bbi.2008.11.004
74. Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun (2007) 21(7):901–12. doi:10.1016/j.bbi.2007.03.011
75. Yamakawa K, Matsunaga M, Isowa T, Kimura K, Kasugai K, Yoneda M, et al. Transient responses of inflammatory cytokines in acute stress. Biol Psychol (2009) 82(1):25–32. doi:10.1016/j.biopsycho.2009.05.001
76. Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A (2003) 100(4):1920–5. doi:10.1073/pnas.0438019100
77. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet (2009) 373(9678):1905–17. doi:10.1016/S0140-6736(09)60326-3
78. Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, et al. Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology (2012) 37(11):1885–95. doi:10.1016/j.psyneuen.2012.03.024
79. Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol (2005) 5(3):243–51. doi:10.1038/nri1571
80. Huang CJ, Acevedo EO, Mari DC, Randazzo C, Shibata Y. Glucocorticoid inhibition of leptin- and lipopolysaccharide-induced interlukin-6 production in obesity. Brain Behav Immun (2013). doi:10.1016/j.bbi.2013.10.004
81. Maranville JC, Micic D, Hanauer SB, Di Rienzo A, Kupfer SS. In vitro sensitivity assays and clinical response to glucocorticoids in patients with inflammatory bowel disease. J Crohns Colitis (2014) 8(11):1539–47. doi:10.1016/j.crohns.2014.06.013
82. Pariante CM. Glucocorticoid receptor function in vitro in patients with major depression. Stress (2004) 7(4):209–19. doi:10.1080/10253890500069650
83. Sauer J, Polack E, Wikinski S, Holsboer F, Stalla GK, Arzt E. The glucocorticoid sensitivity of lymphocytes changes according to the activity of the hypothalamic-pituitary-adrenocortical system. Psychoneuroendocrinology (1995) 20(3):269–80. doi:10.1016/0306-4530(94)00058-I
84. Glaser R. Stress-associated immune dysregulation and its importance for human health: a personal history of psychoneuroimmunology. Brain Behav Immun (2005) 19(1):3–11. doi:10.1016/j.bbi.2004.06.003
85. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A (2010) 107(33):14691–6. doi:10.1073/pnas.1005963107
86. Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, et al. Gut microbiome of the Hadza hunter-gatherers. Nat Commun (2014) 5:3654. doi:10.1038/ncomms4654
87. Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients (2012) 4(8):1095–119. doi:10.3390/nu4081095
88. van Baarlen P, Troost FJ, van Hemert S, van der Meer C, de Vos WM, de Groot PJ, et al. Differential NF-kappaB pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc Natl Acad Sci U S A (2009) 106(7):2371–6. doi:10.1073/pnas.0809919106
89. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341(6150):1241214. doi:10.1126/science.1241214
90. Dinan TG, Cryan JF. Regulation of the stress response by the gut microbiota: implications for psychoneuroendocrinology. Psychoneuroendocrinology (2012) 37(9):1369–78. doi:10.1016/j.psyneuen.2012.03.007
91. Karrow NA. Activation of the hypothalamic-pituitary-adrenal axis and autonomic nervous system during inflammation and altered programming of the neuroendocrine-immune axis during fetal and neonatal development: lessons learned from the model inflammagen, lipopolysaccharide. Brain Behav Immun (2006) 20(2):144–58.
92. Sominsky L, Fuller EA, Bondarenko E, Ong LK, Averell L, Nalivaiko E, et al. Functional programming of the autonomic nervous system by early life immune exposure: implications for anxiety. PLoS One (2013) 8(3):e57700. doi:10.1371/journal.pone.0057700
93. Walker AK, Nakamura T, Hodgson DM. Neonatal lipopolysaccharide exposure alters central cytokine responses to stress in adulthood in Wistar rats. Stress (2010) 13(6):506–15. doi:10.3109/10253890.2010.489977
94. Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol (2004) 558(Pt 1):263–75. doi:10.1113/jphysiol.2004.063388
95. Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun (2011) 25(3):397–407. doi:10.1016/j.bbi.2010.10.023
96. Galley JD, Nelson MC, Yu Z, Dowd SE, Walter J, Kumar PS, et al. Exposure to a social stressor disrupts the community structure of the colonic mucosa-associated microbiota. BMC Microbiol (2014) 14:189. doi:10.1186/1471-2180-14-189
97. Bailey MT, Coe CL. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev Psychobiol (1999) 35(2):146–55. doi:10.1002/(SICI)1098-2302(199909)35:2<146::AID-DEV7>3.3.CO;2-7
98. de Jonge WJ. The gut’s little brain in control of intestinal immunity. ISRN Gastroenterol (2013) 2013:630159. doi:10.1155/2013/630159
99. Bien J, Palagani V, Bozko P. The intestinal microbiota dysbiosis and Clostridium difficile infection: is there a relationship with inflammatory bowel disease? Therap Adv Gastroenterol (2013) 6(1):53–68. doi:10.1177/1756283X12454590
100. Ahrne S, Hagslatt ML. Effect of lactobacilli on paracellular permeability in the gut. Nutrients (2011) 3(1):104–17. doi:10.3390/nu3010104
101. Bergmann KR, Liu SX, Tian R, Kushnir A, Turner JR, Li HL, et al. Bifidobacteria stabilize claudins at tight junctions and prevent intestinal barrier dysfunction in mouse necrotizing enterocolitis. Am J Pathol (2013) 182(5):1595–606. doi:10.1016/j.ajpath.2013.01.013
102. Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Starkel P, et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Natl Acad Sci U S A (2014) 111(42):E4485–93. doi:10.1073/pnas.1415174111
103. Barreau F, Hugot JP. Intestinal barrier dysfunction triggered by invasive bacteria. Curr Opin Microbiol (2014) 17:91–8. doi:10.1016/j.mib.2013.12.003
104. Troeger H, Richter JF, Beutin L, Gunzel D, Dobrindt U, Epple HJ, et al. Escherichia coli alpha-haemolysin induces focal leaks in colonic epithelium: a novel mechanism of bacterial translocation. Cell Microbiol (2007) 9(10):2530–40. doi:10.1111/j.1462-5822.2007.00978.x
105. Moreno-Navarrete JM, Manco M, Ibanez J, Garcia-Fuentes E, Ortega F, Gorostiaga E, et al. Metabolic endotoxemia and saturated fat contribute to circulating NGAL concentrations in subjects with insulin resistance. Int J Obes (2010) 34(2):240–9. doi:10.1038/ijo.2009.242
106. Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr (2008) 87(5):1219–23.
107. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56(7):1761–72. doi:10.2337/db06-1491
108. Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, Fernandez-Real JM, et al. Increase in plasma endotoxin concentrations and the expression of toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care (2009) 32(12):2281–7. doi:10.2337/dc09-0979
109. Herieka M, Erridge C. High-fat meal induced postprandial inflammation. Mol Nutr Food Res (2014) 58(1):136–46. doi:10.1002/mnfr.201300104
110. Deopurkar R, Ghanim H, Friedman J, Abuaysheh S, Sia CL, Mohanty P, et al. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care (2010) 33(5):991–7. doi:10.2337/dc09-1630
111. Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology (2012) 142(5):1100–1e2. doi:10.1053/j.gastro.2012.01.034
112. Kelly CJ, Colgan SP, Frank DN. Of microbes and meals: the health consequences of dietary endotoxemia. Nutr Clin Pract (2012) 27(2):215–25. doi:10.1177/0884533611434934
113. Cani PD, Delzenne NM, Amar J, Burcelin R. Role of gut microflora in the development of obesity and insulin resistance following high-fat diet feeding. Pathol Biol (2008) 56(5):305–9. doi:10.1016/j.patbio.2007.09.008
114. Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol (2011) 589(Pt 7):1643–62. doi:10.1113/jphysiol.2011.205575
115. Simonds SE, Cowley MA. Hypertension in obesity: is leptin the culprit? Trends Neurosci (2013) 36(2):121–32. doi:10.1016/j.tins.2013.01.004
116. Stumpel F, Scholtka B, Jungermann K. Stimulation by portal insulin of intestinal glucose absorption via hepatoenteral nerves and prostaglandin E2 in the isolated, jointly perfused small intestine and liver of the rat. Ann N Y Acad Sci (2000) 915:111–6. doi:10.1111/j.1749-6632.2000.tb05232.x
117. Sander GR, Cummins AG, Henshall T, Powell BC. Rapid disruption of intestinal barrier function by gliadin involves altered expression of apical junctional proteins. FEBS Lett (2005) 579(21):4851–5. doi:10.1016/j.febslet.2005.07.066
118. Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, et al. Gliadin, zonulin and gut permeability: effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol (2006) 41(4):408–19. doi:10.1080/00365520500235334
119. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology (2008) 135(1):194–204e3. doi:10.1053/j.gastro.2008.03.023
120. Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates TNFRI and mediates alcoholic liver disease in mice. Hepatology (2014) 61(3):883–94. doi:10.1002/hep.27489
121. Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol (1987) 4(1):8–14. doi:10.1016/S0168-8278(87)80003-X
122. Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol (1991) 12(2):162–9. doi:10.1016/0168-8278(91)90933-3
123. Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol (2000) 32(5):742–7. doi:10.1016/S0168-8278(00)80242-1
124. Oktedalen O, Lunde OC, Opstad PK, Aabakken L, Kvernebo K. Changes in the gastrointestinal mucosa after long-distance running. Scand J Gastroenterol (1992) 27(4):270–4. doi:10.3109/00365529209000073
125. Ashton T, Young IS, Davison GW, Rowlands CC, McEneny J, Van Blerk C, et al. Exercise-induced endotoxemia: the effect of ascorbic acid supplementation. Free Radic Biol Med (2003) 35(3):284–91. doi:10.1016/S0891-5849(03)00309-5
126. Jeukendrup AE, Vet-Joop K, Sturk A, Stegen JH, Senden J, Saris WH, et al. Relationship between gastro-intestinal complaints and endotoxemia, cytokine release and the acute-phase reaction during and after a long-distance triathlon in highly trained men. Clin Sci (2000) 98(1):47–55. doi:10.1042/CS19990258
127. Pals KL, Chang RT, Ryan AJ, Gisolfi CV. Effect of running intensity on intestinal permeability. J Appl Physiol (1997) 82(2):571–6.
128. Selkirk GA, McLellan TM, Wright HE, Rhind SG. Mild endotoxemia, NF-kappaB translocation, and cytokine increase during exertional heat stress in trained and untrained individuals. Am J Physiol Regul Integr Comp Physiol (2008) 295(2):R611–23. doi:10.1152/ajpregu.00917.2007
129. de Oliveira EP, Burini RC, Jeukendrup A. Gastrointestinal complaints during exercise: prevalence, etiology, and nutritional recommendations. Sports Med (2014) 44(Suppl 1):S79–85. doi:10.1007/s40279-014-0153-2
130. Hart EC, Charkoudian N. Sympathetic neural regulation of blood pressure: influences of sex and aging. Physiology (2014) 29(1):8–15. doi:10.1152/physiol.00031.2013
131. Ghosh S, Lertwattanarak R, Garduno JD, Galeana JJ, Li J, Zamarripa F, et al. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J Gerontol A Biol Sci Med Sci (2014) 70(2):232–46. doi:10.1093/gerona/glu067
132. Lassenius MI, Pietilainen KH, Kaartinen K, Pussinen PJ, Syrjanen J, Forsblom C, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care (2011) 34(8):1809–15. doi:10.2337/dc10-2197
133. Moreno-Navarrete JM, Ortega F, Serino M, Luche E, Waget A, Pardo G, et al. Circulating lipopolysaccharide-binding protein (LBP) as a marker of obesity-related insulin resistance. Int J Obes (2012) 36(11):1442–9. doi:10.1038/ijo.2011.256
134. Romani J, Caixas A, Escote X, Carrascosa JM, Ribera M, Rigla M, et al. Lipopolysaccharide-binding protein is increased in patients with psoriasis with metabolic syndrome, and correlates with C-reactive protein. Clin Exp Dermatol (2013) 38(1):81–4. doi:10.1111/ced.12007
135. Sun L, Yu Z, Ye X, Zou S, Li H, Yu D, et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care (2010) 33(9):1925–32. doi:10.2337/dc10-0340
136. Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, et al. Susceptibility to gut leakiness: a possible mechanism for endotoxemia in non-alcoholic steatohepatitis. Liver Int (2008) 28(7):1026–33. doi:10.1111/j.1478-3231.2008.01723.x
137. Ruiz AG, Casafont F, Crespo J, Cayon A, Mayorga M, Estebanez A, et al. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes Surg (2007) 17(10):1374–80. doi:10.1007/s11695-007-9243-7
138. Al-Attas OS, Al-Daghri NM, Al-Rubeaan K, da Silva NF, Sabico SL, Kumar S, et al. Changes in endotoxin levels in T2DM subjects on anti-diabetic therapies. Cardiovasc Diabetol (2009) 8:20. doi:10.1186/1475-2840-8-20
139. Creely SJ, McTernan PG, Kusminski CM, Fisher FM, Da Silva NF, Khanolkar M, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab (2007) 292(3):E740–7. doi:10.1152/ajpendo.00302.2006
140. Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care (2011) 34(2):392–7. doi:10.2337/dc10-1676
141. Harte AL, Varma MC, Tripathi G, McGee KC, Al-Daghri NM, Al-Attas OS, et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care (2012) 35(2):375–82. doi:10.2337/dc11-1593
142. Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol (2007) 27(6):1433–9. doi:10.1161/ATVBAHA.106.138743
143. Lepper PM, Schumann C, Triantafilou K, Rasche FM, Schuster T, Frank H, et al. Association of lipopolysaccharide-binding protein and coronary artery disease in men. J Am Coll Cardiol (2007) 50(1):25–31. doi:10.1016/j.jacc.2007.02.070
144. Lepper PM, Kleber ME, Grammer TB, Hoffmann K, Dietz S, Winkelmann BR, et al. Lipopolysaccharide-binding protein (LBP) is associated with total and cardiovascular mortality in individuals with or without stable coronary artery disease – results from the Ludwigshafen risk and cardiovascular health study (LURIC). Atherosclerosis (2011) 219(1):291–7. doi:10.1016/j.atherosclerosis.2011.06.001
145. Serrano M, Moreno-Navarrete JM, Puig J, Moreno M, Guerra E, Ortega F, et al. Serum lipopolysaccharide-binding protein as a marker of atherosclerosis. Atherosclerosis (2013) 230(2):223–7. doi:10.1016/j.atherosclerosis.2013.07.004
146. Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, et al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck study. J Am Coll Cardiol (1999) 34(7):1975–81. doi:10.1016/S0735-1097(99)00448-9
147. Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet (1999) 353(9167):1838–42. doi:10.1016/S0140-6736(98)09286-1
148. Pastor Rojo O, Lopez San Roman A, Albeniz Arbizu E, de la Hera Martinez A, Ripoll Sevillano E, Albillos Martinez A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis (2007) 13(3):269–77. doi:10.1002/ibd.20019
149. Funderburg NT, Stubblefield Park SR, Sung HC, Hardy G, Clagett B, Ignatz-Hoover J, et al. Circulating CD4(+) and CD8(+) T cells are activated in inflammatory bowel disease and are associated with plasma markers of inflammation. Immunology (2013) 140(1):87–97. doi:10.1111/imm.12114
150. Gardiner KR, Halliday MI, Barclay GR, Milne L, Brown D, Stephens S, et al. Significance of systemic endotoxemia in inflammatory bowel disease. Gut (1995) 36(6):897–901. doi:10.1136/gut.36.6.897
151. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One (2011) 6(12):e28032. doi:10.1371/journal.pone.0028032
152. Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, et al. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol (2009) 206(1–2):121–4. doi:10.1016/j.jneuroim.2008.09.017
153. Sparks Stein P, Steffen MJ, Smith C, Jicha G, Ebersole JL, Abner E, et al. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer Dement (2012) 8(3):196–203. doi:10.1016/j.jalz.2011.04.006
154. Licht CM, de Geus EJ, Penninx BW. Dysregulation of the autonomic nervous system predicts the development of the metabolic syndrome. J Clin Endocrinol Metab (2013) 98(6):2484–93. doi:10.1210/jc.2012-3104
155. Kazakou P, Kyriazopoulou V, Michalaki M, Ierodiakonou V, Psyrogiannis A, Habeos I. Activated hypothalamic pituitary adrenal axis in patients with metabolic syndrome. Horm Metab Res (2012) 44(11):839–44. doi:10.1055/s-0032-1311632
156. Garg M, Angus PW, Burrell LM, Herath C, Gibson PR, Lubel JS. Review article: the pathophysiological roles of the renin-angiotensin system in the gastrointestinal tract. Aliment Pharmacol Ther (2012) 35(4):414–28. doi:10.1111/j.1365-2036.2011.04971.x
157. Mawdsley JE, Rampton DS. Psychological stress in IBD: new insights into pathogenic and therapeutic implications. Gut (2005) 54(10):1481–91. doi:10.1136/gut.2005.064261
158. Caradonna L, Amati L, Magrone T, Pellegrino NM, Jirillo E, Caccavo D. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and clinical significance. J Endotoxin Res (2000) 6(3):205–14. doi:10.1177/09680519000060030101
Keywords: endotoxemia, endotoxin, inflammation, intestinal permeability, lipopolysaccharide, stress, tight junction
Citation: de Punder K and Pruimboom L (2015) Stress induces endotoxemia and low-grade inflammation by increasing barrier permeability. Front. Immunol. 6:223. doi: 10.3389/fimmu.2015.00223
Received: 13 February 2015; Accepted: 24 April 2015;
Published: 15 May 2015
Edited by:
Heiko Mühl, University Hospital Goethe University Frankfurt, GermanyReviewed by:
Giamila Fantuzzi, University of Illinois at Chicago, USAFulvio D’Acquisto, Queen Mary University of London, UK
Kottarappat N. Dileepan, The University of Kansas Medical Center, USA
Copyright: © 2015 de Punder and Pruimboom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin de Punder, Charité University Medicine Berlin, Hufelandweg 14, 10117 Berlin, Germany,a2FyaW4uZGUtcHVuZGVyQGNoYXJpdGUuZGU=