 Lilly Y. W. Bourguignon
Lilly Y. W. Bourguignon Daniel Bikle
Daniel Bikle- Endocrine Unit (111N2), Department of Medicine, San Francisco Veterans Affairs Medical Center, University of California at San Francisco, San Francisco, CA, USA
Hyaluronan (HA), the major extracellular matrix component, is often anchored to CD44, a family of structurally/functionally important cell surface receptors. Recent results indicate that UV irradiation (UVR)-induced cutaneous squamous cell carcinomas (SCC) overexpress a variety of CD44 variant isoforms (CD44v), with different CD44v isoforms appear to confer malignant SCC properties. UVR also stimulates HA degradation in epidermal keratinocytes. Both large HA polymers and their UVR-induced catabolic products (small HA) selectively activate CD44-mediated cellular signaling in normal keratinocytes and SCC cells, with all of the downstream processes being mediated by RhoGTPases (e.g., Rac1 and Rho). Importantly, we found that the hormonally active form of vitamin D 1,25(OH)2D3 not only prevents the UVR-induced small HA activation of abnormal keratinocyte behavior and SCC progression, but also enhances large HA stimulation of normal keratinocyte activities and epidermal function(s). The aim of this hypothesis and theory article is to question whether matrix HA and its UVR-induced catabolic products (e.g., large and small HA) can selectively activate CD44-mediated cellular signaling such as GTPase (Rac and RhA) activation. We suggested that large HA–CD44 interaction promotes Rac-signaling and normal keratinocyte differentiation (lipid synthesis), DNA repair, and keratinocyte survival function. Conversely, small HA–CD44 interaction stimulates RhoA activation, NFκB/Stat-3 signaling, and miR-21 production, resulting in inflammation and proliferation as well as SCC progression. We also question whether vitamin D treatment displays any effect on small HA–CD44v-mediated RhoA signaling, inflammation, and SCC progression, as well as large HA–CD44-mediated differentiation, DNA repair, keratinocyte survival, and normal keratinocyte function. In addition, we discussed that the topical application of signaling perturbation agents (e.g., Y27623, a ROK inhibitor) may be used to treat certain skin diseases displaying upregulation of keratinocyte proliferation such as psoriasis and actinic keratoses in order to correct the imbalance between Rac and RhoA signaling during various UV irradiation-induced skin diseases in patients. Finally, we proposed that matrix HA/CD44-signaling strategies and matrix HA (HAS vs. HAL or HAS → HAL)-based therapeutic approaches (together with vitamin D) may be used for the treatment of patients suffering a number of UV irradiation-induced skin diseases (e.g., inflammation, skin cancer, and chronic non-healing wounds).
Introduction
The incidence of skin cancer has been increasing rapidly over recent decades. UVB radiation (UVR)-induced squamous cell carcinomas (SCC) is the second most common cancer among Caucasians in the United States, contributing substantially to morbidity among elderly people. Recent studies report that the age-adjusted incidence of SCC has grown by 50–200% over the past 10–30 years. Invasive SCC has the potential to recur locally, tending to invade deeply into subcutaneous structures, fascia, and muscle; and to metastasize, most commonly to regional lymph nodes (1, 2). Several lines of evidence indicate that matrix hyaluronan (HA) plays an important role in regulating inflammation, proliferation, and migration/invasion in the progression of a variety of tumors (3–7). Because little is known about the molecular basis underlying HA effects on influencing skin cancer development, there is currently a need to investigate some key aspects of HA signaling in regulating UVR-induced human SCC progression.
Normal keratinocytes and healthy skin tissues express predominantly one large species of the transmembrane glycoprotein, CD44 (Epican), required for many keratinocyte functions (3, 4). However, UVR-induced cutaneous SCC cells and tissues overexpress a variety of variant isoforms of CD44 (3–9). Different CD44 variant (CD44v) isoforms appear to confer the malignant properties of increased tumor cell growth and cancer progression (5–10). CD44v isoforms bind a number of extracellular matrix (ECM) ligands (e.g., HA), and are known to participate in a variety of both normal keratinocyte and SCC functions (3–10). Our recent results indicate that UVB stimulates HA degradation in normal keratinocytes. Both large HA polymers and their smaller catabolic products selectively activate CD44 isoform-mediated cellular signaling that regulates inflammation, anti-apoptosis, and tumor cell growth, as well as differentiation, DNA repair, and keratinocyte survival function.
Skin Cancer
Recent studies report that the age-adjusted incidence of skin cancer has grown by 50–200% over the past 10–30 years. Exposure to (UVR has well-recognized clinical effects on the skin, including sunburn (inflammation) and keratinocyte transformation, leading to neoplasia and SCC progression. In particular, skin type determines sensitivity to the acute and chronic effects of UVR on the skin. Today, highly aggressive variants of SCC are frequently seen in organ-transplant recipients, as well as in patients who are on immunosuppressive medications or have immunocompromised status for other reasons (1, 2). Human papillomavirus infection has also been associated with some types of cutaneous SCC. Invasive SCC has the potential to recur locally, tending to invade deeply into subcutaneous structures, fascia, and muscle; and to metastasize, primarily to the regional lymph nodes (1, 2). Because little is known about the molecular basis underlying the progression to the invasive phenotype, it is very difficult to predict individual tumor aggressiveness and design effective treatment plans. Thus, there is currently a real need to clarify aspects of tumor biology underlying the clinical behavior of SCC. It is well-known that the tumor-specific phenotype (characteristics such as inflammation, anti-apoptosis, and tumor cell proliferation) is linked to oncogenic signaling. Dissection of the transmembrane pathways controlling cellular signaling and tumor functions should significantly aid in understanding the intracellular events underlying SCC progression.
Hyaluronan and CD44 in Normal Keratinocytes and Squamous Cell Carcinomas
Hyaluronan, the major glycosaminoglycan of ECM component, serves not only as a primary constituent of connective tissue extracellular matrices but also as a bio-regulatory molecule (1, 2). Many studies indicate that HA is also abundant in stratified squamous epithelia, including mammalian epidermis, and that it influences epidermal functions such as skin integrity (10, 11). However, the mechanisms by which HA stimulates keratinocyte functions and regulates tissue integrity are not well understood. HA is synthesized by several HA synthases (12), and its size further modified by hyaluronidases (13). Generally, small size-HA (1 × 105–1 × 104 Da) induces the expression of proinflammatory cytokine/chemokine and proliferative genes as well as cell proliferation and migration; whereas large size-HA (>1 × 106 Da) promotes transcriptional activation and differentiation (14–20). UVR-induced changes in HA production and degradation/fragmentation have also been reported (19, 20). Specifically, while large HA appears to predominate in normal mouse skin, small HA becomes prevalent in tumor tissues (19) and UVR-induced keratinocytes (20). All of these observations are consistent with our hypothesis that both HA production and HA size modifications underline UVR-induced changes associated with onset of keratinocyte transformation.
Both large and small HAs are capable of binding to CD44 (21), which is an ubiquitous, abundant, and functionally important receptor expressed on the surface of many cells, including normal and transformed keratinocytes (23). CD44 is encoded by a single gene which contains 19 exons (22). The most common form, CD44s (CD44 standard form), contains exons 1–5 (N-terminal 150 a.a.), exons 15 and 16 (membrane proximal 85 a.a.), exon 17 (transmembrane domain), and a portion of exons 17 and 19 (cytoplasmic tail, 70 a.a.). Out of the 19 exons, 12 exons can be alternatively spliced. Most often, the alternative splicing occurs between exons 5 and 15 leading to an insertion in tandem of one or more variant exons (exon 6–exon 14; v1–v10) within the membrane proximal region of the extracellular domain (31). For example, in keratinocytes, additional exons v3–v10 are inserted into the CD44s transcripts, and this isoform has been designated as CD44v3-10 (23). Various skin cancer cells and tissues express different CD44 variant (CD44v) isoforms (e.g., CD44v3 and CD44v10) in addition to CD44s and CD44v3-10 (23). These CD44 isoforms have the same amino acid sequences at the two ends of the molecule, but differ in the middle region (v3–v10) within the CD44 membrane proximal domain located at the external side of the membrane (23). Several lines of evidence clearly indicate that CD44 selects unique downstream effectors and coordinates certain downstream, intracellular signaling pathways that influence multiple cellular functions (23).
Hypothesis
In this article, we described the hypothesis that HA and its UVR-induced catabolic products (e.g., large and small HA) selectively activate CD44-mediated Rac and RhoA signaling. Specifically, large HA–CD44 interaction promotes Rac/PKNγ and p38MAPK-dependent normal keratinocyte differentiation (lipid synthesis), DNA repair, and keratinocyte survival function. Conversely, small HA–CD44v isoform interaction stimulates RhoA/ROK-dependent NFκB/Stat-3 signaling and miR-21 production, resulting in inflammation and proliferation as well as SCC progression. Vitamin D treatment inhibits small HA–CD44v-mediated RhoA/ROK signaling, inflammation, and SCC progression, and it also enhances large HA–CD44-mediated differentiation, DNA repair, keratinocyte survival, and normal keratinocyte function. The results of this study will definitely provide a better understanding of the cellular and molecular mechanisms involved in normal keratinocyte-mediated epidermal function and UVR-induced human SCC progression, as well as vitamin D effects on reducing UVR-induced keratinocyte transformation and skin cancer.
Experimental Evidence
Detection of CD44v Isoform Expression in Normal Keratinocytes, SCC Cells, and Skin SCC Tissues
Invasive SCC induced by UVR has the potential to recur locally, tending to invade deeply into subcutaneous structures, fascia, and muscle, and to metastasize, preferably to the regional lymph nodes (1, 2). At the present time, very limited information is available regarding the factors responsible for the onset and progression of skin SCC. One promising candidate in this regard is the CD44 molecule. Our data using anti-CD44 antibody (recognizing the common domain of all CD44 isoforms) confirm that undifferentiated keratinocytes express various CD44 isoforms in abundance (Figure 1-low calcium-treated sample). However, following high Ca2+-induced keratinocyte differentiation, the lower molecular weight forms cease to be expressed leaving Epican as the dominant species (Figure 1). CD44v isoforms are overexpressed in cutaneous SCC cell lines as compared to normal keratinocytes (Figure 1). Furthermore, immunohistochemical staining using anti-CD44v3 antibody and anti-CD44v6 antibody confirms that both CD44v3 and CD44v6 isoforms are overexpressed in human cutaneous SCC tissue (Figure 2). Only a very small amount of CD44v isoforms is detected in normal skin tissue (Figure 2). CD44v3 overexpression can also be detected in mouse skin following chronic (to a lesser extent acute) UVB exposure (Figure 2). These findings clearly indicate a strong correlation between CD44v isoform expression and UVB-induced skin SCC progression.
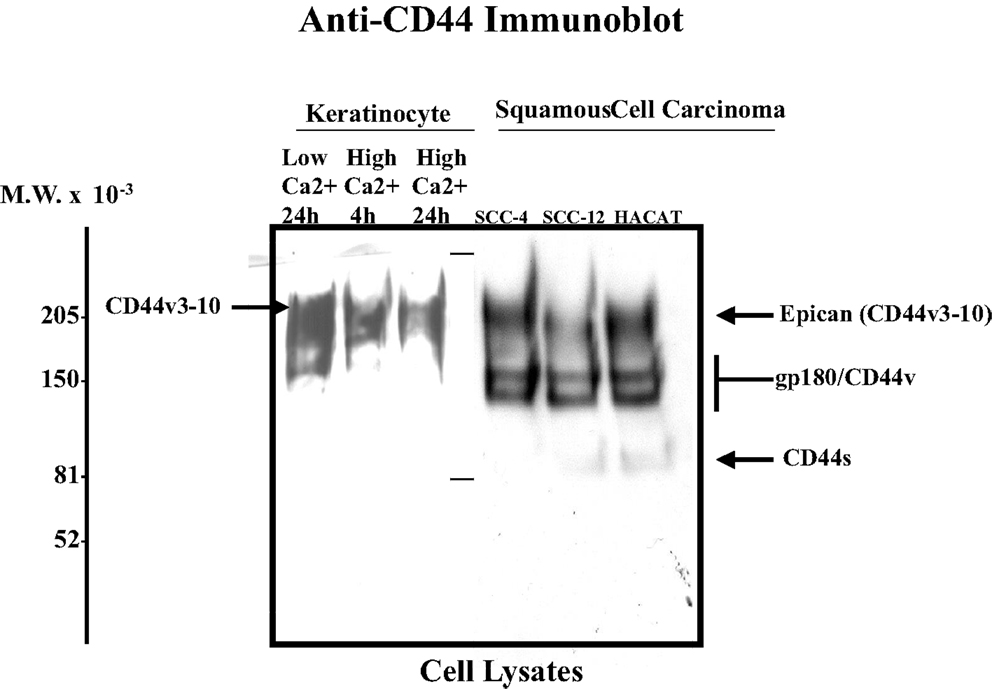
Figure 1. Detection of CD44 isoform expression in normal keratinocytes and SCC cell lines using anti-CD44-mediated immunoblotting techniques [SCC-4, SCC-12 cell lines, and HACAT (a transformed cell line) Epican is designated as a CD44v3-10 form; anti-CD44-mediated immunoblot was used to detect CD44 signal as indicated].
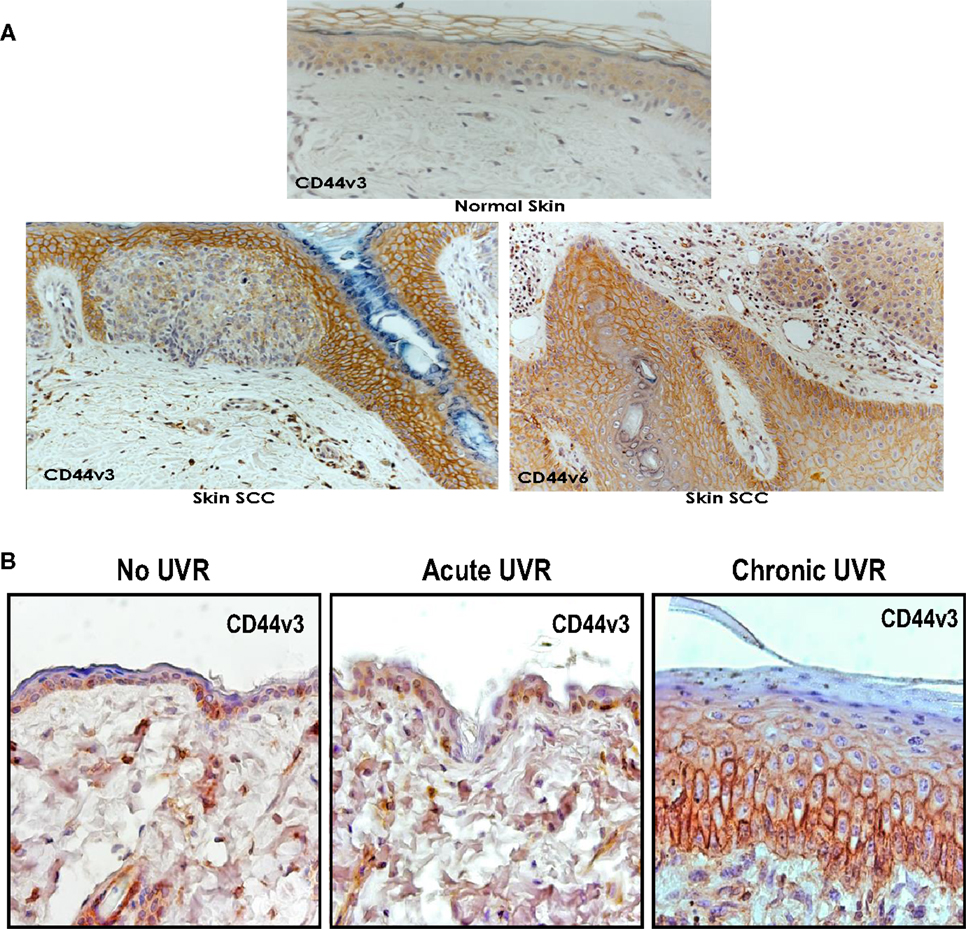
Figure 2. (A) Immunoperoxidase staining of CD44v3 and CD44v6 isoforms in normal and SCC tissues of human skin; and (B) immunoperoxidase staining of CD44v3 isoform in mouse skin following acute or chronic UVR exposure.
UVB-Induced Hyaluronan Fragmentation and Abnormal Keratinocyte Functions
Hyaluronan belongs to one of the major glycosaminoglycan polysaccharide families and plays an important role in the formation/remodeling of ECM, specifically in maintaining the integrity of skin tissues (17, 18). Our data show that HA in normal epidermis (without UVB treatment) exists as a large polymer of approximately 1–2 × 106 Da molecular weight (“large HA”) (Figure 3). These large HA polymers are degraded into smaller HA units (1 × 105–1 × 103 Da; “small HA”) following UVB treatment (Figure 3).
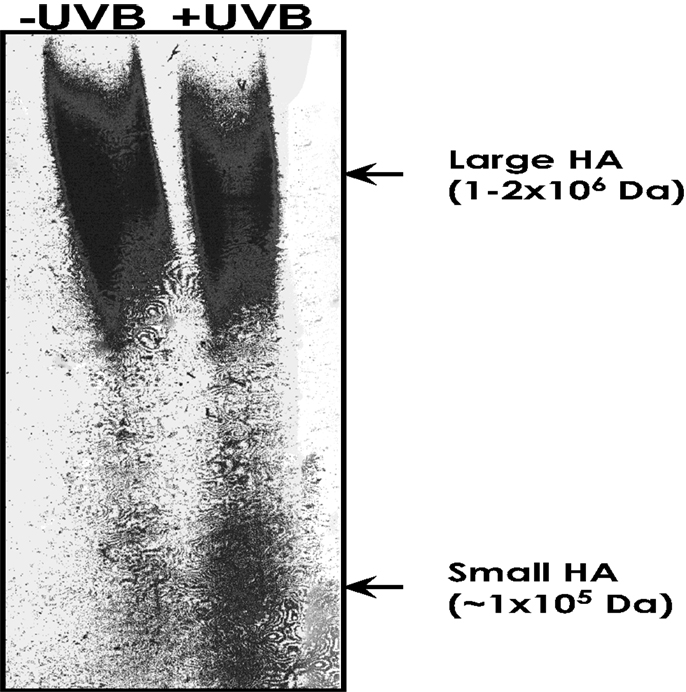
Figure 3. Measurement of HA-size distribution in untreated mouse epidermis vs. acute UVB-treated mouse epidermis. HA from untreated mouse epidermis vs. acute UVB-treated mouse epidermis was isolated and purified using gel filtration column chromatography, and then analyzed by an enzyme-linked binding protein assay, that uses microwells coated with a highly specific HA binding protein (HABP) to capture HA, and an enzyme-conjugated HABP using a commercially available enzyme-linked immunosorbent assay (ELISA)-type test kit. HA was also be analyzed by 0.5% agarose gel followed by Alcian blue and silver staining.
Both large and small HAs are capable of binding to CD44 – a ubiquitous, abundant, and functionally important receptor expressed on the surface of many cells including normal keratinocytes and SCC (Figure 2). Generally, small HA–CD44 interaction activates pro-inflammatory cytokine/chemokine gene expression/production, proliferation, and migration, whereas large HA–CD44 binding promotes certain transcriptional activation and differentiation (19, 20). HA–CD44 interaction induces intracellular RhoGTPase (RhoA and Rac) signaling cascades that regulate cytoskeletal reorganization and cell migration (18). Whether HA/CD44 and RhoGTPase signaling actually cause keratinocyte functions and SCC activation is not presently known, and is addressed below.
HA/CD44-Mediated RhoA and Rac1 Signaling in Cytoskeleton Activation and Keratinocyte and SCC Functions
Members of the Rho subclass of the Ras superfamily [small molecular weight GTPases (e.g., RhoA, Rac1, and Cdc42)] act as molecular switches that alternate between GTP- and GDP-bound states. The “activated” GTP-bound enzymes preferentially interact with downstream effector molecules that modulate other effector activities (24). For example, activation of RhoA and Rac1 signaling has been shown to regulate cytoskeleton-associated functions in normal and transformed keratinocytes (SCC cells) (19, 20).
RhoA-Activated Rho-Kinase Signaling Events
Previous work indicated that HA (mixed sizes) promotes the interaction between CD44 and several Rho-specific guanine nucleotide exchange factors [e.g., p115RhoGEF (25) and LARG (26)], thereby up-regulating RhoA (a member of the Rho subclass of the Ras superfamily), leading to altered cytoskeleton-mediated cell functions (25, 26). For example, activation of RhoA signaling has been shown to be involved in cytoskeleton-associated functions. Several enzymes have been identified as possible downstream targets for RhoGTPases (e.g., RhoA) in regulating cytoskeleton-mediated cell motility (27–38). One such enzyme is Rho-Kinase (ROK, also called Rho-binding kinase), which is a serine–threonine kinase (27–38). ROK interacts with RhoA in a GTP-dependent manner, and phosphorylates a number of cellular substrates including myosin light chain phosphatase, adducin, and LIM kinase (27–38). HA–CD44 interaction promotes RhoA-ROK activation in a number of tumor cells (27–38). Structurally, ROK is composed of a catalytic (CAT), a coiled-coil, a Rho-binding (RB), and a pleckstrin-homology (PH) domain (27–38). Overexpression of either the RB domain or the PH domains (dominant-negative forms) of ROK by transfecting cells with RB cDNA or PH cDNA blocks HA/CD44-specific phenotypic changes (27–38). Also, inhibition of RhoA-activated ROK by Y27632 treatment effectively blocks the HA/CD44-induced cellular signaling and functions (31). These findings suggest that selective activation of CD44 signaling (via small size- vs. large size-HA) induces different RhoA–ROK pathway-specific effects in normal keratinocytes and/or SCC.
Rac1-Activated PKNγ Signaling Events
HA (mixed sizes) also promotes the interaction between CD44 and several Rac1-specific guanine nucleotide exchange factors [e.g., Tiam1 (32) and Vav2 (33)], thereby up-regulating Rac1 (another member of the Rho subclass of the Ras superfamily), leading to altered cytoskeleton-mediated cell functions (32, 33). Rac1 signaling has been shown to play an important role in promoting epidermal stem cells to undergo self-renewal and subsequent terminal differentiation (34). A number of enzymes have been identified as possible downstream effectors for Rac1 signaling. One such enzyme is protein kinase N-γ (PKNγ) (also called PRK2), which belongs to a family of serine–threonine kinases known to interact with Rac1 in a GTP-dependent manner. It also shares a great deal of sequence homology with protein kinase C in the C-terminal region (35–38). The N-terminal region of PKN contains three homologous sequences of approximately 70 aa (relatively rich in charged residues), which form an antiparallel coiled-coil fold (ACC domain) (35–38). This ACC domain has been shown to interact with RhoGTPases such as RhoA and Rac1 (and to a lesser extent with Cdc42) (35–38). The C-terminal region contains the C2-like region, which functions as an auto-inhibitory domain (35–38). Both the ACC and the C2-like domains, together with the catalytic domain, are conserved among the PKN family members (35–38). In keratinocytes, RhoA-activated PKNγ has been found to be involved in Fyn/Src kinase-regulated cell–cell adhesion during Ca2+-induced differentiation (39). Our results indicate that large HA specifically promotes CD44-mediated Rac1-PKNγ kinase signaling (3) and the activation of downstream effectors, including p38MAPK, AP-1 protein (c-Jun), and p63 (Figure 4). This pathway, in turn, regulates keratinocyte differentiation.
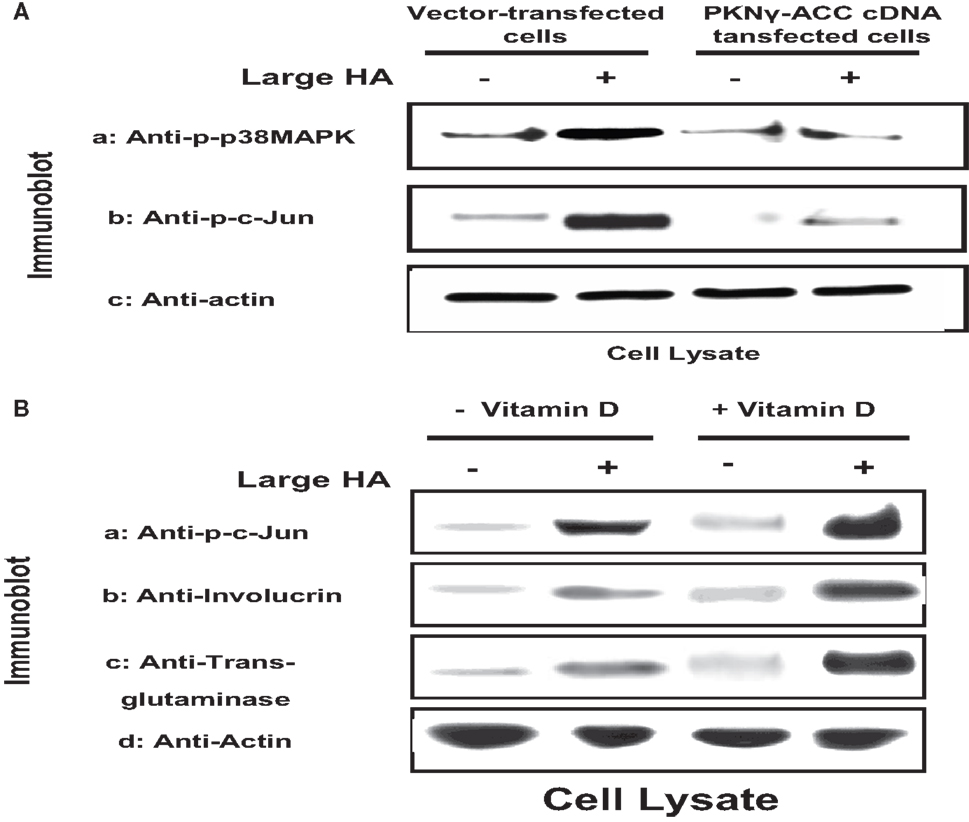
Figure 4. (A) Immunoblot analyses of large HA and PKN-dependent phosphorylation of p38MAPK and AP-1 proteins (e.g., c-Jun) [the PKN-dependent phosphorylation is proven by the effect of PKNγ-ACC cDNA dominant negative mutant], and (B) immunoblot analyses of large HA and vitamin D-induced c-Jun phosphorylation and differentiation marker (involucrin and transglutaminase) expression in cultured keratinocytes.
Hyaluronan, CD44, and RhoGTPases in Normal Keratinocytes and SCC
HA-mediated CD44 signaling has been shown to play an important role in various cellular functions. Our preliminary data indicate that small HA and large HA binding to cultured keratinocytes and SCC-12 cells selectively activates RhoA and Rac1 (Table 1). In particular, small HA (but not large HA) promotes RhoA signaling in cultured SCC-12 cells (to a lesser extent normal keratinocytes) (Table 1). Further analyses indicate that small HA-activated RhoA–ROK is capable of phosphorylating NFκB-p65 (Table 2), PKCε (Table 3), and Stat-3 (Table 3), leading to NFκB-specific (Table 2) and Stat-3-specific transcriptional activation (Table 3), cytokine (IL-6)/chemokine [monocyte-chemoattractant protein-1 (MCP-1)] gene expression (Table 2), and cell proliferation (Table 2). The fact that the ROK inhibitor, Y27632, significantly blocks small HA-induced RhoA–ROK signaling and SCC functions (Tables 2 and 3) suggests that small HA-mediated RhoA–ROK activation plays an important role in regulating oncogenic signaling events and SCC functions.
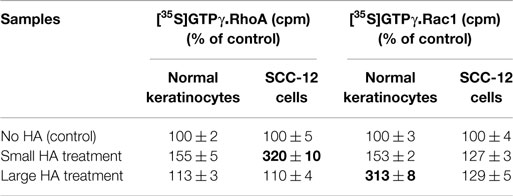
Table 1. HA-induced RhoA and Rac1 activation in cultured keratinocytes and SCC12 cells.
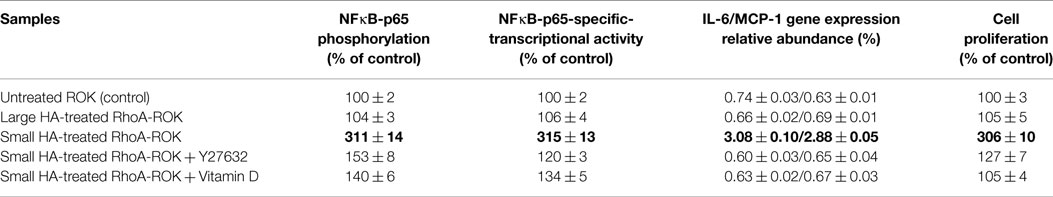
Table 2. Small HA-mediated RhoA–ROK activation of NFκB-p65 and in cultured SCC-12 cells.
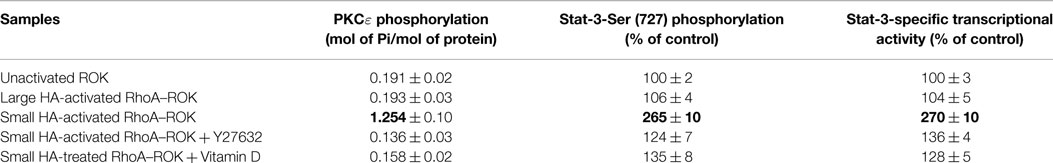
Table 3. HA-mediated RhoA–ROK phosphorylation of PKCε and Stat-3 in cultured SCC-12 cells.
We also observed that the addition of large HA stimulates Rac1 activation in normal keratinocytes and to a lesser extent SCC (Table 1). In contrast, only a low level of Rac1 activation was detected in keratinocytes treated with small HA or no HA (Table 1). These findings suggest that large HA directly promotes Rac1 activation in normal keratinocytes. It is noted that the level of p38MAPK phosphorylation and AP-1 protein (c-Jun) phosphorylation is greatly enhanced in keratinocytes treated with large HA (Figure 4). Transfection of keratinocytes with PKNγ-ACCcDNA (but not vector-transfected keratinocytes) (Figure 4) or treatment of keratinocytes with Vitamin D not only inhibits large HA-mediated p38MAPK phosphorylation but also blocks AP-1 protein (c-Jun) phosphorylation in cultured keratinocytes (Figure 4). These results imply that the ACC fragment of PKNγ (similar as Vitamin D) acts as a dominant-negative mutant that downregulates large HA/CD44-induced Rac1-PKNγ activation and p38MAPK/c-Jun signaling required for normal keratinocyte functions. In addition, we have found that large HA promotes the expression of differentiation markers such as involucrin and transglutaminase in normal keratinocytes, and overexpression of PKNγ-ACC domain (or treatment of Vitamin D) also blocks differentiation marker expression as described previously (3). These findings suggest that large HA-mediated Rac1-PKNγ plays an important role in regulating p38MAPK-AP1 signaling and keratinocyte differentiation.
Selective Small HA-Mediated CD44 Isoform Signaling That Modulates Acute/Chronic UVR-Induced Inflammation in SCC, and Determine the Role of Large HA in Inhibiting Small HA/CD44-Mediated Signaling and Thereby Preventing UVR-Induced SCC Progression
There is compelling evidence that RhoA-activated ROK is involved in the regulation of oncogenesis (18). UVR has acute clinical effects on skin including inflammation. Pro-inflammatory cytokines and chemokines such as IL-6, IL-1β, RANTES, and MCP-1 contribute to inflammatory response in UVR-treated skin (40, 41). Our results indicate that upregulation of cytokine (IL-6) and chemokine (MCP-1) can be detected in mouse skin following acute or chronic UVR exposure (Figure 5). These findings clearly indicate a strong correlation between cytokine/chemokine expression and UVR-induced skin cancer.
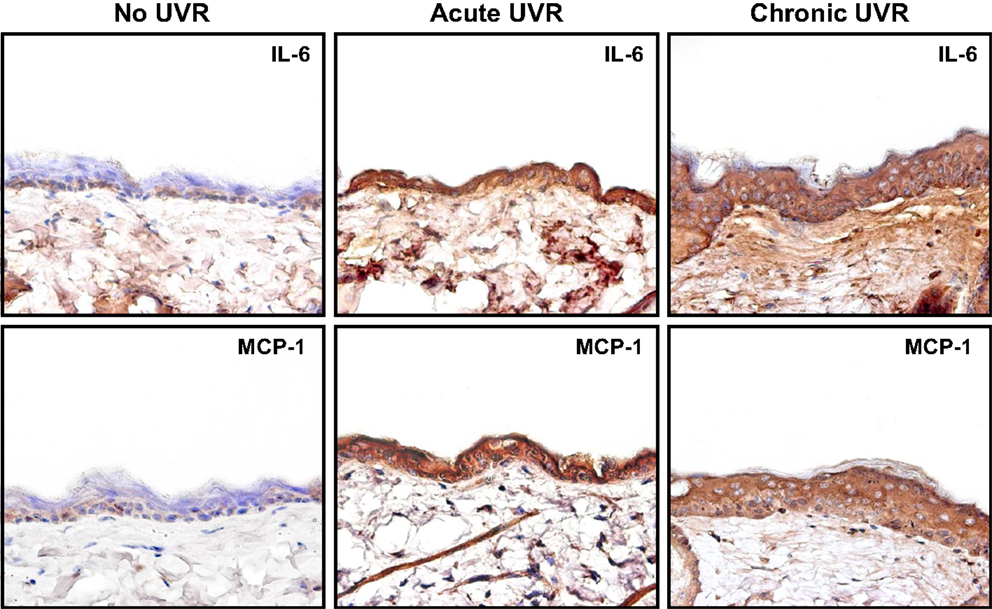
Figure 5. Immunoperoxidase staining of cytokine (IL-6) and chemokine (MCP-1) in mouse skin following acute or chronic UVR exposure.
The transcription factor, NFκB, is known to regulate genes involved in inflammatory responses (42). NFκB also controls cell cycle regulatory genes such as cyclin D1 and promotes cellular transformation (43, 44). Small HA (but not large HA)-mediated CD44 isoform interaction has been shown to play an important role in stimulating NFκB-inducing kinase (NIK). Activation of the inhibitor of κB (IκB) kinase (IKK) complex (IKKα and IKKβ) by NIK causes phosphorylation of the IκBs, targeting them for ubiquitylation and degradation by proteasomes, which liberate NFκB-p65 for nuclear translocation and transactivation of a variety of pro-inflammatory gene expression (21). Small HA also stimulates NFκB, which controls proinflammatory genes (e.g., IL-6, IL-1β, RANTES, or MCP-1 for inducing inflammatory responses) and cell cycle regulatory genes (e.g., cyclin D1 or c-Myc for activating cell proliferation) (42–44). A recent study indicates that RhoA signaling plays a role in activating NFκB signaling (45).
UVR is also known to promote PKCε overexpression (46) and Stat-3 phosphorylation, leading to skin SCC development (47). Our preliminary data indicate that small HA promotes ROK phosphorylation of PKCε and Stat-3, which in turn stimulates Stat-3-specific transcriptional activities (Figure 6; Table 3) in SCC-12 cells. Large HA exerts direct anti-inflammatory and anti-proliferation effects on UVR-induced skin cancers. Whether large HA prevents small HA-mediated RhoA–ROK activation and NFκB-p65 signaling, as well as PKCε-Stat-3 function in transformed keratinocytes/SCC, and reverses small HA/CD44v-mediated tumor-specific behaviors (e.g., pro-inflammatory gene expression, cytokine/chemokine production, and proliferation) in transformed keratinocytes and SCC will be investigated in the future.
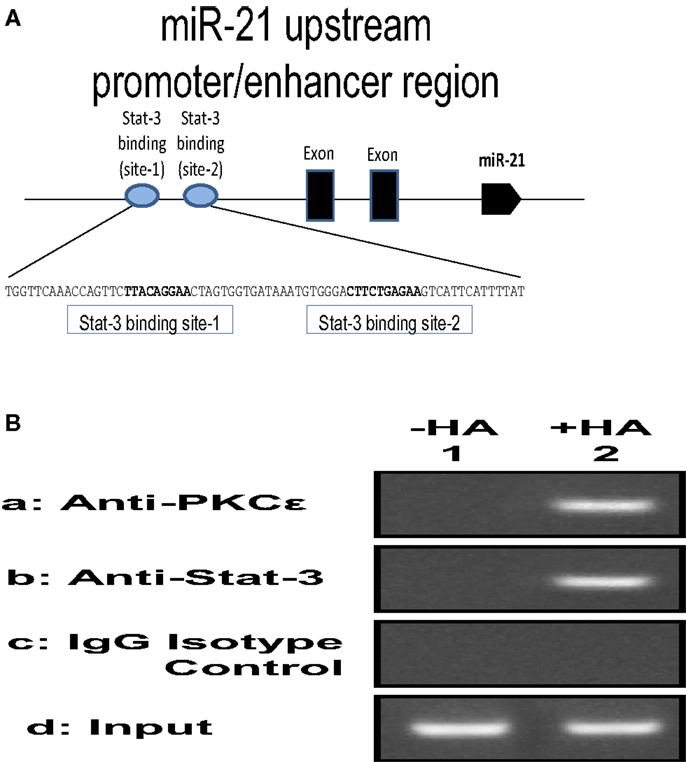
Figure 6. (A) Illustration of 130-bp regions containing two predicted Stat-3 binding sites (site 1 and site 2) upstream of the miR-21 genes; (B) ChIP assay of SCC-12 cells treated with no HA (lane 1) or with small HA (lane 2) using anti-PKCε, anti-Stat-3, or IgG control. Co-immunoprecipitated DNA was amplified by PCR with primers specific for the miR-21 upstream promoter enhancer.
MicroRNA-21 (miR-21) Production and Vitamin D 1,25(OH)2D3 Effect on Mouse Skin Following Acute and/or Chronic UVR Exposure
Dysregulation of microRNAs (miRNA) is observed in many cancers including skin cancer. In particular, miR-21 appears to play an important role in tumor cell growth and SCC progression. Mature miRNA (e.g., miR-21) are detected in UVB-treated cells and skin carcinogenesis. In this study, we investigated the regulation and expression of miR-21 in mouse skin following acute and/or chronic UVR exposure as described below:
Identify HA/CD44-Mediated PKCε-Stat-3-Specific Target Genes
The mechanism of cellular transformation in keratinocytes and SCC is likely to be the results of several genes that are transcriptionally controlled by the PKCε-Stat-3 interaction. Previous studies indicate that activated Stat-3 up-regulates the mRNA levels of many genes, including miR-21. The expression of mature miR-21 is detected in various SCC cell lines and patient specimens. Many studies indicate that miR-21 may function as an oncogene and play a role in anti-apoptosis and tumorigenesis. A recent study indicates that the gene encoding oncogenic miR-21 is regulated by an upstream promoter/enhancer containing two Stat-3 binding sites (as diagramed in Figure 6). In order to investigate whether HA–CD44-induced PKCε-Stat-3 complex is involved in the regulation of miR-21 expression in normal/transformed human keratinocytes (SCC 12F2 and SCC12B2) or UVB-treated murine keratinocytes (from epidermal sheets), we conducted chromatin immunoprecipitation (ChIP) reporter gene assays as described below.
Chromatin Immunoprecipitation Assays
To examine whether the PKCε-Stat-3 complex directly interacts with the upstream promoter/enhancer region of miR-21, ChIP assays were performed in normal/transformed human keratinocytes (SCC12F2 and SCC12B2) or UVB-treated murine keratinocytes (from epidermal sheets) in the presence or absence of small/large HA (50 μg/ml) for 2 h at 37°C using a kit (EZ ChIP) from Millipore Corp., according to the manufacture’s instructions. Cross-linked chromatin lysates were sonicated and diluted with ChIP sonication buffer plus protease inhibitors, divided, and incubated with normal rabbit IgG or rabbit anti-PKCε antibody or rabbit anti-Stat-3 antibody at 4°C overnight, then precipitated with protein G-agarose. Cross-linking will be reversed by overnight 65°C incubation; DNA fragments then will be extracted with PCR purification kit, analyzed by PCR, and quantitated by real-time PCR using primer pair specific for the miR-21 upstream promoter/enhancer region containing the Stat-3 binding sites: forward primer: 5′-CTGGGAGAAACCAAGAGCTG-3′ and reverse primer:AGGGGACAAGTCAGAGAGAGG-3′ on an agarose gel as described previously (48). Results from these proposed experiments will allow us to verify the direct involvement of PKCε and/or Stat-3 in regulating the expression of specific target genes such as miR-21 in UVB-treated normal and transformed keratinocytes. Preliminary data indicate that PKCε and Stat-3 are recruited to the miR-21 upstream promoter enhancer region in SCC-12 cells treated with small HA (Figure 6).
Effect of MicroRNA-21 (miR-21) Production on Mouse Skin Following Acute and/or Chronic UVR Exposure
Our results using RNase protection assays (Figure 7) show that the level of miR-21 production is significantly elevated in cultured keratinocytes treated with UVB (Figure 7, lane 2) as compared to those cells without UVB treatment (Figure 7, lane 1). The expression of Programed Cell Death 4 (PDCD4) gene is strongly induced during apoptosis in a number of cell types (49–51). PDCD4 encodes a tumor suppressor protein whose expression is lost in progressed carcinomas of many solid tumors (49–51). PDCD4 has been identified as one of the tumor suppressor genes regulated by miR-21 (48, 52, 53). It inhibits translation of PDCD4 by forming a complex with the translation initiation factor eIF4A (an RNA helicase) (54–56).
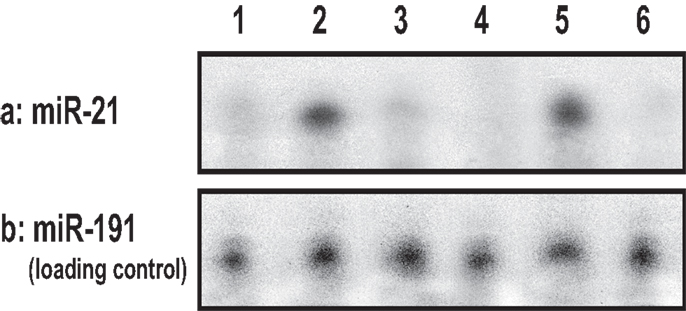
Figure 7. UVB-induced miR-21 production using RNase protection assay in cultured keratinocytes [treated with no UVB (lane 1) or with UVB (lane 2) or treated with ROK inhibitor (Y27632) plus UVB (lane 3) or treated with UVB and vitamin D (lane 4)], and in UVB-induced SCC tumor tissues (lane 5) or normal skin tissues (lane 6).
The “inhibitors of apoptosis family of proteins” (IAPs) constitute a family of at least nine proteins including survivin that block apoptosis by direct binding to caspases (57). Overexpression of IAPs (e.g., survivin) is thought to be linked to chemoresistance by suppressing apoptosis (57, 58). Our data indicate that HA–CD44-mediated Stat-3 activation induces up-regulation of survival proteins (surviving) expression leading to anti-apoptosis and multidrug resistance in epithelial tumor cells. The fact that downregulation of either PKCε or Stat-3 by treating SCC-12 cells with PKCε siRNA or Stat-3 siRNA blocks HA/CD44-mediated cyclin D1/survivin expression suggests that both PKCε and Stat-3 play important roles in the expression of all these proteins.
Vitamin D 1,25(OH)2D3 Effect on Mouse Skin Following Acute and/or Chronic UVR Exposure
Vitamin D 1,25(OH)2D3 and its analogs exert direct anti-inflammatory and anti-proliferation effects on UVR-induced skin cancers (59–62). A previous study indicated that one of the hormonally active vitamin D analogs (BXL-628) inhibits ROK membrane localization and activation in tumor cells (63). Recently, a study also indicates that vitamin D [via vitamin D receptor (VDR) action] was shown to abrogate the ability of NFκB-p65 to transactivate gene transcription and to block cytokine/chemokine gene expression as well as subsequent inflammatory responses and drug resistance in epithelial cells (64, 65). Our data indicate that 1,25(OH)2D3 (similar as large HA) (65) prevents small HA-mediated RhoA–ROK activation and NFκB-p65 signaling, as well as PKCε-Stat-3 function in transformed keratinocytes/SCC, and reverses small HA/CD44v-mediated tumor-specific behaviors (e.g., pro-inflammatory gene expression, cytokine/chemokine production, and proliferation) in transformed keratinocytes and SCC (Tables 2 and 3).
In addition, we found that cultured keratinocytes treated with either ROK inhibitor (Y27632) (Figure 7, lane 3) or vitamin D (Figure 7, lane 4) or normal skin tissues (Figure 7, lane 6) contain significantly less UVB-induced miR-21 [as compared with UVB-induced SCC tumor tissues (Figure 7, lane 5)]. The expression of miR-21 occurs in SCC tumor tissues (Figure 8) (but not in normal skin tissues) (Figure 8). Using digoxigenin (DIG)-labeled miR-21 probe (LNA probe) (and scrambled probe) and in situ hybridization [incubated with an anti-DIG-AP (alkaline phosphatase)-FAB fragment plus NBT/BCIP], we have detected an upregulation of miR-21 in VDR-null mouse skin (to a lesser extent wild-type mouse skin) following chronic UVR exposure. In contrast, very little miR-21 is detected in either VDR-null mice or wild-type mice following acute UVR or no UVR treatment (Figure 8). These findings suggest that VDR has a protective role to attenuate miR-21 production, and downregulation of VDR enhances miR-21 expression leading to tumor cell growth and skin SCC progression. These observations strongly suggest that UVB promotes miR-21 production in both cultured keratinocytes and SCC tumors. Our observations also support our hypothesis that vitamin D (together with ROK inhibition) may serve as a protective agent to impair UVB-induced miR-21 production and skin cancer progression.
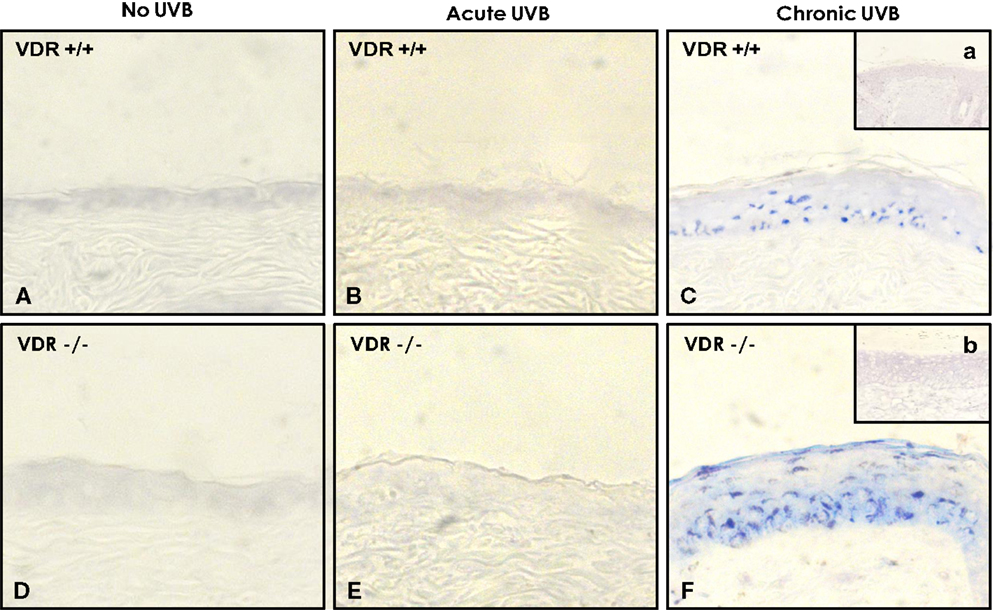
Figure 8. Detection of UVB-induced miR-21 expression in wild-type (VDR+/+) and VDR-null (VDR−/−) mouse skin using DIG-labeled miR-21 probe (LNA probe) (A–F) (and scrambled probe) (a & b) and in situ hybridization [incubated with an anti-DIG-AP (alkaline phosphatase)-FAB fragment plus NBT/BCIP].
Determine How Large HA/CD44 Epican Interactions with Rac1-Activated PKNγ Lead to Modulations in Epidermal Functions, DNA Repair, and Keratinocyte Survival in Mouse Epidermis Following Acute or Chronic UVR-Induced Skin Cancer Development
A previous study showed that HA/CD44-mediated Rac1-PKNγ activation plays an important role in regulating keratinocyte differentiation (8). Our recent work indicates that large HA (but not small HA) stimulates keratinocye signaling (Figure 4). The question of how large HA/CD44-induced Rac1-PKNγ signaling pathway mediates keratinocyte differentiation is not well-understood. In mammalian cells, at least three distinct mitogen-activated kinases (MAPKs) [e.g., ERKs (extracellular signal regulated kinases), the stress-responsive JNK/SAPKs, and p38MAP kinases (p38MAPK)] have been characterized (66, 67). There is compelling evidence that activation of p38MAPK occurs in the membrane (68, 69). Activated p38MAPK has been shown to phosphorylate specific transcription factors and proteins including the transcription factor activator proteins (AP-1) (consisting of members of Jun family of nuclear proteins such as c-Jun, JunB, JunD, Fra-1, Fra-2, c-Fos, and FosB) (70–74). Subsequently, phosphorylated AP-1 proteins bind to DNA elements and induce target gene expression (72, 73). In keratinocytes, the promoter activity of several differentiation-related markers (e.g., involucrin and transglutaminase) and cholesterol synthesis-related proteins (e.g., HMG-CoA synthase and HMG-CoA reductase). Both cholesterol synthesis-related proteins (e.g., HMG-CoA synthase and HMG-CoA reductase) known to be involved in keratinocyte lipid synthesis and differentiation appear to be closely associated with p38MAPK and AP-1 phosphorylation (activated forms) (71–73). Our data indicate that large HA stimulates p38MAPK and c-Jun phosphorylation (Figure 4) as well as keratinocyte differentiation (Figure 4).
UV-induced DNA lesions are known to be repaired by nucleotide excision repair (NER) (74), which operates via either global genomic repair (GGR) or transcription-coupled repair (TCR) (75, 76). Loss of certain NER proteins such as xeroderma pigmentosum group C (XPC) results in a selective impairment of GGR (75, 76). The p38MAPK appears to be involved in activating p53 and p63 – both of which are key regulators of NER (75–80) and have been shown to play an important role in maintenance and induction of VDR and certain key DNA damage recognition proteins (e.g., XPC). Whether large HA/CD44-activated Rac1-PKNγ and p38MAPK are involved in regulating p63/p53 signaling leading to DNA repair and keratinocyte survival following UVR exposure is investigated in this study. Our results indicate that pretreatment of cultured keratinocytes with exogenously added large HA exhibited a 1.7-fold enhancement of cell survival (P < 0.0004) following UVB 708 (J/m2) treatment.
These findings reveal selective activation of Rac1-PKN activation by large HA with pathway-specific effects on normal keratinocyte functions. In particular, we have discovered a novel signaling mechanism showing large HA stimulation of Rac1-PKNγ1 and phosphorylation of p38MAPK in cultured keratinocytes. It is possible that Rac1-PKNγ1 phosphorylated p38MAPK promotes AP-1 (c-Jun) (Figure 4) and p63/p53-mediated transcriptional upregulation (Figure 9), resulting in target gene expression and epidermal functions. We have found that the protein level of certain regulatory molecules such as p38MAPK, p63, VDR, and XPC is greatly enhanced in both cultured keratinocytes/epidermal sheets treated with large HA (but not small HA) (Figure 9). In contrast, the expression level of these key regulatory proteins (e.g., p38MAPK, p63, XPC, or VDR) is relatively low in keratinocytes treated with HA or p38MAPK inhibitor (anti-CD44 antibody and SB203580, respectively) plus large HA (Figure 9). These findings suggest that large HA/PKN-induced p38MAPK signaling plays an important role in both maintenance and induction of tumor suppressor proteins (e.g., p63) and VDR as well as certain key DNA damage recognition proteins (e.g., XPC). These findings support the notion that large HA/CD44 signaling events may interact with DNA repair pathways required for epidermal protection against UVB-induced skin damage.
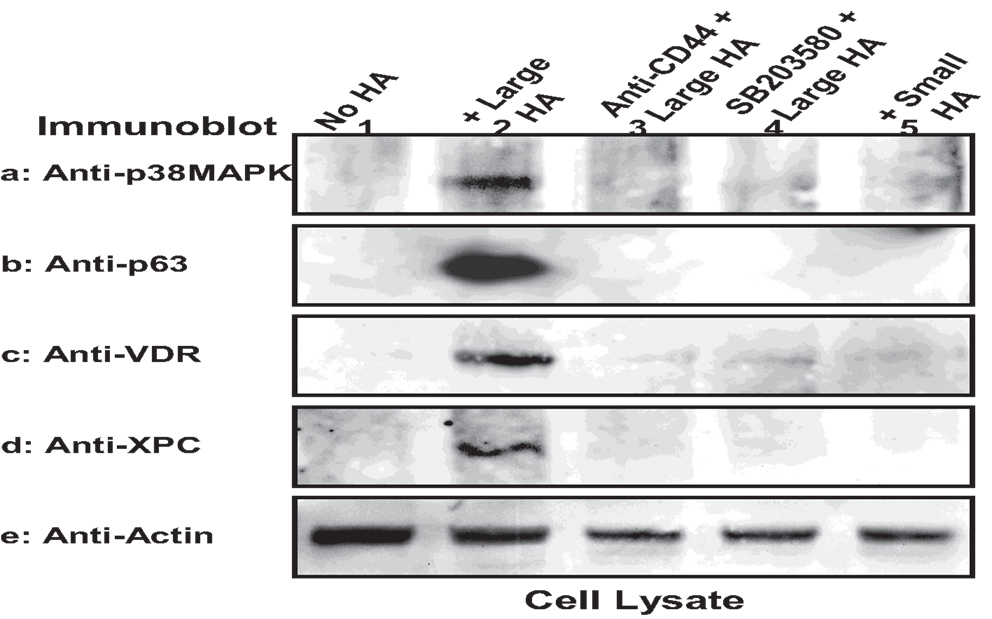
Figure 9. Large HA-mediated upregulation of signaling regulators in keratinocytes. Murine keratinocytes isolated from epidermal sheets were pretreated with HA (50 μg/ml) (lane 2) in the presence or absence of p38MAPK inhibitor (SB203580) for 24 h followed by irradiation with UVB (708 J/m2), harvested and analyzed by immunoblotting using anti-p38MAPK (a) or anti-p63 antibody (b) or anti-VDR antibody (c) or anti-XPC antibody (d) respectively. Actin was probed by anti-actin antibody as a loading control. (Cells were treated with no HA (lane 1) or with large HA (lane 20) or with anti-CD44 plus large HA (lane 3) or with SB203580 plus large HA (lane 4) or with small HA (lane 5) [p63, a p53-like transcription factor; VDR, vitamin D receptor; XPC, DNA repair protein].
In summary, we would like to propose the following model (Figure 10): UVR induced HA degradation and selective HA (small HA vs. large HA)-CD44 interaction with RhoA–ROK and Rac1-PKNγ in regulating transformed and normal keratinocyte signaling leading to cutaneous SCC progression and/or healthy epidermal function. Vitamin D enhances large HA-CD44-mediated differentiation, DNA repair, and keratinocyte survival/normal epidermal function, and it also inhibits small HA/CD44v-mediated RhoA–ROK signaling and SCC-specific tumor behaviors.
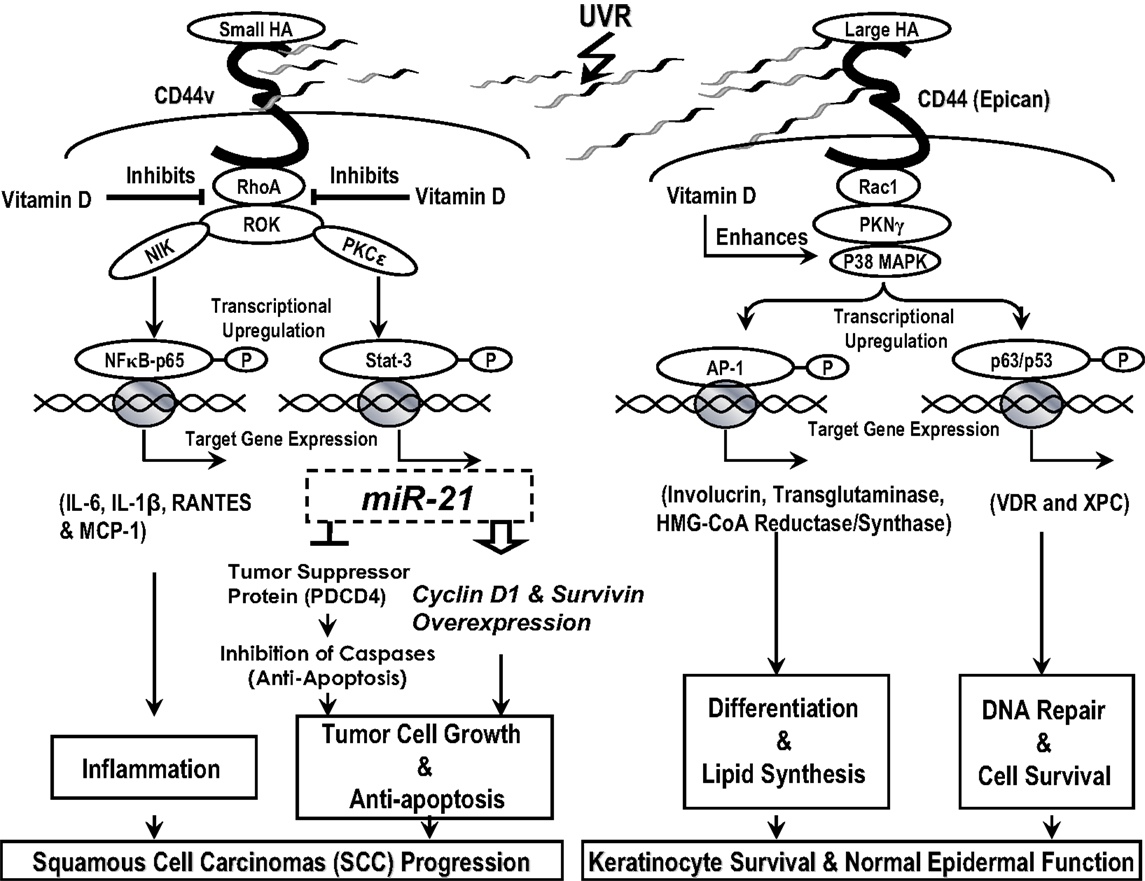
Figure 10. A proposed model for UVR-induced hyaluronan (HA) degradation and selective HA (small HA vs. large HA)-CD44 interaction with RhoA–ROK and Rac1-PKNγ in regulating normal and transformed keratinocyte signaling leading to healthy epidermal function and/or cutaneous squamous cell carcinomas (SCC) progression. Vitamin D enhances large HA-CD44-mediated differentiation, DNA r8epair, and keratinocyte survival/normal epidermal function, and it also inhibits small HA/CD44v-mediated RhoA–ROK signaling and SCC-specific tumor behaviors.
Summary and Outlook
To test our hypothesis, we provided new experimental evidence showing that administration of large HA or vitamin D-related treatments can selectively downregulate the CD44-RhoA/ROK-mediated inflammatory pathways and upregulate CD44-Rac/PKNγ-mediated cell differentiation, DNA repair, and keratinocyte survival function in mouse skin exposed to acute or chronic UVR. The ability for us to identify key HA (small vs. large HA)/CD44-mediated Rho/Rac signaling mechanism(s) by acute or chronic UVR in the regulation of keratincocyte functions will provide valuble insights regarding the possible HA/CD44 involvement of acute or chronic UVR-induced changes in influencing abnormal keratinocyte function, epidermal dysfunction, and tumor formation/SCC progression. In addition, we suggested that signaling perturbation agents (e.g., Y27623, a ROK inhibitor) can be applied to acute or chronic UVR-induced tumor formation/SCC progression displaying upregulation of keratinocyte inflammation and proliferation in order to correct the imbalance between RhoA–ROK signaling and Rac1-PKNγ activation during epidermal dysfunction.
In conclusion, we believe that the information obtained from these studies described in this article will allow us to delineate UVR-induced HA (small vs. large HA)-mediated CD44 signaling mechanisms and subsequent keratinocyte activities, as well as to provide important HA-based and vitamin D-related therapeutic approach regarding the treatment of UVR-induced keratinocyte transformation and skin cancer progression. The overall idea here is to develop innovative approaches to affect skin cancer growth and SCC progression that may be useful for future clinical studies. The new knowledge obtained from our HA/CD44-signaling strategies and HA-based/vitamin D therapeutic approaches should reveal new avenues for possible treatment of UVR-induced skin cancers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the assistance of Dr. Gerard J. Bourguignon in the preparation and review of this manuscript. This work was supported by Veterans Affairs (VA) Merit Review Awards (RR & D-1I01 RX000601 and BLR & D-5I01 BX000628), United States Public Health grants (R01 CA66163), and DOD grant. LB is a VA Senior Research Career Scientist.
References
1. Leffell DJ. The scientific basis of skin cancer. J Am Acad Dermatol (2000) 42:18–22. doi:10.1067/mjd.2000.103340
2. Henson DE, Albores-Saavedra J. Pathology of incipient neoplasia. In: Maize JC editor. Cutaneous Pathology. Philadelphia: Churchill Livingstone (1998).
3. Karvinen S, Kosma VM, Tammi MI, Tammi R. Hyaluronan, CD44 and versican in epidermal keratinocyte tumours. Br J Dermatol (2003) 148:86–94. doi:10.1046/j.1365-2133.2003.05028.x
4. Rodriguez-Rodriguez L, Sancho-Torres I, Gibbon DG, Watelet LF, Mesonero C. Hyaluronan, CD44 and versican in epidermal keratinocyte tumours. Br J Dermatol (2003) 148:86–94. doi:10.1046/j.1365-2133.2003.05028.x
5. Grimme HU, Termeer CC, Bennett KL, Weiss JM, Schopf E, Aruffo A, et al. Colocalization of basic fibroblast growth factor and CD44 isoforms containing the variably spliced exon v3 (CD44v3) in normal skin and in epidermal skin cancers. Br J Dermatol (1999) 141:824–32. doi:10.1046/j.1365-2133.1999.03154.x
6. Dietrich A, Tanczos E, Vanscheidt W, Schopf E, Simon JC. Detection of CD44 splice variants in formalin-fixed, paraffin-embedded specimens of human skin cancer. J Cutan Pathol (1997) 24:37–42. doi:10.1111/j.1600-0560.1997.tb00783.x
7. Heider KH, Sproll M, Susani S, Patzelt E, Beaumier P, Ostermann E, et al. Characterization of a high-affinity monoclonal antibody specific for CD44v6 as candidate for immunotherapy of squamous cell carcinomas. Cancer Immunol Immunother (1996) 43:245–53. doi:10.1007/s002620050329
8. Bourguignon LY, Singleton PA, Diedrich F. Hyaluronan-CD44 interaction with Rac1-dependent protein kinase N-gamma promotes phospholipase Cgamma1 activation, Ca(2+) signaling, and cortactin-cytoskeleton function leading to keratinocyte adhesion and differentiation. J Biol Chem (2004) 279:29654–69. doi:10.1074/jbc.M403608200
9. Bourguignon LY, Ramez M, Gilad E, Singleton PA, Man MQ, Crumrine DA, et al. Hyaluronan-CD44 interaction stimulates keratinocyte differentiation, lamellar body formation/secretion, and permeability barrier homeostasis. J Invest Dermatol (2006) 126:1356–65. doi:10.1038/sj.jid.5700260
10. Lee JY, Spicer AP. Hyaluronan: a multifunctional, megadalton, stealth molecule. Curr Opin Cell Biol (2000) 12:581–6. doi:10.1016/S0955-0674(00)00135-6
11. Maytin EV, Chung HH, Seetharaman VM. Hyaluronan participates in the epidermal response to disruption of the permeability barrier in vivo. Am J Pathol (2004) 165:1331–41. doi:10.1016/S0002-9440(10)63391-3
12. Weigel PH, Hascall VC, Tammi M. Hyaluronan synthases. J Biol Chem (1997) 272:13997–4000. doi:10.1074/jbc.272.22.13997
13. Stern R, Jedrzejas MJ. Hyaluronidase: their genomics, structures, and mechanism of action. Chem Rev (2006) 106:818–39. doi:10.1021/cr050247k
14. Noble PW. Hyaluronan and its catabolic products in tissue injury and repair. Matrix Biol (2002) 25:25–9. doi:10.1016/S0945-053X(01)00184-6
15. Lokeshwar VB, Iida N, Bourguignon LYW. The cell adhesion molecule, GP116 is a new CD44 variant (ex14/v10) involved in hyaluronic acid binding and endothelial cell proliferation. J Biol Chem (1996) 271:23853–64. doi:10.1074/jbc.271.39.23853
16. Bourguignon LYW, Wong G, Xia W, Man MQ, Holleran WM, Elias PM. Selective matrix (Hyaluronan) interaction with CD44 and RhoGTPase signaling promotes keratinocyte functions and overcomes age-related epidermal dysfunction. J Dermatol Sci (2013) 72:32–44. doi:10.1016/j.jdermsci.2013.05.003
17. Bourguignon LYW. Matrix hyaluronan-activated CD44 signaling promotes keratinocyte activities and improves abnormal epidermal functions. Am J Pathol (2014) 184:1912–9. doi:10.1016/j.ajpath.2014.03.010
18. Bourguignon LY. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin Cancer Biol (2008) 18:251–9. doi:10.1016/j.semcancer.2008.03.007
19. Stern R. Complicated hyaluronan patterns in skin: enlightenment by UVB? J Invest Dermatol (2007) 127:512–3. doi:10.1038/sj.jid.5700605
20. Averbeck M, Gebhardt CA, Voigt S, Beilharz S, Anderegg U, Termeer CC, et al. Differential regulation of hyaluronan metabolism in the epidermal and dermal compartments of human skin by UVB irradiation. J Invest Dermatol (2007) 127:687–97. doi:10.1038/sj.jid.5700614
22. Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI. Genomic structure of DNA coding the lymphocyte homing receptor CD44 reveals 12 alternatively spliced exons. Proc Natl Acad Sci U S A (1992) 89:12160–4. doi:10.1073/pnas.89.24.12160
23. Haggerty JG, Bretton RH, Milstone LM. Identification and characterization of a cell surface proteoglycan on keratinocytes. J Invest Dermatol (1992) 99:374–80. doi:10.1111/1523-1747.ep12616087
24. Hall A. Rho GTPases and the actin cytoskeleton. Science (1998) 279:509–14. doi:10.1126/science.279.5350.509
25. Bourguignon LYW, Singleton PA, Zhu H, Diedrich F. Hyaluronan-mediated CD44 interaction with RhoGEF and Rho-Kinase promotes Grb2-associated binding-1 phosphorylation and phosphatidylinositol 3-Kinase signaling leading to cytokine (macrophage-colony stimulating factor) production and breast tumor progression. J Biol Chem (2003) 278:29420–34.
26. Bourguignon LYW, Gilad E, Brightman A, Singleton PA. Hyaluronan-CD44 interaction with leukemia-associated RhoGEF (LARG) and EGF receptor promotes Rho/Ras co-activation and PLCε-Ca2+ signaling leading to cytoskeleton modification and head & neck squamous cell carcinoma progression. J Biol Chem (2006) 281:14026–40. doi:10.1074/jbc.M507734200
27. Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science (1996) 273:245–8. doi:10.1126/science.273.5272.245
28. Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, et al. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J Cell Biol (1999) 145:347–61. doi:10.1083/jcb.145.2.347
29. Amano T, Tanabe K, Eto T, Narumiya S, Mizuno K. LIM-kinase 2 induces formation of stress fibers, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem J (2001) 354:149–59. doi:10.1042/0264-6021:3540149
30. Bourguignon LYW, Zhu H, Shao L, Zhu D, Chen YW. Rho-Kinase (ROK) promotes CD44v3,8-10-ankyrin interaction and tumor cell migration in metastatic breast cancer cells. Cell Motil Cytoskeleton (1999) 43:269–87. doi:10.1002/(SICI)1097-0169(1999)43:4<269::AID-CM1>3.0.CO;2-5
31. Bourguignon LYW, Singleton P, Diedrich F, Stern R, Gilad E. CD44 interaction with Na+/H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 & cathepsin B activation and breast tumor cell invasion. J Biol Chem (2004) 279:26991–7007. doi:10.1074/jbc.M311838200
32. Bourguignon LYW, Zhu H, Shao L, Chen YW. CD44 interaction with Tiam1 promotes Rac1 signaling and hyaluronic acid (HA)-mediated breast tumor cell migration. J Biol Chem (2000) 275:1829–38. doi:10.1074/jbc.275.3.1829
33. Bourguignon LYW, Zhu H, Zhou B, Diedrich F, Singleton PA, Hung MC. Hyaluronan (HA) promotes CD44v3-Vav2 interaction with Grb2-p185HER2 and induces Rac1 & Ras signaling during ovarian tumor cell migration and growth. J Biol Chem (2001) 276:48679–92. doi:10.1074/jbc.M106759200
34. Benitah SA, Frye M, Glogauer M, Watt FM. Stem cell depletion through epidermal deletion of Rac1. Science (2005) 309:933–5. doi:10.1126/science.1113579
35. Mukai H. The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J Biochem (2003) 133:17–27. doi:10.1093/jb/mvg019
36. Vincent S, Settleman J. The PRK2 kinase is a potential effector target of both Rho and Rac GTPase and regulates actin cytoskeletal organization. Mol Cell Biol (1997) 17:2247–56.
37. Quilliam LA, Lambert QT, Mickelson-Young LA, West-wick JK, Sparks AB, Kay BK, et al. Isolation of a NCK-associated kinase, PRK2, an SH3-binding protein and potential effector of Rho protein signaling. J Biol Chem (1996) 271:28772–6. doi:10.1074/jbc.271.46.28772
38. Yoshinaga C, Mukai H, Toshimori M, Miyamoto M, Ono Y. Mutational analysis of the regulatory mechanism of PKN: the regulatory region of PKN contains an arachidonic acid-sensitive autoinhibitory domain. J Biochem (1999) 126:475–84. doi:10.1093/oxfordjournals.jbchem.a022476
39. Calautti E, Grossi M, Mammucari C, Aoyama Y, Pirro M, Ono Y, et al. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell-cell adhesion. J Cell Biol (2002) 156:137–48. doi:10.1083/jcb.200105140
40. Young AR. Acute effects of UVR on human eyes and skin. Prog Biophys Mol Biol (2006) 92:80–5. doi:10.1016/j.pbiomolbio.2006.02.005
41. Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer H-D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr Biol (2007) 17:1140–5. doi:10.1016/j.cub.2007.05.074
42. Baldwin AS. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol (1996) 14:649–83. doi:10.1146/annurev.immunol.14.1.649
43. Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol (1999) 19:5785–99.
44. Duyao MP, Kessler DJ, Spicer DB, Sonenshein GE. Transactivation of the c-myc gene by HTLV-1 tax is mediated by NFkB. Curr Top Microbiol Immunol (1992) 182:421–4.
45. Benitah SA, Valerón PF, Lacal JC. ROCK and nuclear factor-kappaB-dependent activation of cyclooxygenase-2 by Rho GTPases: effects on tumor growth and therapeutic consequences. Mol Biol Cell (2003) 14:3041–54. doi:10.1091/mbc.E03-01-0016
46. Wheeler DL, Martin KE, Ness KJ, Li Y, Dreckschmidt NE, Wartman M, et al. Protein kinase C epsilon is an endogenous photosensitizer that enhances ultraviolet radiation-induced cutaneous damage and development of squamous cell carcinomas. Cancer Res (2004) 64:7756–65. doi:10.1158/0008-5472.CAN-04-1881
47. Aziz MH, Manoharan HT, Verma AK. Protein kinase C epsilon, which sensitizes skin to sun’s UV radiation-induced cutaneous damage and development of squamous cell carcinomas, associates with Stat3. Cancer Res (2007) 67:1385–94. doi:10.1158/0008-5472.CAN-06-3350
48. Bourguignon LYW, Spevak C, Wong G, Xia W, Gilad E. Hyaluronan-CD44 interaction with PKCε promotes oncogenic signaling by the stem cell marker, Nanog and the production of microRNA-21 leading to downregulation of the tumor suppressor protein, PDCD4, anti-apoptosis and chemotherapy resistance in breast tumor cells. J Biol Chem (2009) 284:26533–46. doi:10.1074/jbc.M109.027466
49. Lankat-Buttgereit B, Göke R. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell (2009) 101:309–17. doi:10.1042/BC20080191
50. Göke R, Gregel C, Göke A, Arnold R, Schmidt H, Lankat-Buttgereit B. Programmed cell death protein 4 (PDCD4) acts as a tumor suppressor in neuroendocrine tumor cells. Ann N Y Acad Sci (2004) 14:220–1. doi:10.1196/annals.1294.024
51. Lankat-Buttgereit B, Göke R. Programmed cell death protein 4 (pdcd4): a novel target for antineoplastic therapy? Biol Cell (2003) 95:515–9. doi:10.1016/j.biolcel.2003.09.003
52. Asangani IA, Rasheed SAK, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene (2008) 27:2128–36. doi:10.1038/sj.onc.1210856
53. Wickramasinghe NS, Manavalan TT, Dougherty SM, Riggs KA, Li Y, Klinge CM. Estradiol downregulates miR-21 expression and increases miR-21 target gene expression in MCF-7 breast cancer cells. Nucleic Acids Res (2009) 37:2584–95. doi:10.1093/nar/gkp117
54. Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, et al. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol (2003) 23:26–37. doi:10.1128/MCB.23.1.26-37.2003
55. Suzuki C, Garces RG, Edmonds KA, Hiller S, Hyberts SG, Marintchev A, et al. PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc Natl Acad Sci U S A (2008) 105:3274–9. doi:10.1073/pnas.0712235105
56. Loh PG, Yang HS, Walsh MA, Wang Q, Wang X, Cheng Z, et al. Structural basis for translational inhibition by the tumour suppressor Pdcd4. EMBO J (2009) 28:274–85. doi:10.1038/emboj.2008.278
57. Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis (2007) 12:1543–68. doi:10.1007/s10495-007-0087-3
58. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi:10.1016/S0092-8674(00)81683-9
60. Reichrath J, Rafi L, Rech M, Mitschele T, Meineke V, Gartner BC, et al. Analysis of the vitamin D system in cutaneous squamous cell carcinomas. J Cutan Pathol (2004) 31:224–31. doi:10.1111/j.0303-6987.2003.00183.x
61. Kamradt J, Rafi L, Mitschele T, Meineke V, Gärtner BC, Wolfgang T, et al. Analysis of the vitamin D system in cutaneous malignancies. Recent Results Cancer Res (2003) 164:259–69. doi:10.1007/978-3-642-55580-0_19
62. Bikle DD, Oda Y, Xie Z. Vitamin D and skin cancer: a problem in gene regulation. J Steroid Biochem Mol Biol (2005) 97:83–91. doi:10.1016/j.jsbmb.2005.06.001
63. Morelli A, Vignozzi L, Filippi S, Vannelli GB, Ambrosini S, Mancina R, et al. BXL-628, a vitamin D receptor agonist effective in benign prostatic hyperplasia treatment, prevents RhoA activation and inhibits RhoA/Rho kinase signaling in rat and human bladder. Prostate (2007) 67:234–47. doi:10.1002/pros.20463
64. Tan X, Wen X, Liu Y. Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-kappaB signaling. J Am Soc Nephrol (2008) 19:1741–52. doi:10.1681/ASN.2007060666
65. Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, et al. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature (2013) 499:347–9. doi:10.1038/nature12234
66. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature (2001) 410:37–40. doi:10.1038/35065000
67. Kippenberger S, Bernd A, Loitsch S, Guschel M, Müller J, Bereiter-Hahn J, et al. Signaling of mechanical stretch in human keratinocytes via MAP kinases. J Invest Dermatol (2000) 114:408–12. doi:10.1046/j.1523-1747.2000.00915.x
68. Das M, Cui J, Das DK. Generation of survival signal by differential interaction of p38MAPKalpha and p38MAPKbeta with caveolin-1 and caveolin-3 in the adapted heart. J Mol Cell Cardiol (2007) 42:206–13. doi:10.1016/j.yjmcc.2006.08.118
69. Sugawara Y, Nishii H, Takahashi T, Yamauchi J, Mizuno N, Tago K, et al. The lipid raft proteins flotillins/reggies interact with Galphaq and are involved in Gq-mediated p38 mitogen-activated protein kinase activation through tyrosine kinase. Cell Signal (2007) 19:1301–8. doi:10.1016/j.cellsig.2007.01.012
70. Han JS, Macarak E, Rosenbloom J, Chung KC, Chaqour B. Regulation of Cyr61/CCN1 gene expression through RhoA GTPase and p38MAPK signaling pathways. Eur J Biochem (2003) 270:3408–21. doi:10.1046/j.1432-1033.2003.03723.x
71. Efimova T, LaCelle P, Welter JF, Eckert RL. Regulation of human involucrin promoter activity by a protein kinase C, Ras, MEKK1, MEK3, p38/RK, AP1 signal transduction pathway. J Biol Chem (1998) 273:24387–95. doi:10.1074/jbc.273.38.24387
72. Welter JF, Eckert RL. Differential expression of the fos and jun family members c-fos, fosB, Fra-1, Fra-2, c-jun, junB and junD during human epidermal keratinocyte differentiation. Oncogene (1995) 11:2681–7.
73. Rossi A, Jang SI, Ceci R, Steinert PM, Markova NG. Effect of AP1 transcription factors on the regulation of transcription in normal human epidermal keratinocytes. J Invest Dermatol (1998) 110:34–40. doi:10.1046/j.1523-1747.1998.00071.x
74. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature (2001) 411:366–74. doi:10.1038/35077232
75. Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci U S A (1999) 96:424–8. doi:10.1073/pnas.96.2.424
76. Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci U S A (2002) 99:12985–90. doi:10.1073/pnas.202485699
77. Ferguson-Yates BE, Li H, Dong TK, Hsiao JL, Oh DH. Impaired repair of cyclobutane pyrimidine dimers in human keratinocytes deficient in p53 and p63. Carcinogenesis (2008) 29:70–5. doi:10.1093/carcin/bgm244
78. Finlan LE, Kernohan NM, Thomson G, Beattie PE, Hupp TR, Ibbotson SH. Differential effects of 5-aminolaevulinic acid photodynamic therapy and psoralen + ultraviolet A therapy on p53 phosphorylation in normal human skin in vivo. Br J Dermatol (2005) 153:1001–10. doi:10.1111/j.1365-2133.2005.06922.x
79. Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell (1987) 51:241–9. doi:10.1016/0092-8674(87)90151-6
Keywords: hyaluronan, CD44, RhoGTPase, vitamin D, UVR, miR21, vitamin D, skin cancer
Citation: Bourguignon LYW and Bikle D (2015) Selective hyaluronan–CD44 signaling promotes miRNA-21 expression and interacts with vitamin D function during cutaneous squamous cell carcinomas progression following UV irradiation. Front. Immunol. 6:224. doi: 10.3389/fimmu.2015.00224
Received: 11 December 2014; Accepted: 26 April 2015;
Published: 13 May 2015
Edited by:
David Naor, Hebrew University of Jerusalem, IsraelReviewed by:
Gaurav K. Gupta, Harvard Medical School, USASumit Ghosh, North Dakota State University, USA
Copyright: © 2015 Bourguignon and Bikle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lilly Y. W. Bourguignon, Endocrine Unit (111N2), Department of Medicine, Veterans Affairs Medical Center, University of California at San Francisco, 4150 Clement Street, San Francisco, CA 94121, USA,bGlsbHkuYm91cmd1aWdub25AdWNzZi5lZHU=