 Soledad Mac Keon
Soledad Mac Keon María Sol Ruiz
María Sol Ruiz Silvina Gazzaniga
Silvina Gazzaniga Rosa Wainstok
Rosa Wainstok- 1Laboratorio de Cancerología, Fundación Instituto Leloir, Instituto de Investigaciones Bioquímicas de Buenos Aires IIBBA-CONICET, Buenos Aires, Argentina
- 2Centro de Investigaciones Oncológicas, Fundación para la Investigación, Docencia y Prevención del Cáncer (FUCA), Buenos Aires, Argentina
- 3Laboratorio de Biología Tumoral, Departamento de Química Biológica IQUIBICEN-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina
Dendritic cells (DCs) play a pivotal role in the orchestration of immune responses, and are thus key targets in cancer vaccine design. Since the 2010 FDA approval of the first cancer DC-based vaccine (Sipuleucel-T), there has been a surge of interest in exploiting these cells as a therapeutic option for the treatment of tumors of diverse origin. In spite of the encouraging results obtained in the clinic, many elements of DC-based vaccination strategies need to be optimized. In this context, the use of experimental cancer models can help direct efforts toward an effective vaccine design. This paper reviews recent findings in murine models regarding the antitumoral mechanisms of DC-based vaccination, covering issues related to antigen sources, the use of adjuvants and maturing agents, and the role of DC subsets and their interaction in the initiation of antitumoral immune responses. The summary of such diverse aspects will highlight advantages and drawbacks in the use of murine models, and contribute to the design of successful DC-based translational approaches for cancer treatment.
Introduction
Dendritic cells (DCs) form a network of antigen-presenting cells (APCs) that shape immune responses by linking innate and adaptive immunity. Their ability to induce or suppress the proliferation, activation, and differentiation of specific T-cell subsets renders them an attractive target for cancer immunotherapies.
Tumoral antigens are mostly self-proteins (1). As a result, cancer is associated with T-cell deletion and a defective T-cell memory repertoire, which includes anergic CD8+ T cells (hyporesponsive state upon low co-stimulation or high co-inhibitory stimulation) and exhausted CD8+ T cells (decreased cytokine expression and effector function upon chronic activation), as recently reviewed (2, 3). However, T cells recognizing tumor-specific antigens and tumor-associated antigens (TAAs) that can be targeted by vaccination have been described in the context of cancer (1, 4, 5). Therefore, therapeutic vaccines should be able to prime naïve T cells, but most importantly, induce the transition of existing memory T cells from non-protective to potent effector CD8+ T cells (6). DCs are considered the most efficient APCs (7). In order to instruct naïve T cells into the required functional profile, DCs must present tumor antigens via MHC class I and II molecules (signal 1), express co-stimulatory ligands (signal 2), and inflammatory mediators (signal 3) such as IL-12 or type I interferons (IFNs) (8, 9). Recent works have highlighted the importance of the innate immune system in the tumor microenvironment. This is the site where initial antigen sampling and activation of APCs take place, including different subsets of DCs. It was shown that a T-cell inflamed tumor microenvironment phenotype is associated with clinical responses to therapeutic cancer vaccines (10). Therefore, a complete description of the functional specialization and tissue distribution of DCs deserves special attention in tumor models.
For over 20 years, clinical trials have been performed using DC-based vaccines, and they have proven to be feasible, safe, and to elicit immunological responses (11). Recent reviews that have outlined the clinical effects of DC-based vaccines highlight the discrepancy observed between the immunonogenicity and the therapeutic efficacy in terms of inducing tumor rejection (6, 12, 13). This clinical challenge emphasizes the importance of employing adequate murine models in pre-clinical research. The availability of different genetic backgrounds allows researchers to gain insight into the function and relevance of specific DC subsets, track vaccine cells once injected, evaluate antigen-specific T-cell proliferation and activation, and work with human tumor cells in humanized mouse models. In this review, the knowledge gained through murine models will be summarized, with the aim of helping in the design of efficient DC-based vaccines that can be rapidly translated to human clinical trials.
Murine Models in Cancer Research
Murine models have been an invaluable tool in the study of DCs and cancer immunotherapy. It was in mouse lymph nodes that DCs were first identified by Steinman et al. as a novel cell type with distinct morphological features (14). Regarding tumor immunology, the first evidence of the immune system’s ability to recognize tumoral antigens and reject tumors was obtained using mice with implanted or chemically induced tumors (15) or infected with oncogenic viruses (16). Since then, much has been learned in this field with the aid of wild type and genetically engineered mice (GEM) strains. The approaches used to generate these GEMs and some of their limitations have been reviewed in Ref. (17).
Murine cell lines have been established for a variety of tumor cell types in many genetic backgrounds. The B16 melanoma cell line and its derived sublines B16-F1 and B16-F10 (18) have been used extensively as syngeneic transplants into C57BL/6 mice. B16 cells are poorly immunogenic, and express low levels of MHC class I molecules (19). Therefore, immunotherapeutic strategies that generate tumoral protection in other tumor models do not work as efficiently with B16 tumors (20). For this reason, it is a good model to evaluate the strength of antitumoral immunotherapies.
Although it has proved very useful for the evaluation of anti-melanoma immunotherapies, this model has several features that do not entirely reflect the human disease. Principally, the frequently observed BRAFV600E mutation in human melanomas (21) is absent from B16, B16-F1, and B16-F10 cells, which show a distinct pattern of genetic alterations (22). In this context, GEMs with conditional expression of the BRAFV600Emutation or other mutations frequently observed in human melanomas have been developed. Moreover, human melanomas may take years to become clinically evident, and dormancy periods between primary tumor resection and metastatic disease offer a time-window for therapeutic interventions aiming to prevent future recurrence in high-risk patients. By contrast, all B16 cell lines cause rapid in vivo growth, leading to measurable tumors within 15–20 days of subcutaneous (s.c.) transplantation, and compromising the study of therapeutic approaches. Their rapid s.c. growth does not give cancer cells time to progress into metastatic disease without previous resection of the primary tumor, poorly recapitulating human clinical stages. To study metastatic disease, the more aggressive B16-F10 cell line is usually injected directly intravenously (i.v.). However, stages such as detachment of tumor cells from the primary tumor mass and intravasation into nearby vessels are not represented. GEMs with PTEN deficiency and melanocyte-specific induced BRAFV600E mutation develop malignant melanoma with posterior metastasis to lymph nodes and lung (23), more closely resembling human tumor progression. The main challenge to widespread implementation of GEM models is the technical difficulty and high cost of generating these strains. Recently, there have been BRAFV600E cell lines isolated from transgenic BRAFV600E mice and BRAFV600E PTEN−/− mice (24). These cells recapitulate human BRAFV600E melanoma in vitro, and can be transplanted into syngeneic mice for assessment of host-tumor interactions. Using knock-in technology, mutations can be targeted to the endogenous gene locus of interest, and so innate regulation of gene expression can be achieved. Some examples of GEMs that target oncogenes or tumor suppressor genes and that may be useful for assessing the efficacy of tumor vaccines are those that target KIT, TRP53, BRAF, KRAS, PIK3CA, and EGFR (25).
The ovalbumin (OVA)/OT-I and OT-II murine model have been used to study specific T-cell responses. Briefly, mice are immunized with the OVA antigen (whole protein or peptides), and then OVA-specific CD4+ or CD8+ T-cell restricted responses are assessed using T cells obtained from transgenic animals that recognize OVA epitopes on MHC class I molecules (CD8+ T OT-I cells) or MHC class II molecules (CD4+ T OT-II cells). Though this model has proven very informative in immunology, it diverges critically from the cancer setting in that OVA is an exogenous antigen, and so OT-I and OT-II cells have not been subjected to negative selection or peripheral tolerance. This model can provide information about naïve T-cell distribution and activation but does not reflect the behavior of human tumor-specific clones; so, the results obtained should be evaluated carefully. As will be discussed later, the success of a particular immunotherapeutic strategy may vary depending on whether the antigen is a self-peptide or an exogenous peptide.
An intermediate model between human cell culture and mouse cancer models is the transplantation of human surgical specimens or established cell lines (xenografts) into immunocompromised mice. These have been used successfully to study human cancer cells in an environment that better reflects tumoral complexity and architecture (26). But when these models are used, it is not possible to assess the patient’s immune response to the tumor. This can be solved by the use of “humanized” mice, severely immunodeficient mice with mutations in the IL-2 receptor common γ-chain locus, which can be engrafted with human peripheral blood mononuclear cells (PBMCs) or human stem cells (HSCs) (27). The IL-2 receptor γ-chain is necessary for the binding and signaling several cytokines, and for NK development (28, 29), resulting in the absence of NK cells and improving human cell engraftment and the analysis of antitumoral responses (30). In the NOD-scid Il2Rγ−/−strain, multilineage engraftment including human CD14+ cells, NK cells, and plasmacytoid BDCA-2+ DCs can be observed after HSC transplantation. The drawback is that these mice have severely hypoplastic peripheral lymph nodes, associated with impaired antibody class-switch and affinity maturation (31). They also have proportionally low numbers of human myeloid CD11c+CD86+ DCs, which fail to produce IL12p40 or IFN-γ after CD40 stimulation (30). It is thus difficult to evaluate DC migration to secondary lymphoid organs and their interaction with T cells to trigger adaptive responses. Nonetheless, T-cell responses in vivo using donor-matched DCs have been successfully assessed (32, 33). To facilitate the engraftment of different cell types, GEMs expressing human cytokines (34) or protocols administering such recombinant proteins have been developed, and are reviewed by Drake et al. (35).
Differences between the murine model and the human disease may partially account for the lower efficiency observed in human clinical trials. Hopefully, new models have been designed that better recapitulate human disease or that allow studying immunotherapies utilizing the patient’s own tumor and immune cells. Therefore, researchers should take special care selecting the model that best fits their objectives. The recommended applications and considerations for choosing a murine model for DC-based vaccination in cancer have been summarized in Table 1.
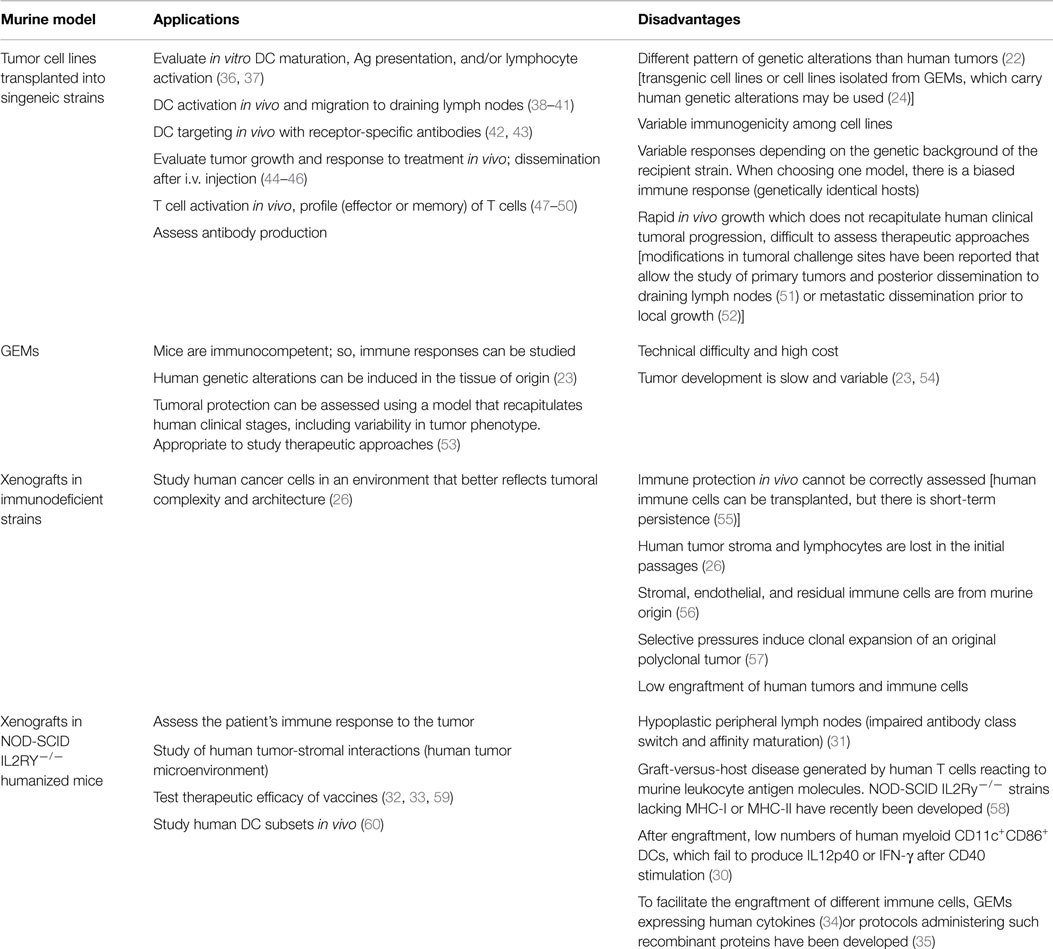
Table 1. Advice for choosing murine models for DC-based tumor immunotherapy.
Lessons Learned from Murine Models
Characterizing DC Subsets
Recent reviews have described at length the ontogeny, phenotype, and transcriptional profile of the heterogeneous population collectively named DCs (61–63). This network relies on the differential expression of a group of transcription factors that determine the specification of the different subsets of DCs (64). Steady-state DCs can be classified into two groups: plasmacytoid DCs (pDCs) and classical/conventional DCs (cDCs). Two further subsets of cDCs can be distinguished in lymphoid tissues: CD8+ and CD11b+cDCs, while in non-lymphoid tissues, cDCs are classified into CD11b−CD103+ and CD11b+CD103−. Langerhans cells (LCs) represent an additional population of DCs that reside in the epidermis, although they can be found at draining lymph nodes both in the steady state and after an inflammatory stimulus. Lastly, during an inflammatory response, monocyte-derived DCs (MoDCs) are induced and recruited to the sites where the response was initiated, and migratory DCs can be found in draining lymph nodes.
Deeper insights at the molecular level have improved the distinction of DCs from other immune population, such as macrophages, by providing a list of transcripts that define a “core cDC signature.” This signature includes the chemokine receptor CCR7, the transcriptional regulator Zbtb46, the Flt3L receptor, and Kit (63). In coming years, transcriptional profiling should be a helpful tool in the difficult task of assigning specific functions to different DC population. So far, functional studies have shown that each subset has particular abilities regarding antigen processing, response to environmental signals, and the induction of naïve T cells into effector lymphocytes (65). The response to environmental signals is mediated by the expression of a set of innate pattern recognition receptors (PRRs) that can bind conserved antigen determinants of virtually all classes of pathogens, which are called pathogen-associated molecular patterns (PAMPs), and also recognize endogenous signals released during a stress or damage response (damage-associated molecular patterns, DAMPs). The pattern of expression of PRRs, scavenger, and lectin receptors on different DC subsets is of great importance to predict their potential activation in different physiological contexts, including the tumor microenvironment. Some of the most relevant phenotypic markers, PRRs, and precursors to each subset are listed in Figure 1. There are controversies regarding the involvement of particular DC subsets in tolerogenic responses to tumors. This section will focus on evidence regarding the observed contributions of specific DC subsets in immune responses elicited by DC-based vaccines in cancer.
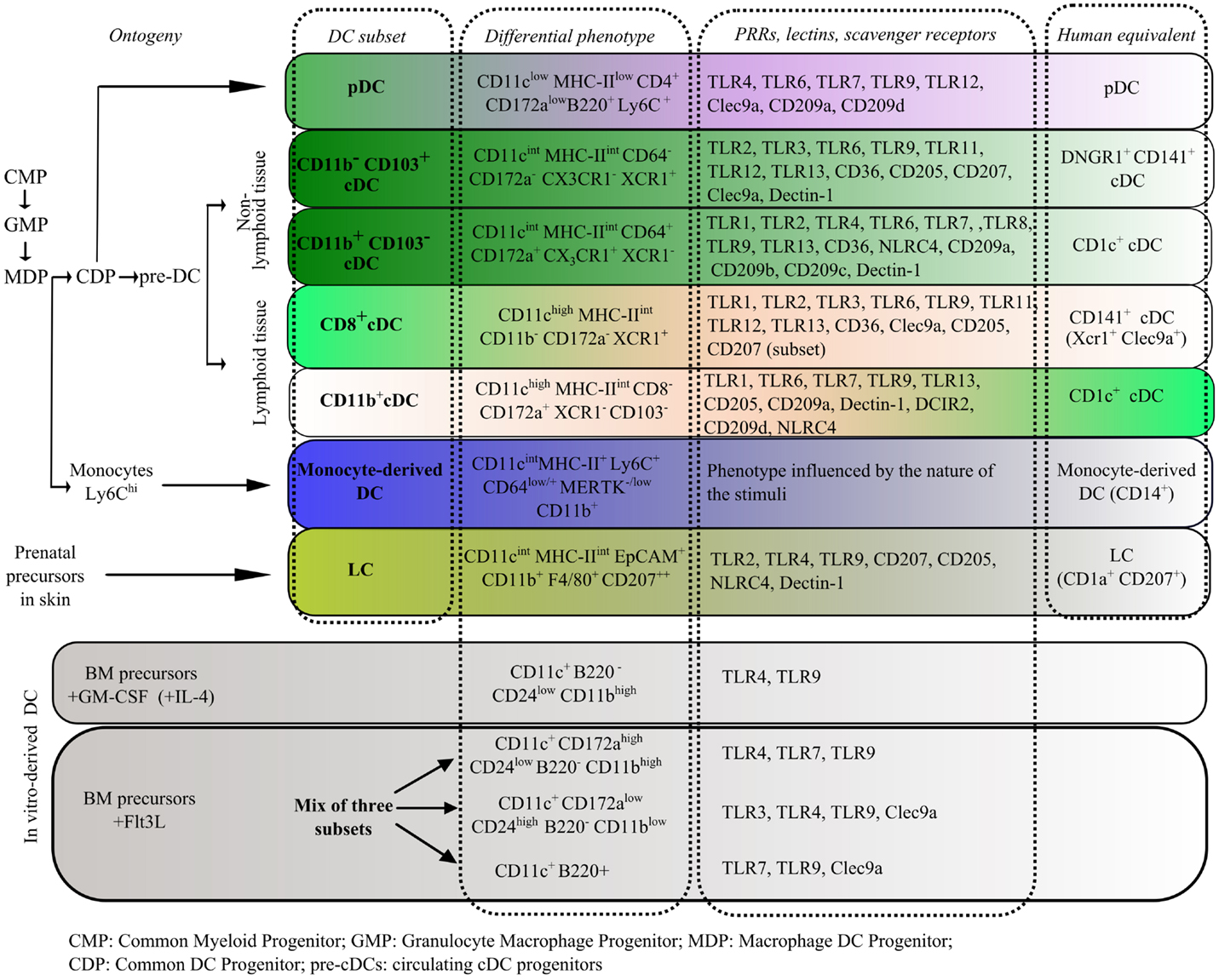
Figure 1. Description of ontogeny, phenotype, and patterns of PRR expression in murine DC subsets in the steady state. Comparison to the equivalent human subsets and to murine in vitro-derived DCs is provided (61, 62, 66–77).
Plasmacytoid DCs
Plasmacytoid DCs are found mainly in the blood and lymphoid tissues. Their unique properties and phenotype separate them from cDCs (Figure 1). Upon encounter with foreign nucleic acids, pDCs produce massive amounts of type I IFNs, which can activate both the innate and the adaptive arms of the immune system. Despite the large body of knowledge acquired about pDC biology, to date, their suitability for DC immunotherapy is only recently being explored. In support of an immunogenic pDC role, imiquimod (TLR7 agonist)-mediated regression of melanoma tumors was shown to be dependent on TLR7 expression on pDCs; neither mice lacking TLR7 nor pDC-depleted mice responded to imiquimod therapy and both saw a reduction in apoptosis within treated tumors (78). Other works have shown that the cross-presentation pathway is under the control of TLR activation (79). Moreover, results obtained from in vitro killing assays suggest that pDCs may exert direct cytotoxic effects on tumor cells by secreting soluble factors in response to imiquimod treatment (78). Recent works have highlighted previously unknown functions of pDCs in antitumor immunity. In work by Guery et al., antigen presentation by pDCs was required for efficient antigen-specific Th17 responses. Mice that were immunized with OVA plus CpG-B (synthetic TLR9 ligand) and possessed pDCs lacking MHC class II molecules showed reduced numbers of IL-17-secreting OT-II cells, exhibited a significant increase in tumor growth, and showed a reduction in the recruitment of tumor-specific cytotoxic T lymphocytes (CTLs) into tumors (80). In a different model, the injection of CpG-activated pDCs after a tumor challenge led to a significant delay in the growth of B16 melanoma and MCA205 sarcoma tumors, and induced the recruitment of antigen-specific CD8+ T cells and activated NK cells into tumors by a mechanism dependent on endogenous cDCs and NK cells. In this work, CpG-activated pDCs were able to stimulate NK cell cytolytic function by secretion of type I IFN (81). This latter works highlight the potential of activated pDCs to activate NK cells and orchestrate a cytotoxic immune response against tumoral antigens.
On the other hand, several evidences in the human setting support an immunosuppressive role for pDCs in the tumor microenvironment: tumor-associated pDCs (TApDC) correlated with unfavorable prognosis in patients with breast cancer (82), where they were found to colocalize and correlate with tumor-associated regulatory T cells (Tregs) (83); ovarian carcinoma ascites-derived pDCs induced IL-10-producing Tregs in vitro (84); tolerogenic pDCs were found infiltrating prostate tumors (85), and together with inducible costimulator (ICOS)-expressing FoxP3+Tregs, pDCs were strong predictors for disease progression in patients with ovarian cancer (86). In mouse models, fewer works have studied the mechanisms of pDCs-induced immunosuppression. Watkins et al. used a transgenic adenocarcinoma of mouse prostate to study tumor infiltrating DCs, and found a predominant pDC population able to induce tolerance in antigen-specific T cells. Furthermore, they studied the role of transcription factor Foxo3 in mediating TApDC-induced suppression (85). In accordance, the depletion of pDCs in mice bearing CpG-treated Lewis lung carcinoma (LLC) tumors correlated with lower numbers of intratumoral Tregs, an increase in the recruitment of mature cDCs, and the arrest of tumor growth (87). Delayed tumor growth was also observed after pDC depletion in an orthotopic mammary tumor model (88). However, in both works, pDC depletion was induced by treatment with the m-927 antibody, which recognizes the antigen BST2. Although this antigen is mostly restricted to pDCs in the steady state, it is upregulated in several cell types following activation. In fact, pDCs and plasma cells are indistinguishable based on BST2 expression after stimulation (89). Therefore, conclusions made about pDC-depletion experiments should take this confounding factor into account. Additional evidence includes indoleamine 2,3-dioxygenase (IDO)-expressing pDCs that were found at draining lymph nodes of tumor-bearing mice. These IDO-pDCs showed ability to activate Tregs in vivo (90). However, pDCs were defined in this system as CD11c+B220+CD19+ cells, which it is still not clear if it represents a rare subset of pDCs that express CD19, or a distinctive B-lymphoid cell type with T-cell regulatory features and phenotypic features of both B cells and DCs (91, 92).
Of main importance for translational research is the possible modulation of murine and human pDC immunosuppressive activity into an effective protective phenotype. In vivo intratumoral administration of TLR7 ligands led to ICOS+ TApDC activation and tumor regression in an orthotopic mammary tumor model (88). In human breast cancer, in vitro neutralization of ICOS molecule blocked pDC-induced Treg expansion and reduced IL-10 secretion by memory TA-CD4+ T cells (93).
Langerhans Cells
Epidermal LCs were originally described as prototype DCs because of their ability to capture antigens in the skin and migrate to draining lymph nodes in response to foreign stimuli. Their strategic location makes them attractive targets for epicutaneous immunization or i.d. vaccination. However, recent discoveries about their ontogeny and functional redundancy have set them apart from other DCs. As opposed to their dermal counterparts, LCs can originate from radio-resistant precursors present in the skin, and their development is independent of Flt3L and Flt3, and dependent on TGF-β1 (94, 95). Since finding that other DCs population express the classical LC marker, langerin (CD207) (96, 97), functions that used to be attributed to LCs are being reevaluated and assigned to dermal CD207+ DCs (65, 98–100). Although CD207+ cells can mediate efficient antigen presentation (100–103), recent works have shown that targeting antigens to LCs via CD207 induces antigen-specific cross-tolerance, while targeting antigens to dermal CD207+ DCs elicits a cytotoxic response and long-lived immunological memory. Surprisingly, the tolerogenic effect mediated by LCs occurred only in the presence of adjuvants, and not in the steady state. It is worth noting that authors observed an initial proliferation response, regardless of the adjuvant used, and only detected a difference between treatments by analyzing the surviving OT-I CD8 T cells, which displayed a phenotype of central memory T cells, at 8 weeks after immunization (47). Such deep analysis of immune responses should be encouraged in all experimental approaches when possible.
Lymphoid Tissue CD8+ and Non-Lymphoid Tissue CD11b− CD103+cDCs
CD8+ and CD103+cDCs share many phenotypic and functional features, including the expression of XCR1 shared by human CD141+cDCs (Figure 1). They also display similar transcriptional profiles (63). Given their superior ability to cross-present antigens to CD8+ T cells, they are especially suitable as inducers of antitumoral responses (66). Mice with defective Batf3 (transcription factor also known as Jun dimerization protein p21SNFT) lack splenic CD11chighCD8+CD205+ cells and show reduced numbers of CD11blow/−CD103+ dermal DCs in skin-draining lymph nodes. Unlike their wild-type controls, Batf3−/− mice failed to reject syngeneic fibrosarcomas, did not develop tumor-specific CTLs, and showed reduced numbers of tumor-infiltrating CD8+ T cells (104). A similar pattern was observed in a model of B16. SIY melanoma, in which CD8+cDCs were critical for the spontaneous priming of tumor-specific CD8+ T cells (105). In this case, the mechanism was shown to be dependent on type I IFN signaling in the CD8+cDCs lineage (105, 106). However, the identity of the specific DC subset of cells producing type I IFNs in response to tumor growth remains to be elucidated, suggesting a collaborative process for induction of effective antitumor T cell priming. In support of these observations, CD8+cDCs pulsed with tumor lysates were able to reduce tumor growth in fibrosarcoma-bearing mice (107).
Lymphoid tissue CD8+cDC subset is a heterogeneous population. CD207+CD8+cDCs were the only splenic DCs capable of cross-presenting a systemic soluble protein antigen in vitro and in vivo to CD8+ T cells, though their role in antigen presentation via MHC class II molecules to CD4+ T cells was limited (108). Furthermore, CD207+CD8+cDCs were the main producers of IL-12 after systemic injection of the iNKT cell ligand α-galactosylceramide (α-GalCer), and contributed to the initial burst of antigen-specific CD8+ T cell proliferation after i.v. injection of the OVA protein plus α-GalCer (108). Accordingly, co-delivery of the protein antigen and α-GalCer to CD8+cDCs by nanoparticles coated with anti-CD205 antibodies elicited potent antitumor responses in a preventive scheme and delayed tumor growth in a therapeutic setting, suggesting that this strategy optimizes iNKT cell-mediated immune response (109). Furthermore, a recent work has identified CD205+ CD8+cDCs as the key APCs for multiple forms of α-GalCer (110). Of translational relevance, these authors showed how the expression of costimulatory and coinhibitory molecules on the surface of CD8+cDCs, such as CD70, Rae-1, PD-L1, and PD-L2, was modulated in response to different iNKT cell agonists. Future experiments on human cells should take into account that, in contrast to the murine system, there are five human isoforms of CD1, with differential ability to bind and present lipidic antigens to T cells (111).
In non-lymphoid tissue, cDCs represent 1–5% of the cells. In the case of the mouse intestine, CD11b−CD103+cDCs are enriched in Peyers’ patches and their putative human counterparts are DNGR-1+CD141+CD11b−DCs (112). CD11b−CD103+CD8α+ have been shown to migrate from intestine to mesenteric lymph nodes, and to be able to induce OVA-specific CD8+ T-cell proliferation in vitro (113). In the lung, CD11b−CD103+ were the only DC subset able to acquire and transport apoptotic cells to draining lymph nodes, and cross-present apoptotic cell-associated antigen to CD8+ T cells (114). Their strategic location makes them suitable targets for mucosal immunization. However, their relevance in tumor models remains poorly understood. In fact, works on mucosal immunization have not addressed the role of different DCs subsets in the outcome of vaccination. In a murine model of human papillomavirus-associated genital cancer, s.c. vaccination route was shown to be superior to mucosal (intranasal and intravaginal) immunization route for inducing regression of established genital tumors (115). By contrast, in a model of orthotopic head and neck cancer, intranasal immunization provided better prophylactic and therapeutic efficacy than intramuscular vaccination (116). In a model of murine spontaneous adenomatous polyps, intrarectal immunization decreased tumor formation and prevented progression to invasive colorectal cancers, inducing potent local cellular and humoral responses (117). Elucidating the role of specific DCs subsets during mucosal immunization in diverse tumor models is required in order to exploit their potential, and represents an exciting field of research.
Much of the research on tumor immunology has focused on the process of antigen presentation at lymph nodes rather than local mechanisms that can occur at the tumor niche, which likely influence the function of tumor CTLs. A recent work by Broz et al. has elegantly dissected the composition of the myeloid tumor microenvironment across a broad range of tumors (118). They found a rare population of immunostimulatory cells at the tumor, corresponding to CD103+cDCs that were capable of inducing TCR signaling in both naïve and previously activated OT-I CD8+ T cells in ectopic and spontaneous mouse tumor models. Furthermore, characterization of different immune population highlights the existence of functional diversity among intratumoral APCs. In a different approach, Woo et al. showed that host APCs in the tumor microenvironment can incorporate tumor-derived DNA, which is proposed to be involved in the activation of the STING pathway, leading to IFN-β production and priming of CD8+ T cells against tumor antigens in vivo (119). These findings highlight that triggering innate immune activation against tumors can be a suitable antitumoral strategy if the adequate APC population is targeted.
Classical CD11b+ CD8−cDCs
Among non-lymphoid tissue cDCs, the transcriptional profile of the CD11b+CD103− subset reveals that the current classification based on phenotypic markers defines a heterogeneous population, with some cells belonging to the cDC lineage and others to the macrophage lineage (63). Furthermore, the phenotypic and functional features of this subset can vary depending on the tissue they reside. In the intestinal lamina propia, CD11b+ CD8−cDCs comprise two population: CD11b+ CD8− CD103+ cDCs express CCR7 and represent the primary DCs subset for antigen sampling and migration to mesenteric lymph nodes, whereas the CD11b+ CD8− CD103− subset represents a non-migratory gut-resident population with slower turnover rates (120).
Classical CD11b+ CD8−cDCs most often predominate the lymphoid-resident cDCs population. They comprise a heterogeneous population of DCs that are preferentially involved in MHC class II-restricted antigen presentation to CD4+ helper T cells (66). At least two subsets can be distinguished in the spleen based on their dependence on Notch signaling for differentiation (121). CD11b+ CD8−cDCs in the spleen can be detected at the red pulp and marginal zone (66), and so they represent a potential target for i.v. immunization. The candidate vaccine CyaA-Tyr (detoxified adenylate cyclase carrying a HLA-A*02-restricted tyrosinase 369–377 CTL epitope), already being evaluated in clinical trials (study identifier: PC1O0VAC02), targets CD11b+ cells (122). Dadaglio et al. showed that i.v. immunization induced antigen-specific CTL responses in mice by a mechanism dependent on activation of the TLR4/TRIF pathway in cDCs. In vivo experiments showed that the vaccine binds mainly to the CD11b+ CD8− DCs subset in the spleen, and induced DCs activation and maturation. Surprisingly, CyA-Tyr administration induced upregulation of CD86, CD40, and MHC class II molecules on both CD8+CD11b− and CD8−CD11b+cDCs, probably by indirect and direct effects, respectively (123).
Monocyte-Derived DCs
This subset, also described as inflammatory DCs, refer to a population of DCs that can be found transiently in response to inflammatory stimuli. MoDCs are phenotypically defined as CD11cintCD11b+Ly6C+ cells (Figure 1); however, their distinction from macrophages has been challenging (124). Ly6Chigh monocytes and early hematopoietic precursors upon TLR engagement can serve as precursors (125, 126). Their putative human equivalent has been recently described as CD11c+ MHC class II+ CD16− CD1c+ cells, with dendritic morphology and robust T-cell stimulatory capacities. When cultured with allogeneic naïve CD4+ T cells, this population was capable of potent stimulation of Th17 responses (127).
Several evidences about the role of MoDCs in models of infection and inflammatory diseases have been recently reviewed (124). Ma et al. have recently highlighted the participation of MoDCs in immune recruitment to the tumor bed in response to chemotherapy. These cells were found infiltrating tumors after anthracycline-based chemotherapy in an ATP-dependent fashion, and efficiently presented tumor antigens to CD8+ T cells in vitro and in vivo (128). However, their relevance in diverse tumor models has been incompletely described, and further research on this subset may provide novel and useful data about their potential modulation for cancer immunotherapies.
In Vitro Bone Marrow-Derived DCs
Given the low number of DCs that can be obtained from primary culture of non-lymphoid and lymphoid tissue, the possibility of obtaining high numbers of DCs from murine progenitors in vitro has greatly contributed to the knowledge of DC biology. There are two main experimental approaches to obtain murine DCs, and they are both based on the culture of bone marrow cells with specific cytokines. However, they give rise to DC population differing in phenotype, morphology, functional properties, and resemblance to in vivo subsets (Figure 1) (67).
In the first approach, bone marrow precursors are cultured with GM-CSF alone, or with the addition of IL-4 (129). These cells display an immature phenotype (low expression of co-stimulatory and MHC class II molecules), express high levels of CD11b, and have a low migrating capacity to draining lymph nodes after s.c. injection (130). They can secrete IL-12p70, TNF-α, IL-10, RANTES, and CCL2 in response to TLR agonists, and confer protective immunity against tumor challenge in the presence of maturation agents such as CD40L or after co-culture with apoptotic/necrotic tumor cells (130, 131). The absence of IL-4 during in vitro culture leads to the presence of additional immune population, such as F4/80+ macrophages and Ly6G+ neutrophils (130, 132).
In the second approach, bone marrow precursors are cultured with Flt3L (133). This culture is comprised of at least three different DCs population that, according to their phenotype and pattern of TLR, chemokine receptor expression, and cytokine production, highly resemble three DCs subsets in vivo: B220+pDCs, CD24highCD11blow DCs (equivalent to CD8+cDCs), and CD24lowCD11bhigh DCs (equivalent to CD8− cDCs). In the absence of adjuvants, only the CD24highCD11blow subset could efficiently cross-present antigens to CD8+ T cells (68). Flt3L-derived DCs can also secrete IL-12p70 and RANTES in response to TLR agonists, while the opposite was found regarding TNF-α, IL-10, and CCL2. Their ability to migrate to draining lymph nodes after s.c. injection was superior to GM-CSF/IL-4-derived DCs (67).
In contrast with the mouse system, in vitro-derived human DCs are usually obtained from peripheral blood monocytes cultured with GM-CSF and IL-4. These cultures render high numbers of cDCs. Other approaches include culturing CD34+ progenitor cells with GM-CSF and TNF-α, or with Flt3L and thrombopoietin (134), or expansion of cord blood Lin− cells followed by a differentiation step with SCF, GM-CSF, IL-4, and Flt3L (135). These latter cultures allow the generation of CD141+ DC subset, equivalent to mouse CD8+ DCs, and XCR1− DCs, equivalent to MoDCs (136). Recently, an improved culturing method has shown to support the development of all the three major types of DCs (CD1c+ cDCs, CD141+ cDCs, and pDCs) used a combination of mouse BM stromal cells and defined human cytokines (Flt3L, SCF, and GM-CSF). These cultures also produced granulocytes, monocytes, NK cells, and B cells, and they resemble their blood-derived counterparts as assessed by gene expression, surface phenotype, and cytokine production (137).
Antigen Transfer Between APCs and Interaction Between DC Subsets
A novel mechanism for the induction of specific CD8+ T cell responses upon vaccination with DCs is antigen transfer between ex vivo-loaded DCs and resident DCs. Recent papers in which selective DC subsets are ablated have allowed authors to identify key players in the induction of immune responses. Petersen et al. made use of transgenic mice in which CD207+ cells can be selectively depleted to show how resident CD207+ CD8α+ DCs are required for efficient induction of CD8+ T cells, but not of the CD4+ T cell counterpart, after the administration of OVA-loaded, bone marrow-derived DCs (108, 138). Antigen transfer has also been reported in endogenous DCs subsets such as LCs and dermal DCs (97, 139).
Using transgenic mice that express a specific MHC class II haplotype under the control of a CD11c promoter, Kleindienst et al. have shown that specific T cell responses following antigen-loaded DC vaccination are enhanced by endogenous DCs that express the correct restriction elements through an antigen transfer mechanism dependent on direct cell-to-cell interaction. In this setting, few injected DCs reached the draining lymph nodes, while most of them were retained at the injection site (140). This observation has been confirmed by several groups (38, 130), and it suggests a putative mechanism in which antigens are taken up in situ by endogenous DC.
Several other works support the notion that injected in vitro-derived DCs have a minor role in the direct priming of T cells in vivo. In a work by Yewdall et al., authors injected an OVA-pulsed, DC vaccine into mice with chimeric bone marrow that lacked the hematopoietic compartment necessary to present the OVA peptide. They showed that both i.v. and s.c. administration of OVA-loaded or OVA-expressing DCs required the transfer of antigens to host DCs for efficient CD8+ T-cell priming (141).
Given these observations, and the complexity of the DCs network, how can we exploit the potential synergy among different immune population? There is compelling evidence that antitumor responses may be enhanced by the interaction between different DCs subsets. In an original and novel approach, recruitment and activation of immune cells in situ were elicited by matrices implanted into s.c. pockets, containing CpG danger signals, GM-CSF, and tumor lysates. Vaccinated mice were challenged with live B16-F10 melanoma tumor cells at day 14 and showed significant protection from tumor-induced lethality (up to 90% survival rate). In this setting, the number of CD8+cDCs and pDCs at the vaccination site correlated strongly with the magnitude of protective antitumor immunity, and a significant expansion of antigen-specific CTLs was observed in the spleens of vaccinated mice. Furthermore, two doses of this vaccine elicited protection in 20–47% of mice in a therapeutic setting of established tumors (142). In a different approach, a peptide-pulsed CpG-activated pDC vaccine induced an antigen-specific CD8+ T cell response directly and also affected cDCs priming capacity, resulting in a synergic antitumor response. Although the detailed mechanism was not described, it required cell-to-cell contact (143).
Cross-dressing is a recently described mechanism for antigen presentation. It involves the transfer of intact peptide-MHC class I or II complexes from dead donor cells to DCs. DCs use these complexes to activate CD8+ or CD4+ T cells that are peptide-specific and restricted to the MHC genotype of the donor cells (144, 145). Although the role of cross-dressing in antitumor immunity requires further study, a recent work by Li et al. has shown that CD8α+ and CD103+ DCs induce proliferation of naïve and memory CD8+ T cells both by cross-dressing and cross-presentation of antigens (146). This novel mechanism could explain some of the observations of antigen transfer between host and injected DCs, or among different resident DCs subsets.
Analysis of Immunogenic Tumoral Antigens
Loading DCs with Tumoral Antigens
Tumor antigens originate from mutated or abnormally expressed endogenous proteins, including differentiation antigens, or are derived from viral proteins. The common strategy to load DCs with antigens is to use short peptides that, independent of the processing machinery, can bind directly to MHC molecules. However, the main drawback is the requirement of specific haplotypes to be identified for efficient antigen presentation. The utilization of longer peptides or whole proteins could overcome this problem and minimize tumor evasion.
Several tumor antigens are produced only by specific proteasomes (1, 147, 148). For example, cells exposed to IFN-γ express different proteasome subunits in addition to the standard counterparts and consequently a different spectrum of peptides with variable immunogenic potency (147). Mature DCs used for immunotherapy express immunoproteasomes and do not express constitutive proteasomes (149). Recent reports show that DC proteasome composition can be manipulated to increase antitumoral responses (150). In a phase I clinical trial, mature melanoma antigen-loaded human DCs were engineered to process antigens through constitutive proteasomes, proving to be superior inducers of antigen-specific T-cell immunity and clinical responses. As in this clinical trial, it becomes relevant to test the outcome of vaccination with functional assays to evaluate whether the induction of peptide-specific CD8+ T cell renders better tumor reactivity (151).
An obvious advantage in employing autologous tumor cells as antigen source for DCs is that they contain patient-specific mutated antigens that have not been subjected to central tolerance. It has been proven that mutated epitopes with single amino acid substitutions can provide tumor control (152). However, these formulations face significant difficulties, such as standardization, scarce autologous tumor cell samples, and technical issues regarding the establishment of successful cell cultures from those tumors. Consequently, the rationale for using allogeneic tumor cell lines instead of autologous tumor cells is that they share several common antigens that could serve as targets, generating comparable responses (153, 154).
Recurrence or relapse after therapies could be due to cancer stem cells being partially or totally untargeted by vaccination. Therefore, one novel and interesting approach is the use of isolated cancer stem cells from the tumor bulk as the source of antigens (155, 156). Mice vaccinated with DCs pulsed with cancer stem cell lysates significantly inhibited tumor growth and displayed higher amounts of lytic IgG antibodies and mononuclear cells (156).
Irradiation of tumor cells provides a safe source of antigens for DC uptake. Loading DCs with a mixture of apoptotic and necrotic cells induces phagocytosis and the upregulation of co-stimulatory molecules on DCs (131, 157), leading to improved protective antitumor immunity compared to other loading approaches (158). The presence of the remaining non-phagocytosed apoptotic/necrotic cells is relevant to the process of DC maturation (38, 131), and probably also for the recruitment of host CD207+ cells to the vaccination site and draining lymph nodes (39). Recently, radiation-mediated antitumor immunity has been found to require a STING DNA-sensing pathway in DCs, which mediates IFN-β DC production (159). The authors hypothesize that DNA from irradiated tumor cells could be delivered to DCs during cell–cell contact processes. Necrosis can be obtained by repeated freeze-thaw cycles but these lysates assayed as source of tumor antigens have provided discrepant results (157, 160–163). It is likely that their utility is influenced by tumor cell type and whether an immunogenic cell death was induced to generate the lysates. Consequently, and in opposition to the past dichotomy between apoptotic and necrotic tumor cells, approaches would have to be reviewed to evaluate whether the applied procedure renders an immunogenic source of antigens through an immunogenic cell death (164). The enhanced immunogenicity can be mediated by DAMPs being recognized by the PRRs expressed on immune cells (165), or by the presence of chaperone or heat shock proteins (HSPs). The intracellular stimulation of HSPs can be induced by various stressors (166, 167), and HSPs derived from stressed tumor cells are able to bind and transport tumor antigens to APCs. This approach turned out to be an interesting possibility to direct antigens to DCs or as an adjuvant therapy to enhance the immune visibility of poorly immunogenic TAAs (157, 162, 166, 168–170). The isolation method and the type of HSP enriched thereafter are crucial factors that may account for efficacy limitations (171). HSP70-peptide complexes derived from DC-tumor fusion cells are enriched in peptides with superior antigenic properties as compared to its tumor cell counterpart (172). Due to this variability, further experimental investigation is required in this promising field. Finally, DCs are also able to acquire antigens from live cells through a mechanism associated with trogocytosis, where individual DCs physically extract plasma membrane from other cells, generating endocytic vesicles up to 1 μm diameter (173). Along this line, authors assayed a vaccine preparation consisting of a 16-h incubation of DCs with live or apoptotic B16-F10 tumor cells, followed by CD11c+ cell enrichment and γ-irradiation before administration (44). Live tumor cells improved cross-presentation by DCs by maintaining antigens in a more native form than apoptotic cells. In a therapeutic assay of B16-F10 lung metastasis, mice were protected upon tumor challenge using the same approach (44, 45). In this study, tumoral protection correlated with lower levels of IL-10 and stronger tumor-specific CD8+ T cells response (45).
The origin of the APCs assayed in the vaccine formulation is also relevant. For this reason, allogeneic DC-based vaccines emerged as an alternative strategy to avoid the potential dysfunction of autologous DCs obtained from cancer patients. Allogeneic DCs can induce a stronger vaccine-specific immune response than syngeneic DCs (174). This is thought to trigger a broader T cell reactive repertoire, in which tumor-reactive T cells are generated by incidental cross-reactivity. The enhanced fraction of helper T cells (in response to alloantigens) leads to a better activation of specific CTLs. Stronger NK activity (175, 176), the presence of Th1-type cytokines, and the absence of the Th2-type cytokines IL-10 and IL-4 (177) were also reported. To simulate a clinical trial, Yasuda et al. analyzed the validity of semi-allogeneic DCs, where some MHC class I and II molecules are likely to be shared by recipients. This approach induced the most effective antitumor immune response in a therapeutic setting. While allo-MHC class II molecules may provide favorable T helper activity, it is likely that partial MHC class I matching is required to induce a CTL response. The flawed aspects of this approach are, on one hand, the possible promotion of regulatory T cell expansion, limiting antitumoral immunity via the suppression of not only T helper cells but also CTLs. On the other, repeated vaccination with allogeneic antigens could potentially elicit an alloreaction, which could blunt its immunizing potential (175). Therefore, a more efficient response will probably be provided by syngeneic cancer cells that express allogeneic MHC molecules, which could act as an adjuvant without counteracting the cancer-specific CTL response. Finally, DC culture conditions can introduce variability in relevant aspect as vaccine quality and immunogenic potency. Trivial conditions as detachment (178) and oxygen percentage (179) are able to alter the maturation state of DCs and antigen-specific CTL activation, respectively.
The fusion of DCs and tumor cells results in a heterokaryon without nuclear fusion, which includes molecules from DCs (MHC class I and II, and co-stimulatory molecules) and abundant TAAs that can be efficiently processed and presented by DC-presentation machinery (180). Several animal studies have demonstrated that DCs fused to tumor cells could be administered as a vaccine, eliciting protection upon challenge with tumor cells and regression of established tumors (181–187). Mice immunized with a fusion cell vaccine induced effective cellular and humoral responses against the antigen MUC1 in MUC1-transgenic mice, whose characteristic is to be unresponsive to the MUC1 antigen without a potent stimulation (188). Moreover, they conferred sufficient antitumor immunity to block or delay mammary tumor development in the same model of transgenic mice (189). When translated to human trials, this vaccine strategy might be improved with strategies to inhibit the immunosuppressive activity of Tregs or by combination with more conventional therapies (180). An interesting feature of these cells that is relevant to the clinical setting is that DC-tumor fusion cells could be efficiently frozen without loss of either antigen presentation potency or T-cell stimulatory capacity inducing polyclonal CTL responses (190).
Target Receptors for In Vivo Antigen Delivery
The development of antigen-coupled APC receptor-specific antibodies, single-chain variable fragments (scFv), and troybodies is another strategy to deliver TAAs to APCs in vivo. The ideal target receptor should present a specific pattern of expression to allow the exclusive targeting of APCs. It should be an endocytic receptor, and its activation should result in the presentation of antigen peptides via MHC molecules. Furthermore, in order to induce a potent immunogenic response to the delivered antigen, the process should lead to APC activation and maturation (191). A variety of target molecules have been assayed for mouse antitumor vaccines (MHC class II, CD11c, DEC205, DCIR2, Dectin-1/2, F4/80-like receptor, CIRE, mannose receptor, CD36, Clec 9A, MadCAM, CD80/CD86, CD40, Siglec-H). This strategy requires a lower antigen dosage than uncoupled peptides to stimulate immune responses in mice (42). The main drawback could be the lack of specificity of expression of the targeted molecule. By targeting XCR1, a chemokine receptor exclusively expressed on murine and human cross-presenting DCs, CD8+ T cell cytotoxicity could be successfully induced (192). This makes this specific and efficient approach, a very interesting candidate for the clinical setting.
Many of the studies were carried out employing proteins like OVA, BSA, or microbial protein or peptides. Further experimental vaccines using targeted delivery of TAAs may contribute to a better understanding of the potential benefit from this approach. One successful example of translational research is the use of HER2/neu as a target for breast cancer immunotherapy. The delivery of the HER2 antigen targets many DC surface molecules (CD80, CD86, CD11c, CD40, mannose, Fc-γ, and DEC-205), and results in potent immunization with significant CD8+ and IFN-γ+CD4+ T cell responses and cytokine secretion (IFN-γ, TNF-α, and IL-2) (43). More recently, a human trial employing an antibody targeted to DEC-205 fused with the tumor antigen NY-ESO-1 plus TLR agonists as adjuvants demonstrated to be a safe vaccine, effective to mount humoral and cellular response. The inclusion of immune checkpoint inhibitors to overcome the immunosuppressive tumor environment points this design as a promising immunotherapeutic strategy (193).
Exploring DC Response to Maturation Antigens and Vaccine-Adjuvant Combinations
Defining DC Maturation Status
Dentritic cell maturation is a very complex process involving diverse signaling pathways. It is characterized by the acquisition of distinctive functional properties involved in antigen processing and presentation, migration, and T cell co-stimulation (194). Costimulatory DC molecules, MHC-II, and CD40 are usually used to assess DC maturation status and immunostimulating potential. Nonetheless, these do not always correlate with the observed in vivo response. Recently, CD70, which binds to CD27 in T cells, has emerged as a very relevant T-cell costimulatory molecule (195, 196). Adoptively transferred CD70-expressing immature DCs were capable of priming CD8+ T cells into effectors, to control B16 melanoma tumor growth, to generate complete tumor rejection, and to induce memory CD8+ T cells (48). These results highlight the relevance of CD70 readout as an antitumoral efficacy DC marker.
On the other hand, without relying on a few specific markers which may not correlate with in vivo DC immunostimulating capacity, DC functionality can be evaluated as a whole by expression profiling. For example, in the steady state, a fraction of cDCs undergo homeostatic (tolerogenic) maturation, upregulating MHC class II molecules to almost the same levels as found under inflammatory conditions (197). Using murine models, it has been possible to isolate different DC subtypes involved in either homeostatic or TLR-induced (immunogenic) maturation, and to characterize the genes involved in these processes (63). Based on bioinformatic analysis, Dalod et al. describe a core set of genes induced in different DC subsets during both homeostatic and TLR-induced maturation (69), suggesting overlapping instructional signals in both maturation processes. On the other hand, there are certain genes that are differentially upregulated (63), and which could account for the difference in DC immunostimulating capacity. Analysis of expression profiles is becoming a useful tool to assess DC functionality, and could eventually lead to predict their immunotherapeutic potential.
Potentiating DC Immunotherapeutic Capacity
Tumor necrosis factor α (TNF-α) was one of the first agents assayed that was able to upregulate CD83 in DCs in vitro, and thereby improve DC T-cell stimulatory capacity (36). At this time, Jonuleit et al. developed a cytokine cocktail containing the proinflammatory cytokines IL-1β, IL-6, and TNF-α, and prostaglandin E2 (PGE2) (198), which was used for many years as the gold standard for DC maturation. Although this cocktail efficiently induced the upregulation of DC maturation markers and DC T-cell priming ability, there were impairments observed in IL-12p70 production by human DCs treated with this cocktail (199). IL-12 production is essential in DC-based cancer immunotherapy because of its important role promoting CD8+ T cell responses (200). It is tightly regulated, and recently it has been observed that for high expression levels both myeloid differentiation factor 88 (MyD88) and TRIF (TIR-domain-containing adapter-inducing IFN-β)-dependent pathways must be triggered simultaneously (195). Thus, other maturation cocktails are being explored to achieve an optimal IL-12-producing DC maturation.
Using murine models, it was shown that IL-12, as well as type I IFNs, induces a complex gene regulation program, involving chromatin remodeling and the induction of the transcription factors Eomes and T-bet, which are important for Th1 differentiation (201). Many DC-based vaccination studies have explored the use of IL-12 (186, 202). There have been dose-limiting toxicities associated with systemic IL-12p70 administration (203), leading to more careful analysis in preclinical models. The use of IL-12 adjuvancy has now advanced to clinical trials (204–206).
The hematopoietic growth factor and immune modulator, GM-CSF, is another of the most evident DC-based vaccine adjuvants, as it was the first cytokine described to efficiently promote DC development in vitro (207). GM-CSF has been shown to increase antitumoral effects when administered or produced locally at the vaccination site (208–210). Both GM-CSF and Flt3L are capable of inducing local and systemic expansion of DCs when used as adjuvants, but the antitumoral efficacy of GM-CSF is significantly higher (211).
The complex combination of TLR expression in different murine DC subsets and monocytes is essential for their functional specialization (63, 69). TLR triggering in DCs induces NF-kβ activation (212) and subsequently, the production of pro-inflammatory cytokines that are important not only for the innate immune response but also for T-cell polarization. TLR3, TLR7, TLR8, and TLR9 are intracellular TLRs recognizing nucleic acids. Nucleic-acid sensing TLRs and TLR4 are able to induce IRF3 or IRF7 activation, leading to type I IFN expression. Type I IFNs can induce Th1 differentiation and IFN-γ production (213), and thus cytotoxic CD8+ T cell responses. In particular, IFN-β signaling in CD8+ DCs has been found to be responsible for spontaneous tumor antigen-specific T cell priming and tumor rejection (105). Triggering TLR3, TLR7, and TLR9 can also enhance antigen cross-presentation by DCs (79, 214). Thus, triggering these nucleic-acid sensing TLRs in DCs is a good strategy to elicit potent antitumor responses, and several combinations and administration schedules are being evaluated.
A synergistic effect was observed when activating both TLR7 and TLR3 in GM-CSF-cultured bone-marrow derived DCs (215). DCs stimulated with R-848 (TLR7/8 agonist) and Poly I:C (TLR3 agonist) were stronger stimulators of specific CD8+ and CD4+ T cells in vitro and induced superior CTL priming in vivo. This was probably due to the simultaneous activation of the MyD88-independent pathway triggered by TLR3 and the MyD88-dependent pathway triggered by TLR7. In another work, Flt3-cultured bone marrow-derived DCs were also stimulated with various concentrations of several TLR agonists (37). Simultaneous stimulation of TLR4, which signals through MyD88 and/or TRIF, and TLR2 or TLR7, which signal through MyD88-dependent pathways, led to higher levels of IL-6 and IL12p70 production. Interestingly, they also observed a synergistically enhanced production of inflammatory cytokines when triggering TLR7 with TLR2 or TLR9, all of which signal through MyD88-dependent signaling pathways. Thus, it is important in order to maximize DC activation, to explore triggering simultaneously different TLRs and signaling pathways.
Due to their DC-stimulating capacity, TLR agonists have been assayed as adjuvants for many cancer immunotherapies. Above all, TLR7 agonists are especially interesting candidates because apart from activating APCs, they induce T-cell, NK, and NKT activation in vitro (46). Oral doses of the TLR7 agonist imiquimod generate antitumoral responses in several murine tumor models as MC-26 colon carcinoma, LLC, and RIF-1 sarcoma (216). Additionally, the combination of DC-based immunotherapies with peritumoral or topical imiquimod has also shown to be a successful approach (46, 217). Topical administration of imiquimod cream induces a strong inflammatory response in the skin and enhances migration of LCs or immature GM-CSF/IL-4-cultured bone marrow-derived DCs to the draining lymph nodes of treated mice (40, 41), inducing a potent CTL response (41). More recent works show that imiquimod skin treatment leads to local recruitment of pDCs and induces melanoma tumor regression (78). It was demonstrated that pDCs are required for the antitumoral imiquimod-mediated effect. Furthermore, the s.c. administration of a GM-CSF gene-transduced tumor vaccine (GVAX) and imiquimod induced the recruitment of activated pDCs to tumor vaccine sites and tumor draining lymph nodes, and elicited the suppression of tumor growth (210).
Sanchez et al. proved in vivo that OVA immunization with a combination of TLR and CD40 agonists generated more potent primary and memory CD8+ T cell responses that either agonist alone (49). They report a synergistic effect when combinations of TLR and CD40 agonists were administered i.p. together. They argue that the increase in classical DC activation markers, such as CD80, CD86, and CD40 induced by TLR agonists is necessary for the initial CD28-dependent CD8+ T cell stimulation, but that CD70 induction by anti-CD40 is needed to stimulate long-term memory. Furthermore, CD70 binding to CD27 in T cells was found to be critical for potent CD8+ T cell responses, since antigen-specific CD8+ T cell expansion in vivo was abrogated by CD70 blockage. The authors observed that the combined stimulation of TLR and CD40 pathways gives maximal CD70 expression. In a promising clinical trial for advanced melanoma, autologous DCs were electroporated with mRNA encoding CD40L, a constitutively active TLR4 and CD70 (TriMix), thereby improving DCs immunostimulatory capacity (218). In another melanoma clinical trial, autologous DCs were activated with CD40L and IFN-y, and a higher level of IL-12p70 production by patient’s DCs correlated with a better clinical outcome. Interestingly, IL-12p35 deficient production by some patient’s DCs could be corrected in vitro by using TLR agonists like poly I:C and R848.
Other TLR-independent innate signaling pathways were found to cooperate with the adaptive signaling CD40 pathway to induce CD70 expression in DCs and a potent CD8+ T cell response in vivo (50). These TLR-independent stimuli include type I IFNs and α-GalCer (219, 220). Maximal CD70 expression and CD8+ T cell memory were found to be induced when α-GalCer or type I IFNs were used in combination with anti-CD40 antibody (50). Therefore, CD40 triggering in combination with TLR agonists or TLR-independent stimuli like type I IFN and α-GalCer is a promising strategy for inducing DC maturation in cancer immunotherapy approaches.
In cancer, vaccine-induced immunity may be dampened by self-regulatory mechanisms, and adjuvants may exacerbate them. Indeed, interesting experiments in a mammary carcinoma murine model have shown that when imiquimod was administered intradermally with the self-antigen IGFBP-2, no antitumor effect was elicited due to the induction of a potent immunosuppressive regulatory response (221). Serum levels of IL-10 and systemic levels of myeloid-derived suppressor cells and Tregs were increased. Furthermore, when GM-CSF was applied as a sole adjuvant, it significantly inhibited tumor growth, but when combined i.d. with imiquimod, the antitumoral effect was abrogated. On the other hand, when imiquimod was used by the same authors in an exogenous OVA-peptide based immunization, DC maturation and mobilization, and OVA-specific CD8+ and CD4+ T cells were detected. This emphasizes the importance of extensive preclinical modeling in cancer vaccine development, and the need of models that do not rely on exogenous antigens in order to detect self-regulatory mechanisms.
Conclusion
Dendritic cell-based vaccination has the potential to make a difference in cancer treatment. It is not sufficient to be a safe approach and to elicit measurable immunological responses. A potent CD8+ T cell effector memory response should be triggered, able to control and eliminate tumors. The rationale to design these vaccines comes from clinical observations and, as depicted in this work, from pertinent investigations performed using murine models. However, each model has specific limitations and biases. Established cell lines, which are easily propagated and studied, transplanted into syngeneic mice have been utilized for many decades. Although they have answered many questions regarding DC functionality and DC use in cancer vaccines, their contribution to approved therapeutics has been limited. This could be due to the fact that heterogeneity and diversity of human cancers are not covered. Though more technically challenging, GEM models, which recapitulate genetic alterations present in human tumors, and humanized mice, which allow the study of human tumors in the context of their own immune system, are useful tools that will contribute to the design of more effective DC-based vaccines.
It is of great importance in order to break immune tolerance to the tumor to induce a proper activation of DCs by triggering several activation pathways. As discussed in this work, this can be done by ex vivo manipulation of DCs, or by using the appropriate adjuvants to boost DC response in vivo. The selection of adjuvant and site of administration will result in the activation of a distinct APC profile. Furthermore, given that synergy and cooperation between DC subsets has been observed, a vaccine design that targets several DC population presents itself as a potent immunization strategy. As shown in this work, DC-based therapies have been efficient in activating tumor-specific CD8+ T cells. In patients, analysis of the tumoral microenvironment has shown T-cell infiltration (222), thus immune failure appears to occur in the effector phase, with a dominant effect of inhibitory mechanisms within the tumor. As has been observed in a recent clinical trial (193), DC-based immunotherapies will surely benefit from combination with recently developed immunomodulatory agents that block negative regulators of T cell immunity, opening a promising field in cancer immunotherapy.
The results presented in this work show that many authors report in vitro immunological and short-term protection results using murine models. We conclude that this is not sufficient to draw relevant conclusions involving the efficacy of DC-based therapies. It is important to perform experiments analyzing long-term tumoral protection and memory CD8+ T-cell profile, and identifying the tumoral antigens that are able to generate long-lasting responses. To help the field move forward, it is vital to reach a consensus in this matter.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the following institutions: CONICET, Universidad de Buenos Aires, Agencia para el Desarrollo Científico y Tecnológico, Fundación SALES, Fundación para la Investigación y Prevención del Cáncer (FUCA), and Fundación María Calderón de la Barca, Argentina. This work benefited from data assembled by the ImmGen Consortium.
References
1. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14(2):135–46. doi:10.1038/nrc3670
2. Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol (2012) 33(7):364–72. doi:10.1016/j.it.2012.02.006
3. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25(2):214–21. doi:10.1016/j.coi.2012.12.003
4. Coulie PG, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med (1994) 180(1):35–42. doi:10.1084/jem.180.1.35
5. Parmiani G, De Filippo A, Novellino L, Castelli C. Unique human tumor antigens: immunobiology and use in clinical trials. J Immunol (2007) 178(4):1975–9. doi:10.4049/jimmunol.178.4.1975
6. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity (2013) 39(1):38–48. doi:10.1016/j.immuni.2013.07.004
7. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi:10.1038/32588
8. Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev (2006) 211:81–92. doi:10.1111/j.0105-2896.2006.00382.x
9. Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, et al. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol (2010) 185(9):4988–92. doi:10.4049/jimmunol.1002042
10. Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol (2015) 33:445–74. doi:10.1146/annurev-immunol-032414-112043
11. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol (2014) 15(7):e257–67. doi:10.1016/S1470-2045(13)70585-0
12. Butterfield LH. Dendritic cells in cancer immunotherapy clinical trials: are we making progress? Front Immunol (2013) 4:454. doi:10.3389/fimmu.2013.00454
13. Galluzzi L, Senovilla L, Vacchelli E, Eggermont A, Fridman WH, Galon J, et al. Trial watch: dendritic cell-based interventions for cancer therapy. Oncoimmunology (2012) 1(7):1111–34. doi:10.4161/onci.21494
14. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med (1973) 137(5):1142–62. doi:10.1084/jem.137.5.1142
15. Gross L. Intradermal immunization of C3H mice against a sarcoma that originated in an animal of the same line. Cancer Res (1943) 3:326–33.
16. Klein G, Klein E. Immune surveillance against virus-induced tumors and nonrejectability of spontaneous tumors: contrasting consequences of host versus tumor evolution. Proc Natl Acad Sci U S A (1977) 74(5):2121–5. doi:10.1073/pnas.74.5.2121
17. Cheon DJ, Orsulic S. Mouse models of cancer. Annu Rev Pathol (2011) 6:95–119. doi:10.1146/annurev.pathol.3.121806.154244
18. Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res (1978) 38(9):2651–60.
19. Nanni P, Colombo MP, De Giovanni C, Lollini PL, Nicoletti G, Parmiani G, et al. Impaired H-2 expression in B16 melanoma variants. J Immunogenet (1983) 10(5):361–70. doi:10.1111/j.1744-313X.1983.tb00348.x
20. Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol (2001) 20:20.1. doi:10.1002/0471142735.im2001s39
21. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417(6892):949–54. doi:10.1038/nature00766
22. Melnikova VO, Bolshakov SV, Walker C, Ananthaswamy HN. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene (2004) 23(13):2347–56. doi:10.1038/sj.onc.1207405
23. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. Braf (V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet (2009) 41(5):544–52. doi:10.1038/ng.356
24. Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, et al. Multiple murine BRaf (V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res (2014) 27(3):495–501. doi:10.1111/pcmr.12220
25. Rappaport A, Johnson L. Genetically engineered knock-in and conditional knock-in mouse models of cancer. Cold Spring Harb Protoc (2014) 2014(9):897–911. doi:10.1101/pdb.top069799
26. Williams SA, Anderson WC, Santaguida MT, Dylla SJ. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest (2013) 93(9):970–82. doi:10.1038/labinvest.2013.92
27. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol (2012) 12(11):786–98. doi:10.1038/nri3311
28. Sugamura K, Asao H, Kondo M, Tanaka N, Ishii N, Ohbo K, et al. The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu Rev Immunol (1996) 14:179–205. doi:10.1146/annurev.immunol.14.1.179
29. Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity (1995) 2(3):223–38. doi:10.1016/1074-7613(95)90047-0
30. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol (2005) 174(10):6477–89. doi:10.4049/jimmunol.174.10.6477
31. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol (2007) 7(2):118–30. doi:10.1038/nri2017
32. Spranger S, Frankenberger B, Schendel DJ. NOD/scid IL-2Rg (null) mice: a preclinical model system to evaluate human dendritic cell-based vaccine strategies in vivo. J Transl Med (2012) 10:30. doi:10.1186/1479-5876-10-30
33. Harui A, Kiertscher SM, Roth MD. Reconstitution of huPBL-NSG mice with donor-matched dendritic cells enables antigen-specific T-cell activation. J Neuroimmune Pharmacol (2011) 6(1):148–57. doi:10.1007/s11481-010-9223-x
34. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol (2014) 32(4):364–72. doi:10.1038/nbt.2858
35. Drake AC, Chen Q, Chen J. Engineering humanized mice for improved hematopoietic reconstitution. Cell Mol Immunol (2012) 9(3):215–24. doi:10.1038/cmi.2012.6
36. Morse MA, Zhou LJ, Tedder TF, Lyerly HK, Smith C. Generation of dendritic cells in vitro from peripheral blood mononuclear cells with granulocyte-macrophage-colony-stimulating factor, interleukin-4, and tumor necrosis factor-alpha for use in cancer immunotherapy. Ann Surg (1997) 226(1):6–16. doi:10.1097/00000658-199707000-00002
37. Mitchell D, Yong M, Schroder W, Black M, Tirrell M, Olive C. Dual stimulation of MyD88-dependent toll-like receptors induces synergistically enhanced production of inflammatory cytokines in murine bone marrow-derived dendritic cells. J Infect Dis (2010) 202(2):318–29. doi:10.1086/653499
38. Mac Keon S, Gazzaniga S, Mallerman J, Bravo AI, Mordoh J, Wainstok R. Vaccination with dendritic cells charged with apoptotic/necrotic B16 melanoma induces the formation of subcutaneous lymphoid tissue. Vaccine (2010) 28(51):8162–8. doi:10.1016/j.vaccine.2010.09.095
39. Ruiz MS, Mac Keon S, Campisano S, Bravo AI, Gazzaniga S, Wainstok R. CD207 (+) cells recruitment to the vaccination site and draining lymph nodes after the administration of DC-Apo/Nec vaccine in mice. Vaccine (2014) 32(11):1229–32. doi:10.1016/j.vaccine.2014.01.025
40. Suzuki H, Wang B, Shivji GM, Toto P, Amerio P, Tomai MA, et al. Imiquimod, a topical immune response modifier, induces migration of Langerhans cells. J Invest Dermatol (2000) 114(1):135–41. doi:10.1046/j.1523-1747.2000.00833.x
41. Nair S, McLaughlin C, Weizer A, Su Z, Boczkowski D, Dannull J, et al. Injection of immature dendritic cells into adjuvant-treated skin obviates the need for ex vivo maturation. J Immunol (2003) 171(11):6275–82. doi:10.4049/jimmunol.171.11.6275
42. Wei H, Wang S, Zhang D, Hou S, Qian W, Li B, et al. Targeted delivery of tumor antigens to activated dendritic cells via CD11c molecules induces potent antitumor immunity in mice. Clin Cancer Res (2009) 15(14):4612–21. doi:10.1158/1078-0432.CCR-08-3321
43. Wang B, Zaidi N, He LZ, Zhang L, Kuroiwa JM, Keler T, et al. Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Res (2012) 14(2):R39. doi:10.1186/bcr3135
44. Matheoud D, Perie L, Hoeffel G, Vimeux L, Parent I, Maranon C, et al. Cross-presentation by dendritic cells from live cells induces protective immune responses in vivo. Blood (2010) 115(22):4412–20. doi:10.1182/blood-2009-11-255935
45. Matheoud D, Baey C, Vimeux L, Tempez A, Valente M, Louche P, et al. Dendritic cells crosspresent antigens from live B16 cells more efficiently than from apoptotic cells and protect from melanoma in a therapeutic model. PLoS One (2011) 6(4):e19104. doi:10.1371/journal.pone.0019104
46. Ma F, Zhang J, Zhang C. The TLR7 agonists imiquimod and gardiquimod improve DC-based immunotherapy for melanoma in mice. Cell Mol Immunol (2010) 7(5):381–8. doi:10.1038/cmi.2010.30
47. Flacher V, Tripp CH, Mairhofer DG, Steinman RM, Stoitzner P, Idoyaga J, et al. Murine Langerin+ dermal dendritic cells prime CD8+ T cells while Langerhans cells induce cross-tolerance. EMBO Mol Med (2014) 6(9):1191–204. doi:10.15252/emmm.201303283
48. Keller AM, Xiao Y, Peperzak V, Naik SH, Borst J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood (2009) 113(21):5167–75. doi:10.1182/blood-2008-03-148007
49. Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J Immunol (2007) 178(3):1564–72. doi:10.4049/jimmunol.178.3.1564
50. McWilliams JA, Sanchez PJ, Haluszczak C, Gapin L, Kedl RM. Multiple innate signaling pathways cooperate with CD40 to induce potent, CD70-dependent cellular immunity. Vaccine (2010) 28(6):1468–76. doi:10.1016/j.vaccine.2009.11.071
51. Eberting CL, Shrayer DP, Butmarc J, Falanga V. Histologic progression of B16 F10 metastatic melanoma in C57BL/6 mice over a six week time period: distant metastases before local growth. J Dermatol (2004) 31(4):299–304. doi:10.1111/j.1346-8138.2004.tb00676.x
52. Bobek V, Kolostova K, Pinterova D, Kacprzak G, Adamiak J, Kolodziej J, et al. A clinically relevant, syngeneic model of spontaneous, highly metastatic B16 mouse melanoma. Anticancer Res (2010) 30(12):4799–803.
53. Xia S, Wei J, Wang J, Sun H, Zheng W, Li Y, et al. A requirement of dendritic cell-derived interleukin-27 for the tumor infiltration of regulatory T cells. J Leukoc Biol (2014) 95(5):733–42. doi:10.1189/jlb.0713371
54. Richmond A, Su Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Model Mech (2008) 1(2–3):78–82. doi:10.1242/dmm.000976
55. Oflazoglu E, Elliott M, Takita H, Ferrone S, Henderson RA, Repasky EA. Adoptively transferred human lung tumor specific cytotoxic T cells can control autologous tumor growth and shape tumor phenotype in a SCID mouse xenograft model. J Transl Med (2007) 5:29. doi:10.1186/1479-5876-5-29
56. Hylander BL, Punt N, Tang H, Hillman J, Vaughan M, Bshara W, et al. Origin of the vasculature supporting growth of primary patient tumor xenografts. J Transl Med (2013) 11:110. doi:10.1186/1479-5876-11-110
57. Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature (2014) 518:422–6. doi:10.1038/nature13952
58. Pino S, Brehm MA, Covassin-Barberis L, King M, Gott B, Chase TH, et al. Development of novel major histocompatibility complex class I and class II-deficient NOD-SCID IL2R gamma chain knockout mice for modeling human xenogeneic graft-versus-host disease. Methods Mol Biol (2010) 602:105–17. doi:10.1007/978-1-60761-058-8_7
59. Mishra S, Lavelle BJ, Desrosiers J, Ardito MT, Terry F, Martin WD, et al. Dendritic cell-mediated, DNA-based vaccination against hepatitis C induces the multi-epitope-specific response of humanized, HLA transgenic mice. PLoS One (2014) 9(8):e104606. doi:10.1371/journal.pone.0104606
60. Meixlsperger S, Leung CS, Ramer PC, Pack M, Vanoaica LD, Breton G, et al. CD141+ dendritic cells produce prominent amounts of IFN-alpha after dsRNA recognition and can be targeted via DEC-205 in humanized mice. Blood (2013) 121(25):5034–44. doi:10.1182/blood-2012-12-473413
61. Malissen B, Tamoutounour S, Henri S. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol (2014) 14(6):417–28. doi:10.1038/nri3683
62. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
63. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol (2012) 13(9):888–99. doi:10.1038/ni.2370
64. Belz GT, Nutt SL. Transcriptional programming of the dendritic cell network. Nat Rev Immunol (2012) 12(2):101–13. doi:10.1038/nri3149
65. Stoecklinger A, Eticha TD, Mesdaghi M, Kissenpfennig A, Malissen B, Thalhamer J, et al. Langerin+ dermal dendritic cells are critical for CD8+ T cell activation and IgH gamma-1 class switching in response to gene gun vaccines. J Immunol (2011) 186(3):1377–83. doi:10.4049/jimmunol.1002557
66. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315(5808):107–11. doi:10.1126/science.1136080
67. Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol (2007) 179(11):7577–84. doi:10.4049/jimmunol.179.11.7577
68. Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, et al. Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol (2005) 174(11):6592–7. doi:10.4049/jimmunol.174.11.6592
69. Dalod M, Chelbi R, Malissen B, Lawrence T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J (2014) 33(10):1104–16. doi:10.1002/embj.201488027
70. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity (2013) 38(5):970–83. doi:10.1016/j.immuni.2013.04.011
71. Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev (2010) 234(1):18–31. doi:10.1111/j.0105-2896.2009.00870.x
72. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206(13):3115–30. doi:10.1084/jem.20091756
73. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol (2004) 5(10):987–95. doi:10.1038/ni1112
74. Mitsui H, Watanabe T, Saeki H, Mori K, Fujita H, Tada Y, et al. Differential expression and function of toll-like receptors in Langerhans cells: comparison with splenic dendritic cells. J Invest Dermatol (2004) 122(1):95–102. doi:10.1046/j.0022-202X.2003.22116.x
75. Sancho D, Mourao-Sa D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest (2008) 118(6):2098–110. doi:10.1172/JCI34584
76. Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, et al. In vivo analysis of dendritic cell development and homeostasis. Science (2009) 324(5925):392–7. doi:10.1126/science.1170540
77. Heng TS, Painter MW. The immunological genome project: networks of gene expression in immune cells. Nat Immunol (2008) 9(10):1091–4. doi:10.1038/ni1008-1091
78. Drobits B, Holcmann M, Amberg N, Swiecki M, Grundtner R, Hammer M, et al. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J Clin Invest (2012) 122(2):575–85. doi:10.1172/JCI61034
79. Mouries J, Moron G, Schlecht G, Escriou N, Dadaglio G, Leclerc C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood (2008) 112(9):3713–22. doi:10.1182/blood-2008-03-146290
80. Guery L, Dubrot J, Lippens C, Brighouse D, Malinge P, Irla M, et al. Ag-presenting CpG-activated pDCs prime Th17 cells that induce tumor regression. Cancer Res (2014) 74(22):6430–40. doi:10.1158/0008-5472.CAN-14-1149
81. Liu C, Lou Y, Lizee G, Qin H, Liu S, Rabinovich B, et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J Clin Invest (2008) 118(3):1165–75. doi:10.1172/JCI33583
82. Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res (2004) 10(22):7466–74. doi:10.1158/1078-0432.CCR-04-0684
83. Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res (2012) 72(20):5188–97. doi:10.1158/0008-5472.CAN-11-3468
84. Wei S, Kryczek I, Zou L, Daniel B, Cheng P, Mottram P, et al. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res (2005) 65(12):5020–6. doi:10.1158/0008-5472.CAN-04-4043
85. Watkins SK, Zhu Z, Riboldi E, Shafer-Weaver KA, Stagliano KE, Sklavos MM, et al. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest (2011) 121(4):1361–72. doi:10.1172/JCI44325
86. Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3 (+) T-regulatory cells. Cancer Res (2012) 72(20):5240–9. doi:10.1158/0008-5472.CAN-12-2271
87. Sorrentino R, Morello S, Luciano A, Crother TR, Maiolino P, Bonavita E, et al. Plasmacytoid dendritic cells alter the antitumor activity of CpG-oligodeoxynucleotides in a mouse model of lung carcinoma. J Immunol (2010) 185(8):4641–50. doi:10.4049/jimmunol.1000881
88. Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res (2013) 73(15):4629–40. doi:10.1158/0008-5472.CAN-12-3058
89. Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol (2006) 177(5):3260–5. doi:10.4049/jimmunol.177.5.3260
90. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest (2007) 117(9):2570–82. doi:10.1172/JCI31911
91. Johnson BA III, Kahler DJ, Baban B, Chandler PR, Kang B, Shimoda M, et al. B-lymphoid cells with attributes of dendritic cells regulate T cells via indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A (2010) 107(23):10644–8. doi:10.1073/pnas.0914347107
92. Vinay DS, Kim CH, Chang KH, Kwon BS. PDCA expression by B lymphocytes reveals important functional attributes. J Immunol (2010) 184(2):807–15. doi:10.4049/jimmunol.0902528
93. Faget J, Bendriss-Vermare N, Gobert M, Durand I, Olive D, Biota C, et al. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer Res (2012) 72(23):6130–41. doi:10.1158/0008-5472.CAN-12-2409
94. Borkowski TA, Letterio JJ, Farr AG, Udey MC. A role for endogenous transforming growth factor beta 1 in Langerhans cell biology: the skin of transforming growth factor beta 1 null mice is devoid of epidermal Langerhans cells. J Exp Med (1996) 184(6):2417–22. doi:10.1084/jem.184.6.2417
95. Nagao K, Ginhoux F, Leitner WW, Motegi S, Bennett CL, Clausen BE, et al. Murine epidermal Langerhans cells and langerin-expressing dermal dendritic cells are unrelated and exhibit distinct functions. Proc Natl Acad Sci U S A (2009) 106(9):3312–7. doi:10.1073/pnas.0807126106
96. Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med (2007) 204(13):3119–31. doi:10.1084/jem.20071724
97. Ginhoux F, Collin MP, Bogunovic M, Abel M, Leboeuf M, Helft J, et al. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med (2007) 204(13):3133–46. doi:10.1084/jem.20071733
98. Douillard P, Stoitzner P, Tripp CH, Clair-Moninot V, Ait-Yahia S, McLellan AD, et al. Mouse lymphoid tissue contains distinct subsets of langerin/CD207 dendritic cells, only one of which represents epidermal-derived Langerhans cells. J Invest Dermatol (2005) 125(5):983–94. doi:10.1111/j.0022-202X.2005.23951.x
99. Flacher V, Douillard P, Ait-Yahia S, Stoitzner P, Clair-Moninot V, Romani N, et al. Expression of langerin/CD207 reveals dendritic cell heterogeneity between inbred mouse strains. Immunology (2008) 123(3):339–47. doi:10.1111/j.1365-2567.2007.02785.x
100. Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med (2010) 207(1):189–206. doi:10.1084/jem.20091964
101. Idoyaga J, Cheong C, Suda K, Suda N, Kim JY, Lee H, et al. Cutting edge: langerin/CD207 receptor on dendritic cells mediates efficient antigen presentation on MHC I and II products in vivo. J Immunol (2008) 180(6):3647–50. doi:10.4049/jimmunol.180.6.3647
102. Stoitzner P, Green LK, Jung JY, Price KM, Tripp CH, Malissen B, et al. Tumor immunotherapy by epicutaneous immunization requires Langerhans cells. J Immunol (2008) 180(3):1991–8. doi:10.4049/jimmunol.180.3.1991
103. Flacher V, Tripp CH, Stoitzner P, Haid B, Ebner S, Del Frari B, et al. Epidermal Langerhans cells rapidly capture and present antigens from C-type lectin-targeting antibodies deposited in the dermis. J Invest Dermatol (2010) 130(3):755–62. doi:10.1038/jid.2009.343
104. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322(5904):1097–100. doi:10.1126/science.1164206
105. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med (2011) 208(10):2005–16. doi:10.1084/jem.20101159
106. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208(10):1989–2003. doi:10.1084/jem.20101158
107. Azadmehr A, Pourfathollah AA, Amirghofran Z, Hassan ZM, Moazzeni SM. Enhancement of Th1 immune response by CD8alpha (+) dendritic cells loaded with heat shock proteins enriched tumor extract in tumor-bearing mice. Cell Immunol (2009) 260(1):28–32. doi:10.1016/j.cellimm.2009.07.003
108. Farrand KJ, Dickgreber N, Stoitzner P, Ronchese F, Petersen TR, Hermans IF. Langerin+ CD8alpha+ dendritic cells are critical for cross-priming and IL-12 production in response to systemic antigens. J Immunol (2009) 183(12):7732–42. doi:10.4049/jimmunol.0902707
109. Macho-Fernandez E, Cruz LJ, Ghinnagow R, Fontaine J, Bialecki E, Frisch B, et al. Targeted delivery of alpha-galactosylceramide to CD8alpha+ dendritic cells optimizes type I NKT cell-based antitumor responses. J Immunol (2014) 193(2):961–9. doi:10.4049/jimmunol.1303029
110. Arora P, Baena A, Yu KO, Saini NK, Kharkwal SS, Goldberg MF, et al. A single subset of dendritic cells controls the cytokine bias of natural killer T cell responses to diverse glycolipid antigens. Immunity (2014) 40(1):105–16. doi:10.1016/j.immuni.2013.12.004
111. Salio M, Silk JD, Cerundolo V. Recent advances in processing and presentation of CD1 bound lipid antigens. Curr Opin Immunol (2010) 22(1):81–8. doi:10.1016/j.coi.2009.12.008
112. Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood (2012) 119(25):6052–62. doi:10.1182/blood-2012-01-406967
113. Cerovic V, Houston SA, Scott CL, Aumeunier A, Yrlid U, Mowat AM, et al. Intestinal CD103 (-) dendritic cells migrate in lymph and prime effector T cells. Mucosal Immunol (2013) 6(1):104–13. doi:10.1038/mi.2012.53
114. Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med (2011) 208(9):1789–97. doi:10.1084/jem.20110538
115. Decrausaz L, Domingos-Pereira S, Duc M, Bobst M, Romero P, Schiller JT, et al. Parenteral is more efficient than mucosal immunization to induce regression of human papillomavirus-associated genital tumors. Int J Cancer (2011) 129(3):762–72. doi:10.1002/ijc.25973
116. Sandoval F, Terme M, Nizard M, Badoual C, Bureau MF, Freyburger L, et al. Mucosal imprinting of vaccine-induced CD8 (+) T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med (2013) 5(172):172ra20. doi:10.1126/scitranslmed.3004888
117. Kim-Schulze S, Kim HS, Wainstein A, Kim DW, Yang WC, Moroziewicz D, et al. Intrarectal vaccination with recombinant vaccinia virus expressing carcinoembronic antigen induces mucosal and systemic immunity and prevents progression of colorectal cancer. J Immunol (2008) 181(11):8112–9. doi:10.4049/jimmunol.181.11.8112
118. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell (2014) 26(5):638–52. doi:10.1016/j.ccell.2014.09.007
119. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41(5):830–42. doi:10.1016/j.immuni.2014.10.017
120. Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, Agace WW, et al. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med (2009) 206(13):3101–14. doi:10.1084/jem.20091925
121. Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity (2011) 35(5):780–91. doi:10.1016/j.immuni.2011.08.013
122. Guermonprez P, Khelef N, Blouin E, Rieu P, Ricciardi-Castagnoli P, Guiso N, et al. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha (M)beta (2) integrin (CD11b/CD18). J Exp Med (2001) 193(9):1035–44. doi:10.1084/jem.193.9.1035
123. Dadaglio G, Fayolle C, Zhang X, Ryffel B, Oberkampf M, Felix T, et al. Antigen targeting to CD11b+ dendritic cells in association with TLR4/TRIF signaling promotes strong CD8+ T cell responses. J Immunol (2014) 193(4):1787–98. doi:10.4049/jimmunol.1302974
124. Segura E, Amigorena S. Inflammatory dendritic cells in mice and humans. Trends Immunol (2013) 34(9):440–5. doi:10.1016/j.it.2013.06.001
125. Dominguez PM, Ardavin C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev (2010) 234(1):90–104. doi:10.1111/j.0105-2896.2009.00876.x
126. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity (2006) 24(6):801–12. doi:10.1016/j.immuni.2006.04.008
127. Segura E, Amigorena S. Identification of human inflammatory dendritic cells. Oncoimmunology (2013) 2(5):e23851. doi:10.4161/onci.23851
128. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38(4):729–41. doi:10.1016/j.immuni.2013.03.003
129. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med (1992) 176(6):1693–702. doi:10.1084/jem.176.6.1693
130. Labeur MS, Roters B, Pers B, Mehling A, Luger TA, Schwarz T, et al. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J Immunol (1999) 162(1):168–75.
131. Goldszmid RS, Idoyaga J, Bravo AI, Steinman R, Mordoh J, Wainstok R. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol (2003) 171(11):5940–7. doi:10.4049/jimmunol.171.11.5940
132. Campisano S, Mac Keon S, Gazzaniga S, Ruiz MS, Traian MD, Mordoh J, et al. Anti-melanoma vaccinal capacity of CD11c-positive and -negative cell populations present in GM-CSF cultures derived from murine bone marrow precursors. Vaccine (2013) 31(2):354–61. doi:10.1016/j.vaccine.2012.10.114
133. Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood (2000) 96(9):3029–39.
134. Chen W, Antonenko S, Sederstrom JM, Liang X, Chan AS, Kanzler H, et al. Thrombopoietin cooperates with FLT3-ligand in the generation of plasmacytoid dendritic cell precursors from human hematopoietic progenitors. Blood (2004) 103(7):2547–53. doi:10.1182/blood-2003-09-3058
135. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207(6):1261–71. doi:10.1084/jem.20092618
136. Balan S, Ollion V, Colletti N, Chelbi R, Montanana-Sanchis F, Liu H, et al. Human XCR1+ dendritic cells derived in vitro from CD34+ progenitors closely resemble blood dendritic cells, including their adjuvant responsiveness, contrary to monocyte-derived dendritic cells. J Immunol (2014) 193(4):1622–35. doi:10.4049/jimmunol.1401243
137. Lee J, Breton G, Oliveira TY, Zhou YJ, Aljoufi A, Puhr S, et al. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J Exp Med (2015) 212(3):385–99. doi:10.1084/jem.20141442
138. Petersen TR, Sika-Paotonu D, Knight DA, Simkins HM, Hermans IF. Exploiting the role of endogenous lymphoid-resident dendritic cells in the priming of NKT cells and CD8+ T cells to dendritic cell-based vaccines. PLoS One (2011) 6(3):e17657. doi:10.1371/journal.pone.0017657
139. Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity (2006) 25(1):153–62. doi:10.1016/j.immuni.2006.04.017
140. Kleindienst P, Brocker T. Endogenous dendritic cells are required for amplification of T cell responses induced by dendritic cell vaccines in vivo. J Immunol (2003) 170(6):2817–23. doi:10.4049/jimmunol.170.6.2817
141. Yewdall AW, Drutman SB, Jinwala F, Bahjat KS, Bhardwaj N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One (2010) 5(6):e11144. doi:10.1371/journal.pone.0011144
142. Ali OA, Emerich D, Dranoff G, Mooney DJ. In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci Transl Med (2009) 1(8):8ra19. doi:10.1126/scitranslmed.3000359
143. Lou Y, Liu C, Kim GJ, Liu YJ, Hwu P, Wang G. Plasmacytoid dendritic cells synergize with myeloid dendritic cells in the induction of antigen-specific antitumor immune responses. J Immunol (2007) 178(3):1534–41. doi:10.4049/jimmunol.178.3.1534
144. Dolan BP, Gibbs KD Jr, Ostrand-Rosenberg S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J Immunol (2006) 177(9):6018–24. doi:10.4049/jimmunol.177.9.6018
145. Dolan BP, Gibbs KD Jr, Ostrand-Rosenberg S. Tumor-specific CD4+ T cells are activated by “cross-dressed” dendritic cells presenting peptide-MHC class II complexes acquired from cell-based cancer vaccines. J Immunol (2006) 176(3):1447–55. doi:10.4049/jimmunol.176.3.1447
146. Li L, Kim S, Herndon JM, Goedegebuure P, Belt BA, Satpathy AT, et al. Cross-dressed CD8alpha+/CD103+ dendritic cells prime CD8+ T cells following vaccination. Proc Natl Acad Sci U S A (2012) 109(31):12716–21. doi:10.1073/pnas.1203468109
147. Groettrup M, Kirk CJ, Basler M. Proteasomes in immune cells: more than peptide producers? Nat Rev Immunol (2010) 10(1):73–8. doi:10.1038/nri2687
148. Guillaume B, Stroobant V, Bousquet-Dubouch MP, Colau D, Chapiro J, Parmentier N, et al. Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J Immunol (2012) 189(7):3538–47. doi:10.4049/jimmunol.1103213
149. Macagno A, Gilliet M, Sallusto F, Lanzavecchia A, Nestle FO, Groettrup M. Dendritic cells up-regulate immunoproteasomes and the proteasome regulator PA28 during maturation. Eur J Immunol (1999) 29(12):4037–42. doi:10.1002/(SICI)1521-4141(199912)29:12<4037::AID-IMMU4037>3.0.CO;2-T
150. Dannull J, Haley NR, Archer G, Nair S, Boczkowski D, Harper M, et al. Melanoma immunotherapy using mature DCs expressing the constitutive proteasome. J Clin Invest (2013) 123(7):3135–45. doi:10.1172/JCI67544
151. Butterfield LH, Palucka AK, Britten CM, Dhodapkar MV, Hakansson L, Janetzki S, et al. Recommendations from the iSBTc-SITC/FDA/NCI workshop on immunotherapy biomarkers. Clin Cancer Res (2011) 17(10):3064–76. doi:10.1158/1078-0432.CCR-10-2234
152. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res (2012) 72(5):1081–91. doi:10.1158/0008-5472.CAN-11-3722
153. Casey DG, Lysaght J, James T, Bateman A, Melcher AA, Todryk SM. Heat shock protein derived from a non-autologous tumour can be used as an anti-tumour vaccine. Immunology (2003) 110(1):105–11. doi:10.1046/j.1365-2567.2003.01726.x
154. Li B, Simmons A, Du T, Lin C, Moskalenko M, Gonzalez-Edick M, et al. Allogeneic GM-CSF-secreting tumor cell immunotherapies generate potent anti-tumor responses comparable to autologous tumor cell immunotherapies. Clin Immunol (2009) 133(2):184–97. doi:10.1016/j.clim.2009.07.008
155. Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res (2012) 72(7):1853–64. doi:10.1158/0008-5472.CAN-11-1400
156. Teitz-Tennenbaum S, Wicha MS, Chang AE, Li Q. Targeting cancer stem cells via dendritic-cell vaccination. Oncoimmunology (2012) 1(8):1401–3. doi:10.4161/onci.21026
157. Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med (2000) 191(3):423–34. doi:10.1084/jem.191.3.423
158. Fry TJ, Shand JL, Milliron M, Tasian SK, Mackall CL. Antigen loading of DCs with irradiated apoptotic tumor cells induces improved anti-tumor immunity compared to other approaches. Cancer Immunol Immunother (2009) 58(8):1257–64. doi:10.1007/s00262-008-0638-7
159. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity (2014) 41(5):843–52. doi:10.1016/j.immuni.2014.10.019
160. Schnurr M, Scholz C, Rothenfusser S, Galambos P, Dauer M, Robe J, et al. Apoptotic pancreatic tumor cells are superior to cell lysates in promoting cross-priming of cytotoxic T cells and activate NK and gammadelta T cells. Cancer Res (2002) 62(8):2347–52.
161. Inzkirweli N, Guckel B, Sohn C, Wallwiener D, Bastert G, Lindner M. Antigen loading of dendritic cells with apoptotic tumor cell-preparations is superior to that using necrotic cells or tumor lysates. Anticancer Res (2007) 27(4B):2121–9.
162. Yoon TJ, Kim JY, Kim H, Hong C, Lee H, Lee CK, et al. Anti-tumor immunostimulatory effect of heat-killed tumor cells. Exp Mol Med (2008) 40(1):130–44. doi:10.3858/emm.2008.40.1.130
163. Kawahara M, Takaku H. Intradermal immunization with combined baculovirus and tumor cell lysate induces effective antitumor immunity in mice. Int J Oncol (2013) 43(6):2023–30. doi:10.3892/ijo.2013.2125
164. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
165. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12(12):860–75. doi:10.1038/nrc3380
166. Todryk S, Melcher AA, Hardwick N, Linardakis E, Bateman A, Colombo MP, et al. Heat shock protein 70 induced during tumor cell killing induces Th1 cytokines and targets immature dendritic cell precursors to enhance antigen uptake. J Immunol (1999) 163(3):1398–408.
167. Calderwood SK, Theriault JR, Gong J. How is the immune response affected by hyperthermia and heat shock proteins? Int J Hyperthermia (2005) 21(8):713–6. doi:10.1080/02656730500340794
168. Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med (2000) 6(4):435–42. doi:10.1038/74697
169. Wang XY, Yi H, Yu X, Zuo D, Subjeck JR. Enhancing antigen cross-presentation and T-cell priming by complexing protein antigen to recombinant large heat-shock protein. Methods Mol Biol (2011) 787:277–87. doi:10.1007/978-1-61779-295-3_21
170. Jiang J, Xie D, Zhang W, Xiao G, Wen J. Fusion of Hsp70 to Mage-a1 enhances the potency of vaccine-specific immune responses. J Transl Med (2013) 11:300. doi:10.1186/1479-5876-11-300
171. McNulty S, Colaco CA, Blandford LE, Bailey CR, Baschieri S, Todryk S. Heat-shock proteins as dendritic cell-targeting vaccines – getting warmer. Immunology (2013) 139(4):407–15. doi:10.1111/imm.12104
172. Enomoto Y, Bharti A, Khaleque AA, Song B, Liu C, Apostolopoulos V, et al. Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells. J Immunol (2006) 177(9):5946–55. doi:10.4049/jimmunol.177.9.5946
173. Harshyne LA, Watkins SC, Gambotto A, Barratt-Boyes SM. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J Immunol (2001) 166(6):3717–23. doi:10.4049/jimmunol.166.6.3717
174. Toes RE, Blom RJ, van der Voort E, Offringa R, Melief CJ, Kast WM. Protective antitumor immunity induced by immunization with completely allogeneic tumor cells. Cancer Res (1996) 56(16):3782–7.
175. Yasuda T, Kamigaki T, Kawasaki K, Nakamura T, Yamamoto M, Kanemitsu K, et al. Superior anti-tumor protection and therapeutic efficacy of vaccination with allogeneic and semiallogeneic dendritic cell/tumor cell fusion hybrids for murine colon adenocarcinoma. Cancer Immunol Immunother (2007) 56(7):1025–36. doi:10.1007/s00262-006-0252-5
176. Merrick A, Diaz RM, O’Donnell D, Selby P, Vile R, Melcher A. Autologous versus allogeneic peptide-pulsed dendritic cells for anti-tumour vaccination: expression of allogeneic MHC supports activation of antigen specific T cells, but impairs early naive cytotoxic priming and anti-tumour therapy. Cancer Immunol Immunother (2008) 57(6):897–906. doi:10.1007/s00262-007-0426-9
177. Suzuki T, Fukuhara T, Tanaka M, Nakamura A, Akiyama K, Sakakibara T, et al. Vaccination of dendritic cells loaded with interleukin-12-secreting cancer cells augments in vivo antitumor immunity: characteristics of syngeneic and allogeneic antigen-presenting cell cancer hybrid cells. Clin Cancer Res (2005) 11(1):58–66.
178. Herzog GI, Solgi G, Wiegmann DS, Nienhaus C, Schrezenmeier H, Yildiz T, et al. Quality of tumor lysates used for pulsing dendritic cells is influenced by the method used to harvest adherent tumor cells. BMC Res Notes (2011) 4:153. doi:10.1186/1756-0500-4-153
179. Olin MR, Andersen BM, Zellmer DM, Grogan PT, Popescu FE, Xiong Z, et al. Superior efficacy of tumor cell vaccines grown in physiologic oxygen. Clin Cancer Res (2010) 16(19):4800–8. doi:10.1158/1078-0432.CCR-10-1572
180. Koido S, Homma S, Okamoto M, Namiki Y, Takakura K, Uchiyama K, et al. Strategies to improve the immunogenicity of anticancer vaccines based on dendritic cell/malignant cell fusions. Oncoimmunology (2013) 2(9):e25994. doi:10.4161/onci.25994
181. Tanaka Y, Koido S, Ohana M, Liu C, Gong J. Induction of impaired antitumor immunity by fusion of MHC class II-deficient dendritic cells with tumor cells. J Immunol (2005) 174(3):1274–80. doi:10.4049/jimmunol.174.3.1274
182. Ishida A, Tanaka H, Hiura T, Miura S, Watanabe S, Matsuyama K, et al. Generation of anti-tumour effector T cells from naive T cells by stimulation with dendritic/tumour fusion cells. Scand J Immunol (2007) 66(5):546–54. doi:10.1111/j.1365-3083.2007.02012.x
183. Tamai H, Watanabe S, Zheng R, Deguchi K, Cohen PA, Koski GK, et al. Effective treatment of spontaneous metastases derived from a poorly immunogenic murine mammary carcinoma by combined dendritic-tumor hybrid vaccination and adoptive transfer of sensitized T cells. Clin Immunol (2008) 127(1):66–77. doi:10.1016/j.clim.2007.12.001
184. Guo GH, Chen SZ, Yu J, Zhang J, Luo LL, Xie LH, et al. In vivo anti-tumor effect of hybrid vaccine of dendritic cells and esophageal carcinoma cells on esophageal carcinoma cell line 109 in mice with severe combined immune deficiency. World J Gastroenterol (2008) 14(8):1167–74. doi:10.3748/wjg.14.1167
185. Yamamoto M, Kamigaki T, Yamashita K, Hori Y, Hasegawa H, Kuroda D, et al. Enhancement of anti-tumor immunity by high levels of Th1 and Th17 with a combination of dendritic cell fusion hybrids and regulatory T cell depletion in pancreatic cancer. Oncol Rep (2009) 22(2):337–43. doi:10.3892/or_00000442
186. Tan C, Dannull J, Nair SK, Ding E, Tyler DS, Pruitt SK, et al. Local secretion of IL-12 augments the therapeutic impact of dendritic cell-tumor cell fusion vaccination. J Surg Res (2013) 185(2):904–11. doi:10.1016/j.jss.2013.06.045
187. Chen X, Liu Z, Huang Y, Li R, Zhang H, Dong S, et al. Superior anti-tumor protection and therapeutic efficacy of vaccination with dendritic cell/tumor cell fusion hybrids for murine Lewis lung carcinoma. Autoimmunity (2014) 47(1):46–56. doi:10.3109/08916934.2013.850080
188. Tanaka Y, Koido S, Chen D, Gendler SJ, Kufe D, Gong J. Vaccination with allogeneic dendritic cells fused to carcinoma cells induces antitumor immunity in MUC1 transgenic mice. Clin Immunol (2001) 101(2):192–200. doi:10.1006/clim.2001.5112
189. Chen D, Xia J, Tanaka Y, Chen H, Koido S, Wernet O, et al. Immunotherapy of spontaneous mammary carcinoma with fusions of dendritic cells and mucin 1-positive carcinoma cells. Immunology (2003) 109(2):300–7. doi:10.1046/j.1365-2567.2003.01656.x
190. Koido S, Hara E, Homma S, Torii A, Mitsunaga M, Yanagisawa S, et al. Streptococcal preparation OK-432 promotes fusion efficiency and enhances induction of antigen-specific CTL by fusions of dendritic cells and colorectal cancer cells. J Immunol (2007) 178(1):613–22. doi:10.4049/jimmunol.178.1.613
191. Caminschi I, Shortman K. Boosting antibody responses by targeting antigens to dendritic cells. Trends Immunol (2012) 33(2):71–7. doi:10.1016/j.it.2011.10.007
192. Hartung E, Becker M, Bachem A, Reeg N, Jakel A, Hutloff A, et al. Induction of potent CD8 T cell cytotoxicity by specific targeting of antigen to cross-presenting dendritic cells in vivo via murine or human XCR1. J Immunol (2015) 194(3):1069–79. doi:10.4049/jimmunol.1401903
193. Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med (2014) 6(232):232ra51. doi:10.1126/scitranslmed.3008068
194. Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell (2001) 106(3):255–8. doi:10.1016/S0092-8674(01)00449-4
195. Van Brussel I, Berneman ZN, Cools N. Optimizing dendritic cell-based immunotherapy: tackling the complexity of different arms of the immune system. Mediators Inflamm (2012) 2012:690643. doi:10.1155/2012/690643
196. Taraban VY, Rowley TF, Al-Shamkhani A. Cutting edge: a critical role for CD70 in CD8 T cell priming by CD40-licensed APCs. J Immunol (2004) 173(11):6542–6. doi:10.4049/jimmunol.173.11.6542
197. Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity (2004) 21(2):279–88. doi:10.1016/j.immuni.2004.06.014
198. Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol (1997) 27(12):3135–42. doi:10.1002/eji.1830271209
199. Zobywalski A, Javorovic M, Frankenberger B, Pohla H, Kremmer E, Bigalke I, et al. Generation of clinical grade dendritic cells with capacity to produce biologically active IL-12p70. J Transl Med (2007) 5:18. doi:10.1186/1479-5876-5-18
200. Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol (2003) 171(10):5165–71. doi:10.4049/jimmunol.171.10.5165
201. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol (2009) 183(3):1695–704. doi:10.4049/jimmunol.0900592
202. Zapala L, Wolny R, Wachowska M, Jakobisiak M, Lasek W. Synergistic antitumor effect of JAWSII dendritic cells and interleukin 12 in a melanoma mouse model. Oncol Rep (2013) 29(3):1208–14. doi:10.3892/or.2012.2193
203. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood (1997) 90(7):2541–8.
204. Carreno BM, Becker-Hapak M, Huang A, Chan M, Alyasiry A, Lie WR, et al. IL-12p70-producing patient DC vaccine elicits Tc1-polarized immunity. J Clin Invest (2013) 123(8):3383–94. doi:10.1172/JCI68395
205. Peterson AC, Harlin H, Gajewski TF. Immunization with Melan-A peptide-pulsed peripheral blood mononuclear cells plus recombinant human interleukin-12 induces clinical activity and T-cell responses in advanced melanoma. J Clin Oncol (2003) 21(12):2342–8. doi:10.1200/JCO.2003.12.144
206. Hamid O, Solomon JC, Scotland R, Garcia M, Sian S, Ye W, et al. Alum with interleukin-12 augments immunity to a melanoma peptide vaccine: correlation with time to relapse in patients with resected high-risk disease. Clin Cancer Res (2007) 13(1):215–22. doi:10.1158/1078-0432.CCR-06-1450
207. Inaba K, Steinman RM, Pack MW, Aya H, Inaba M, Sudo T, et al. Identification of proliferating dendritic cell precursors in mouse blood. J Exp Med (1992) 175(5):1157–67. doi:10.1084/jem.175.5.1157
208. Dranoff G. GM-CSF-secreting melanoma vaccines. Oncogene (2003) 22(20):3188–92. doi:10.1038/sj.onc.1206459
209. Ojima T, Iwahashi M, Nakamura M, Matsuda K, Naka T, Nakamori M, et al. The boosting effect of co-transduction with cytokine genes on cancer vaccine therapy using genetically modified dendritic cells expressing tumor-associated antigen. Int J Oncol (2006) 28(4):947–53. doi:10.3892/ijo.28.4.947
210. Narusawa M, Inoue H, Sakamoto C, Matsumura Y, Takahashi A, Inoue T, et al. TLR7 ligand augments GM-CSF-initiated antitumor immunity through activation of plasmacytoid dendritic cells. Cancer Immunol Res (2014) 2(6):568–80. doi:10.1158/2326-6066.CIR-13-0143
211. Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, Dranoff G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res (2000) 60(12):3239–46.
212. Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med (2007) 13(11):460–9. doi:10.1016/j.molmed.2007.09.002
213. Prchal M, Pilz A, Simma O, Lingnau K, von Gabain A, Strobl B, et al. Type I interferons as mediators of immune adjuvants for T- and B cell-dependent acquired immunity. Vaccine (2009) 27(Suppl 6):G17–20. doi:10.1016/j.vaccine.2009.10.016
214. Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, et al. A subset of toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol (2003) 170(8):4102–10. doi:10.4049/jimmunol.170.8.4102
215. Warger T, Osterloh P, Rechtsteiner G, Fassbender M, Heib V, Schmid B, et al. Synergistic activation of dendritic cells by combined toll-like receptor ligation induces superior CTL responses in vivo. Blood (2006) 108(2):544–50. doi:10.1182/blood-2005-10-4015
216. Sidky YA, Borden EC, Weeks CE, Reiter MJ, Hatcher JF, Bryan GT. Inhibition of murine tumor growth by an interferon-inducing imidazoquinolinamine. Cancer Res (1992) 52(13):3528–33.
217. Lee JR, Shin JH, Park JH, Song SU, Choi GS. Combined treatment with intratumoral injection of dendritic cells and topical application of imiquimod for murine melanoma. Clin Exp Dermatol (2007) 32(5):541–9. doi:10.1111/j.1365-2230.2007.02453.x
218. Wilgenhof S, Van Nuffel AM, Corthals J, Heirman C, Tuyaerts S, Benteyn D, et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother (2011) 34(5):448–56. doi:10.1097/CJI.0b013e31821dcb31
219. Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med (2004) 199(12):1607–18. doi:10.1084/jem.20040317
220. Taraban VY, Martin S, Attfield KE, Glennie MJ, Elliott T, Elewaut D, et al. Invariant NKT cells promote CD8+ cytotoxic T cell responses by inducing CD70 expression on dendritic cells. J Immunol (2008) 180(7):4615–20. doi:10.4049/jimmunol.180.7.4615
221. Dang Y, Wagner WM, Gad E, Rastetter L, Berger CM, Holt GE, et al. Dendritic cell-activating vaccine adjuvants differ in the ability to elicit antitumor immunity due to an adjuvant-specific induction of immunosuppressive cells. Clin Cancer Res (2012) 18(11):3122–31. doi:10.1158/1078-0432.CCR-12-0113
Keywords: cancer immunotherapy, dendritic cell-based vaccines, dendritic cells, dendritic cell maturation, dendritic cell subsets, adjuvants
Citation: Mac Keon S, Ruiz MS, Gazzaniga S and Wainstok R (2015) Dendritic cell-based vaccination in cancer: therapeutic implications emerging from murine models. Front. Immunol. 6:243. doi: 10.3389/fimmu.2015.00243
Received: 17 December 2014; Accepted: 06 May 2015;
Published: 20 May 2015
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Muriel Moser, Université Libre de Bruxelles, BelgiumMarc Dalod, Centre National de la Recherche Scientifique, France
Daniel Benitez-Ribas, CIBERehd, Spain
Copyright: © 2015 Mac Keon, Ruiz, Gazzaniga and Wainstok. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Wainstok, Laboratorio de Biología Tumoral, Departamento de Química Biológica IQUIBICEN-CONICET, Universidad de Buenos Aires (UBA), Intendente Güiraldes 2160, Buenos Aires C1428EGA, Argentina,cndhaW5zQHFiLmZjZW4udWJhLmFy