 Marian Nassef Kadry Naguib Roufaiel
Marian Nassef Kadry Naguib Roufaiel James W. Wells
James W. Wells Raymond J. Steptoe
Raymond J. Steptoe- The University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, Brisbane, QLD, Australia
Tumors can escape immune destruction through the development of antigen loss variants and loss of antigen processing/presentation pathways, thereby rendering them invisible to T cells. Alternatively, mechanisms of peripheral T-cell tolerance that would normally be important for protection from the development of autoimmunity may also be co-opted to (i) generate an immuno-inhibitory tumor environment, (ii) promote development of regulatory cell populations, or (iii) cell-intrinsically inactivate tumor-specific T cells. Emerging evidence suggests that T-cell function is impaired in hematological malignancies, which may manifest from cognate interactions between T cells and the tumor. The immunological synapse forms the cognate T-cell and antigen-presenting cell interaction and is the site where key signalling events, including those delivered by co-inhibitory receptors, that determine the fate of T cells occur. Here, we review evidence that events at the immune synapse between T cells and malignant B cells and alterations in immune synapse function may contribute to loss of T-cell function in B-cell malignancies.
B-Cell Lymphoma and T-Cell Responses
Lymphomas are a range of lymphoid tissue malignancies arising principally from B cells, but a minority (<10%) are derived from T cells (1). Broadly, two categories of B-cell lymphoma are recognized. Hodgkin’s lymphoma (HL), characterized by multinucleated Reed–Sternberg cells in affected sites, and non-Hodgkin’s lymphoma (NHL), which are highly diverse malignancies constituting up to 90% of lymphoma cases in developed countries. It was estimated that in the United States, lymphoma would represent approximately 5% of newly diagnosed cancers and account for approximately 18,990 deaths in 2014 (2, 3). In B-cell chronic lymphocytic leukemia (B-CLL), which is the most common chronic leukemia, malignant B cells accumulate in blood and bone marrow. While classified as different diseases, similar treatment challenges exist for B-cell lymphoma and B-CLL.
Modulation of T-Cell Responses by B-Cell Malignancies
A key risk factor for development of B-cell lymphoma is immunodeficiency or immune suppression (4). Patients with HIV-1 and AIDS and children with primary immunodeficiency diseases have elevated rates of B-lymphoma, and an aggressive form of EBV+ve lymphoma, post-transplant lymphoproliferative disease (PTLD), is common in immunosuppressed transplant recipients (5–7). These observations strongly suggest that effective T-cell responses are required for prevention and control of B-cell malignancies. Emergence of antigen loss variants and disruption of antigen processing/presentation pathways can contribute to immune escape of B-lymphomas (8–11). In addition, metabolically hostile and immune-suppressive tumor microenvironments (12–15), and/or expansion or induction of immunosuppressive cells, such as regulatory T cells (16), myeloid-derived suppressor cells, and immunosuppressive macrophages (17), may also play a role. However, compelling evidence suggests that B-cell malignancies induce T-cell intrinsic alterations resulting in loss of T-cell function. In EBV+ve lymphoma, T-cell responses to EBV proteins serve as surrogates for tumor-specific responses and are likely indicators of the response of T-cells to non-viral lymphoma antigens. CD8+ T-cell responses to latency-phase proteins expressed by EBV+ve lymphomas, such as EBNA-1, are reduced in patients with endemic Burkitt’s lymphoma (BL), whereas responses to lytic or latency phase proteins not expressed by the tumors are preserved (18). Similarly, expression of T-cell “exhaustion” markers in Hodgkin lymphoma (HL) is associated with loss of function in EBV-specific CD8+ T cells (19). In patients with EBV+ve nasopharyngeal carcinoma, the absolute frequency of LMP1-, LMP2-, and EBNA-1-specific CD8+ T cells is reduced in blood (20, 21), and these T cells appear to be functionally inactivated at the tumor site (22). These all suggest strong inhibition of T-cell responses specific for B lymphoma antigens. Tumor-induced T-cell exhaustion is well-characterized in many solid tumors [reviewed in Ref. (23, 24)], leading to similar patterns of tumor-specific T-cell dysfunction. Taking melanoma as an example, tumor infiltrating T cells appear to be rendered unresponsive locally in the tumor bed (25), and this can be associated with poor responses to adoptive immunotherapy (26), substantial expression of co-inhibitory molecules by tumor-specific T cells (27, 28), and transcriptional profiles consistent with exhaustion (29). Overall, these observations are consistent with tumor-associated T-cell dysfunction and tumor-specific tolerance.
B Cells as Tolerogenic Antigen-Presenting Cells
Effector and memory differentiation of T cells results when co-stimulatory receptors (CD28, CD27, 41BB etc) are ligated by the high levels of ligands on activated antigen-presenting cells (APC) during cognate activation. But, in the absence of activation by pathogen- or danger-associated signals, APC provide insufficient signals for full T-cell activation and the outcome is peripheral T-cell tolerance. While DC are well recognized as potently tolerogenic cells in the steady-state (30, 31), reports extending back to the 1990s imply B cells are also tolerogenic (32–34). Some reports describe a direct role for B cells in peripheral deletion of naive CD8+ T cells, possibly by CD95-mediated effects (35). Others demonstrate contributions from both deletion and inactivation for naïve CD8+ T cells (36) and abortive proliferation appears to be a key requirement for tolerance induction (33, 36). More recent evidence suggests B cells may also inactivate memory CD4+ T cells (37). Moreover, we have shown that B cells expressing cognate antigens rapidly inactivate memory CD8+ T cells and CTL (38). Although “regulation” [e.g., through induction of regulatory T cells (39–41)] may be induced by B cells under certain conditions, T-cell intrinsic deletion and induction of unresponsiveness (anergy) are prominent when T cells interact with tolerogenic B cells. A subpopulation of IL-10-producing regulatory B cells (Breg) has also been described [reviewed in Ref. (42)]. These observations all suggest that B-cell lymphomas are potentially highly tolerogenic.
Preclinical B-lymphoma models in mice typically employ A20 lymphoma cells or transgenic Eμ-driven oncogenes such as c-Myc, and may express model neo-antigens like OVA in order to permit analysis of the impact on T cells (43). Although innate immune mechanisms impact on B-lymphoma in pre-clinical models, it is clear that inactivation of CD4+ and CD8+ T cells specific for tumor-expressed antigens also occurs (44–49). In models described to date, tolerance mechanisms are similar to those described for “tolerogenic B cells” with deletion and induction of unresponsiveness playing key roles (43–49). Whether the tumor cells themselves are tolerogenic or whether other APC presenting tumor-derived antigens are the proximal APC has been addressed. Whereas, host BM-derived APC appear to be required for CD4+ T cell tolerance in lymphoma models (44–46); it appears CD8+ T cells directly interact with antigen-expressing tumor cells (47–49). In fact, for CD8+ T cells, lymphoma cells appear to be the proximal APC for tolerance induction (49). Remarkably, OVA-specific CTL adoptively transferred into mice bearing OVA-expressing Eμ-myc lymphoma cells are rapidly deleted or rendered unresponsive (47, 49). This is remarkably similar to our own findings when CTL are transferred into mice where OVA is expressed within non-malignant B cells (38), which might suggest that this is an intrinsic outcome following the interaction between B cells and CTL.
Immunological Synapse Structure and Function
A critical component of the interaction between T-cells and APC is formation of the immunological synapse (IS), defined as the contact area between a T cell and an APC presenting a peptide ligand. Various aspects of IS structure and function have been reviewed in detail (50–53), but a brief introduction will be provided here. Upon TCR ligation by pMHC, nanoclusters containing TCR begin to assemble around the initial site of APC/T-cell contact and these increase in size to form microclusters (MC) of TCR and associated molecules such as co-stimulatory receptors (e.g., CD28), tyrosine kinases (Lck and ZAP70), serine kinases [protein kinase C (PKC-θ)], and adaptor molecules (LAT, SLP76) (54). TCR MC begin to move toward the center of the ring of contact between the APC and T cell (Figure 1) where they aggregate to form a central supramolecular activation cluster (cSMAC) (55).
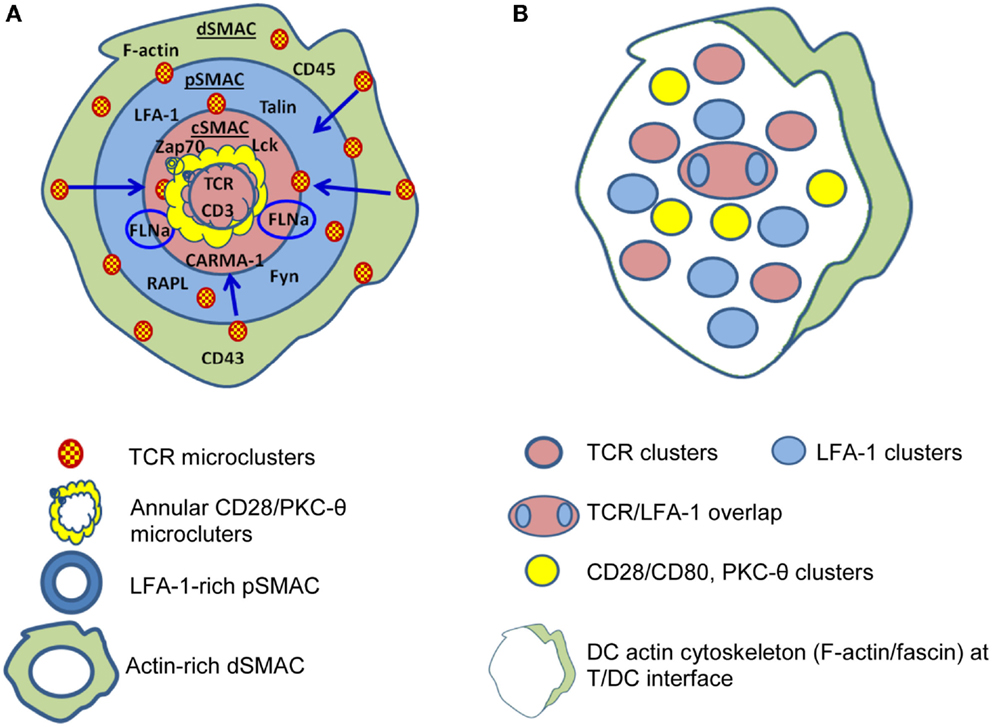
Figure 1. Differences are apparent between immunological synapses formed by B cells and dendritic cells (DC). (A) B cells, B cell tumors, and lipid bilayers form classical “bulls-eye” immunological synapses. TCR-containing microclusters form in the dSMAC, contain Lck and ZAP70, protein kinase C (PKC-θ), LAT, SLP76 etc., and migrate centripetally through the LFA-1-rich pSMAC to the cSMAC. The cSMAC is segregated into a central CD3hi region where CD3 accumulates and TCR is internalized to resulting in termination of TCR signaling and an outer CD3lo region where the signaling molecules accumulate either in conjugation [annular CD28/PKC-θ conjugates and PKC-θ/filaminA (FLNa) clusters] or separately. (B) DC typically form “multifocal” synapses where TCR-containing clusters are segregated from CD28/PKC-θ containing clusters and no clear “ring” of LFA-1 is formed. TCR signaling stabilizes the multifocal structure, particularly the CD28/PKC-θ containing clusters. A prominent polarization of the DC cytoskeleton is often present at the periphery. Based on Ref. (51, 53, 55–57) (A); (58–60) (B).
The cSMAC is surrounded by a ring of lymphocyte function associated antigen-1/intercellular adhesion molecule (LFA-1/ICAM) making up the peripheral SMAC (pSMAC) (55, 61, 62). After initial antigen recognition, the IS is stabilized by TCR-induced increases in LFA-1 affinity (63, 64). The pSMAC also contains LFA-1-associated proteins that regulate LFA-1 adhesion (55, 65) and LFA-1 here serves a crucial role in T-cell function by integrating internal cytoskeletal dynamics with the external environment (64, 66). This is mediated through, among other pathways, the actions of talin, an actin adapter protein, and RAPL, a Rap1 effector (55, 65) that modulate LFA-1 adhesion. In addition to its adhesive function, LFA-1 may be important by promoting pMHC/TCR localization to, and CD45 exclusion from, the cSMAC (67).
The pSMAC is surrounded by a more distal ring (dSMAC) containing membrane proteins with large ectodomains such as CD43 and CD45 (50, 68). The dSMAC appears to be the site of initial pMHC/TCR MC formation and, once formed, MC move centripetally through the pSMAC, facilitated by the concurrent centripetal movement of LFA-1/ICAM (69), to accumulate in the cSMAC (54, 70). Ca++ mobilization studies indicate TCR signaling commences with TCR MC formation in the dSMAC, and as MC move toward the cSMAC associations with ZAP70, Lck, LAT, and SLP76 are lost suggesting that by the time MC arrive at the cSMAC signaling capacity is lost (70). Additionally, MC in the cSMAC co-localize with markers of protein degradation and ubiquitinylation including Cbl-b (54, 71), a known inhibitor of TCR signaling. The cSMAC is also the site of TCR internalization for degradation (54, 72). Consistent with these observations, there is growing recognition that the cSMAC is a site for signal termination rather than stabilization of TCR signaling as originally thought [reviewed in Ref. (73)]. TCR signaling is initiated by the CD4 or CD8 co-receptors binding to the MHC molecules presenting cognate peptide, which activates the co-receptor-associated tyrosine kinase Lck. This in turn phosphorylates ITAM motifs within CD3-ζ. The tandem SH2-domains of ZAP-70 become engaged by the bi-phosphorylated ITAMs of CD3-ζ, and this then arranges ZAP-70 in a way that leads to phosphorylation of the transmembrane protein linker of activated T cells (LAT). Phosphorylated LAT, in turn, serves as a docking site to which a number of signaling proteins bind including SLP-76, which leads to signaling by the Ras-Erk pathway, and Ca++ flux [reviewed in Ref. (74)] and, ultimately, transcription of a range of gene products including those of immediate/early genes c-Fos, c-myc, c-jun, NF-AT, and NF-κB that ultimately lead to expression of IL-2, IL-2R, and other molecules that allow T cells to proliferate, differentiate, and exert effector function (75, 76).
An important point when considering the IS is that our understanding has been largely defined using in vitro models, some employing “artificial” APC, and hence, differences may exist between these and in vivo settings. For example, substantial differences in IS structure exist between different T-cell/APC combinations [reviewed in Ref. (53)]. Whereas classic “bulls-eye” IS are formed for T cell/B cell contacts (55, 77) and have been considered the “archetypal” IS, multifocal IS are characteristic of the interactions of DC with naive and activated CD4+ and CD8+ T cells, for example Ref. (58–60). Additionally, T-cell/DC conjugates develop in the absence of antigen (78) whereas T-cell/B-cell interactions do not (79). Interestingly, the antigen requirement for cytoskeletal rearrangement differs between T cells and DC. Naive CD4+ cytoskeletal polarization occurs during DC/T interactions in the absence of antigen, DC cytoskeletal polarization, and the formation of fully developed “multifocal” IS requires the presence of cognate pMHC (58, 80), suggesting that rearrangements in DC may be driven by the T cell.
B-Lymphoma Induced Alterations in IS Formation
The “bulls-eye” IS formed between T cells and B cells (77) or B cell tumors (55) potentially favors damping of TCR signaling (73), but it is possible that altered IS formation by malignant B cells could contribute to perturbations of T-cell function. Indeed, altered IS formation between T cells and superantigen-pulsed malignant or healthy B cells has been observed in follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL) (81), and in B-CLL (82, 83) as well as a mouse model of B-CLL (84). From these studies, it appears that several critical steps during and subsequent to IS formation are altered.
Events Occurring within the cSMAC and Signaling Zone
Phosphorylation of ZAP-70 is crucial for signaling downstream of TCR. In the absence of ZAP-70 activity, formation of TCR/CD3ζ clusters and exclusion of CD43 from the cSMAC proceeds, but TCR-induced microtubule organizing center (MTOC) polarization and overall actin cytoskeletal changes and recruitment of signaling molecules such as PKC-θ and LAT to the T-cell/APC interface are impaired (85, 86). Interestingly, alterations in IS formation by CD4+ or CD8+ T cells from FL, DLBCL, and B-CLL (81–84) resemble those that occur in the absence of ZAP-70 activity (85, 86). For example, T cell/B cell conjugate formation rate is reduced and F-actin polymerization at the IS substantially impaired in CD4+ and CD8+ T cells isolated from tumor sites or the blood of leukemic-phase FL patients compared to healthy T cells or circulating T cells from non-leukemic phase FL (81). Disruptions in actin-based motility and cytoskeleton polarization have also been observed in acute myeloid leukemia (AML) (87).
Immunological synapse defects appear to be induced by tumor cells themselves, as impaired IS formation is induced in healthy allogeneic T lymphocytes by direct contact with FL, DLBCL, or B-CLL cells tumor cells (81, 82). Exposure to malignant B cells resulted in reduced recruitment of LFA-1 (particularly the high-affinity form), Lck, tyrosine-phosphorylated protein, Itk, filamin-A, and Rab27A to T-cell/APC contact sites (82), and these changes were apparent on re-culture with healthy B cells. Associated with this, functional alterations extended to reduced IL-2 production and CTL activity in T cells exposed to FL, DLBCL, or B-CLL cells (81, 82). Cell–cell contact was required and prevention of cell adhesion during the primary exposure to malignant B cells eliminated the effect (81, 82). These data suggest that interaction with malignant B cells could induce long-lived changes in T cells and, consistent with this, altered gene expression patterns have been detected in CD4+ and CD8+ T cells recovered from B-CLL patients and in tumor-infiltrating lymphocytes in FL (83, 88). Interestingly, the immunomodulatory drug lenalidomide, which shows effectiveness in B-lymphoma alone (89–91) or combined with Rituximab (92–94), could reinstate F-actin polymerization and signaling at the IS (81, 82).
Co-Inhibitory Molecules within the IS
CTLA4 and PD-1 are co-inhibitory receptors that negatively regulate T-cell activation and act within the IS (Figure S1 in Supplementary Material). Their actions at the IS level may differ depending on the state of T-cell differentiation and the extent and site of ligand expression (95). If ligated during the initial activation of naive T cells by professional APC, co-inhibitory receptors can impart long-lived inhibitory effects on T-cell function (96, 97). While the effects of CTLA4 ligation may be most profound during the initial development of a T-cell response when priming is occurring in lymphoid tissues, PD-1 in addition to effects during priming, powerfully modulates effector responses in an apparently reversible manner (98). CTLA4 is normally stored in secretory granules but traffics to the cSMAC upon TCR activation (99) and accumulation at the cSMAC is required for its inhibitory function (100). CTLA4 has a higher affinity for CD80 and CD86 than CD28 and competes with CD28 resulting in termination of PKC-θ-mediated NF-κB signaling (100, 101), mainly through the prevention of the recruitment of the downstream scaffolding signaling protein, CARMA-1 to the cSMAC, which is critical for the NF-κB signaling pathway activation (102). Unlike CD28, CTLA-4 trafficking to the IS is directly related to the strength of TCR signaling, with higher levels occurring when TCR signal strength is greatest (99). CTLA4 has been reported to be strongly expressed by T cells in HL (103), and may contribute to damping of T-cell function. In line with this, administration of CTLA4-blocking antibodies such as Ipilimumab appears to have antitumor activity in DLBCL and FL patients (104) and following HSCT for HL and mantle cell lymphoma (105). Although testing of anti-CTLA4 appears limited, it is currently being tested in combination with anti-PD-1. Interestingly, polymorphisms of CTLA4 have been associated with increased susceptibility to NHL in some populations (106).
PD-1 is expressed by antigen-stimulated T cells and, in chronic viral infection, contributes to T-cell “exhaustion” (98, 107), where blockade can reinvigorate T-cell function, allowing expansion and production of effector cytokines (108, 109). Other co-inhibitory molecules appear to work in a similar way and PD-1 can act in conjunction with other co-inhibitory receptors (23). Expression of co-inhibitory receptor ligands such as PD-L1 by tumors is associated with poor prognosis (110–113). For example, PD-L1 is over-expressed in DLBCL and may contribute to poor outcomes (114, 115). In mantle cell lymphoma, PD-L1 expression inhibits T-cell proliferation and T-cell lytic activity (116). Similar results have been reported in a murine AML model (117). Engagement of PD-1 concurrently with TCR ligation impairs TCR-induced phosphorylation of CD3ζ, ZAP-70, and PKC-θ (118). PD-1 expressed on the surface of effector T cells is recruited to TCR MC upon their formation and is translocated to the cSMAC within the MC (119) and the higher the ligand availability, the more localization of PD-1 at the IS (120). During this process, SHP-2 is recruited to the immunoreceptor tyrosine-based switch motif (ITSM) of PD-1, which in turn causes dephosphorylation of TCR proximal signaling molecules within MC (119) impairing TCR-induced “stop” signals required for T-cell activation (121). Blockade of PD-1 ligation partially restores IS formation between healthy T cells and CLL cells (122). Blockade of co-inhibitory receptor/ligand interactions through a PD-1 antibody promotes T-cell function and immune-clearance of solid tumors (123, 124), and early indications suggest a similar effect in FL (125) and HSCT for relapsed or refractory DLBCL (126). The use of anti-PD-1 antibody has been extended and combined with the anti-CD20 Ab Rituximab in relapsed FL (127). Generally, the use of PD-1 blockage has shown promising outcomes in the case of lymphomas (128).
Events Outside the cSMAC
Malignant B cell-induced alterations in the IS extend beyond the cSMAC (Figure 2). Stabilization of pSMAC LFA-1/ICAM-1 interactions are impaired in T cells from FL and B-CLL patients (81, 82). Alterations in Rho-GTPase signaling that likely underlie these IS alterations (83, 129) also appear to perturb LFA-1 mediated migration. Perhaps, more pertinent for the topic under consideration, effective LFA-1/ICAM interactions are required for memory T-cell differentiation. In the absence of ICAM-1-mediated stabilization of the IS, long-lived T-cell/DC conjugates are reduced in frequency (130). While this has little effect on T-cell activation, proliferation, and cytotoxicity, a key outcome is failure of activated T cells to develop effective memory populations and clonal deletion of activated T cells (130). It is plausible if perturbed LFA-1/ICAM-1 interactions led to similar outcomes in human T-cells, this could underlie the reduction in frequency and loss of responsiveness of EBV-reactive T cells in EBV+ve lymphoma.
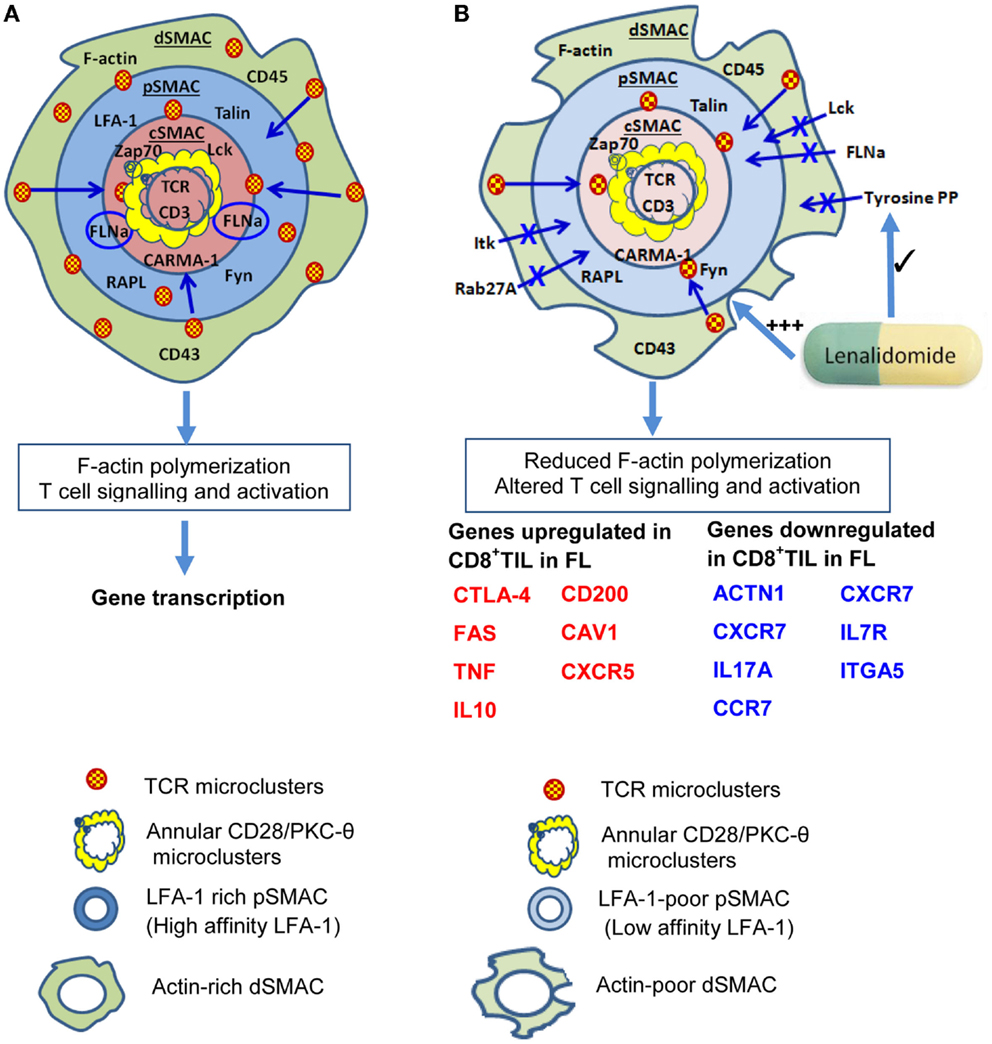
Figure 2. Impaired immunological synapse formation at the T-cell/B-lymphoma interface. (A) The stylized, classical “bulls-eye” IS as depicted in Figure 1. (B) During interaction with B lymphoma cells, IS formation is altered and a reduction in actin cytoskeletal remodeling is evident in the dSMAC. This likely leads to reduced formation and centripetal migration of TCR/CD28 microclusters. As a result of altered cytoskeletal remodeling, dSMAC formation is incomplete, and Lck, Itk, filaminA (FLNa), tyrosine-phosphorylated protein, and Rab27A recruitment to the IS are also reduced. Consequently, the pSMAC contains reduced amounts of LFA-1 and this is primarily the low affinity form which destabilizes T-cell: B-lymphoma conjugate formation. This leads to reduced tyrosine phosphorylation and TCR signaling leading to altered downstream gene transcription. Reduced TCR signaling could also perpetuate impaired IS formation through reduced ZAP-70 signaling. Increased transcription of genes encoding some molecules that inhibit T-cell activity or regulate motility or division (e.g., CTLA-4,FAS, TNF, IL10, CD200) occurs in CD8+ tumor-infiltrating lymphocytes (CD8+ TIL) as well as reduced transcription of some molecules that contribute to efficient IS formation [e.g., actinin-1 (ACTN1)], IL7R, CCR7, ITGA5. Lenalidomide acts to restore F-actin polymerization, rho-GTPase signaling, recruitment of tyrosine-phosphorylated protein (tyrosine PP), and also improves conjugate formation between T cells and B lymphoma cells. Based on Ref. (51, 53, 55–57) (A); (81, 88) (B).
Future Directions
Understanding the mechanistic origins of IS alterations in lymphoma is an area that could significantly advance therapy. Transcriptional profiling has provided insight into pathways through which altered IS structure and function are potentially established and downstream effects mediated (83, 88). Several areas of investigation are likely to be fruitful, but fundamental questions remain. We have principally discussed the role of lymphoma cells as APC for T-cell activation, but clearance of B-cell malignancies also requires CTL recognition of malignant cells. This is an understudied area, and dissecting the role of malignant B cells as “activating APC” for CTL will require further sophisticated studies.
It is intriguing, however, to consider whether antigen-specific tolerance mechanisms contribute and whether this could be a cause or consequence of altered IS function. An outstanding question is whether functional alterations observed in T cells is a global effect or the consequence of cognate tumor interactions that affects only tumor-antigen specific T cells. For instance, does impaired IS formation occur during the primary interactions of T cells with malignant B cells in a way that programs subsequent outcomes for those T cells? Impaired priming of T cells to a model antigen in a mouse model of B-CLL (84) suggests global effects, and clinical (82) and mouse studies (38, 47, 49) suggest tumor burden is an underlying determinant of the effect. Rituximab treatment restores immune responsiveness in FL in keeping with a suggestion that reduction in tumor burden may reduce the effect on T-cell dysfunction (131). On the other hand, some mouse studies indicate that T-cell dysfunction is restricted specifically to T-cells that display specificity for lymphoma cells (49), indicating tumor antigen-specificity of the effect, and T-cell dysfunction in B-lymphoma shows some evidence of specificity for tumor-derived antigens (18–22). Many of the IS alterations reported for T cells from lymphoma patients could be caused by proximal defects in TCR signaling (38, 132) found in tolerant T cells. Tolerant T-cells demonstrate impaired translocation of ZAP70, LAT, and phospholipase C γ1, into the IS and IS formation (133–137). Further investigation may reveal whether antigen dose/affinity effects on ZAP-70 signaling and TCR damping molecule recruitment (71, 138, 139) or modulation of lipid rafts (140–142) underlie some of the effects observed. In vitro visualization of the defects of the IS and testing the capacity of pharmacological agents such as lenalidomide (89–91) or co-inhibitory receptor blockade to modulate this, using live cell and confocal microscopy, might be a promising transitional step for a more advanced understanding.
Summary
It is apparent many processes are perturbed at the IS in B-lymphoma. Several of these processes may act in concert to inhibit generation of effective T-cell responses to malignant B cells. Alternatively, a small number of processes with widespread influences may underlie the changes observed. Further characterization is required to determine whether “defects” observed are “downstream” of other tumor effects or whether the alteration in IS function described is the primary cause of failure of effective T-cell immunosurveillance. This is an area that could provide useful insights for the development of more effective therapies for B-cell and other malignancies.
Author Contributions
MR, JW, and RS wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RS is a recipient of Australian Research Council Future Fellowship (FT110100372). JW was supported in part by grants from the University of Queensland and Cancer Council Queensland, and by a Perpetual Trustees Fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2015.00258/abstract
References
1. Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer (2005) 5:251–62. doi: 10.1038/nrc1589
2. National Cancer Institute Non-Hodgkin Lymphoma. Available from: http://www.cancer.gov/cancertopics/types/non-hodgkin (accessed October 27 2014)
3. National Cancer Institute. A Snapshot of Lymphoma. Available from: http://www.cancer.gov/researchandfunding/snapshots/lymphoma (accessed December 22 2014).
5. Goedert JJ, Cote TR, Virgo P, Scoppa SM, Kingma DW, Gail MH, et al. Spectrum of AIDS-associated malignant disorders. Lancet (1998) 351:1833–9. doi:10.1016/S0140-6736(97)09028-4
6. Hadden JW. Immunodeficiency and cancer: prospects for correction. Int Immunopharmacol (2003) 3:1061–71. doi:10.1016/s1567-5769(03)00060-2
7. Bollard CM, Rooney CM, Heslop HE. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol (2012) 9:510–9. doi:10.1038/nrclinonc.2012.111
8. Guy K, Krajewski AS, Dewar AE. Expression of MHC class II antigens in human B-cell leukaemia and non-Hodgkin’s lymphoma. Br J Cancer (1986) 53:161–73. doi:10.1038/bjc.1986.31
9. Moller P, Lammler B, Herrmann B, Otto HF, Moldenhauer G, Momburg F. The primary mediastinal clear cell lymphoma of B-cell type has variable defects in MHC antigen expression. Immunology (1986) 59:411–7.
10. Booman M, Szuhai K, Rosenwald A, Hartmann E, Kluin-Nelemans H, de Jong D, et al. Genomic alterations and gene expression in primary diffuse large B-cell lymphomas of immune-privileged sites: the importance of apoptosis and immunomodulatory pathways. J Pathol (2008) 216:209–17. doi:10.1002/path.2399
11. Rimsza LM, Roberts RA, Miller TP, Unger JM, LeBlanc M, Braziel RM, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood (2004) 103:4251–8. doi:10.1182/blood-2003-07-2365
12. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72:2746–56. doi:10.1158/0008-5472.can-11-1272
13. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. doi:10.1038/nrc1478
14. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202:931–9. doi:10.1084/jem.20050715
15. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi:10.1038/nm934
16. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6:295–307. doi:10.1038/nri1806
17. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
18. Moormann AM, Heller KN, Chelimo K, Embury P, Ploutz-Snyder R, Otieno JA, et al. Children with endemic Burkitt lymphoma are deficient in EBNA1-specific IFN-gamma T cell responses. Int J Cancer (2009) 124:1721–6. doi:10.1002/ijc.24014
19. Gandhi MK, Lambley E, Duraiswamy J, Dua U, Smith C, Elliott S, et al. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood (2006) 108:2280–9. doi:10.1182/blood-2006-04-015164
20. Li J, Zeng XH, Mo HY, Rolén U, Gao YF, Zhang XS, et al. Functional inactivation of EBV-specific T-lymphocytes in nasopharyngeal carcinoma: implications for tumor immunotherapy. PLoS One (2007) 2:e1122. doi:10.1371/journal.pone.0001122
21. Fogg MH, Wirth LJ, Posner M, Wang F. Decreased EBNA-1-specific CD8+ T cells in patients with Epstein-Barr virus-associated nasopharyngeal carcinoma. Proc Natl Acad Sci U S A (2009) 106:3318–23. doi:10.1073/pnas.0813320106
22. Frisan T, Sjoberg J, Dolcetti R, Boiocchi M, De Re V, Carbone A, et al. Local suppression of Epstein-Barr virus (EBV)-specific cytotoxicity in biopsies of EBVpositive Hodgkin’s disease. Blood (1995) 86:1493–501.
23. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
24. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25:214–21. doi:10.1016/j.coi.2012.12.003
25. Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Lienard D, Lejeune F, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res (2004) 64:2865–73. doi:10.1158/0008-5472.CAN-03-3066
26. Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol (2005) 175:6169–76. doi:10.4049/jimmunol.175.9.6169
27. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi:10.1182/blood-2008-12-195792
28. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res (2012) 72:887–96. doi:10.1158/0008-5472.can-11-2637
29. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest (2011) 121:2350–60. doi:10.1172/jci46102
30. Steptoe RJ, Ritchie JM, Wilson NS, Villadangos JA, Lew AM, Harrison LC. Cognate CD4+ help elicited by resting dendritic cells does not impair the induction of peripheral tolerance in CD8+ T cells. J Immunol (2007) 178:2094–103. doi:10.4049/jimmunol.178.4.2094
31. Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med (2002) 196:1627–38. doi:10.1084/jem.20021598
32. Buhlmann JE, Foy TM, Aruffo A, Crassi KM, Ledbetter JA, Green WR, et al. In the absence of a CD40 signal, B cells are tolerogenic. Immunity (1995) 2:645–53. doi:10.1016/1074-7613(95)90009-8
33. Townsend SE, Goodnow CC. Abortive proliferation of rare T cells induced by direct or indirect antigen presentation by rare B cells in vivo. J Exp Med (1998) 187:1611–21. doi:10.1084/jem.187.10.1611
34. Eynon EE, Parker DC. Parameters of tolerance induction by antigen targeted to B lymphocytes. J Immunol (1993) 151:2958–64.
35. Bennett SR, Carbone FR, Toy T, Miller JF, Heath WR. B cells directly tolerize CD8(+) T cells. J Exp Med (1998) 188:1977–83. doi:10.1084/jem.188.11.1977
36. Werner-Klein M, Dresch C, Marcon IP, Brocker T. Transcriptional targeting of B cells for induction of peripheral CD8 T cell tolerance. J Immunol (2007) 178:7738–46. doi:10.4049/jimmunol.178.12.7738
37. Dalai SK, Mirshahidi S, Morrot A, Zavala F, Sadegh-Nasseri S. Anergy in memory CD4+ T cells is induced by B cells. J Immunol (2008) 181:3221–31. doi:10.4049/jimmunol.181.5.3221
38. Kenna TJ, Waldie T, McNally A, Thomson M, Yagita H, Thomas R, et al. Targeting antigen to diverse APCs inactivates memory CD8+ T cells without eliciting tissue-destructive effector function. J Immunol (2010) 184:598–606. doi:10.4049/jimmunol.0900032
39. Morlacchi S, Soldani C, Viola A, Sarukhan A. Self-antigen presentation by mouse B cells results in regulatory T-cell induction rather than anergy or clonal deletion. Blood (2011) 118:984–91. doi:10.1182/blood-2011-02-336115
40. Lee KM, Stott RT, Zhao G, SooHoo J, Xiong W, Lian MM, et al. TGF-beta-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol (2014) 44:1728–36. doi:10.1002/eji.201344062
41. Nouel A, Pochard P, Simon Q, Segalen I, Le Meur Y, Pers JO, et al. B-Cells induce regulatory T cells through TGF-beta/IDO production in A CTLA-4 dependent manner. J Autoimmun (2015) 59:53–60. doi:10.1016/j.jaut.2015.02.004
42. Candando KM, Lykken JM, Tedder TF. B10 cell regulation of health and disease. Immunol Rev (2014) 259:259–72. doi:10.1111/imr.12176
43. Donnou S, Galand C, Touitou V, Saut‘es-Fridman C, Fabry Z, Fisson S. Murine models of B-Cell lymphomas: promising tools for designing cancer therapies. Adv Hematol (2012) 2012:701704. doi:10.1155/2012/701704
44. Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, et al. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc Natl Acad Sci U S A (1998) 95:1178–83. doi:10.1073/pnas.95.3.1178
45. Horna P, Cuenca A, Cheng F, Brayer J, Wang HW, Borrello I, et al. In vivo disruption of tolerogenic cross-presentation mechanisms uncovers an effective T-cell activation by B-cell lymphomas leading to antitumor immunity. Blood (2006) 107:2871–8. doi:10.1182/blood-2005-07-3014
46. Sotomayor EM, Borrello I, Rattis FM, Cuenca AG, Abrams J, Staveley-O’Carroll K, et al. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood (2001) 98:1070–7. doi:10.1182/blood.V98.4.1070
47. Prato S, Mintern JD, Lahoud MH, Huang DC, Villadangos JA. Induction of antigen-specific effector-phase tolerance following vaccination against a previously ignored B-cell lymphoma. Immunol Cell Biol (2011) 89:595–603. doi:10.1038/icb.2010.131
48. Gerbitz A, Sukumar M, Helm F, Wilke A, Friese C, Fahrenwald C, et al. Stromal interferon-γ signaling and cross-presentation are required to eliminate antigen-loss variants of B cell lymphomas in mice. PLoS One (2012) 7:e34552. doi:10.1371/journal.pone.0034552
49. Prato S, Zhan Y, Mintern JD, Villadangos JA. Rapid deletion and inactivation of CTLs upon recognition of a number of target cells over a critical threshold. J Immunol (2013) 191:3534–44. doi:10.4049/jimmunol.1300803
50. Dustin ML. A dynamic view of the immunological synapse. Semin Immunol (2005) 17:400–10. doi:10.1016/j.smim.2005.09.002
51. Yokosuka T, Saito T. The immunological synapse, TCR microclusters, and T cell activation. Curr Top Microbiol Immunol (2010) 340:81–107. doi:10.1007/978-3-642-03858-7_5
52. Dustin ML, Chakraborty AK, Shaw AS. Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol (2010) 2:a002311. doi:10.1101/cshperspect.a002311
53. Thauland TJ, Parker DC. Diversity in immunological synapse structure. Immunology (2010) 131:466–72. doi:10.1111/j.1365-2567.2010.03366.x
54. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity (2006) 25:117–27. doi:10.1016/j.immuni.2006.04.010
55. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature (1998) 395:82–6. doi:10.1038/25764
56. Hayashi K, Altman A. Filamin A is required for T cell activation mediated by protein kinase C-theta. J Immunol (2006) 177:1721–8. doi:10.4049/jimmunol.177.3.1721
57. Yokosuka T, Kobayashi W, Sakata-Sogawa K, Takamatsu M, Hashimoto-Tane A, Dustin ML, et al. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity (2008) 29:589–601. doi:10.1016/j.immuni.2008.08.011
58. Brossard C, Feuillet V, Schmitt A, Randriamampita C, Romao M, Raposo G, et al. Multifocal structure of the T cell – dendritic cell synapse. Eur J Immunol (2005) 35:1741–53. doi:10.1002/eji.200425857
59. Fisher PJ, Bulur PA, Vuk-Pavlovic S, Prendergast FG, Dietz AB. Dendritic cell microvilli: a novel membrane structure associated with the multifocal synapse and T-cell clustering. Blood (2008) 112:5037–45. doi:10.1182/blood-2008-04-149526
60. Tseng SY, Waite JC, Liu M, Vardhana S, Dustin ML. T cell-dendritic cell immunological synapses contain TCR-dependent CD28-CD80 clusters that recruit protein kinase C theta. J Immunol (2008) 181:4852–63. doi:10.4049/jimmunol.181.7.4852
61. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285:221–7. doi:10.1126/science.285.5425.221
62. Sims TN, Dustin ML. The immunological synapse: integrins take the stage. Immunol Rev (2002) 186:100–17. doi:10.1034/j.1600-065X.2002.18610.x
63. Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature (1989) 341:619–24. doi:10.1038/341619a0
64. Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol (2012) 24:107–15. doi:10.1016/j.ceb.2011.10.004
65. Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat Immunol (2003) 4:741–8. doi:10.1038/ni950
66. Smith A, Stanley P, Jones K, Svensson L, McDowall A, Hogg N. The role of the integrin LFA-1 in T-lymphocyte migration. Immunol Rev (2007) 218:135–46. doi:10.1111/j.1600-065X.2007.00537.x
67. Graf B, Bushnell T, Miller J. LFA-1-mediated T cell costimulation through increased localization of TCR/class II complexes to the central supramolecular activation cluster and exclusion of CD45 from the immunological synapse. J Immunol (2007) 179:1616–24. doi:10.4049/jimmunol.179.3.1616
68. Leupin O, Zaru R, Laroche T, Muller S, Valitutti S. Exclusion of CD45 from the T-cell receptor signaling area in antigen-stimulated T lymphocytes. Curr Biol (2000) 10:277–80. doi:10.1016/S0960-9822(00)00362-6
69. Hartman NC, Nye JA, Groves JT. Cluster size regulates protein sorting in the immunological synapse. Proc Natl Acad Sci U S A (2009) 106:12729–34. doi:10.1073/pnas.0902621106
70. Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol (2005) 6:1253–62. doi:10.1038/ni1272
71. Vardhana S, Choudhuri K, Varma R, Dustin ML. Essential role of ubiquitin and TSG101 protein in formation and function of the central supramolecular activation cluster. Immunity (2010) 32:531–40. doi:10.1016/j.immuni.2010.04.005
72. Valitutti S, Muller S, Salio M, Lanzavecchia A. Degradation of T cell receptor (TCR)-CD3-zeta complexes after antigenic stimulation. J Exp Med (1997) 185:1859–64. doi:10.1084/jem.185.10.1859
73. Alarcon B, Mestre D, Martinez-Martin N. The immunological synapse: a cause or consequence of T-cell receptor triggering? Immunology (2011) 133:420–5. doi:10.1111/j.1365-2567.2011.03458.x
74. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol (2009) 27:591–619. doi:10.1146/annurev.immunol.021908.132706
75. Marrack P, Mitchell T, Hildeman D, Kedl R, Teague TK, Bender J, et al. Genomic-scale analysis of gene expression in resting and activated T cells. Curr Opin Immunol (2000) 12:206–9. doi:10.1016/S0952-7915(99)00075-8
76. Rangel C, Angus J, Ghahramani Z, Lioumi M, Sotheran E, Gaiba A, et al. Modeling T-cell activation using gene expression profiling and state-space models. Bioinformatics (2004) 20:1361–72. doi:10.1093/bioinformatics/bth093
77. Reichardt P, Dornbach B, Rong S, Beissert S, Gueler F, Loser K, et al. Naive B cells generate regulatory T cells in the presence of a mature immunologic synapse. Blood (2007) 110:1519–29. doi:10.1182/blood-2006-10-053793
78. Revy P, Sospedra M, Barbour B, Trautmann A. Functional antigen-independent synapses formed between T cells and dendritic cells. Nat Immunol (2001) 2:925–31. doi:10.1038/ni713
79. Delon J, Bercovici N, Raposo G, Liblau R, Trautmann A. Antigen-dependent and -independent Ca2+ responses triggered in T cells by dendritic cells compared with B cells. J Exp Med (1998) 188:1473–84. doi:10.1084/jem.188.8.1473
80. Al-Alwan MM, Liwski RS, Haeryfar SM, Baldridge WH, Hoskin DW, Rowden G, et al. Cutting edge: dendritic cell actin cytoskeletal polarization during immunological synapse formation is highly antigen-dependent. J Immunol (2003) 171:4479–83. doi:10.4049/jimmunol.171.9.4479
81. Ramsay AG, Clear AJ, Kelly G, Fatah R, Matthews J, Macdougall F, et al. Follicular lymphoma cells induce T-cell immunologic synapse dysfunction that can be repaired with lenalidomide: implications for the tumor microenvironment and immunotherapy. Blood (2009) 114:4713–20. doi:10.1182/blood-2009-04-217687
82. Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest (2008) 118:2427–37. doi:10.1172/jci35017
83. Gorgun G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest (2005) 115:1797–805. doi:10.1172/jci24176
84. Gorgun G, Ramsay AG, Holderried TA, Zahrieh D, Le Dieu R, Liu F, et al. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc Natl Acad Sci U S A (2009) 106:6250–5. doi:10.1073/pnas.0901166106
85. Blanchard N, Di Bartolo V, Hivroz C. In the immune synapse, ZAP-70 controls T cell polarization and recruitment of signaling proteins but not formation of the synaptic pattern. Immunity (2002) 17:389–99. doi:10.1016/S1074-7613(02)00421-1
86. Morgan MM, Labno CM, Van Seventer GA, Denny MF, Straus DB, Burkhardt JK. Superantigen-induced T cell:B cell conjugation is mediated by LFA-1 and requires signaling through Lck, but not ZAP-70. J Immunol (2001) 167:5708–18. doi:10.4049/jimmunol.167.10.5708
87. Le Dieu R, Taussig DC, Ramsay AG, Mitter R, Miraki-Moud F, Fatah R, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood (2009) 114:3909–16. doi:10.1182/blood-2009-02-206946
88. Kiaii S, Clear AJ, Ramsay AG, Davies D, Sangaralingam A, Lee A, et al. Follicular lymphoma cells induce changes in T-cell gene expression and function: potential impact on survival and risk of transformation. J Clin Oncol (2013) 31:2654–61. doi:10.1200/jco.2012.44.2137
89. Wiernik PH, Lossos IS, Tuscano JM, Justice G, Vose JM, Cole CE, et al. Lenalidomide monotherapy in relapsed or refractory aggressive non-Hodgkin’s lymphoma. J Clin Oncol (2008) 26:4952–7. doi:10.1200/jco.2007.15.3429
90. Witzig TE, Wiernik PH, Moore T, Reeder C, Cole C, Justice G, et al. Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin’s Lymphoma. J Clin Oncol (2009) 27:5404–9. doi:10.1200/jco.2008.21.1169
91. Zinzani PL, Vose JM, Czuczman MS, Reeder CB, Haioun C, Polikoff J, et al. Long-term follow-up of lenalidomide in relapsed/refractory mantle cell lymphoma: subset analysis of the NHL-003 study. Ann Oncol (2013) 24:2892–7. doi:10.1093/annonc/mdt366
92. Wang M, Fowler N, Wagner-Bartak N, Feng L, Romaguera J, Neelapu SS, et al. Oral lenalidomide with rituximab in relapsed or refractory diffuse large cell, follicular and transformed lymphoma: a phase II clinical trial. Leukemia (2013) 27:1902–9. doi:10.1038/leu.2013.95
93. Ahmadi T, Chong EA, Gordon A, Aqui NA, Nasta SD, Svoboda J, et al. Combined lenalidomide, low-dose dexamethasone, and rituximab achieves durable responses in rituximab-resistant indolent and mantle cell lymphomas. Cancer (2014) 120:222–8. doi:10.1002/cncr.28405
94. Wang M, Fayad L, Wagner-Bartak N, Zhang L, Hagemeister F, Neelapu SS, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol (2012) 13:716–23. doi:10.1016/s1470-2045(12)70200-0
95. Blake SJ, Ching ALH, Kenna TJ, Galea R, Large J, Yagita H, et al. Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS One (2015) 10:e0119483. doi:10.1371/journal.pone.0119483
96. Goldberg MV, Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM, et al. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood (2007) 110:186–92. doi:10.1182/blood-2006-12-062422
97. Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol (1999) 162:5813–20.
98. Barber DL, Wherry EJ, Masupost D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8+ T cells during chronic viral infection. Nature (2006) 439:682–7. doi:10.1038/nature04444
99. Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity (2002) 16:23–35. doi:10.1016/S1074-7613(01)00259-X
100. Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity (2010) 33:326–39. doi:10.1016/j.immuni.2010.09.006
101. Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, et al. The interaction properties of costimulatory molecules revisited. Immunity (2002) 17:201–10. doi:10.1016/S1074-7613(02)00362-X
102. Huse M. The T-cell-receptor signaling network. J Cell Sci (2009) 122:1269–73. doi:10.1242/jcs.042762
103. Xerri L, Devilard E, Hassoun J, Olive D, Birg F. In vivo expression of the CTLA4 inhibitory receptor in malignant and reactive cells from human lymphomas. J Pathol (1997) 183:182–7. doi:10.1002/(sici)1096-9896(199710)183:2<182:aid-path918>3.0.co;2-i
104. Ansell SM, Hurvitz SA, Koenig PA, LaPlant BR, Kabat BF, Fernando D, et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res (2009) 15:6446–53. doi:10.1158/1078-0432.ccr-09-1339
105. Bashey A, Medina B, Corringham S, Pasek M, Carrier E, Vrooman L, et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood (2009) 113:1581–8. doi:10.1182/blood-2008-07-168468
106. Khorshied MM, Gouda HM, Khorshid OM. Association of cytotoxic T-lymphocyte antigen 4 genetic polymorphism, hepatitis C viral infection and B-cell non-Hodgkin lymphoma: an Egyptian study. Leuk Lymphoma (2014) 55:1061–6. doi:10.3109/10428194.2013.820294
107. Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med (2006) 203:2223–7. doi:10.1084/jem.20061800
108. West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest (2013) 123:2604–15. doi:10.1172/jci67008
109. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature (2009) 458:206–10. doi:10.1038/nature07662
110. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
111. Thompson RH, Kuntz SM, Leibovich BC, Dong H, Lohse CM, Webster WS, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res (2006) 66:3381–5. doi:10.1158/0008-5472.can-05-4303
112. Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res (2005) 11:2947–53. doi:10.1158/1078-0432.ccr-04-1469
113. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer (2010) 116:1757–66. doi:10.1002/cncr.24899
114. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood (2010) 116:3268–77. doi:10.1182/blood-2010-05-282780
115. Rossille D, Gressier M, Damotte D, Maucort-Boulch D, Pangault C, Semana G, et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia (2014) 28:2367–75. doi:10.1038/leu.2014.137
116. Wang L, Qian J, Lu Y, Li H, Bao H, He D, et al. Immune evasion of mantle cell lymphoma: expression of B7-H1 leads to inhibited T-cell response to and killing of tumor cells. Haematologica (2013) 98:1458–66. doi:10.3324/haematol.2012.071340
117. Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood (2009) 114:1545–52. doi:10.1182/blood-2009-03-206672
118. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett (2004) 574:37–41. doi:10.1016/j.febslet.2004.07.083
119. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209:1201–17. doi:10.1084/jem.20112741
120. Pentcheva-Hoang T, Chen L, Pardoll DM, Allison JP. Programmed death-1 concentration at the immunological synapse is determined by ligand affinity and availability. Proc Natl Acad Sci U S A (2007) 104:17765–70. doi:10.1073/pnas.0708767104
121. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol (2009) 10:1185–92. doi:10.1038/ni.1790
122. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood (2012) 120:1412–21. doi:10.1182/blood-2012-02-411678
123. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol (2014) 32:1020–30. doi:10.1200/jco.2013.53.0105
124. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
125. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res (2008) 14:3044–51. doi:10.1158/1078-0432.ccr-07-4079
126. Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol (2013) 31:4199–206. doi:10.1200/jco.2012.48.3685
127. Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol (2014) 15:69–77. doi:10.1016/s1470-2045(13)70551-5
128. Bachy E, Coiffier B. Anti-PD1 antibody: a new approach to treatment of lymphomas. Lancet Oncol (2014) 15:7–8. doi:10.1016/s1470-2045(13)70587-4
129. Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood (2013) 121:2704–14. doi:10.1182/blood-2012-08-448332
130. Scholer A, Hugues S, Boissonnas A, Fetler L, Amigorena S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity (2008) 28:258–70. doi:10.1016/j.immuni.2007.12.016
131. Hilchey SP, Hyrien O, Mosmann TR, Livingstone AM, Friedberg JW, Young F, et al. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood (2009) 113:3809–12. doi:10.1182/blood-2008-10-185280
132. Teague RM, Greenberg PD, Fowler C, Huang MZ, Tan X, Morimoto J, et al. Peripheral CD8+ T cell tolerance to self-proteins is regulated proximally at the T cell receptor. Immunity (2008) 28:662–74. doi:10.1016/j.immuni.2008.03.012
133. Choi S, Schwartz RH. Impairment of immunological synapse formation in adaptively tolerant T cells. J Immunol (2011) 187:805–16. doi:10.4049/jimmunol.1003314
134. Carlin LM, Yanagi K, Verhoef A, Nolte-’t Hoen EN, Yates J, Gardner L, et al. Secretion of IFN-gamma and not IL-2 by anergic human T cells correlates with assembly of an immature immune synapse. Blood (2005) 106:3874–9. doi:10.1182/blood-2005-03-0996
135. Ise W, Nakamura K, Shimizu N, Goto H, Fujimoto K, Kaminogawa S, et al. Orally tolerized T cells can form conjugates with APCs but are defective in immunological synapse formation. J Immunol (2005) 175:829–38. doi:10.4049/jimmunol.175.2.829
136. Zambricki E, Zal T, Yachi P, Shigeoka A, Sprent J, Gascoigne N, et al. In vivo anergized T cells form altered immunological synapses in vitro. Am J Transplant (2006) 6:2572–9. doi:10.1111/j.1600-6143.2006.01517.x
137. Doherty M, Osborne DG, Browning DL, Parker DC, Wetzel SA. Anergic CD4+ T cells form mature immunological synapses with enhanced accumulation of c-Cbl and Cbl-b. J Immunol (2010) 184:3598–608. doi:10.4049/jimmunol.0902285
138. Dumont C, Blanchard N, Di Bartolo V, Lezot N, Dufour E, Jauliac S, et al. TCR/CD3 down-modulation and zeta degradation are regulated by ZAP-70. J Immunol (2002) 169:1705–12. doi:10.4049/jimmunol.169.4.1705
139. Jenkins MR, Tsun A, Stinchcombe JC, Griffiths GM. The strength of T cell receptor signal controls the polarization of cytotoxic machinery to the immunological synapse. Immunity (2009) 31:621–31. doi:10.1016/j.immuni.2009.08.024
140. Gombos I, Detre C, Vamosi G, Matko J. Rafting MHC-II domains in the APC (presynaptic) plasma membrane and the thresholds for T-cell activation and immunological synapse formation. Immunol Lett (2004) 92:117–24. doi:10.1016/j.imlet.2003.11.022
141. Anderson HA, Hiltbold EM, Roche PA. Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat Immunol (2000) 1:156–62. doi:10.1038/77842
Keywords: lymphoma, immunological synapse, T-cell tolerance, T-cell function
Citation: Nassef Kadry Naguib Roufaiel M, Wells JW and Steptoe RJ (2015) Impaired T-cell function in B-cell lymphoma: a direct consequence of events at the immunological synapse? Front. Immunol. 6:258. doi: 10.3389/fimmu.2015.00258
Received: 16 February 2015; Accepted: 11 May 2015;
Published: 02 June 2015
Edited by:
Sacha Gnjatic, Ludwig Institute for Cancer Research, USAReviewed by:
Alan G. Ramsay, Kings College London, UKChris Schmidt, Queensland Institute of Medical Research, Australia
Copyright: © 2015 Nassef Kadry Naguib Roufaiel, Wells and Steptoe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raymond J. Steptoe, UQ Diamantina Institute, The University of Queensland, Level 6, TRI, 37 Kent Street, Woolloongabba, Brisbane, QLD 4102, Australia,ci5zdGVwdG9lQHVxLmVkdS5hdQ==