 Silvia A. Fuertes Marraco
Silvia A. Fuertes Marraco Natalie J. Neubert
Natalie J. Neubert Grégory Verdeil
Grégory Verdeil Daniel E. Speiser
Daniel E. Speiser- 1Ludwig Cancer Research Center, University of Lausanne, Lausanne, Switzerland
- 2Department of Oncology, Lausanne University Hospital (CHUV), Lausanne, Switzerland
Inhibitory receptors (iRs) are frequently associated with “T cell exhaustion”. However, the expression of iRs is also dependent on T cell differentiation and activation. Therapeutic blockade of various iRs, also referred to as “checkpoint blockade”, is showing unprecedented results in the treatment of cancer patients. Consequently, the clinical potential in this field is broad, calling for increased research efforts and rapid refinements in the understanding of iR function. In this review, we provide an overview on the significance of iR expression for the interpretation of T cell functionality. We summarize how iRs have been strongly associated with “T cell exhaustion” and illustrate the parallel evidence on the importance of T cell differentiation and activation for the expression of iRs. The differentiation subsets of CD8 T cells (naïve, effector, and memory cells) show broad and inherent differences in iR expression, while activation leads to strong upregulation of iRs. Therefore, changes in iR expression during an immune response are often concomitant with T cell differentiation and activation. Sustained expression of iRs in chronic infection and in the tumor microenvironment likely reflects a specialized T cell differentiation. In these situations of prolonged antigen exposure and chronic inflammation, T cells are “downtuned” in order to limit tissue damage. Furthermore, we review the novel “checkpoint blockade” treatments and the potential of iRs as biomarkers. Finally, we provide recommendations for the immune monitoring of patients to interpret iR expression data combined with parameters of activation and differentiation of T cells.
Definition of Inhibitory Receptors
Immune cell function is tightly controlled and fine-tuned via co-stimulatory and co-inhibitory molecules. Co-stimulatory receptors were originally described to share a common sequence motif called immunoreceptor tyrosine-based activation motif (ITAM) consisting of two YxxL sequences separated by 7–12 amino acids (1, 2). This motif binds Zap70/Syk protein tyrosine kinases via their src homology 2 domain (SH2) (3). A few years later, an inhibitory motif (immunoreceptor tyrosine-based inhibitory motif, ITIM) was discovered in the cytoplasmic tail of Fcγ receptor IIB, a single chain low-affinity receptor for the Fc portion of IgG (4, 5). This receptor could inhibit immune activatory signals by dimerizing with the respective co-activatory receptors on mast cells, T cells, and B cells (6, 7). Like the ITAM motif, the ITIM motif interacts with SH2 domains (8–11). Thereafter, proteins that featured such ITIM motifs and that related to the immune synapse were classified as co-inhibitory molecules or (co-)inhibitory receptors (hereafter referred to as iRs).
The iRs that are nowadays studied in T cells were first described in natural killer cells in the early 90s (12, 13) and were defined as follows (3, 5, 12, 14, 15): (a) they possess an ITIM that, (b) when phosphorylated, (c) can recruit SHP1 and possibly SHP2 (1, 2, 9), which (d) in turn interfere with activating receptors to inhibit downstream activatory pathways (3, 16). At that time, two families of iRs were known: the immunoglobulin superfamily (IgSF) and the c-type lectin superfamily (17–20).
Two of the probably most well-studied T cell-related iRs are programed cell death 1 (PDCD1, also called PD1) and cytotoxic T lymphocyte-associated Antigen 4 (CTLA-4). PD1 was originally discovered by screening for genes that are involved in classical programed cell death in a mouse lymphoid hybridoma and a mouse lymphoid/myeloid progenitor cell line (21). The sequence of its human counterpart was described shortly after (22). CTLA-4 was discovered already in 1987 (23) as an immunoglobulin receptor, but its function was only determined in the mid-90s.
Today, the definition of iRs has changed from the above-mentioned features to a rather functional definition: most surface receptors that are classified as iRs contain an ITIM, such as B and T lymphocyte associated (BTLA) and PD1. But in addition, receptors that lack an ITIM are recognized as iRs, making the first characteristic above (a) no obligation. A prominent example is CTLA-4, which contains a Tyr–Xaa–Xaa–Met motif that is thought to have inhibitory function (12, 24). Also, lymphocyte-activation gene 3 (LAG3) does not feature an ITIM (25). It belongs to the CD4 family and binds to MHC class II with high affinity (26). The most frequent alternative inhibitory motif is the immune receptor tyrosine-based switch motif (ITSM), which also interacts with SH2-domain containing phosphatases such as SHIPs (27–30). ITSMs can be found in the cytoplasmic tail of PD1 and BTLA among other iRs (and in addition to ITIMs). A summary of the inhibitory sequence motifs on iRs including less frequent motifs can be found in the review by Odorizzi and Wherry (31).
The new functional definition describes iRs as co-inhibitory molecules that negatively interfere with T cell activation and function. iRs can inhibit T cell functions at several levels (31): (i) through competition with co-stimulatory receptors for binding to shared ligands or interference in the formation of microclusters and lipid rafts (32–34), (ii) by interfering with downstream signals from co-activatory and T cell receptors (TCRs), and (iii) by upregulating genes that are involved in T cell dysfunction (35). Over the past two decades, the IgSF has grown to include far more than 10 members divided into 8 receptor subfamilies (31, 36). A comprehensive overview of T cell co-stimulatory and co-iRs and their molecular mechanisms can be found in a review by Chen and Flies (36).
iRs as Hallmarks in “T Cell Exhaustion”
Inhibitory receptors were set at the forefront of research interest with the description of their involvement in the phenomenon of “T cell exhaustion”. The phenomenal clinical impact of the therapeutic blockade of iRs to restore T cell function in cancer (as outlined in the last parts of our review) has greatly contributed to the widespread acknowledgment of the functional importance of iRs. “T cell exhaustion” was coined to describe the functional state of CD8 T cells that persist but show poor effector functions in mice chronically infected with LCMV Clone 13 (37). The molecular signature of exhausted CD8 T cells was later characterized in this prototypic mouse model of LCMV infection, comparing the phenotype and functionality of CD8 T cells in chronic (Clone 13 strain) versus acute (Armstrong strain) infections (38). As opposed to acute infection, where effector CD8 T cells give rise to memory cells once the pathogen is cleared, persistent antigen stimulation and inflammation in the chronic infection setting leads to progressive loss of function in CD8 T cells, termed “T cell exhaustion”. At the center of the molecular signature of exhausted CD8 T cells was the upregulation of iRs, including PD1, CTLA-4, CD160, and LAG3, setting iRs as hallmarks of “T cell exhaustion” (38, 39). In the field of “T cell exhaustion”, scientific advances were strongly promoted by mouse studies using the prototypic LCMV model (acute versus chronic infection). By contrast, the profiling of exhausted CD8 T cells in cancer was first done in humans, demonstrating that Melan-A-specific T cells in melanoma patients share many molecular features with exhausted T cells in chronic infection (40). Over the last decade, several studies have further linked expression of iRs (e.g. PD1, CTLA-4, TIM3, LAG3, CD160, BTLA, and 2B4) to the phenomenon of “T cell exhaustion” both in mouse models and in patients, in chronic infections, including HIV, hepatitis C virus, EBV, malaria, as well as autoimmune disorders such as systemic lupus erythematosus and in several cancers (41–50). In these various pathological scenarios, expression of multiple and different combinations of iRs has been associated with the exhausted phenotype of CD8 T cells, implying that iRs are not only diverse but also co-regulate “CD8 T cell exhaustion” (51, 52). Consequently, iRs have been generally referred to as “exhaustion markers” (53–55). Importantly, this knowledge on “T cell exhaustion” and the implication of iRs has had full impact on therapeutic strategies, with the breakthrough of monoclonal antibodies that block iRs (also referred to as “checkpoint blockade”) to restore T cell function and yielding unprecedented clinical improvements on overall survival of cancer patients. This therapeutic breakthrough of monoclonal antibodies against iRs and “checkpoint blockade” is discussed in the final sections of this review.
Understanding iR Function: Tribulations through the “T Cell Exhaustion” Field (Authors’ Path, Part I)
In the framework of the characterization of CD8 T cells in cancer, we and others discovered that CD8 T cells in metastasis of melanoma patients had increased levels of iRs, as compared to CD8 T cell counterparts in circulation (40, 44, 48). This increase of iRs at the tumor site correlated with the previously observed decreased functional properties of CD8 T cells in metastasis, as opposed to blood-derived CD8 T cells (40, 56). Therefore, these studies demonstrated that “T cell exhaustion” (increased iR levels and diminished T cell function) also occurs in the context of chronic antigen stimulation and inflammation in the tumor microenvironment (TME) (involving self antigens), similarly to chronic viral infections (involving non-self antigens). “T cell exhaustion” at the tumor site constitutes thus the third stumbling block, in addition to the poor naïve repertoire of self antigen-specific CD8 T cells (low affinity and precursor frequency: first stumbling block) and the poor priming capacity against tumors (inefficient tumor antigen presentation and co-stimulation by tumors: second stumbling block) [reviewed in Ref. (57)].
As mentioned above, multiple studies showed that CD8 T cells in chronic antigen stimulation settings (cancer or viral infections) display diminished function associated with increased iR levels. However, this association (high iR = low function) was neither a direct proof that an iR per se provoked lower functionality nor did this association provide mechanistic insights into the function of iRs. In fact, there is as yet limited knowledge concerning iR function and the signaling of the various iRs: what are the precise molecular pathways, the signaling cascades and events downstream of the interactions of iRs with their respective iR ligands? Further to structural considerations whereby iRs contain inhibitory motifs (described above), the evidence on signaling mechanisms is summarized in the reviews by Chen and Flies (36), Baitsch et al. (57), and Odorizzi and Wherry (31).
In order to assess directly the impact of iRs on T cell function, we setup an in vitro system to study T cells that express iRs and are exposed to TCR activation surrounded by iR ligands. To control the presence and dose of each iR ligand, and to avoid uncontrolled secondary events from the antigen presenting cell (APC), we made use of artificial APCs (aAPC), namely, beads that could be coated with the desired dose and composition of iR ligands (58). These were cell-sized beads (4.5 μm diameter) covered with epoxy groups that covalently attach any protein (or protein mix). We used anti-CD3 antibody (OKT3 clone) to activate T cells together with combinations of recombinant iR ligands, including human PD-L1:Fc (PD1 ligand), HLA-DR (LAG3 ligand), and HVEM:Fc (ligand of BTLA and CD160). We initially found that beads coated with anti-CD3 and any combination of iR ligands barely activated CD8 T cell clones or primary CD8 T cells to produce cytokines in a 4-h assay, as opposed to beads coated with anti-CD3 only, pointing toward strong inhibition by the presence of iR ligands. However, we performed quality controls of the APC beads and discovered that the procedure used to coat the beads (based on standard protocols) lead to the out-competition of anti-CD3 from the surface of the beads upon co-incubation with iR ligands, leading the artifactual “inhibition” of T cell function by iR ligands (in fact, due to less anti-CD3 antibody coated on the beads in presence of iR ligands). After optimization of aAPC bead preparation to obtain beads with equivalent doses of anti-CD3 in absence or presence of iR ligands, the repetition of the experiments revealed that the presence of iR ligands did not result in reduced CD8 T cell function (in clones nor primary cells), neither in 4-h assays of cytokine production nor in proliferation assays for up to 4 days. It is possible that the functional impact of iRs differs depending on the context, for instance, different T cell types may have different susceptibilities to iR-mediated inhibition (“exhausted” CD8 T cells from tumor metastasis may be more susceptible than primary cells from blood of healthy individuals). Several previous studies had investigated iR function using aAPC beads prepared with standard procedures without explicit quality control on the bead coating; our experiments using quality-controlled aAPC beads showed that the mere presence of iR ligands such as PD-L1 did not lead to inhibition of T cell activation (58).
Notwithstanding, in addition to the use of beads coated with T cell-stimulatory antibodies and iR ligands, several other experimental strategies exist to assess iR function. These include the use of T cells over-expressing iRs, stimulated with APC over-expressing respective iR ligands, as well as the T cell functional assays in presence of iR-blocking antibodies.
For instance, the mechanism of PD1 action has been addressed by various experimental means. Wei et al. over-expressed high or intermediate levels of PD1 in primary human T cells by RNA electroporation and stimulated these with aAPC (K562 or T2) over-expressing PD-L1: the different T cell functions tested (cytokine secretion, Ca2+ flux, proliferation) were differentially sensitive to PD1 expression (59). Similarly, using mouse T cells over-expressing PD1 and planar bilayers containing stimulatory molecules in combination with PD-L1, Yokosuka et al. showed that PD1 forms microclusters with the TCR and recruits SHP2 to negatively regulate TCR signaling (60). A multitude of studies have shown that blockade of PD1 and other iRs leads to improved T cell functionality, including cytotoxicity, proliferation, and cytokine production [e.g., Ref. (44, 49)]. Interestingly, in the context of the LCMV infection model of “T cell exhaustion”, PD1-blockade may reverse dysfunction in PD1int- but not in PD1high-expressing T cells (61), emphasizing again that the levels of iR (high or intermediate) are crucial to determine the impact and strength of its inhibitory action (59). In the case of CTLA-4, several mechanisms of action have been proposed, including inhibitory events downstream CTLA-4 and interference with TCR signaling as well as T cell-extrinsic mechanisms [reviewed in Ref. (62, 63)]. For example, it is known that CTLA-4 is competing with co-stimulatory B7 ligands for binding to CD28. Two models have been suggested, the first in which CTLA-4 increases the threshold for T cell activation and the second in which CTLA-4 attenuates T cell expansion (64). More recently, the efficacy of CTLA-4 blockade in anti-tumor therapy has been at least partly attributed to another T cell-extrinsic mechanism, namely antibody-dependent cell-mediated cytotoxicity (ADCC), whereby regulatory T cells (Tregs) expressing high levels of CTLA-4 are tagged by the CTLA-4-blocking antibody for ADCC by macrophages and depleted within the TME (65, 66). Moreover, an iR might even have opposite functions depending on the molecules that participate to the immune synapse, as is the case for TIM3, which is inhibitory if it interacts with CEACAM-1 and activatory in absence of CEACAM-1 (67). This example shows that it is crucial to consider the remainder of interacting partners, whether co-stimulatory and co-inhibitory, when assessing iR function and CD8 T cell functionality. It also impacts on the design of strategies to block iRs and reverse T cell function, as it might be more relevant to interfere with other interacting partners such as CEACAM-1 rather than blocking TIM3. Altogether, the precise mechanisms whereby each iR works are as yet not fully defined.
Finally, a commonly practiced strategy to address iR function is to assess the functionality of T cells that show positive expression of iRs and compare them to iR-negative counterparts. Several studies have reported that positive expression of iRs (including PD1, CTLA-4, TIM3, LAG3, 2B4, CD160, and BTLA) marked cells that expressed less cytokines (44, 50, 68–72). It is in this comparative exercise that we have found that the consideration of T cell differentiation and activation status is crucial, as will be explained in full detail in the following section.
iR Expression is Driven by T Cell Activation and Differentiation (Authors’ Path, Part II)
Further to the association of iR expression with “T cell exhaustion” in pathological settings, important insights on the significance of iR expression for the functionality of T cells have been gained from the study of iR expression in CD8 T cells from healthy individuals.
Analysis of PBMC from healthy individuals shows, first, that circulating CD8 T cells express various iRs in steady-state, and second, that iR levels clearly vary depending on T cell differentiation (73, 74) (Figure 1). For instance, iRs such as 2B4, KLRG1, and CD160 are expressed at higher levels with increased differentiation. By contrast, BTLA is high on naïve cells and decreases with differentiation. PD1 is particularly expressed in effector memory (EM) cells. TIM3 is present at low levels on naïve T cells. Other iRs such as CTLA-4 and LAG3 are not detectable in steady-state in circulating CD8 T cells from healthy donors.
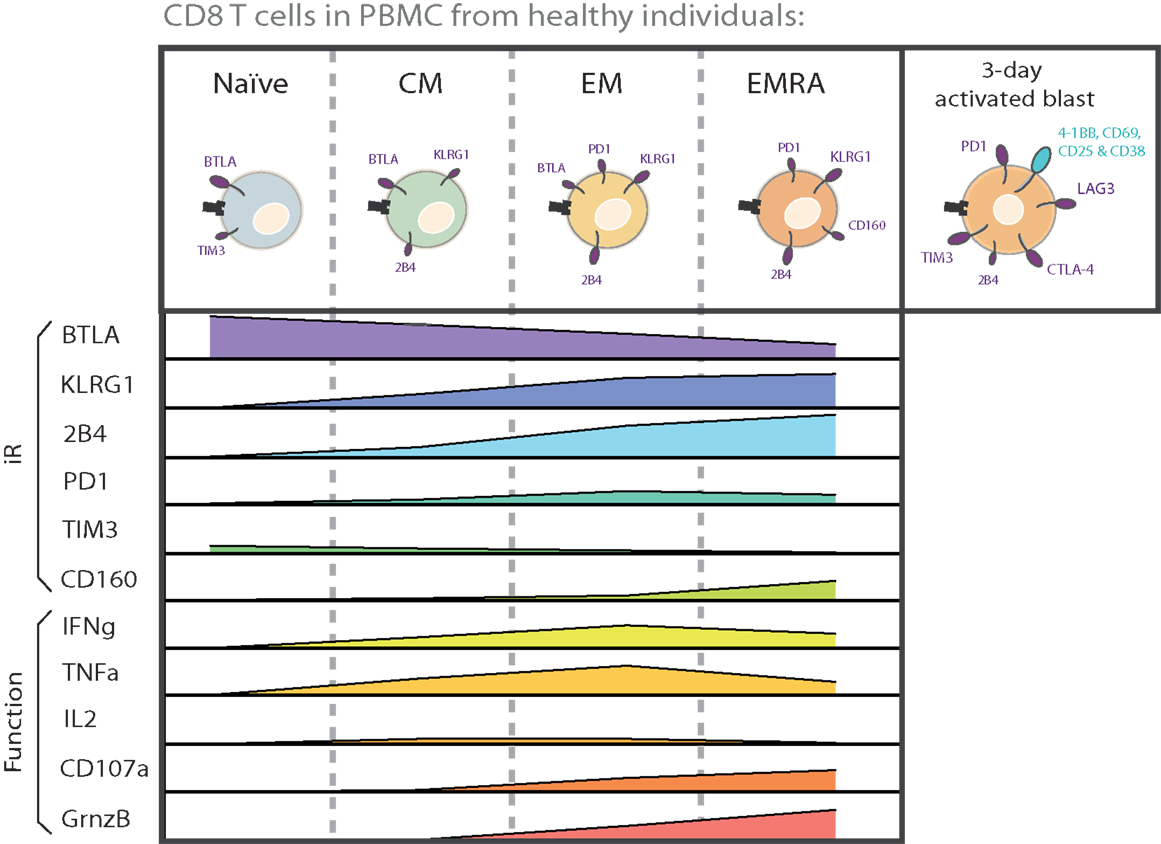
Figure 1. CD8 T cell subsets are inherently different in iR expression and effector functions. The various differentiation subsets that are found in circulation in healthy donors are depicted with the iRs that they predominantly express. The relative levels of each iR are indicated below each subset, in parallel to the capacity for cytokine secretion and CD107a translocation. The graphs are based on data from a 6-h intracellular cytokine production assay using PBMC from healthy donors (75). Also shown is iR expression in activated cells (“activated blast”), together with activation markers, based on a 3-day stimulation assay on CD8 T cells from healthy donors (75). The size of the receptor depicted on cells reflects the relative expression of iRs.
Importantly, the functional capacities of CD8 T cells also largely vary according to their differentiation stage. For instance, upon stimulation with anti-CD3 and anti-CD28, naïve cells do not produce cytokines, while EM cells are most effective at producing IFNγ and TNFα, central memory (CM) and EM cells at producing IL-2, and EM RA+ (EMRA) cells at expressing Granzyme B and at degranulating (CD107a translocation) [Ref. (75), Figure 1]. The expression of a multitude of other molecules and functions involved in cytokine signaling, cytotoxicity, migration, proliferation, apoptosis, senescence, and stemness are also well known to vary according to T cell differentiation (76–78). There are thus inherent differences in functionality as well as iR expression among CD8 T cell subsets. Therefore, in the study of iR expression and T cell function, it is crucial to keep in consideration that iR expression is tightly linked to the differentiation status, and that this link is already observable in healthy individuals, where it is not related to a particular pathological state nor “T cell exhaustion” (this is further described below in the Section “Physiological Role of iRs to Regulate T Cells in Health and Immune Homeostasis”).
KEY CONCEPT 1. Inherent Differences
CD8 T cell subsets are inherently different in the expression of a wide array of molecules and functionalities, including iR expression. iR expression is tightly linked to the differentiation status, and this link is present in healthy immune homeostasis.
In addition to the conventional differentiation subsets Naïve, CM, EM, and EMRA, expression of several iRs has also been assessed in the more recently described Naïve-like stem cell-like memory T cells (SCM) (77, 79). For instance, in the context of Yellow Fever vaccination, we recently showed that human SCM CD8 T cells express high levels of BTLA, very low levels of 2B4, no PD1, and intermediate levels of KLRG1 (79). This iR profile would place them in between Naïve and CM cells in the differentiation gradient shown in Figure 1, as is the case for several other functions that have been addressed by gene expression profiling (77, 79).
The importance of considering T cell differentiation is best depicted by the exercise where we compared cytokine production by iR-positive CD8 T cells versus iR-negative counterparts, either considering the total CD8 T cells (all subsets mixed) or following the separation of subsets (75) (Figure 2). To this end, we used stimulation with anti-CD3 and anti-CD28, importantly in absence of added iR ligands, in order to assess the functionality of iR-positive cells and not the impact of stimulating the iR per se. Given that naïve cells do not produce cytokines upon anti-CD3 and anti-CD28 stimulation, the analysis of total CD8 T cells showed highly significant differences between iR- positive versus iR-negative cells in the case of iRs that are inherently expressed at clearly distinct levels between naïve versus differentiated subsets. Yet, these differences were diminished or disappeared when cytokine production was analyzed by comparing iR-positive versus iR-negative cells within each individual differentiation subset (75) (Figure 2). This applies to BTLA (high in naïve) as well as 2B4 and KLRG1 (high in differentiated cells). Interestingly, in general, iR-positive cells were still largely capable to produce cytokines and did not show major impairments as compared to iR-negative counterparts, in agreement with previous reports analyzing PD1 in CD8 T cells from healthy donors (74).
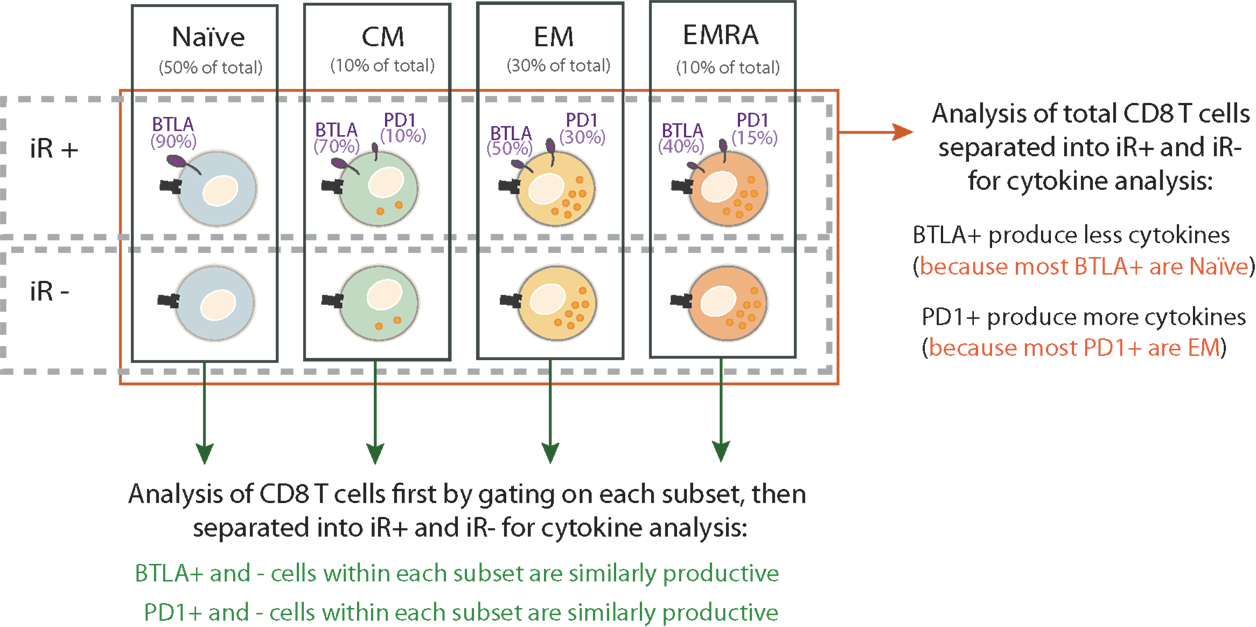
Figure 2. Discriminating differentiation subsets is the minimal analysis to assess the link between iR expression and functionality of CD8 T cells. The significance of iR expression on CD8 T cell function can be assessed by analyzing cytokine production by iR-positive versus iR-negative cells. The relative capacity for cytokine production in the various subsets is depicted by the numbers of orange intracellular dots. As an example, the expression of two iRs are shown: BTLA and PD1. The percentage below each subset name indicates the relative frequency of that subset within total CD8 T cells. The percentages next to each receptor indicate the fraction of cells within a given subset that express the receptor (the size of the receptor depicted on cells also reflects relative expression of iRs). These percentages are estimated based on average values in PBMC of healthy donors (75). When iR-positive cells are compared to iR-negative cells within the total CD8 T cells (red arrow), artifactual conclusions can be driven due to the inherent differences in iR expression and effector function of the various subsets. For example, BTLA+ cells in total CD8 T may show less cytokine secretion than their BTLA− counterparts, but this is due to the enrichment of Naïve cells in BTLA+ cells, which do not produce cytokines as compared to the enrichment of differentiated cells in BTLA−. With each subset, BTLA+ and BTLA− are comparable cytokine producers. Therefore, it is not “cytokine production capacity” (=artifactual conclusion) but “differentiation status” what accounts for differences in comparing BTLA+ and BTLA− cells. As a minimal requirement, analysis of individual subsets is necessary to adequately address the link between iR expression and effector function.
Altogether, this comparative exercise showed that the direct link between positive iR expression and lower cytokine production is generally absent or weak, or only applies to a limited number of iRs in certain CD8 T cell subsets. Moreover, a failure to consider the differentiation status can lead to artifactual conclusions on functional differences between iR-positive and -negative cells.
KEY CONCEPT 2. Artifactual conclusions
A failure to consider differentiation status can lead to conclusions that are drawn from wrong argumentation (artifactual) on functional differences between iR-positive and -negative cells. This is illustrated in Figure 2.
We also compared iR expression and function in CD8 T cells from tumor-infiltrated lymph nodes (TILN) in melanoma patients with similar results, in agreement with previous studies where we showed that PD1+ CD8 T cells from PBMC from melanoma patients are not necessarily impaired (80). In the TME (e.g., TILN), however, CD8 T cells can express CTLA-4 and high levels of PD1, and positive expression may correlate with lower cytokine production (69, 75). In our settings, it is technically difficult to compare cytokine levels in iR-positive versus negative cells due to the low cytokine production of CD8 T cells from TILN, although a trend was seen for less cytokines in PD1+, TIM3+, and CTLA-4+ CD8 T cells in TILN (75). Therefore, even in the absence of added inhibitory ligands (only stimulation with anti-CD3 and anti-CD28), the impact of iR expression may be context-dependent, e.g. showing no effect in CD8 T cells in circulation but negative effects in cells from the TME.
In addition, stimulation of healthy donor CD8 T cells for several days with anti-CD3 and anti-CD28 showed that certain iRs are strongly up-regulated, including PD1, CTLA-4, TIM3, and LAG3. Furthermore, such iR upregulation positively correlates with the expression of several activation markers, including 4-1BB, CD69, CD38, and CD25. This indicated that not only differentiation but also the activation status can dictate the levels of iR expression in CD8 T cells.
Of note, the broad inter-individual variability in T cell subset composition, function, and iR expression (75) further aggravates the artifacts in analyzing total CD8 T cells instead of individual subsets separately. This is also aggravated in the case where different samples are compared (e.g., different tissues, infections, time-points) that inherently display different composition of CD8 T cell subsets and where analyses based on comparison of total CD8 T cells undermines variability in differentiation or activation status.
Significance of iR Expression Beyond “T Cell Exhaustion”
Based on the aforementioned analyses of CD8 T cells in healthy individuals, it became clear that both T cell differentiation and activation were major drivers of iR expression. Consequently, iR expression may, in fact, be an indication of ongoing or recent activation in CD8 T cells as well effective T cell differentiation, in contrast to the association of iR expression with “T cell exhaustion”.
Various lines of evidence exist that iRs participate in T cell activation and differentiation, in a “tide model” where iRs can be up-regulated in order to counterbalance co-stimulatory signals following the peak of activation [reviewed in Ref. (36, 81)]. PD1, for instance, was described already in 1996 as a protein that was up-regulated in T and B lymphocytes upon activation in vitro (well before it was linked to “T cell exhaustion”) (82). In fact, in the LCMV model of “T cell exhaustion”, iRs are expressed at high levels in the effector phase in both settings, whether in the acute or chronic infection setting (38). Yet, only in the acute infection, the pathogen is cleared leading to contraction of effector cells into memory cells, with the consequent down-modulation of iRs, as opposed to the chronic infection, where the expression of iRs remains elevated. In addition to T cell differentiation, it is important to highlight this “effector/acute” component of elevated iR expression (Figure 3), which has been neglected in the depiction of “T cell exhaustion” models (39) or only recently addressed (36, 41).
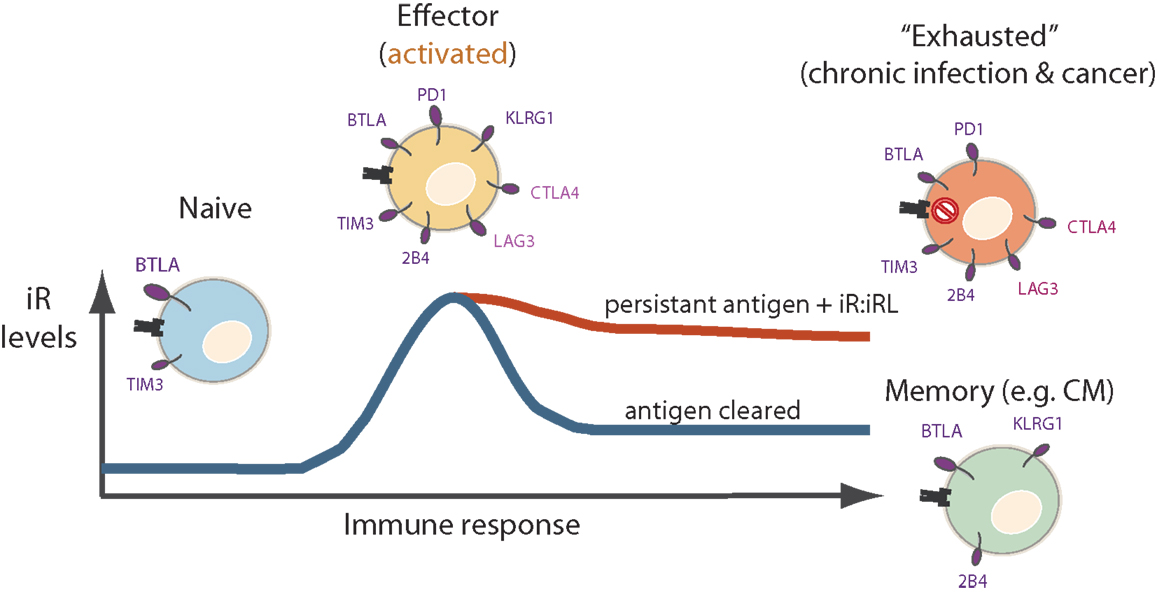
Figure 3. Levels of iR may peak at the effector phase, and may further modulate differently during acute versus chronic immune responses. Naïve cells express mainly BTLA and low levels of TIM3. Effector cells express a wider variety of iRs. The levels of certain iRs such as PD1, CTLA-4, LAG3, and TIM3 may peak at the effector phase. Thereafter, iR expression differs in chronically stimulated cells (“exhausted cells”) where iRs are relatively maintained, as opposed to memory cells after clearance of an acute infection where iRs are down-modulated.
In the functional characterization of CD8 T cells, iRs have been used as surrogate markers for T cell dysfunction, in a “guilty by association” reflex where iR expression would mark exhausted cells, even referring to iRs as “exhaustion markers” (53–55). However, multiple studies have directly or indirectly shown that expression of one or the other iR is not always a sign of “T cell exhaustion” but is rather associated with markers or transcription factors involved in T cell differentiation and activation.
KEY CONCEPT 3. Sign of “T cell exhaustion”
The phenomenon of “T cell exhaustion” is characterized by T cell dysfunction with elevated levels of iRs. However, iR expression on CD8 T cells is not always a sign of T cell dysfunction and does not necessarily correspond to reduced T cell functions, but may rather mark activated and/or different subsets of functional CD8 T cells.
For instance, high co-expression of PD1, 2B4, CD160, and KLRG1 on HCV-specific CD8 T cells is linked to a CD127 low phenotype (i.e., late differentiation) and recurrent antigen triggering (low epitope diversity) (83). PD1, 2B4, and CD160 are also co-expressed in Flu-, EBV-, and CMV-specific CD8 T cells, where CD160+ but not PD1+ cells are less functional (84). Similarly, in HIV infection, CD160 and PD1 double positive cells are a dysfunctional subset of CD8 T cells, as opposed to cells positive only for PD1 that are rather in an activated state (85). TIM3 expression is linked to an EM phenotype and stronger effector responses in tuberculosis (86). PD1 levels are increased in Acute Friend virus infection yet PD1+ cells are cytotoxic and control infection (87). PD1 correlates with activation markers 41BB (88) in breast cancer and CD38 in HIV (53). In the AT-3 tumor model of breast cancer, PD1high TIM3+ CD8 T cells also express 4-1BB, as well as granzyme B, Ki67, and IFNγ, and persist following radiotherapy (88).
PD1 has also been shown to regulate the development of CM cells following acute vaccinia virus infection in mice: in PD1−/− mice, primary and secondary responses are enhanced and CD8 T cells have a phenotype with high CD62L, CD27, CCR7, and IL-2, which supports that a skewing toward the CM phenotype occurs in absence of PD1 (89). Along the lines of the role of PD1 in differentiation, PD1high CD8 T cells in chronic LCMV infection express high levels of EOMES, in contrast to PD1int that rather express high levels of Tbet (90). Interestingly, the transcription factor FoxO1 sustains high PD1 expression, promoting survival and differentiation of CD8 T cells in chronic LCMV infection (91). The increased levels of PD1, CD160, and 2B4 in total CD8 T cells during chronic HIV infection are also associated with Tbetdim and EOMEShigh expression (92).
Altogether, in the midst of the implication of iR in “T cell exhaustion”, there is thus also diverse evidence that iRs primarily participate in T cell activation and differentiation.
This has lead to re-evaluations of the significance of iR as “exhaustion markers”, for instance, in the context of SIV infection [where PD1 expression was associated with a CCR7− CCR5+ phenotype rather than dysfunction per se, questioning the value of PD1 as a marker of “immune exhaustion” (93)], as well as in cancer [where BTLA and PD1 are proposed to mark cells in a “heightened state of T cell activation” (94)].
Physiological Role of iRs to Regulate T Cells in Health and Immune Homeostasis
Expression of iRs is physiologically associated with T cell-regulatory events that are not linked to pathological processes. In the context of “healthy” immune homeostasis, iRs co-evolved with co-stimulatory molecules to form a multi-component system of positive and negative regulatory signals surrounding TCR stimulation, which supports the fine-tuning of T cell activation. Possibly, the initial recognition of the target cell via the TCR receptor is priming T cells for response and signaling through co-stimulatory or co-iRs determines the direction and intensity of the response.
KEY CONCEPT 4. Co-evolved with co-stimulatory receptors
iRs co-evolved with co-stimulatory receptors to allow for the fine-tuning of T cell responses – as such, iRs are not only present in “T cell exhaustion” but also have a primary role in the regulation of immune homeostasis and preservation of healthy tissue from autoimmunity.
Thus, the expression of co-stimulatory and co-iRs is not mutually exclusive. In the “tide model” suggested by Zhu et al. (81), the rise and fall of waves of co-stimulatory versus co-inhibitory signaling tightly regulates and fine tunes the immune response. The diversity of co-stimulatory receptors and iRs on T cells allows to control the T cell response at every step, from T cell priming, T cell expansion, and contraction, to the various functions of memory T cells. In addition, different co-signaling receptors are important in CD8 versus CD4 T cell subsets.
Pivotal studies in mice have allowed establishing the principles of iR function and their importance for the regulation of the immune response using transgene and (conditional) knock-out technologies, as well as transplantation or cell transfer experiments. Most of the basic knowledge about iRs has been generated and confirmed with such models. For instance, CTLA-4-deficient mice develop lymphoproliferative disease, which leads to tissue damage and ultimately to death within the first month of age (95, 96). In addition to its important constitutive expression and function in Treg cells (97), CTLA-4 is up-regulated following T cell activation (98), and is involved in late stages of T cell priming and systemic activation of T cell responses [reviewed in Ref. (99)].
Like CTLA-4, PD1 expression is induced on T cells by activation (82). PD1-deficient mice develop autoimmune diseases, such as lupus-like diseases or dilated cardiomyopathy (100, 101). When infected with chronic pathogens such as LCMV strain 13 or with Mycobacterium tuberculosis, PD1-deficient mice develop strong immunopathological reactions and succumb within days or weeks (100, 102). However, in contrast to CTLA-4 in priming, the PD1-pathway rather plays an important role at the sites of effector T cell activity, by limiting self-tissue damage (101, 103, 104). Upon exposure to target antigen, PD1-deficient T cells show an increased proliferation compared to PD1-expressing T cells (104). This suggests that PD1 is playing a role at later stages of the immune responses, when T cells are already activated.
As described in more detail below, “checkpoint blockade” can provoke immune-related adverse events (IRAE). CTLA-4 blockade for treatment of melanoma patients provoked grade 3 and grade 4 IRAE including autoimmune damages in colon, liver, and hormonal glands (105). The immunological side effects are reminiscent of those in mice that lack Tregs. Indeed, anti-CTLA-4 antibody treatment can lead to Treg depletion via antibody-dependent cellular cytotoxicity (65, 66).
These evidences taken together, it becomes clear that each iR has a specific role at specific events during the tightly controlled immune response of T cells. The plasticity of the immune response requires an interplay of co-stimulatory and co-inhibitory signals in order be executed in a beneficial manner for its host and without causing unwanted damage of healthy tissue. The autoimmune disorders in iR knock-out mice and the IRAE in “checkpoint blockade”-treated patients show that iRs such as PD1 and CTLA-4 are fundamental in the maintenance of a healthy immune homeostasis.
Breakthrough in Immunotherapy: “Checkpoint Blockade”
During recent years, novel therapies with iR-specific antibodies (referred to as “checkpoint blockade”) have brought major breakthroughs for patients with solid cancers, including melanoma and carcinomas of various organs. This unprecedented success in immunotherapy of cancer is changing the therapeutic principles in clinical oncology, based on the conceptual proof that the immune system of many patients bears the potential to combat malignant disease up to clinically significant levels.
The initial report of clinical benefit for melanoma patients by targeting CTLA-4 (106) was followed by rapid publications confirming and extending these findings through targeting CTLA-4 or PD1 pathways. Two phase 1 clinical trials suggested even further improvements for melanoma patients, by combination therapies with PD1 and CTLA-4 blocking monoclonal antibodies (107, 108). This particular combination causes more frequent autoimmune toxicities than the single agent therapies (109). Nevertheless, the increased clinical benefit for patients with metastatic disease justifies the further development of “checkpoint blockade”. Today, immunotherapy has reached a high level of clinical usefulness. Often, clinical responses in melanoma patients are durable, and some patients are still disease free after many years (110–113). Progress is evident not only in melanoma patients but also in patients with lung and kidney cancer, and more recently bladder and head and neck cancer patients. Currently, there are more than 300 clinical trials registered (www.clinicaltrials.gov) that target iRs. Most of them are using CTLA-4 and PD1/PD-L1 specific antibodies. But other similar approaches are on their way: antibodies against killer cell immunoglobulin-like receptors (KIR) and LAG3 are in early phase clinical studies of novel cancer therapies (NCT01968109, clinicaltrials.gov), and further reagents specific for e.g. TIM3 and BTLA are in preclinical development (114, 115).
iRs and iR Ligands as Biomarkers
Biomarkers that may Correlate with Disease Outcome
Major research efforts are made with the aim to identify biomarkers that may help evaluating patient’s prognosis. For many years, tumor immunity parameters were rarely considered as biomarkers for patients with solid tumors. Rather, research focused on other factors related to hormonal or metabolic mechanisms. Even in melanoma, a disease that is since long considered as “immunogenic”, tumor immunity candidate biomarkers played a minor role (116). Now, this has changed, thanks to the new awareness of the importance of immunological mechanisms. Many studies focus on immune cells and their functions. Tumor infiltrating CD8 T cells are of prime interest, because they are frequently associated with better prognosis in the majority of human cancers (117). Currently, there are large multicenter efforts ongoing to determine whether tumor infiltration by activated CD8 T cells can be reliably assessed by standardized methods, and systematically evaluated for eventual routine staging of patients with colorectal cancers (118). The implementation of routine application represents a major challenge. In fact, the vast majority of candidate biomarkers never become routine tools.
During recent years, large efforts have been taken to characterize inhibitory immune receptors and their ligands in cancer patients. Analysis of their expression in the TME revealed highly interesting results [reviewed in Ref. (119, 120)], with much attention having been paid to PD1. In renal cell carcinoma (121, 122), follicular lymphoma (123), and soft tissue sarcomas (124) enhanced expression of PD1 by tumor infiltrating lymphocytes (TILs) was found to be associated with advanced tumor stages and reduced overall survival. Other studies, however, reported that PD1 expression was not associated with clinical outcome [e.g., in melanoma (125)], or correlated with a favorable outcome [e.g., in HPV-associated head and neck cancer patients (126)]. Yet, bad prognostic PD1 expression in one renal cell carcinoma study was linked to Foxp3 and Tregs in TIL (122), showing it is important to concomitantly determine which cell type (and T cell subset) expresses a given iR. Many studies also analyzed the expression of PD1 ligand 1 (PD-L1). Similar to PD1, the results are discrepant. In patients with mismatch-repair-proficient colon cancers, intratumoral expression of PD-L1 was associated with good prognosis (127). A similar finding was made for melanoma (128). In fact, intratumoral PD-L1 expression has been co-localized to infiltrating activated CD8 T cells (129), showing that presence of iR ligands may also reflect ongoing immune responses. However, Massi et al. (130) found that melanoma patients with high-intratumoral levels of PD-L1 expression have a significantly shorter overall survival. Several studies simultaneously evaluated multiple biomarkers. Kim et al. reported that PD1-positive lymphocytes and the expression of PD-L1 predicted poor clinical outcome of patients with soft tissue sarcoma (124). For LAG3, its enhanced expression was found to be associated with poor prognosis of patients with colorectal cancers (131). A similar association was found for patients with chronic lymphocytic leukemia (132). In the case of TIM3, associations with poor prognosis were found for patients with prostate cancer (133), renal cancer (134), gastric cancer (135), and cervical cancer (136). By contrast, TIM3 expression by tumor infiltrating T cells was associated with increased recurrence-free survival in patients with usual vulvar intraepithelial neoplasia (137).
Biomarkers that may Help Predicting Therapy Outcome
Besides the search for associations with prognosis, many of the above-mentioned biomarkers were also evaluated with respect to therapy outcome prediction. The “predictive value” of a given biomarker is high when it correlates strongly with the subsequent response to a specific therapy. This is of particular interest for “personalized medicine”, because better predictions indicate that more efficient and less toxic treatments can be selected for individual patients or groups of highly defined patients. Biomarkers may not only suggest the outcome of a given treatment but can also support the improvement of drug toxicity management.
A major focus is on the role of PD-L1 expression in patients receiving anti-PD1 or anti-PD-L1 antibodies. Several studies reported that clinical responses to PD1-pathway blockade are more likely when tumor cells and/or immune cells express PD-L1 at baseline (138, 139). However, clinical responses to these therapies can also occur in patients where PD-L1 expression is absent or only very low at baseline (140), indicating that they should also have a chance to benefit from PD1-pathway blockade. In fact, currently, there is no biomarker that can be used for patient selection for treatments with antibodies for iRs (140).
Simultaneous assessment of multiple biomarkers may also be useful for predicting therapy outcome. In melanoma samples before treatment with the PD1 specific antibody pembrolizumab, several immune features were associated with subsequent clinical responses, i.e. enrichments of CD8-, PD1-, and PD-L1-expressing cells inside tumors and at invasive margins, with close proximity of PD1/PD-L1, and increased TCR diversity (141).
In humans, the possibilities for assessing functional roles are limited. Nevertheless, cellular assays allow determining functional roles at the level of individual cells, or groups of highly defined cells. Gros et al. found that tumor-reactive T cells in situ show increased PD1 expression, more so than expression of TIM3, LAG3, or CD137 (142). Fourcade et al. demonstrated that the combined expression of PD1 and TIM3 identified T cells with diminished functional capabilities (143). However, functional studies in absence of the cells’ natural environment (i.e. outside of the organisms) may give misleading results. As outlined above, T cells that express certain iRs may show reduced function because the receptor indeed mediates inhibition, or because the iR-positive cells are in a different functional state, with different activation and/or differentiation (72).
Triggering of expression of many immune genes is promoted by IFNγ, which is produced by activated immune cells in tumors. PD-L1 is a typical example, and may be taken as a “marker” for immune activation and ongoing CTL activity in situ (129). However, PD-L1 can also be constitutively expressed by tumor cells, such as found in a minority of melanoma patients. The combined assessment of PD-L1 expression and CD8 T cells in tumors is currently being used to distinguish scenarios of constitutive versus induced PD-L1 expression, whereby the quantification of CD8 T cells serves as parameter of immune cell activation in situ (142, 144).
Overall, it remains difficult to draw conclusions from human studies. iR expression in the TME can be a good sign when anti-tumor immune responses are nevertheless taking place and thus contribute to favorable clinical outcomes. As mentioned, iR expression is frequently up-regulated by T cell activation and may be thus a sign of ongoing immune responses. In turn, iR expression may be associated with unfavorable clinical course when occurring in (relative) absence of anti-tumor T cell responses, for example, when tumors constitutively express immune inhibitory molecules (e.g. PD-L1) and fundamentally avoid and or block (almost) any effective immune response against the cancer, even under immunotherapy.
KEY CONCEPT 5. Favorable clinical outcomes
The expression of multiple iRs can reflect recent or ongoing T cell activation – iRs may, in fact, mark the cells that responded to a given stimulus (including therapy) and be good prognostic indicators.
Recommendations for Immune Monitoring of T Cell Function
Biological readouts do often not allow conclusions for individual patients. It remains challenging to identify biomarkers, as most of them end up not being useful for routine clinical management of individual patients. Nevertheless, biomarkers research is often very worthwhile, through their contributions for identifying disease mechanisms.
When analyzing immune responses from patients, caution is required for the interpretation of results. In view of the increasing clinical importance of iRs, patient parameters are now frequently quantified by various methods such as flow cytometry or molecular techniques. From the literature as reviewed here, it becomes clear that the quantification of iR expression by T cells does not allow to conclude that they have impaired functions. Besides considering exhaustion and the TME, it is crucial and possible to analyze parameters of T cell differentiation and activation. Subsets at different stages of differentiation can be distinguished by analyzing T cells with regard to lymph node homing receptors (such as CCR7 and CD62L), and differentiation markers (e.g. KLRG-1 and CD45 isotypes). The separate analysis of naïve cells is most important, but memory cells should also be distinguished from effector cells, as they may also differ largely for iR expression and functions. As mentioned in the previous sections, shifts in iR expression of total cells are often due to changes in differentiation, rather than changes of iR expression within a particular differentiation subset. Similarly, changes in T cell activation are associated with altered iR expression. Therefore, activation markers should also be included, to evaluate this possibility as driver for iR expression.
Regarding the analyses of functional properties, the question remains open whether the available in vitro methods allow concluding on in vivo T cell functions. The high degree of context dependency and the fact that multiple receptors may simultaneously be involved in controlling T cell function in vivo should be taken into account. In vitro assays often fail to reproduce the reality. Great care should be taken for drawing conclusions. Nevertheless, in conjunction with results from increasingly sophisticated and realistic mouse models, it is worthwhile to perform functional in vitro experiments with human cells, particularly also for the identification of synergistic or antagonistic effects between different iRs and activatory receptors, for example, 4-1BB. It may be useful to characterize the co-expression of multiple iRs, together with other receptors. The recent discovery that TIM3 mediates different functions depending on co-expression of CEACAM-1 (67) represents a typical example for the need to identify possible receptor pairing with functional relevance.
It is necessary to elucidate the contradictory results obtained from studies analyzing iRs and their ligands, and their associations with favorable or unfavorable clinical outcome. Distinguishing tumors in presence versus absence of anti-tumor T cells may lead to a better understanding. Possibly, up-regulation of iRs and iR ligands in the TME in presence of anti-cancer T cells may be favorable indicators because they reflect ongoing anti-cancer immune responses. By contrast, in absence of immune responses, the expression of iRs and iR ligands point to a constitutive immune blockade, which is often associated with unfavorable prognosis and non-responsiveness to immunotherapy.
For the future, advances in basic science are necessary for improved understanding and interpretation of clinical data. For the time being, detailed characterization of T cell activation and differentiation represents already a significant step forward toward a comprehensive characterization and interpretation of iR expression, providing insight in patient’s immune status.
Concluding Remarks
The success of “checkpoint blockade” therapies in clinical oncology is probably the best proof that iRs can play crucial roles in the regulation of cellular immune responses. However, the mechanisms of action of these novel therapies are still poorly understood, and probably involve multiple cell types and functions. iRs do not only inhibit the effector T cells that express them but can also act early in the generation of an immune response, and/or promote inhibitory functions of immune suppressive cell types such as Treg cells or myeloid cells. Importantly, the sole expression of iRs does not allow to directly conclude on the functional status of T cells. Historically, iR expression was associated with T cell “exhaustion,” i.e. those T cells with reduced effector functions found in chronic infection and in the TME. iRs were considered “guilty by association”, without systematic critical evaluation of their functional impact. Importantly, iRs are also frequently expressed by highly functional T cells such as the ones that predominate in acute infections. This is explained by the fact that T cell activation and T cell differentiation lead to strong upregulation of many iRs, as part of the physiological balance of T cell activation, even though these cells remain highly functional. Clearly, the impact of iRs on T cell function is context-dependent, as the result of the sum of activatory and inhibitory signals. For research and the immune monitoring of patients, it is important to primarily study immune cell functions in vivo, and to carefully consider T cell activation and differentiation by subset analyses for the interpretation of iR expression data.
KEY CONCEPT 6. Consider T cell activation and differentiation
T cell differentiation and activation are major drivers of iR expression. For research and immune monitoring of patients, it is an absolute need to analyze parameters of differentiation (cell type and subset analysis) and activation markers to interpret iR expression data and functionality of T cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our colleagues for collaboration and support. We apologize for not mentioning and citing numerous studies due to space limitations. This work was supported by the Cancer Research Institute (USA), Ludwig Cancer Research (USA), the Cancer Vaccine Collaborative (USA), Atlantic Philanthropies (USA), the Wilhelm Sander-Foundation (Germany), a Swiss Cancer Research grant (3507-08-2014), a SwissTransMed grant (KIP 18), and two Swiss National Science Foundation grants (320030_152856, CRSII3_141879).
Author Biography

References
2. Cambier JC. New nomenclature for the Reth motif (or ARH1/TAM/ARAM/YXXL). Immunol Today (1995) 16:110. doi:10.1016/0167-5699(95)80105-7
3. Iwashima M, Irving BA, van Oers NS, Chan AC, Weiss A. Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science (1994) 263:1136–9. doi:10.1126/science.7509083
4. Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature (1994) 368:70–3. doi:10.1038/368070a0
5. Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science (1992) 256:1808–12. doi:10.1126/science.1535455
6. Daëron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, et al. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity (1995) 3:635–46. doi:10.1016/1074-7613(95)90134-5
7. Daëron M, Malbec O, Latour S, Arock M, Fridman W-H. Regulation of high-affinity IgE receptor-mediated mast cell activation by murine low-affinity IgG receptors. J Clin Invest (1995) 95:577–85. doi:10.1172/JCI117701
8. Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. SH2 domains recognize specific phosphopeptide sequences. Cell (1993) 72:767–78. doi:10.1016/0092-8674(93)90404-E
9. Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, et al. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity (1996) 4:77–85. doi:10.1016/S1074-7613(00)80300-3
10. Burshtyn DN, Yang W, Yi T, Long EO. A novel phosphotyrosine motif with a critical amino acid at position -2 for the SH2 domain-mediated activation of the tyrosine phosphatase SHP-1. J Biol Chem (1997) 272:13066–72. doi:10.1074/jbc.272.20.13066
11. Bléry M, Kubagawa H, Chen CC, Vély F, Cooper MD, Vivier E. The paired Ig-like receptor PIR-B is an inhibitory receptor that recruits the protein-tyrosine phosphatase SHP-1. Proc Natl Acad Sci U S A (1998) 95:2446–51. doi:10.1073/pnas.95.5.2446
12. Schneider H, Prasad KV, Shoelson SE, Rudd CE. CTLA-4 binding to the lipid kinase phosphatidylinositol 3-kinase in T cells. J Exp Med (1995) 181:351–5. doi:10.1084/jem.181.1.351
13. Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature (1992) 358:66–70. doi:10.1038/358066a0
14. Lanier LL. NK cell receptors. Annu Rev Immunol (1998) 16:359–93. doi:10.1146/annurev.immunol.16.1.359
15. Vély F, Olivero S, Olcese L, Moretta A, Damen JE, Liu L, et al. Differential association of phosphatases with hematopoietic co-receptors bearing immunoreceptor tyrosine-based inhibition motifs. Eur J Immunol (1997) 27:1994–2000. doi:10.1002/eji.1830270825
16. Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol (1999) 17:875–904. doi:10.1146/annurev.immunol.17.1.875
17. Pérez-Villar JJ, Carretero M, Navarro F, Melero I, Rodríguez A, Bottino C, et al. Biochemical and serologic evidence for the existence of functionally distinct forms of the CD94 NK cell receptor. J Immunol (1996) 157:5367–74.
18. Wagtmann N, Biassoni R, Cantoni C, Verdiani S, Malnati MS, Vitale M, et al. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity (1995) 2:439–49. doi:10.1016/1074-7613(95)90025-X
19. Vivier E, Daëron M. Immunoreceptor tyrosine-based inhibition motifs. Immunol Today (1997) 18:286–91. doi:10.1016/S0167-5699(97)80025-4
20. Cambier JC. Inhibitory receptors abound? Proc Natl Acad Sci U S A (1997) 94:5993–5. doi:10.1073/pnas.94.12.5993
21. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11:3887–95.
22. Shinohara T, Taniwaki M, Ishida Y, Kawaichi M, Honjo T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics (1994) 23:704–6. doi:10.1006/geno.1994.1562
23. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily – CTLA-4. Nature (1987) 328:267–70. doi:10.1038/328267a0
24. Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol (2003) 3:544–56. doi:10.1038/nri1131
25. Workman CJ, Vignali D. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol (2003) 33:970–9. doi:10.1002/eji.200323382
26. Sierro S, Romero P, Speiser DE. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin Ther Targets (2011) 15:91–101. doi:10.1517/14712598.2011.540563
27. Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol (2004) 4:336–47. doi:10.1038/nri1349
28. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A (2001) 98:13866–71. doi:10.1073/pnas.231486598
29. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173:945–54. doi:10.4049/jimmunol.173.2.945
30. Shlapatska LM, Mikhalap SV, Berdova AG, Zelensky OM, Yun TJ, Nichols KE, et al. CD150 association with either the SH2-containing inositol phosphatase or the SH2-containing protein tyrosine phosphatase is regulated by the adaptor protein SH2D1A. J Immunol (2001) 166:5480–7. doi:10.4049/jimmunol.166.9.5480
31. Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol (2012) 188:2957–65. doi:10.4049/jimmunol.1100038
32. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27:111–22. doi:10.1016/j.immuni.2007.05.016
33. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174:561–9. doi:10.1084/jem.174.3.561
34. Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity (2004) 21:401–13. doi:10.1016/j.immuni.2004.06.017
35. Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med (2010) 16:1147–51. doi:10.1038/nm.2232
36. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13:227–42. doi:10.1038/nri3405
37. Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature (1993) 362:758–61. doi:10.1038/362758a0
38. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27:670–84. doi:10.1016/j.immuni.2007.09.006
40. Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest (2011) 121:2350–60. doi:10.1172/JCI46102
41. Youngblood B, Wherry EJ, Ahmed R. Acquired transcriptional programming in functional and exhausted virus-specific CD8 T cells. Curr Opin HIV AIDS (2012) 7:50–7. doi:10.1097/COH.0b013e32834ddcf2
42. Vali B, Jones RB, Sakhdari A, Sheth PM, Clayton K, Yue F-Y, et al. HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression. Eur J Immunol (2010) 40:2493–505. doi:10.1002/eji.201040340
43. Sakhdari A, Mujib S, Vali B, Yue F-Y, MacParland S, Clayton K, et al. Tim-3 negatively regulates cytotoxicity in exhausted CD8+ T cells in HIV infection. PLoS One (2012) 7:e40146. doi:10.1371/journal.pone.0040146
44. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8+ T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res (2012) 72:887–96. doi:10.1158/0008-5472.CAN-11-2637
45. Larsen M, Sauce D, Deback C, Arnaud L, Mathian A, Miyara M, et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PLoS Pathog (2011) 7:e1002328. doi:10.1371/journal.ppat.1002328
46. Horne-Debets JM, Faleiro R, Karunarathne DS, Liu XQ, Lineburg KE, Poh CM, et al. PD-1 dependent exhaustion of CD8+ T cells drives chronic malaria. Cell Rep (2013) 5:1204–13. doi:10.1016/j.celrep.2013.11.002
47. Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol (2010) 22:223–30. doi:10.1016/j.coi.2010.02.005
48. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207:2175–86. doi:10.1084/jem.20100637
49. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A (2002) 99:12293–7. doi:10.1073/pnas.192461099
50. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A (2010) 107:7875–80. doi:10.1073/pnas.1003345107
51. Crawford A, Wherry EJ. The diversity of costimulatory and inhibitory receptor pathways and the regulation of antiviral T cell responses. Curr Opin Immunol (2009) 21:179–86. doi:10.1016/j.coi.2009.01.010
52. Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology (2010) 129:474–81. doi:10.1111/j.1365-2567.2010.03255.x
53. Khaitan A, Unutmaz D. Revisiting immune exhaustion during HIV infection. Curr HIV/AIDS Rep (2011) 8:4–11. doi:10.1007/s11904-010-0066-0
54. Henao-Tamayo M, Irwin SM, Shang S, Ordway D, Orme IM. T lymphocyte surface expression of exhaustion markers as biomarkers of the efficacy of chemotherapy for tuberculosis. Tuberculosis (Edinb) (2011) 91:308–13. doi:10.1016/j.tube.2011.04.001
55. Shin E-C, Park S-H, Nascimbeni M, Major M, Caggiari L, de Re V, et al. The frequency of CD127(+) hepatitis C virus (HCV)-specific T cells but not the expression of exhaustion markers predicts the outcome of acute HCV infection. J Virol (2013) 87:4772–7. doi:10.1128/JVI.03122-12
56. Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Lienard D, Lejeune F, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res (2004) 64:2865–73. doi:10.1158/0008-5472.CAN-03-3066
57. Baitsch L, Fuertes Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol (2012) 33:364–72. doi:10.1016/j.it.2012.02.006
58. Fuertes Marraco SA, Baumgaertner P, Legat A, Rufer N, Speiser DE. A stepwise protocol to coat aAPC beads prevents out-competition of anti-CD3 mAb and consequent experimental artefacts. J Immunol Methods (2012) 385(1–2):90–5. doi:10.1016/j.jim.2012.07.017
59. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A (2013) 110(27):E2480–9. doi:10.1073/pnas.1305394110
60. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209:1201–17. doi:10.1084/jem.20112741
61. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A (2008) 105:15016–21. doi:10.1073/pnas.0801497105
62. Walker LSK, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol (2011) 11:852–63. doi:10.1038/nri3108
63. Walker LSK, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol (2015) 36:63–70. doi:10.1016/j.it.2014.12.001
64. Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol (2002) 3:611–8. doi:10.1038/ni0702-611
65. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. doi:10.1084/jem.20130579
66. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A (2015) 112:6140–5. doi:10.1073/pnas.1417320112
67. Huang Y-H, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi:10.1038/nature13848
68. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2008) 10:29–37. doi:10.1038/ni.1679
69. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi:10.1182/blood-2008-12-195792
70. Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol (2009) 83:9122–30. doi:10.1128/JVI.00639-09
71. Merino J, Ramírez N, Moreno C, Toledo E, Fernández M, Sánchez-Ibarrola A. BY55/CD160 cannot be considered a cytotoxic marker in cytomegalovirus-specific human CD8(+) T cells. Clin Exp Immunol (2007) 149:87–96. doi:10.1111/j.1365-2249.2007.03387.x
72. Derré L, Rivals J-P, Jandus C, Pastor S, Rimoldi D, Romero P, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest (2010) 120:157–67. doi:10.1172/JCI40070
73. Baitsch L, Legat A, Barba L, Fuertes Marraco SA, Rivals J-P, Baumgaertner P, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One (2012) 7:e30852. doi:10.1371/journal.pone.0030852
74. Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J Immunol (2011) 186:4200–12. doi:10.4049/jimmunol.1001783
75. Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “exhaustion” of human CD8 T cells. Front Immunol (2013) 4:455. doi:10.3389/fimmu.2013.00455
76. Appay V, van Lier RAW, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A (2008) 73:975–83. doi:10.1002/cyto.a.20643
77. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17:1290–7. doi:10.1038/nm.2446
78. Roychoudhuri R, Lefebvre F, Honda M, Pan L, Ji Y, Klebanoff CA, et al. Transcriptional profiles reveal a stepwise developmental program of memory CD8(+) T cell differentiation. Vaccine (2015) 33:914–23. doi:10.1016/j.vaccine.2014.10.007
79. Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Maillard SA, et al. Long-lasting stem cell-like memory CD8+ T cells with a naïve-like profile upon yellow fever vaccination. Sci Transl Med (2015) 7:282ra48. doi:10.1126/scitranslmed.aaa3700
80. Utzschneider DT, Legat A, Fuertes Marraco SA, Carrié L, Luescher I, Speiser DE, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol (2013) 14:603–10. doi:10.1038/ni.2606
81. Zhu Y, Yao S, Chen L. Cell surface signaling molecules in the control of immune responses: a tide model. Immunity (2011) 34:466–78. doi:10.1016/j.immuni.2011.04.008
82. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol (1996) 8:765–72. doi:10.1093/intimm/8.5.765
83. Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog (2010) 6:e1000947. doi:10.1371/journal.ppat.1000947
84. Viganò S, Banga R, Bellanger F, Pellaton C, Farina A, Comte D, et al. CD160-associated CD8 T-cell functional impairment is independent of PD-1 expression. PLoS Pathog (2014) 10:e1004380. doi:10.1371/journal.ppat.1004380
85. Peretz Y, He Z, Shi Y, Yassine-Diab B, Goulet J-P, Bordi R, et al. CD160 and PD-1 co-expression on HIV-specific CD8 T cells defines a subset with advanced dysfunction. PLoS Pathog (2012) 8:e1002840. doi:10.1371/journal.ppat.1002840
86. Qiu Y, Chen J, Liao H, Zhang Y, Wang H, Li S, et al. Tim-3-expressing CD4+ and CD8+ T cells in human tuberculosis (TB) exhibit polarized effector memory phenotypes and stronger anti-TB effector functions. PLoS Pathog (2012) 8:e1002984. doi:10.1371/journal.ppat.1002984
87. Zelinskyy G, Myers L, Dietze KK, Gibbert K, Roggendorf M, Liu J, et al. Virus-specific CD8+ T cells upregulate programmed death-1 expression during acute friend retrovirus infection but are highly cytotoxic and control virus replication. J Immunol (2011) 187:3730–7. doi:10.4049/jimmunol.1101612
88. Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res (2012) 72:3163–74. doi:10.1158/0008-5472.CAN-12-0210
89. Allie SR, Zhang W, Fuse S, Usherwood EJ. Programmed death 1 regulates development of central memory CD8 T cells after acute viral infection. J Immunol (2011) 186:6280–6. doi:10.4049/jimmunol.1003870
90. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (2012) 338:1220–5. doi:10.1126/science.1229620
91. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity (2014) 41:802–14. doi:10.1016/j.immuni.2014.10.013
92. Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaëlsson J, et al. T-bet and eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog (2014) 10:e1004251. doi:10.1371/journal.ppat.1004251
93. Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Re-evaluation of PD-1 expression by T cells as a marker for immune exhaustion during SIV infection. PLoS One (2013) 8:e60186. doi:10.1371/journal.pone.0060186
94. Haymaker C, Wu R, Bernatchez C, Radvanyi L. PD-1 and BTLA and CD8(+) T-cell “exhaustion” in cancer: “exercising” an alternative viewpoint. Oncoimmunology (2012) 1:735–8. doi:10.4161/onci.20823
95. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270:985–8. doi:10.1126/science.270.5238.985
96. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi:10.1016/1074-7613(95)90125-6
97. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi:10.1126/science.1160062
98. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1:405–13. doi:10.1016/1074-7613(94)90071-X
99. Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res (2013) 19:5300–9. doi:10.1158/1078-0432.CCR-13-0143
100. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2005) 439:682–7. doi:10.1038/nature04444
101. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med (2012) 209:2485–99. doi:10.1084/jem.20121015
102. Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U S A (2010) 107:13402–7. doi:10.1073/pnas.1007394107
103. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291:319–22. doi:10.1126/science.291.5502.319
104. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51. doi:10.1016/S1074-7613(00)80089-8
105. Minor DR, Chin K, Kashani-Sabet M. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm (2009) 24:321–5. doi:10.1089/cbr.2008.0607
106. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
107. Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369:134–44. doi:10.1056/NEJMoa1305133
108. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369:122–33. doi:10.1056/NEJMoa1302369
109. Riley JL. Combination checkpoint blockade – taking melanoma immunotherapy to the next level. N Engl J Med (2013) 369:187–9. doi:10.1056/NEJMe1305484
110. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet (2014) 384:1109–17. doi:10.1016/S0140-6736(14)60958-2
111. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol (2014) 32:1020–30. doi:10.1200/JCO.2013.53.0105
112. Wolchok JD, Weber JS, Maio M, Neyns B, Harmankaya K, Chin K, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol (2013) 24:2174–80. doi:10.1093/annonc/mdt161
113. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol (2015) 33:1889–94. doi:10.1200/JCO.2014.56.2736
114. Naidoo J, Page DB, Wolchok JD. Immune modulation for cancer therapy. Br J Cancer (2014) 111:2214–9. doi:10.1038/bjc.2014.348
115. Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol (2015) 33:23–35. doi:10.1016/j.coi.2015.01.006
116. Karagiannis P, Fittall M, Karagiannis SN. Evaluating biomarkers in melanoma. Front Oncol (2014) 4:383. doi:10.3389/fonc.2014.00383
117. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12:298–306. doi:10.1038/nrc3245
118. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the “immunoscore” in the classification of malignant tumours. J Pathol (2014) 232:199–209. doi:10.1002/path.4287
119. Mahoney KM, Atkins MB. Prognostic and predictive markers for the new immunotherapies. Oncology (Williston Park) (2014) 28(Suppl 3):39–48.
120. Kim JW, Eder JP. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology (Williston Park) (2014) 28(Suppl 3):15–28.
121. Thompson RH, Dong H, Lohse CM, Leibovich BC, Blute ML, Cheville JC, et al. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res (2007) 13:1757–61. doi:10.1158/1078-0432.CCR-06-2599
122. Kang MJ, Kim KM, Bae JS, Park HS, Lee H, Chung MJ, et al. Tumor-infiltrating PD1-positive lymphocytes and FoxP3-positive regulatory T cells predict distant metastatic relapse and survival of clear cell renal cell carcinoma. Transl Oncol (2013) 6:282–9. doi:10.1593/tlo.13256
123. Richendollar BG, Pohlman B, Elson P, Hsi ED. Follicular programmed death 1-positive lymphocytes in the tumor microenvironment are an independent prognostic factor in follicular lymphoma. Hum Pathol (2011) 42:552–7. doi:10.1016/j.humpath.2010.08.015
124. Kim JR, Moon YJ, Kwon KS, Bae JS, Wagle S, Kim KM, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS One (2013) 8:e82870. doi:10.1371/journal.pone.0082870
125. Krönig H, Julia Falchner K, Odendahl M, Brackertz B, Conrad H, Muck D, et al. PD-1 expression on Melan-A-reactive T cells increases during progression to metastatic disease. Int J Cancer (2012) 130:2327–36. doi:10.1002/ijc.26272
126. Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res (2013) 73:128–38. doi:10.1158/0008-5472.CAN-12-2606
127. Droeser RA, Hirt C, Viehl CT, Frey DM, Nebiker C, Huber X, et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur J Cancer (2013) 49:2233–42. doi:10.1016/j.ejca.2013.02.015
128. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer (2010) 116:1757–66. doi:10.1002/cncr.24899
129. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med (2012) 4:ra37–127. doi:10.1126/scitranslmed.3003689
130. Massi D, Brusa D, Merelli B, Ciano M, Audrito V, Serra S, et al. PD-L1 marks a subset of melanomas with a shorter overall survival and distinct genetic and morphological characteristics. Ann Oncol (2014) 25:2433–42. doi:10.1093/annonc/mdu452
131. Chen J, Chen Z. The effect of immune microenvironment on the progression and prognosis of colorectal cancer. Med Oncol (2014) 31:82. doi:10.1007/s12032-014-0082-9
132. Kotaskova J, Tichy B, Trbusek M, Francova HS, Kabathova J, Malcikova J, et al. High expression of lymphocyte-activation gene 3 (LAG3) in chronic lymphocytic leukemia cells is associated with unmutated immunoglobulin variable heavy chain region (IGHV) gene and reduced treatment-free survival. J Mol Diagn (2010) 12:328–34. doi:10.2353/jmoldx.2010.090100
133. Piao Y-R, Piao L-Z, Zhu L-H, Jin Z-H, Dong X-Z. Prognostic value of T cell immunoglobulin mucin-3 in prostate cancer. Asian Pac J Cancer Prev (2013) 14:3897–901. doi:10.7314/APJCP.2013.14.6.3897
134. Yuan J, Jiang B, Zhao H, Huang Q. Prognostic implication of TIM-3 in clear cell renal cell carcinoma. Neoplasma (2014) 61:35–40. doi:10.4149/neo_2014_006
135. Jiang J, Jin M-S, Kong F, Cao D, Ma H-X, Jia Z, et al. Decreased galectin-9 and increased Tim-3 expression are related to poor prognosis in gastric cancer. PLoS One (2013) 8:e81799. doi:10.1371/journal.pone.0081799
136. Cao Y, Zhou X, Huang X, Li Q, Gao L, Jiang L, et al. Tim-3 expression in cervical cancer promotes tumor metastasis. PLoS One (2013) 8:e53834. doi:10.1371/journal.pone.0053834
137. van Esch EMG, van Poelgeest MIE, Kouwenberg S, Osse EM, Trimbos JBMZ, Fleuren GJ, et al. Expression of coinhibitory receptors on T cells in the microenvironment of usual vulvar intraepithelial neoplasia is related to proinflammatory effector T cells and an increased recurrence-free survival. Int J Cancer (2015) 136:E95–106. doi:10.1002/ijc.29174
138. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28:3167–75. doi:10.1200/JCO.2009.26.7609
139. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
140. Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol (2014) 4:385. doi:10.3389/fonc.2014.00385
141. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi:10.1038/nature13954
142. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124:2246–59. doi:10.1172/JCI73639
143. Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res (2014) 74:1045–55. doi:10.1158/0008-5472.CAN-13-2908
Keywords: inhibitory receptors, T cell exhaustion, activation, differentiation, immune monitoring, immunotherapy, checkpoint blockade
Citation: Fuertes Marraco SA, Neubert NJ, Verdeil G and Speiser DE (2015) Inhibitory receptors beyond T cell exhaustion. Front. Immunol. 6:310. doi: 10.3389/fimmu.2015.00310
Received: 27 April 2015; Accepted: 30 May 2015;
Published: 26 June 2015
Edited by:
Vincenzo Bronte, Verona University, ItalyReviewed by:
Luca Gattinoni, National Cancer Institute, USAViktor Umansky, German Cancer Research Center (DKFZ), Germany
Copyright: © 2015 Fuertes Marraco, Neubert, Verdeil and Speiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:ZG9jQGRzcGVpc2VyLmNo