 Alaa Kassim Ali
Alaa Kassim Ali Neethi Nandagopal
Neethi Nandagopal Seung-Hwan Lee
Seung-Hwan Lee- Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
Among numerous cytokines modulating natural killer (NK) cell function, interleukin 15 (IL-15) exerts a broad range of effect from development and homeostasis, to activation of mature NK cells during infection. Its significance is further highlighted by clinical trials in which IL-15 is being used to boost the proliferation and anti-tumor response of NK cells. Among the signal transduction pathways triggered by the engagement of IL-15 receptor with its ligand, the PI3K–AKT–mTOR pathway seems to be critical for the IL-15-mediated activation of NK cells, therefore being responsible for efficient anti-viral and anti-tumor responses. This review provides an overview of the role of IL-15 at multiple stages of NK cell life journey. Understanding the pathway by which IL-15 conveys critical signals for the generation of NK cells with efficient effector functions, in combination with established protocols for NK cell expansion ex vivo, will undoubtedly open new avenues for therapeutic applications for immunomodulation against infections and cancers.
Introduction
Natural killer (NK) cells play critical roles in innate immune responses mainly against intracellular viruses and tumors (1–3). The term NK was originally derived from the observation that they are poised to kill target cells without prior sensitization or further differentiation (4, 5). Even though recognition receptors that target infected cells are germline-encoded with restricted recognition compared to T and B cell receptors, the mechanism of NK cell cytotoxicity against virus-infected cells is similar to that of CD8+ cytotoxic T lymphocytes (CTL). NK cells induce perforin/granzyme-dependent apoptosis in target cells and thereby eliminate reservoirs of virus replication (2, 3, 6). For this, NK cells express an array of inhibitory and activating receptors, which receive multiple stimuli from virus-infected cells. The balance between signals that are generated from activating receptors and inhibitory receptors mainly determines the immediate cytotoxic activation. NK cell activation also induces cytokine secretion such as IFN-γ and TNF-α. These cytokines enhance the phagocytic function of macrophages and their antimicrobial activity, and augment the adaptive immune response via up-regulation of antigen presentation by antigen presenting cells such as dendritic cells (DCs) (2, 7).
In addition to signals from both NK cell activating and inhibitory receptors, signals from cytokine receptors on NK cells greatly modulate the development and activation of NK cells. Even though cytokines can exhibit pleiotropic effects depending on the responding cell’s condition, IL-2, -12, -15, -18, and -21 are regarded as being stimulators for NK cell function (8, 9) whereas TGF-β and IL-10 are known as negative regulators (9). Notably, IL-15 is the most potent among them by orchestrating the entire life of NK cells and holds a great potential for immunotherapy in cancer and infectious research. Here, we will provide an overview of our knowledge regarding the PI3K–AKT–mTOR pathway as a major IL-15-mediated pathway driving the development, proliferation, and effector functions of NK cells.
KEY CONCEPT 1. mTOR Pathway.
The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that functions as a sensor of environmental nutrient availability, and translates the information into cellular signals that regulate ribosomal biogenesis, protein translation, cell growth, and cell proliferation.
Interleukin-15
IL-15 drew much attention due to its similarity to IL-2 in its cytokine receptor biology. IL-15R is a heterotrimeric receptor consisting of a unique α chain, a shared β subunit with IL-2 (CD122) and a common γ subunit (CD132) shared with several cytokines (10), implying similar biological activities of IL-2 and IL-15. IL-2 was the first cytokine used in clinical cancer trials due to its great ability to expand NK cells and stimulate NK cell functions (11). However, severe toxicity associated with the administration of IL-2 limited its application. Recently, the clinical interest of using IL-15 was further strengthened because IL-2 administration also expanded regulatory T cells, which suppress the anti-tumor response of NK cells and effector T cells (12–14). On the other hand, IL-15 therapy resulted in low toxicity with expansion of NK cells without expansion of Tregs in vivo, in a study of non-human primates (15).
KEY CONCEPT 2. IL-15
IL-15 is a key cytokine exhibiting a pleiotropic effect on the development, proliferation and activation of natural killer cells, as well as the proliferation and activation in CD8+ T-cell, therefore being considered as one of most promising molecules for anti-cancer immune therapy.
Studies have shown that IL-15-/- mice exhibit a selective loss of memory phenotype CD8+ T cells, NKT cells, and NK cells (10, 16, 17). A unique aspect of IL-15 is that it employs trans-presentation in which IL-15-producing cells present the cytokine in the context of IL-15 receptor α (IL-15Rα) chain on the cell surface. A pivotal role of dendritic cell (DC)-expressed IL-15Rα for trans-presenting IL-15 to NK cells has been demonstrated (18, 19). Engagement of IL-15R on NK cells causes the auto-phosphorylation and activation of Janus Kinases (JAK1 and JAK3), which induces at least three parallel signaling cascades: Ras–Raf–MAPK, signal transduction and activation of transcription (STAT) 5 and PI3K–AKT–mTOR pathways (10, 20). The indispensable role of IL-15–STAT5 pathway for NK cell development and homeostasis was also exemplified in mice having defects in the signaling pathway in which the number of mature NK cells was dramatically reduced (21–23). Data from Stat5-deficient and NK cell-specific Stat5-deficient mice showed that NK cells are absent in peripheral lymphoid organs, suggesting a crucial role of the IL-15–STAT5 pathway in NK cell development (21–23). In addition, similarly to STAT5 knock-out mice, a severe reduction in NK cell numbers was found in a patient carrying the STAT5b mutation (24).
PI3K–AKT–mTOR Pathway
In addition to the IL-15–STAT5 pathway, accumulating data pointed out that the PI3K–AKT–mTOR pathway is essential for modulating the development, differentiation, and activation of immune cells including NK cells. Signaling triggered by cytokines employing the JAK–STAT pathway generally stimulates the PI3K/AKT signaling pathway in immune cells (25). PI3K, phosphatidylinositol 3-kinase, is conserved in all mammalian cells and is known to control diverse processes including cell proliferation, survival, differentiation, activation of effector functions, and metabolism (26, 27). Among three classes (I, II, and III), the class I PI3Ks, which are heterodimeric enzymes consisting of a regulatory subunit (p85) and a catalytic subunit (p110), predominately regulate downstream signals emanating from cytokine receptor activation. Upon cytokines binding to their receptors, receptor tyrosine kinases activate PI3K, which generates phosphatidylinositol trisphosphate (PIP3) from plasma membrane-associated phosphatidylinositol bisphosphate (PIP2). PIP3 has an affinity for pleckstrin homology (PH) domain-containing molecules such as AKT and phosphoinositide-dependent protein kinase (PDK1) on the inner leaflet of the plasma membrane. At the plasma membrane, the interaction between the PH domain of AKT and PIP3 induces important conformational changes in AKT, which allow subsequent modifications of AKT at threonine 308 by PDK1. mTORC2 also can phosphorylate AKT at serine 473 for further activation (28).
Activated AKT phosphorylates crucial targets and contributes to cell survival by inhibiting pro-apoptotic members of the Bcl-2 family. One of the important downstream effectors for the PI3K/AKT signaling is mTOR, which is a serine/threonine protein kinase required for the translation of proteins that promote cell survival and proliferation. mTOR exists as two complexes, mTORC1 and mTORC2. Even though mTORC2 can activate mTORC1 by AKT phosphorylation, a metabolic reprograming which supports effector T cell proliferation and functions has been mainly investigated in the context of mTORC1 complex. mTORC1 is negatively regulated by a heterodimeric protein complex called tuberous sclerosis complex (TSC) 1 and 2. The TSC inhibits mTORC1 by suppressing the conversion of Rheb-GDP to Rheb-GTP, a small GTPase, required for mTORC1 activation. PI3K–AKT signaling results in the phosphorylation and inactivation of TSC2, which increases Rheb-GTP and mTORC1 kinase activity (29–32). mTORC1 promotes the translation machinery through the phosphorylation of the translation-initiation factor eIF4E-binding protein (4EBP1), and the S6 ribosomal kinase (S6K). Upon phosphorylation, the translation repressor protein 4EBP1 is dissociated from eIF4E, leading to the subsequent formation of the translation initiation complex. S6K directly phosphorylates several proteins implicated in protein translation including eukaryotic initiation factors and ribosomal protein S6 (33). In addition, mTORC1 increases the rate of glycolysis by inducing the expression of HIF-1α and c-Myc and nutrient transporters (30).
PI3K–AKT–mTOR Pathway for NK Cell Development
Mature NK cells are differentiated from common lymphoid progenitors (CLPs). Even though NK cells can develop in extra-medullary sites such as the thymus and liver, the developmental program from CLPs to mature NK cells mainly occurs in the bone marrow (34, 35). CLPs differentiate into NK cell progenitors which are defined as Lin- NK1.1- CD122+ cells (36) and the acquisition of IL-15R-β chain (CD122) is a critical step allowing the progenitor cells to become responsive to IL-15 in the bone marrow compartment (Figure 1). Interestingly, NK cell progenitors display high proliferative potentials which are dependent on IL-15. Several studies from immune cell-specific deficient mice or in vitro NK cell differentiation identified factors responsible for the IL-15-mediated development process (35, 37).
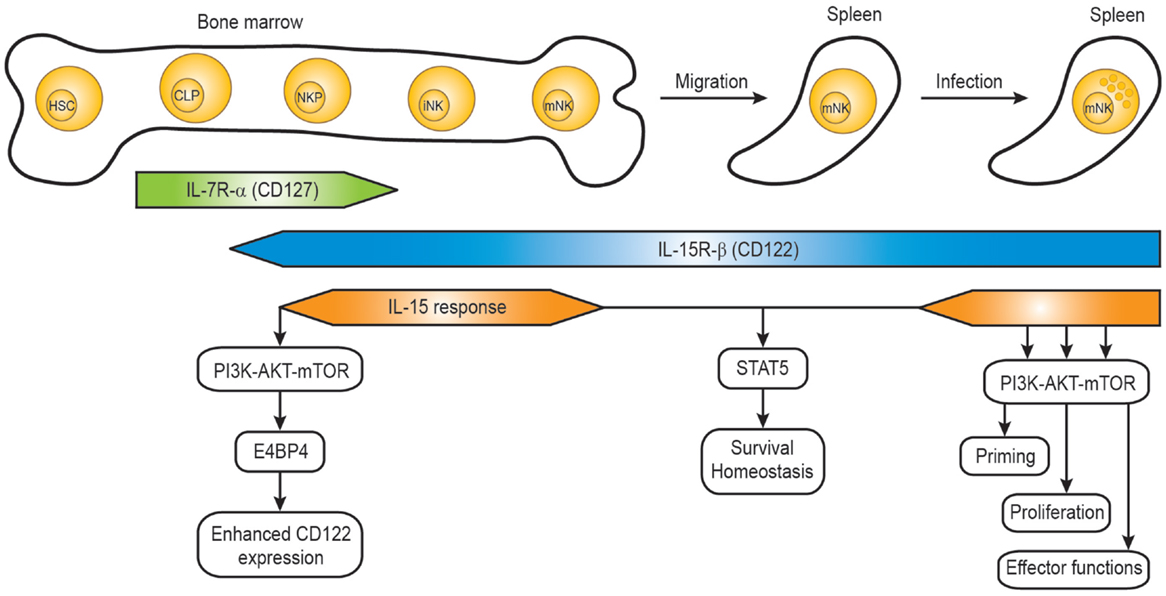
Figure 1. IL-15 response during natural killer cell development. The developmental stages of mouse NK cells in the bone marrow and periphery are shown, together with the IL-15R expression and IL-15 response. HSC, hematopoietic stem cell; CLP, common lymphoid progenitor; NKP, NK precursor; iNK, immature NK cell; mNK, mature NK cell.
Several identified factors are required for the acquisition and maintenance of CD122 on NK cell progenitors. The T-box transcription factor Eomes (also known as Eomesodermin) was shown to bind the CD122 promoter region, and the expression of CD122 on NK cells and memory CD8+ T cells from Eomes-deficient mice was significantly lower resulting in reduced responsiveness to IL-15 (38). The basic leucine zipper transcription factor E4BP4 (also known as Nfil3) seems to function upstream of Eomes, so that E4BP4 deficiency caused severe defects in NK cell development (39, 40). A recent paper demonstrated that PDK1, a kinase downstream of PI3K and upstream of mTOR, functions as a critical component in the positive feedback loop (41). Rescue of the defect of PDK1 by ectopic expression of E4BP4 or Eomes suggests that PDK1 signaling is critical for NK cell development via induction of E4BP4 and Eomes. Therefore, the IL-15 positive feedback loop might involve IL-15–PDK1–mTOR–E4BP4-Eomes–high levels of CD122-enhanced IL-15 signaling (Figure 1). The role of the metabolic checkpoint kinase mTOR on controlling NK cell proliferation in the bone marrow was also demonstrated in NK cell-specific mTOR-deficient mice (Ncr1-Cre × Mtorlox) (42). Consistently, a severe defect in early NK cell development was also observed in mice deficient in both the p110γ and p110δ catalytic subunits of PI3K (43), suggesting that PDK1 is a kinase linking IL-15–PI3K with the activation of AKT by phosphorylation.
PI3K–AKT–mTOR Pathway for NK Priming Effect
The priming effect is based on the fact that exposure of IL-15 sensitizes NK cells to secondary stimuli, thereby resulting in a heightened response. It is a physiologically relevant process because NK cells are recruited to lymph nodes where they are first activated by trans-presentation of IL-15 by IL-15Rα expressed on DCs (44) during inflammation and stimulated by subsequent cytokines or activating ligands. A well-known “priming” is exemplified by an exaggerated IFN-γ response when NK cells are co-stimulated with IL-12, IL-18, and IL-15 ex vivo (44–46). In our previous work, we demonstrate that the priming effect by IL-15 can be extended to stimulation by most cytokines employing the JAK–STAT pathway and ligands of NK cell activation receptors (47). When the priming effect is evaluated by measuring the phosphorylation of respective STATs, the response to type I IFN, IL-21, IL-12, IL-2, and IL-4 stimulation in NK cells primed by IL-15 was found greatly enhanced compared to naïve NK cells. In addition, the IFN-γ expression in primed NK cells after stimulation through Ly49H activation receptor was increased.
KEY CONCEPT 3. Priming Effect
The priming effect of cytokines refers to a phenomenon in which one cytokine can increase the sensitivity of cells to stimulation with other cytokines, resulting in an exaggerated response.
Several mechanisms have been proposed to explain the priming effect. Stimulation with IL-12 or IL-15 can induce the up-regulation of IL-18R transcript in human naive CD56+ NK cells, therefore enhancing the response to subsequent IL-18 exposure and IFN-γ production (45, 48). In our study, PI3K–AKT–mTOR pathway was identified as a principal pathway inducing the priming effect. Blocking of individual components of the pathway using cell-permeable reversible inhibitors severely abrogates the IL-15-mediated priming effects, as tested for, at least, IL-12-induced phosphorylation of STAT4, IL-21-induced phosphorylation of STAT3, and Ly49H-induced IFN-γ production. One caveat of using pharmacological inhibitors is that some inhibitors could modulate off-targets. In particular, Ly294002, a common PI3K inhibitor, has been known to induce non-specific effects, including the direct inhibition of mTOR’s catalytic function (49). Therefore, data obtained from experiments using Ly294002 should be taken cautiously. Nonetheless, inhibitors used for blocking AKT and mTOR in our study were generally regarded as highly specific. It is noteworthy that the requirement of mTOR for JAK–STAT activation might be extended to T cell regulation. Similar to the diminished levels of phosphorylated STATs in rapamycin-treated IL-15-primed NK cells, mTOR-deficient CD4 T cells fail to differentiate into Th1, Th2, and Th17 cells and this defect was largely due to impaired phosphorylation of respective STAT molecules required for each lineage differentiation (50). Mechanisms by which the PI3K–AKT–mTOR pathway crosstalks with the JAK–STAT pathway for inducing the priming effect are still not clear. It would be interesting to investigate the role of PI3K–AKT–mTOR pathway in the regulatory pathways of general JAK–STAT signaling such as suppressor of cytokine signaling (SOCS), protein inhibitors of activated STAT (PIAS), and protein tyrosine phosphatases (PTPs) (51).
Recently, a new form of priming using IL-15, IL-12, and IL-18 was suggested to generate memory-like NK cells which show enhanced effector functions upon restimulation following a 3-week resting period (52). This memory-like phenotype was found in proliferating NK cells. Due to the long-lasting and enhanced effector phenotype, studies that are trying to use adoptive transfer of the memory-like NK cells pre-activated with IL-12/-15/-18 for cancer immunotherapy (8, 53, 54) are being carried out, and one group showed that adoptive transfer of those pre-activated NK cells greatly increases the numbers and effector functions of NK cells, resulting in pronounced tumor regression in a mouse model of established tumor (53). One of the mechanisms responsible for this prolonged and enhanced tumor surveillance was mediated through IL-2Rα chain (CD25) up-regulation induced by IL-12 and IL-18 stimulation, thereby allowing NK cells to be sustained under the low physiological levels of IL-2 (53, 55). Nonetheless, whether PI3K–AKT–mTOR pathway is required to induce memory-like NK cells has not been tested.
PI3K–AKT–mTOR Pathway for NK Cell Proliferation during Virus Infection
During their differentiation in the bone marrow, NK cells hold a great potential of proliferation. However, NK cells become gradually quiescent as they fully mature (Figure 1) (56, 57). Ample data support that IL-15–JAK1/3-STAT5 is required for the homeostasis and survival of peripheral mature NK cells (10, 58, 59). Since IL-15 is essential for NK cell survival, cells deprived of IL-15 undergo considerable death. When adoptively transferred to IL-15-/- mice, mature splenic NK cells failed to be maintained, mainly due to the inability to sustain sufficient levels of the anti-apoptotic Bcl-2 expression in vivo (60, 61). Moreover, in vivo antibody blocking of IL-15R signaling also resulted in a severe loss of splenic NK cells within a week. Consistent with the previously identified indispensable role of IL-15-JAK1/3–STAT5, blocking of JAK and STAT5 pathways resulted in significant cell death, similar to when NK cells are deprived of IL-15. Notably, NK cells treated with inhibitors for the PI3K–AKT–mTOR were still able to maintain their viability similar to controls (cells without any inhibitor treatment) in the presence of IL-15, suggesting that the pathway is dispensable to support the survival of mature NK cells (47). The dispensable role of mTOR for survival was further supported in the study showing intact prosurvival signals provided by IL-15 in mTOR-deficient NK cells (21, 42).
Mature peripheral NK cells can rapidly and intensively proliferate during viral infections. The evidence for NK cells proliferation during virus infections was first recognized by Biron and colleagues three decades ago when they observed transient NK cell blastogenesis characterized by increased cell mass and proliferation at the onset of lymphocytic choriomeningitis virus (LCMV) and murine cytomegalovirus (MCMV) infections (62–64). Early on during the course of MCMV infection, blastogenesis and proliferation of NK cell is dependent on IL-15 (65, 66) (Figure 2). The IL-15 trans-presentation and the resulting activation of NK cells are dependent on type I IFN mainly induced by TLR recognition of viral PAMP (44). Our study pointed out that the PI3K–AKT–mTOR pathway is essential for the proliferation of mature NK cells upon IL-15 stimulation (47). Ex vivo IL-15 stimulation of splenic NK cell in the presence of inhibitors for the pathway resulted in a dramatic reduction of proliferation. The defect was consistently observed in a broad range of IL-15 concentrations. To test the role of the pathway in vivo, we decided to evaluate NK cell proliferation in mice treated with rapamycin to block mTOR pathway during MCMV infection, where NK cell proliferation at early infection is well reported. During MCMV infection, NK cells show a peak proliferation response at days 2.5–3 post infection. In line with ex vivo data, proliferation of NK cells as measured by BrdU incorporation was severely reduced in rapamycin-treated mice during MCMV infection. This mTOR-dependent NK cell proliferation has been reproduced in an independent study showing that early proliferation during MCMV infection was greatly impaired in NK cell-specific mTOR-deficient mice (42).
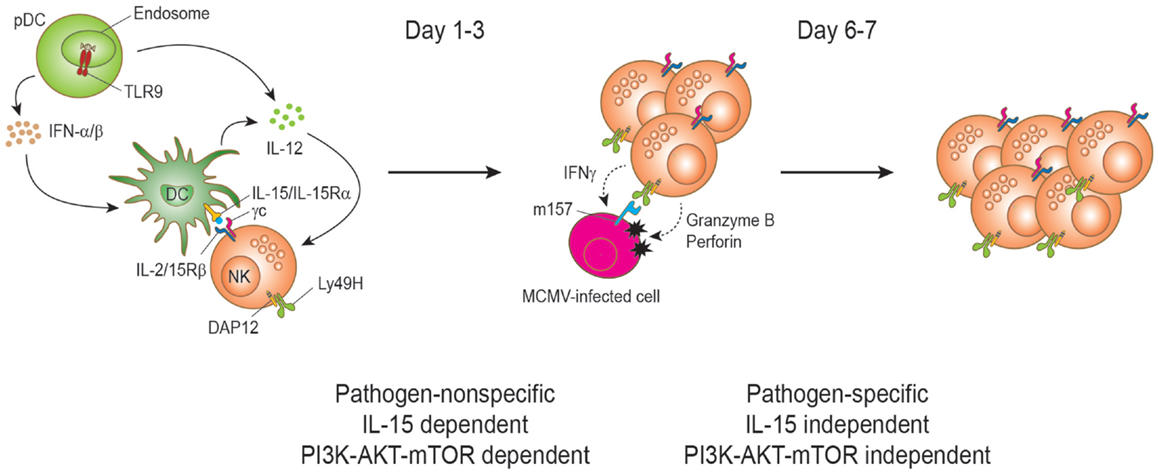
Figure 2. Natural killer cell proliferation during murine cytomegalovirus infection. During MCMV infection, two stages of NK cell proliferation showing pathogen non-specific and specific responses have been proposed. IL-15 drives NK cell proliferation during the early phase of MCMV infection. Upon recognition of CpG motifs from MCMV viral DNA by TLR9, plasmacytoid dendritic cells (pDCs) secrete type I interferons (IFNα/β) and interleukin-12 (IL-12) cytokines. DC-derived IL-12 stimulates NK cells to produce IFN-γ. IFNα/β is transiently produced and reaches a peak level at day 1.5 post-infection and the production is important to induce the expression of IL-15. IL-15 is trans-presented by DCs to NK cells to induce proliferation and enhanced effector functions of NK cells. Activated NK cells can induce perforin/granzyme-mediated apoptosis of MCMV-infected cells by recognizing the viral m157 protein on the cell surface. This direct recognition depends on the activating receptor Ly49H. Proliferation at this stage is dependent on the PI3K–AKT–mTOR pathway. The interaction between Ly49H and the MCMV-encoded protein m157 drives the proliferation and expansion of Ly49H+ NK cells on days 6–7 post infection. Notably, this proliferation can occur in IL-15- and IL-15Rα-deficient mice during MCMV infection and is independent of the PI3K–AKT–mTOR pathway.
Yokoyama and colleagues proposed two stages of NK cell proliferation consisting of pathogen-non-specific and -specific proliferation during MCMV infection (57) (Figure 2). Following the early IL-15-induced NK cell proliferation (first stage), the proliferation can be extended to a later stage of preferential proliferation of NK cells expressing the specific activating receptor that recognizes a viral ligand on the infected cells (second stage). The main example is the Ly49H-activating receptor that recognizes the MCMV-encoded m157 glycoprotein on MCMV-infected cells (67–71). This recognition induces perforin/granzyme-mediated killing of the infected cells and confers NK cell-mediated resistance to MCMV (2). During MCMV infection, Ly49H+ NK cells display preferential proliferation and the expansion is blocked by α-Ly49H antibody treatment, indicating that a stimulatory signal through the Ly49H is responsible for the proliferation. This pathogen-specific NK cell proliferation and expansion is comparable to that of antigen-specific T cells during virus infection and shows a peak response on days 6 and 7 post infection (72). Analogous NK cell expansion has also been found in other virus infections such as hantavirus, HCMV, and HIV (73–75). Notably, NK cells in NK cell-specific mTOR-deficient mice displayed substantial Ly49H-dependent proliferation at day 6.5, even though less proliferation was observed compared to mTOR-sufficient NK cells (42). Therefore, mTOR pathway is critical in IL-15-driven proliferation, but has a minimal role on activating receptor-driven proliferation. Such IL-15-independent and Ly49H-mediated NK cell proliferation has previously been reported (76).
PI3K–AKT–mTOR Pathway for NK Cell Effector Function during Virus Infection
Natural killer cells were originally regarded as “ready to kill” cells that can immediately destroy virus-infected or transformed cells (5, 77). However, unlike human NK cells, naïve mouse NK cells derived from laboratory strains of mice housed in a specific pathogen-free environment are devoid of perforin and granzyme B cytotoxic granules and exhibit minimal effector functions (44, 78, 79). A report demonstrated that IL-15 can induce rapid translation of pre-existing perforin and granzyme B mRNAs in NK cells in order to be fully equipped for action (78). Accordingly, splenic NK cells are induced to express high levels of granzyme B upon stimulation with IL-15 for 24 h. Notably, the granzyme B induction was abrogated in the presence of inhibitors blocking the PI3K–AKT–mTORC1 pathway (47). Recent reports have demonstrated that mTORC1 plays a major role in IL-15-induced NK cell activation. mTORC1 activity is greatly increased in mice treated with poly IC (a mimic of virus infection and inducer of IL-15 expression) or infected with MCMV, as exemplified by the drastic increase in the mTORC1-mediated phosphorylation of S6 ribosomal protein, a downstream target (42, 47, 80). Both pharmacological inhibition and genetic ablation demonstrated that mTORC1 is a non-redundant metabolic regulator for NK cell activation, dictating proliferation, and effector functions upon poly IC treatment (42, 80). Ablation of mTORC1 signaling reduced blastogenesis, and inhibited the expression of effector molecules such as IFN-γ and granzyme B.
To translate the importance of PI3K–mTOR pathway for NK cell functions during virus infection, we treated mice with rapamycin to block mTOR kinase activity downstream of PI3K (30, 81). The treatment of rapamycin on NK cell functions during MCMV infection also resulted in severe defects in IFN-γ and granzyme B productions in addition to defects in proliferation. Similar impairments were also observed in NK cells treated ex vivo with inhibitors for activating kinases upstream of mTORC1 such as PI3K and AKT (47). It is well established that granzymes and IFN-γ are effector molecules required for the efficient anti-viral activity of NK cells (82–85). Therefore, in vivo treatment with the mTORC1 inhibitor rapamycin during MCMV infection abrogated NK cell cytotoxicity toward YAC-1 tumor cells and resulted in elevated viral loads in the infected organs (47). This was accompanied by defective NK cell effector functions, thereby coupling this important metabolic sensor to NK cell anti-viral responses. Taken together, the results demonstrated the central role of PI3K–AKT–mTORC1 pathway in IL-15-induced NK cell activation of effector functions during virus infection.
During virus infection, activated NK cells must increase their metabolic uptake to support blastogenesis and the accelerated cell cycles. It includes elevated production of biomolecules such as nucleic acids, proteins, and lipids accompanied by the increased uptake of nutrients such as glucose and increased oxygen consumption. The major role of IL-15 in the metabolic changes in NK cells during virus infection was supported by the observation that up-regulated genes associated with metabolic pathways are largely shared in microarray data of NK cells from mice infected with MCMV in vivo or of NK cell treated with IL-15 in vitro (42). NK cells from both conditions induce increased glucose uptake and increased expression of the nutrient receptors CD71 (transferrin receptor) and CD98 (a component of the l-amino acid transporter). So how does IL-15 fulfill such heightened metabolic demands in NK cells during virus infection? Among the pathways activated upon IL-15 engagement, it seems that the IL-15–PI3K–mTOR pathway is critical for the metabolic reprograming. Whereas mTOR senses environmental nutrient availability and translates the information into cellular responses (50, 86, 87), it has become evident that mTOR-regulated cellular metabolism plays a fundamental role in dictating immune cell differentiation and function (88, 89). mTOR-regulated glycolysis is also linked to the differentiation of activated CD4+ T cells into anti-microbial Th1 and Th17 cells (50, 90, 91). In TCR-stimulated CD8+ T cells, the PI3K–mTORC1 pathway was also suggested to activate lipid synthesis for membrane biosynthesis during blastogenesis (92).
Concluding Remarks
In this review, we have discussed how the IL-15–PI3K–mTOR signaling pathway controls distinct stages of NK cell life cycle. Accumulated evidence demonstrated that the signaling axis of IL-15–PI3K–mTOR in NK cells is important for their development, cellular proliferation, responsiveness to cytokine stimulations, and cytotoxic functions. mTOR is a key regulator of cell growth and metabolism and also provides a link between the metabolism and effector functions of immune cells. IL-2/IL-15 share the receptor subunits IL-2/IL-15R-β and -γ chains and both are being widely used for ex vivo expansion of NK cells in immunotherapy. Their promising therapeutic capacity for a variety of human malignancies has stimulated an interest in using NK cells for anti-cancer treatments (93, 94). Recently, IL-15 rather than IL-2 is the preferred cytokine for expanding NK cells in vivo due to the minimal concern about toxicity and expansion of Treg (95). Therefore, understanding the molecular mechanisms by which IL-15 primes and activates NK cells will allow the manipulation of IL-15 signaling for improving NK cell-based therapeutic strategies against cancers and infectious diseases.
Interestingly, even though the IL-15–PI3K–AKT–mTOR pathway regulates multiple processes for NK cell biology from development in the bone marrow to activation in the periphery, some factors such as E4BP4 and PDK1 required for IL-15-mediated development stages of NK cells are dispensable for differentiation, survival, or effector function of mature NK cells (39, 95, 96). Therefore, it suggests that additional signaling modules might need to incorporate with the common IL-15–PI3K–AKT–mTOR pathway in order to induce stage-specific processes in NK cells.
The NK cell proliferation, cytokine production, and cytotoxicity are dependent on the metabolic induction of mTOR pathway during virus infection (42, 47, 80). Therefore, understanding the metabolic program utilized by NK cells will guide the development of optimal NK cell-mediated therapeutic interventions. Such metabolic modulations on immune cells have been used to induce enhanced CD8+ T cell memory responses. For instance, treatment of rapamycin reduces glycolysis and promotes the lipid metabolic program (50, 97), which is favorable for the generation of memory CD8+ T cells (98–100); therefore suggesting its potential application to enhance vaccine efficacy against viruses. The study of the metabolic regulation of NK cells is still in its infancy compared to works established in T cells. Our current knowledge has been mainly limited to cell culture experiments in vitro; therefore translation of these findings for in vivo immunotherapy warrants caution and careful interpretation. Without any doubt, modulating the immune responses by manipulating cellular metabolic pathways may provide an innovative option for cancer immunotherapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funding from J. P. Bickell Foundation and Canadian Institutes of Health Research (MOP-130385) to S-H Lee. S-H Lee holds a Canada Research Chair in Viral Infection and Immunity. We thank Dr. Morgan Fullerton for his advice and critical reading of this manuscript.
Authors Biography

References
1. Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol (1999) 17:189. doi: 10.1146/annurev.immunol.17.1.189
2. Lee SH, Miyagi T, Biron CA. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol (2007) 28(6):252–9. doi:10.1016/j.it.2007.04.001
3. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi:10.1038/ni1582
4. Kiessling R, Petranyi G, Karre K, Jondal M, Tracey D, Wigzell H. Killer cells: a functional comparison between natural, immune T-cell and antibody-dependent in vitro systems. J Exp Med (1976) 143(4):772–80. doi:10.1084/jem.143.4.772
5. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16(2):216–29. doi:10.1002/ijc.2910160204
6. Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol (2008) 9(5):495–502. doi:10.1038/ni1581
7. Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol (2005) 5(2):112–24. doi:10.1038/nri1549
8. Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo) (2014) 2014:205796. doi:10.1155/2014/205796
9. Marcais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol (2013) 4:450. doi:10.3389/fimmu.2013.00450
10. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol (2006) 24:657–79. doi:10.1146/annurev.immunol.24.021605.090727
11. Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol (2006) 6(8):595–601. doi:10.1038/nri1901
12. Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood (2006) 107(6):2409–14. doi:10.1182/blood-2005-06-2399
13. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365(22):2055–66. doi:10.1056/NEJMoa1108188
14. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108(5):1571–9. doi:10.1182/blood-2006-02-004747
15. Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood (2009) 114(12):2417–26. doi:10.1182/blood-2008-12-189266
16. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med (2000) 191(5):771–80. doi:10.1084/jem.191.5.771
17. Fehniger TA, Caligiuri MA. Interleukin 15: biology and relevance to human disease. Blood (2001) 97(1):14–32. doi:10.1182/blood.V97.1.14
18. Koka R, Burkett P, Chien M, Chai S, Boone DL, Ma A. Cutting edge: murine dendritic cells require IL-15R alpha to prime NK cells. J Immunol (2004) 173(6):3594–8. doi:10.4049/jimmunol.173.6.3594
19. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity (2002) 17(5):537–47. doi:10.1016/S1074-7613(02)00429-6
20. Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev (2004) 202:67–83. doi:10.1111/j.0105-2896.2004.00203.x
21. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood (2011) 117(5):1565–73. doi:10.1182/blood-2010-06-291633
22. Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity (1999) 10(2):249–59. doi:10.1016/S1074-7613(00)80025-4
23. Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med (1998) 188(11):2067–74. doi:10.1084/jem.188.11.2067
24. Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics (2006) 118(5):e1584–92. doi:10.1542/peds.2005-2882
25. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675–704. doi:10.1146/annurev-immunol-032712-095946
26. Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol (2013) 4:20. doi:10.3389/fimmu.2013.00020
27. Oak JS, Fruman DA. Role of phosphoinositide 3-kinase signaling in autoimmunity. Autoimmunity (2007) 40(6):433–41. doi:10.1080/08916930701464780
28. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307(5712):1098–101. doi:10.1126/science.1106148
29. Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol (2011) 12(9):888–97. doi:10.1038/ni.2068
30. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi:10.1016/j.cell.2012.03.017
31. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev (2003) 17(15):1829–34. doi:10.1101/gad.1110003
32. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell (2007) 25(6):903–15. doi:10.1016/j.molcel.2007.03.003
33. Huang K, Fingar DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol (2014) 36:79–90. doi:10.1016/j.semcdb.2014.09.011
34. Di Santo JP. Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol (2006) 24:257–86. doi:10.1146/annurev.immunol.24.021605.090700
35. Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol (2007) 7(9):703–14. doi:10.1038/nri2154
36. Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Di Santo JP. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol (2001) 31(6):1900–9. doi:10.1002/1521-4141(200106)31:6<1900::AID-IMMU1900>3.0.CO;2-M
37. Male V, Brady HJ. Transcriptional control of NK cell differentiation and function. Curr Top Microbiol Immunol (2014) 381:173–87. doi:10.1007/82_2014_376
38. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi:10.1038/ni1268
39. Kamizono S, Duncan GS, Seidel MG, Morimoto A, Hamada K, Grosveld G, et al. Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J Exp Med (2009) 206(13):2977–86. doi:10.1084/jem.20092176
40. Male V, Nisoli I, Kostrzewski T, Allan DS, Carlyle JR, Lord GM, et al. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med (2014) 211(4):635–42. doi:10.1084/jem.20132398
41. Yang M, Li D, Chang Z, Yang Z, Tian Z, Dong Z. PDK1 orchestrates early NK cell development through induction of E4BP4 expression and maintenance of IL-15 responsiveness. J Exp Med (2015) 212(2):253–65. doi:10.1084/jem.20141703
42. Marcais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol (2014) 15(8):749–57. doi:10.1038/ni.2936
43. Tassi I, Cella M, Gilfillan S, Turnbull I, Diacovo TG, Penninger JM, et al. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity (2007) 27(2):214–27. doi:10.1016/j.immuni.2007.07.014
44. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26(4):503–17. doi:10.1016/j.immuni.2007.03.006
45. Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, et al. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol (1999) 162(8):4511–20.
46. Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, et al. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med (1994) 180(4):1395–403. doi:10.1084/jem.180.4.1395
47. Nandagopal N, Ali AK, Komal AK, Lee SH. The critical role of IL-15-PI3K-mTOR pathway in natural killer cell effector functions. Front Immunol (2014) 5:187. doi:10.3389/fimmu.2014.00187
48. Kunikata T, Torigoe K, Ushio S, Okura T, Ushio C, Yamauchi H, et al. Constitutive and induced IL-18 receptor expression by various peripheral blood cell subsets as determined by anti-hIL-18R monoclonal antibody. Cell Immunol (1998) 189(2):135–43. doi:10.1006/cimm.1998.1376
49. Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC Jr, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J (1996) 15(19):5256–67.
50. Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity (2010) 33(3):301–11. doi:10.1016/j.immuni.2010.09.002
51. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol (2003) 3(11):900–11. doi:10.1038/nri1226
52. Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood (2012) 120(24):4751–60. doi:10.1182/blood-2012-04-419283
53. Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med (2012) 209(13):2351–65. doi:10.1084/jem.20120944
54. Berrien-Elliott MM, Wagner JA, Fehniger TA. Human cytokine-induced memory-like natural killer cells. J Innate Immun (2015). doi:10.1159/000382019
55. Lee SH, Fragoso MF, Biron CA. Cutting edge: a novel mechanism bridging innate and adaptive immunity: IL-12 induction of CD25 to form high-affinity IL-2 receptors on NK cells. J Immunol (2012) 189(6):2712–6. doi:10.4049/jimmunol.1201528
56. Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, et al. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol (2002) 3(6):523–8. doi:10.1038/ni796
57. Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol (2004) 22:405–29. doi:10.1146/annurev.immunol.22.012703.104711
58. Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity (1995) 3(6):771–82. doi:10.1016/1074-7613(95)90066-7
59. Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci U S A (2006) 103(4):1000–5. doi:10.1073/pnas.0507350103
60. Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood (2002) 100(10):3633–8. doi:10.1182/blood-2001-12-0293
61. Ranson T, Vosshenrich CA, Corcuff E, Richard O, Muller W, Di Santo JP. IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood (2003) 101(12):4887–93. doi:10.1182/blood-2002-11-3392
62. Biron CA, Turgiss LR, Welsh RM. Increase in NK cell number and turnover rate during acute viral infection. J Immunol (1983) 131(3):1539–45.
63. Biron CA, Welsh RM. Blastogenesis of natural killer cells during viral infection in vivo. J Immunol (1982) 129(6):2788–95.
64. McIntyre KW, Natuk RJ, Biron CA, Kase K, Greenberger J, Welsh RM. Blastogenesis of large granular lymphocytes in nonlymphoid organs. J Leukoc Biol (1988) 43(6):492–501.
65. Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol (2002) 169(8):4279–87. doi:10.4049/jimmunol.169.8.4279
66. Biron CA, Sonnenfeld G, Welsh RM. Interferon induces natural killer cell blastogenesis in vivo. J Leukoc Biol (1984) 35(1):31–7.
67. Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science (2002) 296(5571):1323–6. doi:10.1126/science.1070884
68. Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A (2002) 99(13):8826–31. doi:10.1073/pnas.092258599
69. Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, et al. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science (2001) 292(5518):934–7. doi:10.1126/science.1060042
70. Daniels KA, Devora G, Lai WC, O’Donnell CL, Bennett M, Welsh RM. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med (2001) 194(1):29. doi:10.1084/jem.194.1.29
71. Lee SH, Girard S, Macina D, Busa M, Zafer A, Belouchi A, et al. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat Genet (2001) 28(1):42–5. doi:10.1038/ng0501-42
72. Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, Yokoyama WM. Specific and nonspecific NK cell activation during virus infection. Nat Immunol (2001) 2(10):951–6. doi:10.1038/ni714
73. Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol (2009) 83(13):6798–805. doi:10.1128/JVI.00256-09
74. Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104(12):3664–71. doi:10.1182/blood-2004-05-2058
75. Bjorkstrom NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med (2011) 208(1):13–21. doi:10.1084/jem.20100762
76. Sun JC, Ma A, Lanier LL. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol (2009) 183(5):2911–4. doi:10.4049/jimmunol.0901872
77. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5(2):112–7. doi:10.1002/eji.1830050208
78. Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity (2007) 26(6):798–811. doi:10.1016/j.immuni.2007.04.010
79. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107(1):159–66. doi:10.1182/blood-2005-04-1351
80. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol (2014) 193(9):4477–84. doi:10.4049/jimmunol.1401558
81. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene (2006) 25(48):6416–22. doi:10.1038/sj.onc.1209888
82. Loh J, Chu DT, O’Guin AK, Yokoyama WM, Virgin HW IV. Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol (2005) 79(1):661–7. doi:10.1128/JVI.79.1.661-667.2005
83. Orange JS, Wang B, Terhorst C, Biron CA. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J Exp Med (1995) 182(4):1045–56. doi:10.1084/jem.182.4.1045
84. Tay CH, Welsh RM. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J Virol (1997) 71(1):267–75.
85. van Dommelen SL, Sumaria N, Schreiber RD, Scalzo AA, Smyth MJ, Degli-Esposti MA. Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity (2006) 25(5):835–48. doi:10.1016/j.immuni.2006.09.010
86. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi:10.1146/annurev-immunol-020711-075024
87. Xu X, Ye L, Araki K, Ahmed R. mTOR, linking metabolism and immunity. Semin Immunol (2012) 24(6):429–35. doi:10.1016/j.smim.2012.12.005
88. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
89. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev (2012) 249(1):14–26. doi:10.1111/j.1600-065X.2012.01155.x
90. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38(4):633–43. doi:10.1016/j.immuni.2013.04.005
91. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342(6155):1242454. doi:10.1126/science.1242454
92. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14(5):489–99. doi:10.1038/ni.2570
93. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12(4):239–52. doi:10.1038/nri3174
94. Kalinski P, Mailliard RB, Giermasz A, Zeh HJ, Basse P, Bartlett DL, et al. Natural killer-dendritic cell cross-talk in cancer immunotherapy. Expert Opin Biol Ther (2005) 5(10):1303–15. doi:10.1517/14712598.5.10.1303
95. Gascoyne DM, Long E, Veiga-Fernandes H, de Boer J, Williams O, Seddon B, et al. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol (2009) 10(10):1118–24. doi:10.1038/ni.1787
96. Kashiwada M, Levy DM, McKeag L, Murray K, Schroder AJ, Canfield SM, et al. IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc Natl Acad Sci U S A (2010) 107(2):821–6. doi:10.1073/pnas.0909235107
97. Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol (2009) 19(22):R1046–52. doi:10.1016/j.cub.2009.09.058
98. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460(7251):108–12. doi:10.1038/nature08155
99. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi:10.1038/nature08097
Keywords: natural killer cells, IL-15, PI3K, AKT, mTOR, proliferation, effector functions, virus infection
Citation: Ali AK, Nandagopal N and Lee S-H (2015) IL-15–PI3K–AKT–mTOR: a critical pathway in the life journey of natural killer cells. Front. Immunol. 6:355. doi: 10.3389/fimmu.2015.00355
Received: 12 March 2015; Accepted: 30 June 2015;
Published: 20 July 2015
Edited by:
Charles Dudley Mills, BioMedical Consultants, USAReviewed by:
Laurel L. Lenz, University of Colorado School of Medicine, USAClaire Anne Chougnet, Cincinnati Children’s Hospital Medical Center Research Foundation, USA
David K. Finlay, Trinity College Dublin, Ireland
Copyright: © 2015 Ali, Nandagopal and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:c2V1bmdsZWVAdW90dGF3YS5jYQ==
†Present address: Neethi Nandagopal, Institute for Research in Immunology and Cancer (IRIC), Université de Montréal, Montréal, QC, Canada