 Bennett Davenport
Bennett Davenport Yuan Li1,2
Yuan Li1,2 Justin W. Heizer
Justin W. Heizer Anne-Laure Perraud
Anne-Laure Perraud- 1Department of Biomedical Research, National Jewish Health, Denver, CO, USA
- 2Department of Immunology and Microbiology, University of Colorado Denver, Denver, CO, USA
Although the concept of Ca2+ as a universal messenger is well established, it was assumed that the regulatory mechanisms of Ca2+-signaling were divided along the line of electric excitability. Recent advances in molecular biology and genomics have, however, provided evidence that non-excitable cells such as immunocytes also express a wide and diverse pool of ion channels that does not differ as significantly from that of excitable cells as originally assumed. Ion channels and transporters are involved in virtually all aspects of immune response regulation, from cell differentiation and development to activation, and effector functions such as migration, antibody-secretion, phagosomal maturation, or vesicular delivery of bactericidal agents. This comprises TRP channel family members, voltage- and Ca2+-gated K+- and Na+-channels, as well as unexpectedly, components of the CaV1-subfamily of voltage-gated L-type Ca2+-channels, originally thought to be signature molecules of excitability. This article provides an overview of recent observations made in the field of CaV1 L-type channel function in the immune context, as well as presents results we obtained studying these channels in B-lymphocytes.
Introduction
Virtually all biological processes, including immune responses, require at some stage the carefully orchestrated elevation of cytoplasmic Ca2+ by various molecular mechanisms, which provide crucial cellular signals. When it comes to molecular components of ion flow regulation, it was commonly assumed that excitable cells such as neurons or cardiac muscle cells possess a panoply of unique ion channels and transporters representing the molecular tools of electrical excitability. In contrast, non-excitable cells such as immunocytes were thought to mainly, if not even in some cases exclusively, rely on store-operated Ca2+-entry (SOCE) to generate Ca2+-signals. This pathway, also called capacitative Ca2+-entry (1), is triggered by the activation of cell surface receptors and subsequent activation of phospholipases that generate the soluble second messenger IP3 (Inositol 3-Phosphate). Through binding to IP3-receptors at the surface of the endoplasmic reticulum (ER), IP3 elicits the depletion of the ER Ca2+-stores resulting in a first phase of Ca2+-elevation. In response to this store-depletion event, Ca2+-permeable channels at the plasma membrane get in turn activated, leading to Ca2+-entry from the extracellular space and to a sustained phase of cytosolic Ca2+-elevation and replenishment of the stores. In recent years, the molecular identity of the proteins underlying SOCE has been unveiled. The STIM proteins were identified as the ER Ca2+-sensors, which in response to ER store depletion interact with, and open the pore of the ORAI ion channels in the plasma membrane (2–4). Extensive functional studies in various populations of immune cells have become available. Unexpectedly, genetic deletion of these molecular components of store-operated Ca2+-entry has revealed that immune cell development and activation is not as reliant on this Ca2+-entry pathway as originally hypothesized. For example, although Ca2+-signals are known to be generated during development and selection of thymocytes, neither humans with genetic deficiencies in ORAI and STIM proteins, nor genetically engineered mice lacking SOCE show impaired T-cell development and selection (5–7). The same is true of B-cell development as no anomalies were found in B-cell populations in the bone marrow and secondary lymphoid organs of patients with mutations in the Orai1 or Stim1 genes (6, 7), or in mice with defects in these same molecules or STIM2 (5, 8), despite deficient B-cell receptor (BCR)-mediated Ca2+-signaling. On the other hand, T-Lymphocyte activation is SOCE-dependent, as illustrated by STIM1/ORAI1 deficient humans who exhibit lymphoproliferative defects and severe combined immunodeficiency (SCID), a phenotype consistent with SOCE-deficient mouse models, although murine STIM/ORAI proteins show a higher level of functional redundancy (9).
Following the crucial characterization of STIM and ORAI and the availability of expression datasets in various immune cell populations, it has become increasingly clear that beyond SOCE, non-excitable immune cells possess a large and diverse pool of ion channels involved in all aspects of immune response regulation. This includes numerous members of the TRP channel family of cationic channels, voltage- and Ca2+-gated K+-channels, and also, surprisingly, voltage-gated Sodium channels (10), and components of the CaV1 subfamily of L-type voltage-gated Ca2+-channels (VGCC), originally thought to be signature molecules of excitability (11–14).
In excitable cells, voltage-dependent Ca2+-entry has been extensively characterized biophysically and pharmacologically. These currents were subdivided into several subclasses based on these electrophysiological and pharmacological properties (15, 16). Molecules mediating “Long-lasting” L-type currents are commonly described as high voltage-activated channels with comparatively slow activation and rapid deactivation. Another important hallmark of L-type channels in the excitable context is the strong Ca2+-dependence of their inactivation, and their inhibition by 1,4-dihydropyridines (DHPs). L-type Ca2+-channels are often mentioned as signature channels of excitability since they couple excitation to contraction in skeletal, cardiac, and smooth muscle cells. They are also present in neurons and endocrine cells where they participate in a wide range of biological processes from cell death to transcriptional regulation or hormone secretion. Although immune cells are not known to undergo massive membrane depolarization, and lack the typical voltage-activated Ca2+-entry linked to L-type channels in the excitable context, there is mounting evidence that pore-forming L-type VGCC α1 subunits, as well as accessory β-subunits, are functionally expressed in various types of immunocytes, including B- and T-lymphocytes, but also in cells of the myeloid lineage (12–14, 17, 18). L-type channel blockers are commonly used to treat cardiovascular conditions such as high blood pressure. Understanding the role of these channels in the context of immunity and inflammation is therefore also relevant therapeutically. Before reviewing the current knowledge about the presence and potential involvement of L-type channels in the immune system, a brief overview of their structure, regulation, and biology will be given.
Topology, Nomenclature, and Regulation of Voltage-Gated Ca2+ Channels
The α1 pore-forming subunits of VGCCs are predicted to contain a total of 24 transmembrane (TM) spans arranged in four groups of six spans where the fourth one functions as a voltage sensor, and the loop between the fifth and sixth span is part of the channel’s ionic selectivity filter (Figure 1). This overall topology is common to several other families of cationic channels, such as TRP (transient receptor potential), Kv (voltage-gated K+) or CNG (cyclic nucleotide-gated) channels, that all harbor the same TM architecture. One main difference to VGCCs is, however, that in all these other channels the four groups of six TM spans are expressed as single independent entities that tetramerize to form a complete pore, allowing for the heteromultimerization of several members of the same channel family. Although the pore-forming subunit of VGCCs is in one continuous polypeptide chain, VGCCs are also multi-subunit complexes where the pore-forming α1 subunit interacts with regulatory/auxiliary subunits designated β, α2/δ, and γ, which are playing an essential role in regulating trafficking and assembly, but also in shaping channel activity features such as kinetics of activation or inactivation. Ten distinct genes subdivided into three phylogenetic subfamilies have been found to encode α1 subunits in mammals. In an effort to reorganize the nomenclature, VGCCs have been renamed CaV (for voltage-gated Ca2+), followed by the subfamily number (1–3), and the particular member number. L-type currents are mediated by four different α1 subunits, now called CaV1.1 to 1.4 (formerly α1S, α1C, α1D, and α1F, gene names are cacna1s, cacna1c, cacna1d, and cacna1f, respectively) (19). Multiple splice variants from all subunits listed above further contribute to the amazing molecular plasticity of these channels [reviewed in Ref. (16, 20)]. It was, for example, described that for the gene encoding human CaV1.2, at least 20 from the 50 exons can be subjected to splicing (16). In T-lymphocytes, two splice variants of CaV1.4 (α1F) have been found, one lacking major parts of the fourth group of TM spans (IVS3–S6), including the voltage sensor and the pore loop, and another variant lacking part of the extracellular loop connecting IVS3 with S4 (21) (Figure 1). As a result of the deletion of exon 37, both these T-cell-specific splice variants exhibit a novel and shorter C-terminal end that surprisingly shows significant homology to CaV1.1. As might have been expected because of the lack of typical voltage-gated Ca2+-entry, it therefore appears that T-cell-specific versions of CaV1 channels exist, although they have not yet been functionally characterized. A similar CaV1.3 variant had been described several years prior in a rat hepatoma cell line, and was also assumed to be voltage-insensitive, and thus gated via different mechanisms than in excitable cells (22). In addition to the extensive mRNA splicing, multiple reports have documented the proteolytic cleavage of the C-terminal portions of CaV1.2 (α1C) and CaV1.3 (α1D), resulting in truncated channels with altered biophysical properties (23–25). Recent studies have shown that the CaV1.2 C-terminal fragment translocates in a Ca2+-dependent manner into the nucleus, where it acts as a positive and negative regulator of transcription (26, 27). This finding further emphasizes the complex and multilayered physiological functions of these molecules. Importantly, the predicted topology shown in Figure 1 results in both protein termini being cytoplasmic, provided the channel is inserted into the plasma membrane. The gating of these channels is regulated at multiple levels, and often involves the interaction of their cytoplasmic regions with gating agents, as well as with other modulatory proteins that adjust the extent and kinetics of channel activation to cellular needs. Several molecules can bind to the same region of a given channel, like calmodulin (CaM), CaBP1, and CaMKII, which all interact with the N-terminal tail of CaV1.2, potentially resulting in a competition between these proteins to regulate channel kinetics (28). One extensively studied example is the modulation of voltage-gated Ca2+ channels by the Ca2+-sensor CaM [reviewed in Ref. (29)].
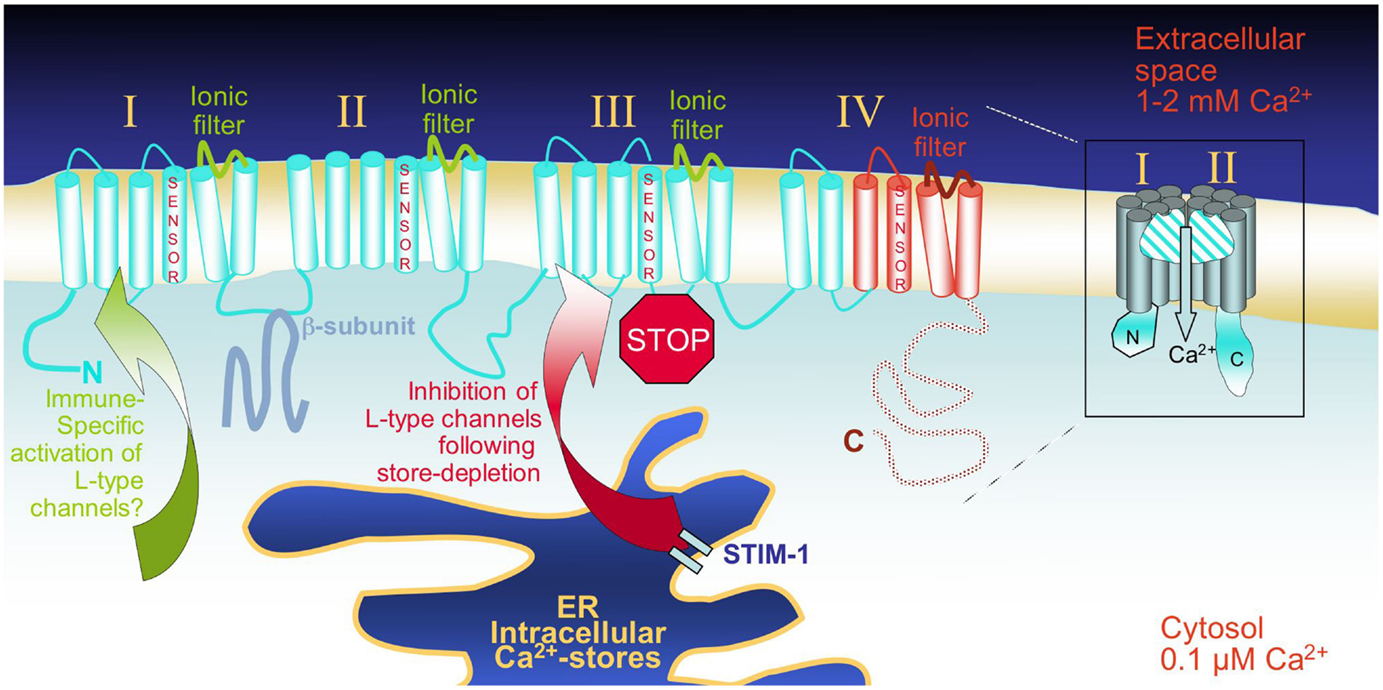
Figure 1. Schematic representation of the overall predicted topology and of the pore forming CaVα1 subunits: The 24 transmembrane (TM) regions of a typical CaV1 pore-forming unit are depicted in a linear fashion. In red are the C-terminal protein segments that were found to be affected by alternative splicing in T-cells, and might as a result be partly lacking in these cells. On the right, one channel is represented as a “half-pore” cross section through the plasma membrane. CaVα1 subunits are assumed to show the same fold as other cationic channel families, which is characterized by the formation of a pore through four groups (I–IV) of six TM domains that line the ionic selectivity filter. The voltage-gated sensor that is essential for gating in excitable cells is located on TM4 of each group. L-type channels are currently discussed as “store-inhibited channels,” representing a novel mechanism of Ca2+-signal coordination – upon depletion of the ER Ca2+-stores, the ER Ca2+-sensing STIM proteins activate store-operated Orai-channels, but at the same time, repress L-type channels. The mode of activation of L-type channel family members in various immune cell types remains to be determined.
Store-Operated vs. L-Type Channel-Mediated Ca2+-Entry: Unexpected Reciprocity
An unforeseen consequence of identifying the molecular components of store-operated Ca2+-entry – the ER Ca2+ sensor proteins STIM1/2, and the STIM-activated Orai (also CRACM1) plasma membrane channels – has been the realization that VGCCs are “store-inhibited” channels (30). As several studies have documented, there appears to be a reciprocal relationship between store-operated and VGCC-mediated Ca2+-entry pathways ensuring a coordinated activation pattern of these two major Ca2+-signaling pathways. Two simultaneous reports in 2010 described how the ER Ca2+-sensor STIM1 not only activates Orai-channels, but also suppresses the activation of CaV1.2 (31, 32). As a similar relationship was recently discovered between STIM1 and T-type Ca2+-channels (33), this reciprocity might represent a common mode of Ca2+-signal coordination. Moreover, beyond IP3 receptors, STIM proteins have also been shown to colocalize with the intracellular Ca2+ release ryanodine receptors (RyR) in T-cells, indicating the functional coupling of RyR and SOCE (34). A tight relationship also exists between the SOCE machinery and the SERCA pumps that are responsible for refilling the ER stores (35). Thus, STIM is emerging as a molecular hub for cellular Ca2+ homeostasis regulation. This could have important implications when considering the physiological and potentially pathological consequences of STIM function alterations.
Evidence for L-Type Channels in the Immune Context
The presence of L-type channels in immune cells is mostly discussed in lymphocytes, but several studies also describe their occurrence in cells of the myleoid lineage. As mentioned before, the DHP sensitivity of L-type channels has been extensively used to assess the role of these channels under various circumstances and in diverse cell types. Since these drugs have been applied therapeutically already for decades and are “valuable and widely used agents in the management of essential hypertension and angina” (36), researchers early on have investigated the possible effect of these therapeutics on lymphocytes, and found that Ca2+-flux and cell proliferation in response to activation are diminished in lymphocytes exposed to these compounds. The potential immunosuppressive action of DHPs, in particular at the comparatively low concentrations used clinically, remains however uncertain. A possible synergy between DHPs and cyclosporine A, a combination frequently used in transplantation patients, has been described (37).
An important clue to the potential role of L-type channels in immunity comes from patients with rare genetic diseases affecting CaV1 ion channels, and who also suffer from immune impairments. Because cardiac issues are usually severe and lead to the most acute and life-threatening health crises in these children, the known immune defects they present have only been characterized superficially. Timothy syndrome (TS) is a complex disorder caused by point mutations in the CaV1.2 gene. The mutated channels were found to generate sustained inward Ca2+-currents originating from an almost complete loss of voltage-dependent channel inactivation (38). Severe infections, particularly bronchial and sinus infections, show a higher incidence and severity in individuals with TS, suggesting a significant role for CaV1.2 in mounting a potent immune response in humans (39). In the following, an overview of the current knowledge about L-type channel function in major immune cell types will be given.
L-Type Channels in T-Lymphocytes
Most of our knowledge about the involvement of CaV channels in the immune context was acquired in T-lymphocytes. As several excellent contributions in a recent special issue of Frontiers Immunology entitled “The Regulation of Calcium Homeostasis in T Lymphocytes” have provided in-depth reports about L-type channels in T-lymphocytes (13, 14, 40), we will only give a succinct review of this topic.
Although earlier reports had suggested the presence of members of the L-type family of ion channels in T-cells (22), it is mostly over the past decade that this unexpected finding has been more closely investigated. The group from Wilfred Jefferies published the first detailed evidence of CaV involvement in human T-cells by showing expression of cacna1f (CaV1.4) in Jurkat T-cells in addition to primary peripheral blood CD4 and CD8 T-cells (11). The function of cacna1f expression was confirmed through a series of experiments utilizing L-type-specific DHP agonist and antagonist. These studies revealed that pharmacological manipulation of CaV channels in T-cells modulated T-cell receptor (TCR)-dependent Ca2+ flux, influenced phosphorylation and translocation of key TCR signaling pathways, regulated production of the proinflammatory cytokine IL-2 and upregulation of the IL-2R (CD25), and blunted TCR-induced proliferation. It has since been shown that both CD4+ and CD8+ T-cells express unique patterns of L-type CaV channels, dependent upon their lineage differentiation and activation status (13).
Useful insights have been gained from studies conducted in mouse models with alterations in genes encoding CaV-channel subunits (41–43). Mice deficient in the cytoplasmic β-subunits β3 (knock-out), or β4 (spontaneous mutation), show normal intrinsic T-cell development – previously described anomalies in thymus development in the β4 mutant (44) are likely due to the onset of neuropathy exhibited in these mice at 2 weeks of age (41). It was further concluded through bone marrow chimera experiments that the β4 mutant contained a normal assortment of peripheral CD4 and CD8 T-cells, and a preserved naïve T-cell phenotype (CD44loCD62Lhi, CD25−CD69−). In contrast, β3-deficiency resulted in a dramatic reduction of splenic CD8+ T-cells, of which a significant portion presented with an activated phenotype (CD44hiCD62Llo) (42). TCR-mediated Ca2+ responses in CD4+ T-cells are diminished in both CaVβ3- and CaVβ4-deficient mice, directly contributing to impaired nuclear translocation of the Ca2+-sensitive transcription factor NFAT, leading to reduced production of the proinflammatory cytokines IFNγ and IL-4 (41, 42). Although CD4+ and CD8+ T-cells express unique profiles of β subunits (β4/β3 and β2/β3, respectively), it has yet to be clarified how these patterns directly regulate T-cell biology. Auxiliary subunits of CaV channels come in many different variants, increasing the molecular diversity of these channels. Functionally, auxiliary subunits enhance membrane expression of the channels, influence current properties, and shape the composition of signaling complexes associated with the channels. As subunit interactions of L-type channels are highly promiscuous, cell-specific expression patterns are crucial to define the type of channel complex being functionally expressed (45). Thus, it is plausible that the unique characteristics of each β subunit (subcellular localization coordination, phosphorylation status, complex formation with other adaptor/signaling molecules) could be an additional layer of T-cell regulation (42, 45).
Interestingly, CD8+ T-cells show a state-dependent expression pattern for CaV1 channels. Although the CD8+ T-cell population as a whole expresses mRNA transcripts for all four members of the CaV1 family, CaV1.1 and CaV1.4 exhibit differential regulation. Whereas CaV1.4 protein is found in naïve but not in activated CD8+ T-cells, CaV1.1 expression is the opposite during TCR stimulation, showing prominent expression in activated CD8+ T-cells, but not naïve. In addition, only the β3 regulatory subunit was shown to be prominently expressed in CD8+ T-cells (42). In this same study, utilizing β3-deficient mice, it was observed that peripheral naïve CD8+ T-cell homeostasis was dramatically altered. In the absence of β3, although thymic T-cell development was not perturbed, peripheral CD8+ T-cell numbers were greatly diminished. It was shown that CD8+ T-cells in β3−/− mice exhibit a skewed activated phenotype (CD44hiCD62Llo) in the absence of any simulation/immunization, as well as increased activation-induced cell death, and impaired TCR-induced Ca2+ flux, proliferation, and effector cell function (as evident by lack of proinflammatory IFNγ, TNFα, IL-2 and granzyme B production).
The CD8+ T-cell phenotype and functional capacity in β3−/− mice is partly reminiscent of exhausted T-cells in chronic viral infection as shown by the activated phenotype, impaired proliferative capacity and diminished cytokine production, although the cardinal marker for exhaustion PD-1 is not upregulated in β3−/− CD8+ T-cells (42, 43, 46). In depth analysis of the molecular signatures of exhausted antiviral CD8+ T-cells revealed, among other things, an absence in NFAT nuclear translocation (46, 47). In addition to the absence of nuclear NFAT in exhausted CD8+ T-cells, alteration in the balance of NFAT and its binding partner AP-1 has dramatic effects on the targeted transcriptional program (48). When NFAT fails to complex with AP-1, the genes targeted are associated with anergy, tolerance, and exhaustion (48–50). Although the phenotypic and functional discrepancies observed in CD8+ T-cells in β3−/− mice do not completely mirror those of exhausted antiviral CD8+ T-cells, it is convincing to predict that the culmination of TCR-mediated Ca2+ flux and coordinated regulation of NFAT nuclear translocation mediated by the CaV1 β3 subunit has the potential to regulate the onset of CD8+ T-cell exhaustion.
It was also shown that the absence of β3 regulatory subunit resulted in a complete loss of cellular CaV1.4 protein. The authors concluded from their observations that the CaV1.4-β3 complex in naïve CD8+ T-cells contributes to antigen independent, MHC-triggered Ca2+ responses that are required for tonic signaling and the survival of these cells. This finding was confirmed and further expanded when the phenotype of mice constitutively lacking CaV1.4 was characterized. Recapitulating previous investigations, these studies demonstrated that CaV1.4 (cacna1f) is needed for the survival of peripheral naïve CD4 and CD8 T-cells. They further showed that CaV1.4 deficiency significantly reduced both CD4+ and CD8+ antigen-specific T effector cell responses. When challenged with ovalbumin-expressing Listeria, cacna1f−/− mice exhibited a significantly reduced expansion in antigen-specific CD4+ and CD8+ T effector cells, in addition to impaired CD8+ T-cell-mediated cytotoxicity (43).
Expression analyses of CaV1 pore-forming subunits in different subsets of CD4+ Th cells have shown that CaV1.2 and 1.3 are a hallmark of Th2 cells where they promote cytokine production (51, 52). Administration by airway inhalation of a combination of CaV1.2 and CaV1.3 antisense oligonucleotides provided protection against the development of Th2-dependent airway inflammation and hyperreactivity in mice (53), pointing at the therapeutic potential of airway-targeted inhibition of CaV-channels for the treatment of asthma. In humans, gene expression analyses in circulating blood cells revealed expression of CaV1.2, 1.3, and 1.4, as well as of accessory subunits required to form functional CaV channels (54). Comparably to mice, CaV1.4 was detected in Th1 and Th17 cells, whereas CaV1.2 is selectively expressed in Th2 cells. Although CaV1.4 was down-regulated following TCR-stimulation of Th1 cells, this was not the case for CaV1.2 in Th2 cells. Accordingly, pharmacological inhibition of TCR-mediated cytokine production was specifically reduced in Th2, but not in Th1, cells (54).
The differential expression profile of expressed CaV subunits in T lymphocytes is therefore expected to be an important factor in generating the diversity of Ca2+ signals required for T-cell development, homeostasis, and effector functions.
L-Type Channels in B-Lymphocytes
The generation of Ca2+-signals in B-lymphocytes appears to be diversified and not as dependent upon STIM/Orai-mediated store-operated Ca2+-entry as originally anticipated (55). In mice lacking both ER Ca2+-sensor proteins Stim1 and 2 selectively in B-cells (Mb1-Cre-mediated deletion), B-cell development was normal, although SOCE was shown to be largely deficient. Antibody production, as well as LPS- and anti CD40-dependent B-cell proliferation, are also intact in these mice. However, BCR-mediated proliferation is strongly diminished, and B-cell-derived production of the anti-inflammatory cytokine IL-10 is severely reduced as a result of defective activation of the Ca2+-regulated transcription factor NFAT, leading to enhanced Th1-driven experimental autoimmune encephalomyelitis (EAE) (8).
Because many Ca2+-regulated B-cell functions remain intact in the absence of functional SOCE, it suggests that other Ca2+-signaling mechanisms must exist in these cells. Comparatively little is known about the role of L-type channels in B-lymphocytes. Several pharmacological studies have demonstrated that blocking L-type channels in human B-cells reduces BCR-induced Ca2+-entry (17, 56). In freshly isolated rat B-cells, an antibody against CaV1.3 was found to block IgD-mediated Ca2+-responses (56). In this same study, it was confirmed that depolarization does not result in a Ca2+-response in B-lymphocytes, however, a cGMP-dependent protein kinase involvement was discovered. In the human L3055 B-cell line, gene and protein expression of CaV1.2α1 and β1 was demonstrated, whereby the CaV1.2α1 version expressed in these cells appeared to be non-voltage-gated and truncated in comparison to the version present in cardiac tissues (17). In this same study, an antibody raised against the extracellular region of CaV1.2 was found to trigger a Ca2+-flux, indicating functional expression of this channel at the plasma membrane of L3055 B-cells. A prior report about a CaV1.2α1 version expressed in murine erythroleukemia cells document the occurrence of putatively voltage-insensitive versions of the CaV channels expressed in non-excitable cells. This channel can be blocked by nifedipine, which partially inhibits differentiation of these cells. This CaV1.2 variant lacks the first four segments of domain I of the pore-forming subunit, and does not produce measurable currents when expressed by itself in Xenopus oocytes (57). Therefore, the gating mechanism of this CaV1.2 variant remains unknown. A study performed on subsets of sorted splenic mouse B-cells mentions that BCR-mediated Ca2+-mobilization in transitional T1 B-cells is more sensitive to the L-type channel blocker verapamil than that in T2 cells (58). From our review of the literature, there is currently no answer to the question under which circumstances and in which particular subsets of B-cells particular L-type channel subunits are expressed in vivo.
Studying L-Type Channel Function in DT40 B-Cells
Gene and Protein Expression of CaV1 Subunits in DT40 Cells
To further address the potential role of L-type Ca2+ channels in B-lymphocytes, we chose to utilize the chicken DT40 B-cell line that allows for the comparatively easy genomic disruption of genes of interest due to an unusually high rate of homologous recombination (59). DT40s have been shown to express an IgM isotype BCR at the cell surface, and BCR-stimulation by anti IgM leads to PLCγ2 activation and subsequent cytosolic [Ca2+] elevation. Signaling components of the avian BCR-pathway show very high conservation to their mammalian counterparts, and the DT40 system has been extensively used to characterize this pathway [reviewed in Ref. (60)].
We first aimed at defining the expression pattern of individual pore-forming subunits of the CaV1 L-type family of channels in DT40 wild-type cells. After identifying in the NCBI database the chicken homologs of the mammalian CaV1.1, 1.2, 1.3, and 1.4, we performed RT-PCR experiments. We found all CaV1 genes except CaV1.1 to be expressed in DT40 cells (Figure 2A). The amplified ~1.5 kb from the 3′ end of CaV1.2 and 1.3 DT40 cDNA were cloned and sequenced, and are encoding the complete cytoplasmic C-terminal domains of these two channels. In order to confirm that also proteins from these channels can be detected in DT40s, we conducted simple cell fractionation experiments, and used the excitable INS-1 rat pancreatic beta-cell line as a positive control. To visualize the channels, we used a PAN-antibody recognizing an epitope present in all L-type α1 pore-forming subunits (aa1506-1524 of rat CaV1.1), which is conserved in the chicken version as well. In the DT40 cell membrane fraction, a band of the same size as a strong signal obtained in the INS-1 cells (~200 kDa) was detected, indicating that full-length α-subunits are expressed in DT40 B-cells (Figure 2B). Lower molecular weight bands were also seen, suggesting degradation or shorter splice variants. As these results were obtained using a PAN anti-CaV1-antibody, we cannot conclude which combination of the three subunits whose genes are expressed, CaV1.2, 1.3, or 1.4, is present at protein level. Because individual antibodies against each of the subunits are recognizing epitopes that are not well conserved in chicken, this question could not be further addressed using existing reagents.
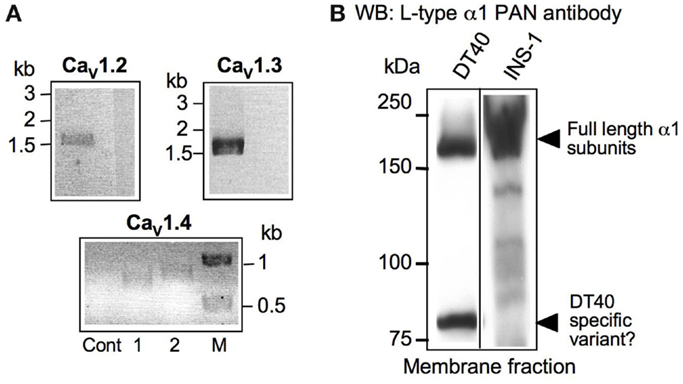
Figure 2. Gene and Protein expression of CaV1α1 family members in DT40 B-cells. (A) RT-PCR using primer combinations specific for the indicated chicken version of CaV1.2 (1.5 kb,), 1.3 (1.6 kb), and 1.4 (656 and 761 bp, lanes 1 and 2, respectively). M = marker. All PCR-fragments have been cloned and sequenced for verification. The negative control lanes (Cont) are reactions containing RNA as template. (B) Western blot from DT40 membrane fraction lysates developed using L-type α1 PAN antibody. As a positive control, the excitable pancreatic rat beta cell line INS-1 was used.
Effect of L-Type Inhibitor on BCR-Mediated Ca2+-Signals in DT40s
As mentioned previously, several pharmacological studies using inhibitors of L-type channels such as nifedipine or diltiazem have shown a decrease of the Ca2+-response following receptor ligation in lymphocytes. We confirmed this finding in wild-type DT40 cells by showing a dose-dependent diltiazem-mediated inhibition of the Ca2+-signal following BCR-stimulation (Figure 3A). However, diltiazem had no effect on the pharmacological depletion of intracellular ER stores and subsequent activation of store-operated Ca2+-entry using the SERCA-pump inhibitor thapsigargin, suggesting that diltiazem targets a BCR-specific event that is not triggered by thapsigargin (Figure 3B). The anti-IgM-mediated depletion of the Ca2+-stores in the absence of extracellular Ca2+ was not inhibited by diltiazem (Figure 3C). Solely the second phase of Ca2+-elevation requiring entry from the extracellular space was affected, and the amplitude of Ca2+-elevation following exposure to thapsigargin was again unchanged by the addition of diltiazem. Noticeably, a similar pattern was observed in murine CD4+ T-cells lacking the CaVβ3-subunit required for proper trafficking of the CaV1 subunits; a decrease in the amplitude of the Ca2+-response was only observed after cross-linking of the TCR, but not following the application of thapsigargin (41). In this context, it is interesting that a recent study characterizing CaV1.4-deficient (Cacna1f−/−) mice has shown that whereas naïve CD44lo CD4+ T-cells show a CaV1.4 dependence for both their TCR- and thapsigargin-mediated Ca2+-responses, more mature CD44hi CD4+ T-cells exhibit a pattern similar to the diltiazem effect in DT40 B-cells with only their TCR-mediated, but not their thapsigargin-induced Ca2+-response relying on CaV1.4 (43). The molecular basis for this differential CaV-dependence of pharmacological store-depletion vs. immunoreceptor-mediated Ca2+-signals in certain immune cell subsets remains to be elucidated.
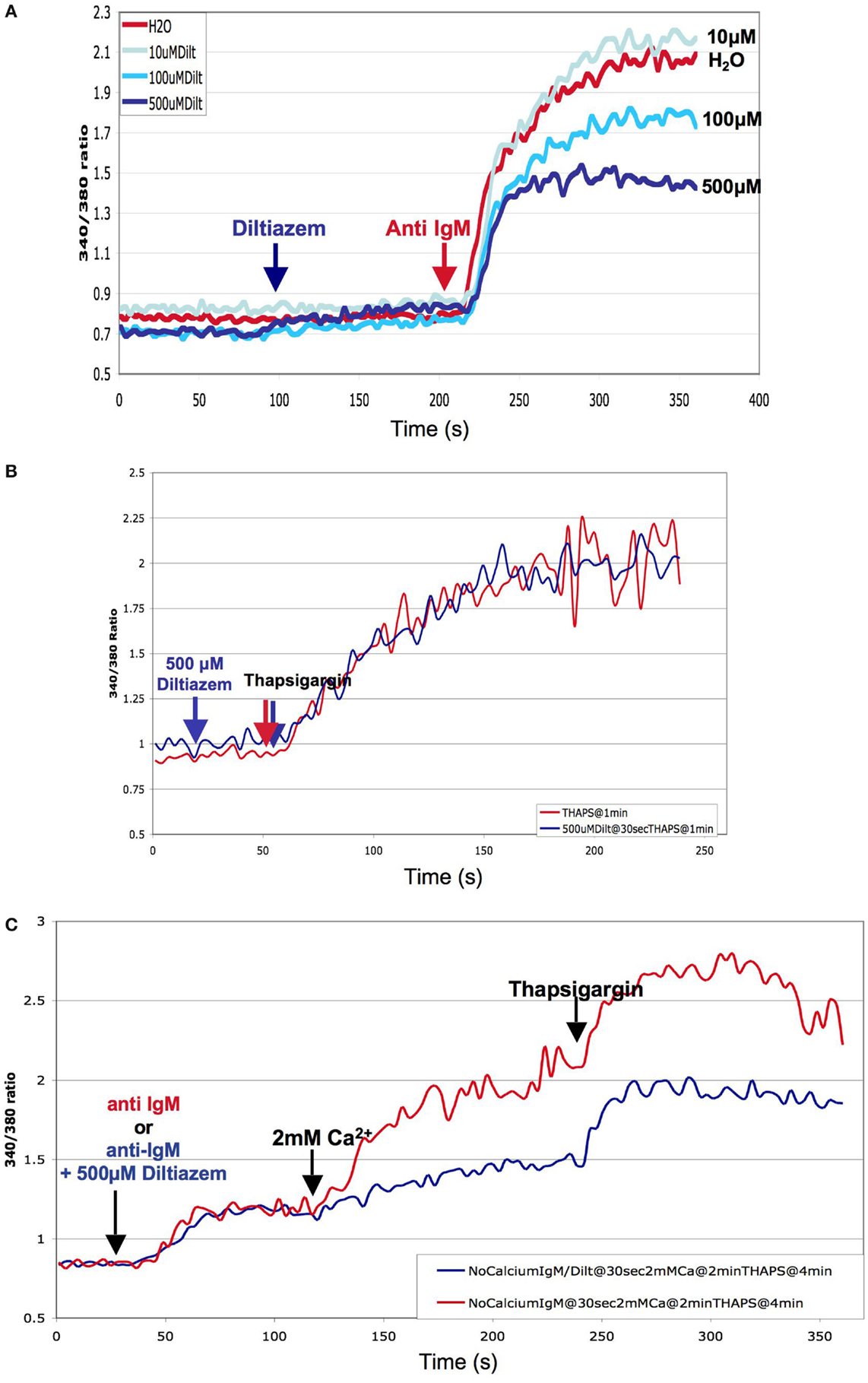
Figure 3. Pharmacological inhibition of BCR-mediated Ca2+-response by the L-type channel blocker Diltiazem: DT40 cells were loaded with the fluorescent Ca2+-dye Fura-2 and the cells stimulated either through the addition of chicken anti-IgM (BCR-ligation) or thapsigargin (pharmacological store-depletion) into the cuvette. The measurements were performed using a spectrofluorometer (Photon Technology International) at 37°C. (A) Dose dependence of the inhibitory effect of diltiazem on BCR-mediated cytosolic Ca2+-mobilization. (B) Effect of diltiazem on the store-operated Ca2+-response elicited by thapsigargin in DT40 cells (C) Diltiazem inhibition of the influx of extracellular Ca2+ following BCR-ligation. DT40 cells were loaded with the fluorescent Ca2+-dye Fura-2 in Ca2+-free buffer, and the cells stimulated through the sequential addition of chick anti IgM, 2 mM Ca2+, and thapsigargin into the cuvette.
Because DHPs need to be applied in the higher μM range to elicit an effect on the Ca2+-response of immunocytes, their specificity under these conditions has been questioned since an inhibitory effect on other channels such as K+ channels (KV and KCa) was shown in some studies. However, as discussed by Kotturi et al. (11), in these cases the reported effects were not consistent with the observations made in immune cells. It is therefore very probable that the Ca2+-entry pathway inhibited by diltiazem in DT40 cells originates from CaV variants that appear to be less sensitive toward this compound than their counterparts expressed in the excitable context.
Inducible Deletion of CaV1.3α1 in DT40 B-Cells
The results of the gene-expression and pharmacological studies in DT40s presented above confirmed the presence and potential functional relevance of L-type channels in this B-cell line. We therefore decided to utilize the high genomic plasticity of DT40 cells to generate DT40 lines in which genes encoding L-type channels are disrupted, beginning with CaV1.3 α1. To this aim, we chose a targeting strategy that resulted in the deletion of exons encoding several TM regions, in particular S4, S5, and S6 that are including the putative pore-forming loop between S5 and S6 (see also Figure 1). We opted to delete the exons encoding S4–S6 of the chicken cacna1d gene within the first six TM group (IS4–S6). We designed a “conventional” targeting construct allowing for the exchange of the CaV1.3 region of interest against a “recyclable” puromycin resistance cassette (flanked by loxP sites, generous gift from Dr. Jean-Marie Buerstedde). DT40 cells stably expressing a tamoxifen (TX)-inducible version of the Cre recombinase [MerCreMer, kindly provided by Dr. Michael Reth (61)] for potential inducible Cre-mediated genomic deletions were transfected with the targeting construct depicted in Figure 4A. We identified by Southern blot (Figure 4B) multiple DT40 clones with targeted integration of the puromycin resistance gene into the gene encoding CaV1.3 with an efficiency of over 25% (5 from 19 analyzed clones). We then tried to target the second allele using a different drug resistance cassette (against histidinol), and unexpectedly did not obtain any double-targeted DT40 clones, despite screening over 50 independent cell clones, a number that in our experience is largely sufficient to isolate the desired doubly targeted mutant cells. We thus concluded that the deletion of CaV1.3 is deleterious to DT40 cells, and designed an alternative targeting construct allowing for the inducible deletion of the same IS4–S6 encoding genomic region using the flox/Cre recombinase system (Figure 4A, bottom construct). Using this strategy, we were able to obtain TX-inducible deletion of the targeted CaV1.3 genomic region. We determined by RT-PCR that following TX-treatment and excision of the floxed cacna1d region, the complete CaV1.3 transcript appears to be missing in these DT40 cells (Figure 4C). The analysis of total CaV1 proteins detected with the PAN antibody by immunoblotting showed that the high-molecular band (>200 kDa), which putatively represents all full-length CaV1 channels in DT40s, is still present after deletion of CaV1.3. This result might have been expected since our gene expression studies have demonstrated that CaV1.2 and CaV1.4 are also present in DT40s (Figure 2A). Noticeably, a smaller (~90 kDa) band seems to disappear in CaV1.3−/− DT40 cells (Figure 4D). Although we need to confirm this result by cloning the CaV1.3 version(s) expressed in DT40 cells in the future, a possible interpretation is that CaV1.3 is not (solely) expressed as a full-length ion channel in DT40s. This is consistent with findings described above that both in B- and T-lymphocytes, L-type variants shorter than their excitable counterparts can be identified. The characterization of immune variants of L-type channels will be essential to elucidate the role of these proteins in this context. As discussed in section topology, nomenclature, and regulation of voltage-gated Ca2+ channels of this article, immune splice variants are known which lack some of the TM spans containing the voltage sensor, consistent with the observation that depolarization does not appear to activate Ca2+-signals in immunocytes. It is also conceivable that some of the truncated CaV versions might actually not function as ion transport pathways, and instead fulfill other cellular roles, as illustrated by studies mentioned previously showing the ability of C-terminal portions of CaV proteins to translocate into the nucleus to act as transcriptional regulators (26, 27).
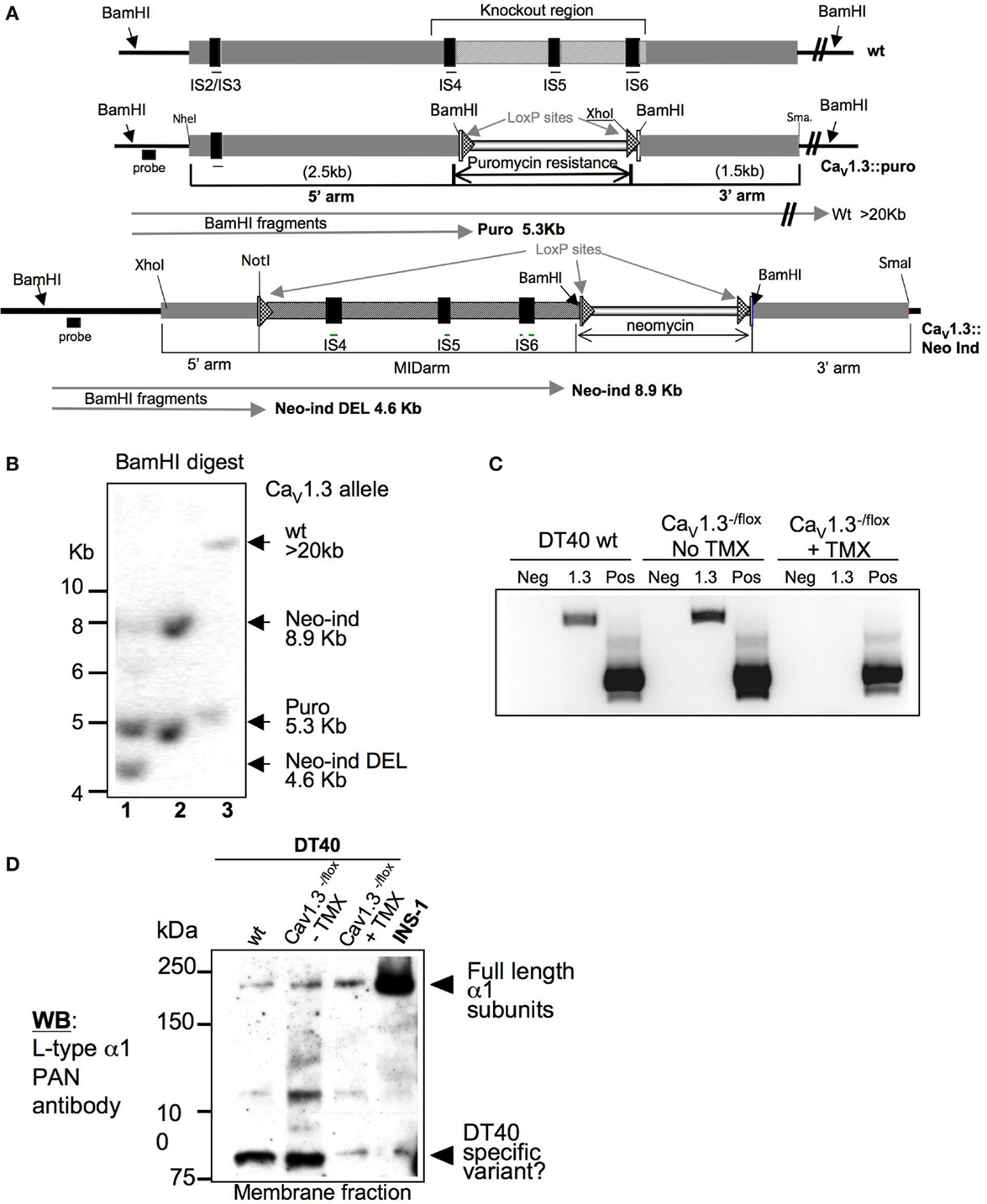
Figure 4. Targeting strategy of the CaV1.3 encoding gene cacna1d, and validation of the tamoxifen-inducible cacna1d disruption in the DT40 cell line: (A) Schematic representation of the magnified genomic organization of the region containing the exons encoding TM spans IS4-S6 of the chicken CaV1.3 channel that was deleted is shown. The constitutive and inducible targeting constructs are depicted underneath. The expected BamHI fragment sizes by Southern are indicated by arrows. (B) Representative Southern blot showing one of the CaV1.3± clones and of the CaV1.3flox/− clones before (lane 1) and after tamoxifen (lane 2)-induced deletion of the cacna1d region of interest in comparison to a cacna1d± clone (lane 3). (C) RT-PCR of wild-type DT40 cells and of one CaV1.3flox/− clone before and after tamoxifen (TMX)-induced deletion of the cacna1d region of interest using cacna1d-specific primers. The unrelated chicken nudT9 gene was used as a positive control (Pos) to verify the quality of the cDNA. Neg control is RNA only. (D) Western blot of membrane fraction lysates of wild-type DT40s and excitable INS-1 rat pancreatic cells in comparison to one CaV1.3flox/− clone before and after tamoxifen-induced cacna1d deletion.
We found that following deletion of CaV1.3, DT40 cells show a marked decrease in growth rate (Figure 5A), which is potentially causal to our failure to obtain DT40 clones constitutively deficient in CaV1.3 using a conventional targeting strategy. Based on simple counts of dead cell bodies, it does not appear that cell death is substantially increased in the CaV1.3−/− DT40s, perhaps pointing at a defect in cell proliferation, rather than a deregulation of apoptotic, necrotic, or cell survival pathways (Figure 5A, dashed lines), although this will need to be more carefully investigated in the future. We analyzed the effect of the CaV1.3 deletion on the Ca2+-response triggered by anti-IgM and thapsigargin treatment, and did not observe any change in the extent or shape of the Ca2+-responses under these conditions, nor did we see a difference in the effect of diltiazem on the Ca2+-increase (Figure 5B). Although speculative, this result might suggest that CaV1.3′s biological effect is not mediated by Ca2+ influx, which might be further corroborated by the observation discussed above that a protein substantially smaller than full-length CaV1.3 is seeing as missing in the CaV1.3−/− cells. Alternatively, it might be that CaV1.3 is not activated downstream of BCR ligation, and that we need to test other triggers, for example, through GPCRs. We have indications that CaV1.2 deletion in DT40s also requires an inducible strategy, suggesting that CaV1.2 and CaV1.3 fulfill non-redundant roles in DT40s. Ultimately, for a more complete picture of the function of these channels in DT40s to be drawn, a full set of CaV1-deficient DT40 lines will need to be generated, including cell lines with combined deletions of CaV1.2, CaV1.3, and CaV1.4. These cell lines will represent a useful set of reagents to assess the respective contribution of these different pore-forming CaV subunits in the context of BCR-signaling.
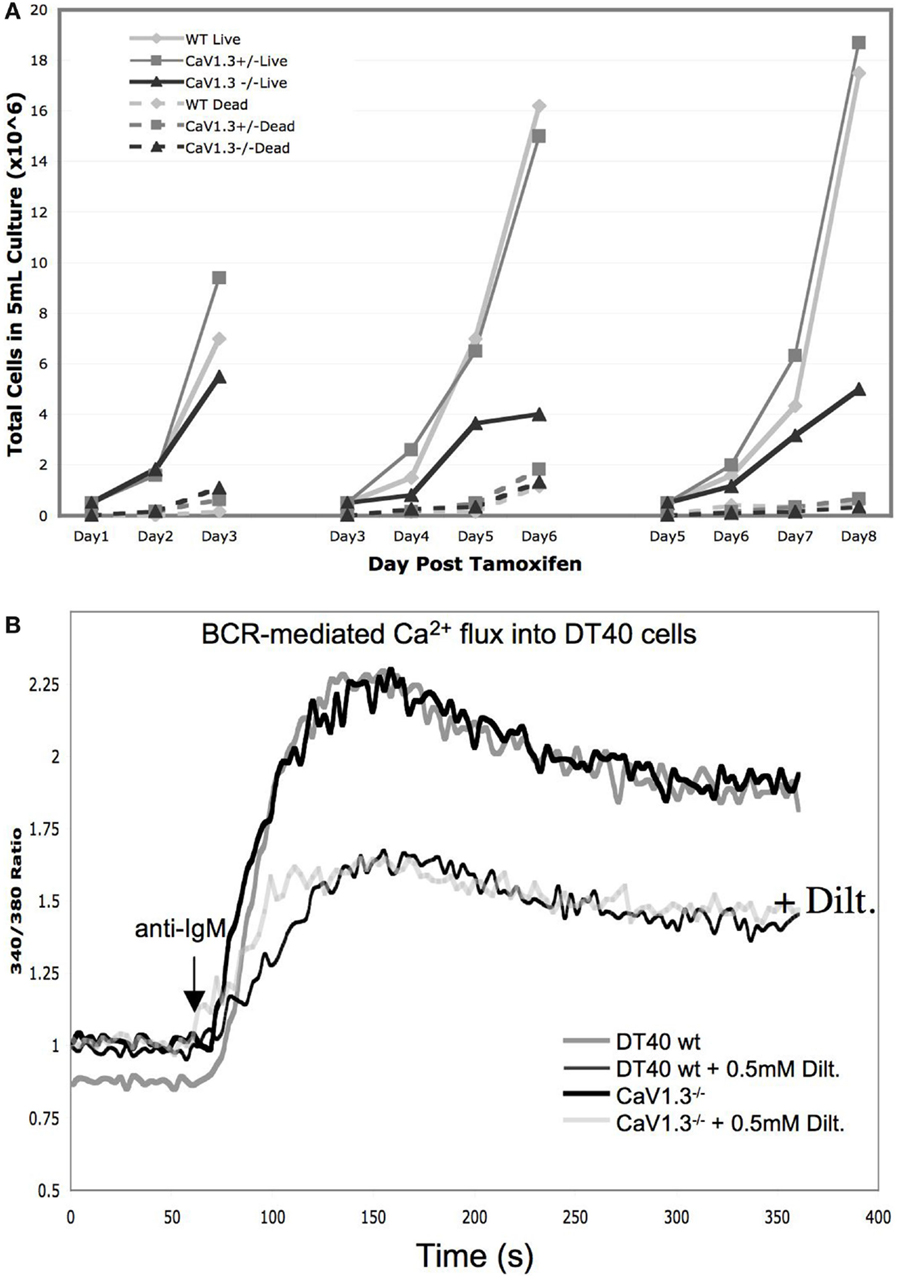
Figure 5. Phenotypic characterization of the CaV1.3-deficient DT40 cells: (A) Growth curves of DT40 wild-type cells (light gray) in comparison to the CaV1.3−/flox cells before (dark gray) and after deletion (black) of the floxed region following tamoxifen treatment. Dashed lines represent the corresponding numbers of dead cells as determined by trypan blue in the same cultures. Cells were split back every 3 days to 0.5 × 106/ml. (B) Ca2+-transients in wt vs. CaV1.3-deficient DT40 cells following BCR-activation with or without L-type channel blocker diltiazem.
L-Type Channels in Immune Cells of the Myeloid Lineage
Insights into the possible involvement of L-type channels in the development, homeostasis, or biological function of cells of the myleoid lineage are few. It is, however, known that L-type channel blockers such as diltiazem, which are clinically widely used to treat hypertension, also have anti-inflammatory effects (62). This could have clinical implications as pointed out in a recent study demonstrating the beneficial effect of diltiazem to prevent aneurysm formation in a mouse model through the inhibition of inflammatory cytokine production by monocytic cells (18).
In neutrophils, pharmacological inhibitors of L-type channels have been reported to reduce the Ca2+ response of human polymorphonuclear neutrophils (PMNs) to a neuropeptide (63), and to reduce the release of elastases and the production of ROS from these cells via diminished cytosolic Ca2+ mobilization and PKC activation (64). In monocytes, it was similarly reported that nifedipine dampens superoxide production and that in addition it directly contributes to reducing PKC activity (65). It has also been proposed that the anti-inflammatory effects of L-type channel blockers is reinforced by the effects of these drugs on suppressing the participation of plasminogen leading to the inhibition of macrophage emigration through tissues (66, 67). In peripheral blood-derived human dendritic cells (DCs), an early study found that apoptotic body engulfment and IL-12 production are inhibited by nifedipine (68). More recently a group proposed a model in which similar to the situation in cardiac and skeletal muscles, membrane depolarization in immature human DCs results in CaV1.2 activation, which in turn triggers intracellular Ca2+ release via ryanodine receptor 1 (RyR1), resulting in the rapid delivery of MHC class II molecules to the plasma membrane (69). The role of RyR1 in DCs is further supported by the finding that mice carrying a gain of function mutation in RyR1 exhibit enhanced DC function (70).
Collectively, theses results suggest that beyond lymphocytes, L-type channels also play a significant role in immune cells of the myeloid lineage, which will be important to further shed light on.
Concluding Remarks
Calcium signals in non-excitable cells such as immune cells are as diverse as the biological processes they regulate. In order to accommodate this need, immune cells rely on an equally diverse set of ion channels that unexpectedly includes molecules thought to be signature molecules of excitability, such as the L-type channels discussed in this article. Many questions remain to be addressed, such as the nature of the immune-specific L-type channel variants expressed in defined subsets of immunocytes, or the important but still elusive mechanism of activation of these immune CaV1 variants since classic depolarization-mediated activation like in excitable cells do not seem to be a major factor in immune cells. In this context, it might be relevant that a voltage-gated sodium channel was recently reported to be crucial for positive selection of CD4+ T-lymphocytes (10), opening up the possibility that during specific biological processes, and in some distinct cell types that harbor the appropriate molecular equipment, voltage-gating of L-type channels might be an option, although this remains speculative at this time.
Despite these gaps in knowledge it seems now well established and accepted in the field that L-type channels are a force to be reckoned with in the context of immunity and inflammation. As the activity of these channels can be manipulated for therapeutic purposes, and the medical community has ample experience with drugs targeting L-type channels in the context of hypertension and cardiac conditions, there is a very real and promising potential to utilize these compounds for immunomodulatory and anti-inflammatory purposes in the future.
Materials and Methods
Cell Culture
The DT40 cell lines were cultured in RPMI supplemented with Pen/Strep, 10% FBS, and 1% chicken serum (Sigma). DT40 wt cells stably expressing the Tet-repressor were transfected with the targeting constructs for chicken CaV1.3 described under “Generation of a CaV1.3-deficient DT40 B-cell line.”
Generation of a Cre-Inducible CaV1.3-Deficient DT40 B-Cell Line
Chicken cacna1d (CaV1.3) genomic DNA was obtained by screening NCBI chicken genome. The conventional targeting vectors [cacna1d::puromycin (puro)] were constructed by replacing the genomic fragment containing exons encoding the structurally essential TM regions IS4 to IS6 of the CaV1.3 pore-forming subunit with puromycin cassettes. This cassette was flanked by 2.5 and 1.5 kb of chicken cacna1d genomic sequence on the 5′ and 3′ sides, respectively. The predicted DNA sequences of all constructs were verified by sequencing. The linearized targeting construct, cacna1d:puromycin was introduced into DT40 cells by electroporation and the cells were subsequently set under selection in serial dilutions to ensure the obtention of single clones.
Genomic DNA for Southern blot was isolated from drug-resistant DT40 cell clones, and digested with BamHI. Restriction enzyme sites, probe for Southern blot analysis (solid bar) and targeted exons are indicated in Figure 4. BamHI fragments detected by the probe are shown for wild-type and mutated alleles. The first allele targeting using the cacna1d:puro construct resulted in homologous recombination with a frequency of 20%. The targeting of the second allele using a different drug resistance (histidinol), however, failed. We therefore concluded that constitutional homozygous disruption of both cacna1d alleles is deleterious to DT40 cells. We thus designed and generated an inducible targeting construct allowing for the Cre-recombinase-mediated deletion of the floxed cacana1d region of interest (Figure 4A). The MerCreMer hybrid protein we used was kindly provided by Dr. Michael Reth (Freiburg, Germany), and allows for TX-inducible activation of the Cre-recombinase activity. The successful integration of the cacna1d:Neo-ind construct was verified by Southern blot, as well as the Cre-mediated excision of the deleted region following addition of TX to the media (Figure 4B).
Expression Analysis by RT-PCR
RT-PCR was performed with chicken DT40 WT RNA for the reverse transcription using a Superscript III kit ssDNA synthesis kit from Invitrogen following the manufacturer’s protocol. The PCR was performed with the Advantage pcDNA Polymerase Mix from Clontech, and the following chicken specific oligonucleotides were used:
- CaV1.2: ACTTCAGATGGGCCAAAACTCTTCC
CACCTCTTGGAGGCACAGGAGTGAAGG
- CaV1.3: TACAGGAATGGCACACAGCATCGC
CAATGAAGCACGTCCATCTTTTGGC
PCR (125 ng of single-strand DNA per reaction) was performed using standard techniques, a 2-step program for 35 cycles of 94°C for 20 s, and 60°C for 45 s. The DNA bands have been visualized onto an ethidium bromide stained 1% agarose gel with a gel documentation system (Bio-Rad). The fragments were cloned and sequenced.
Immunoblotting
0.1–5 × 106 DT40 wt or mutant cells were plated, cells lysed and proteins of the cell membrane fraction were separated by SDS/PAGE using 8% polyacrylamide gels, and transferred to a PVDF membrane. The membranes were analyzed using L-type α1 PAN antibody from Alomone Labs.
Calcium Measurements
Cytosolic [Ca2+] was evaluated in the indicated DT40 cell lines using the fluorescent Ca2+-sensitive dye fura-2. 4 × 106 cells were loaded with 1 μg/ml Fura-2 (Invitrogen) for 30 min at 25°C in Ringer buffer and analyzed using a bulk assay in a spectrofluorometer (Photon Technology International) as previously described (71). L-type channel inhibitors were ordered from sigma.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by an R21 Award from the NIH (NIAID, 1R21AI078400) to A-LP, and by an NIH R01 grant to CS (NIGMS & Office of Dietary Supplements, 5R01GM090123). We would like to thank Fabienne Gally and Deviyani Rao (National Jewish Health) for carefully reviewing the manuscript.
References
1. Putney JW Jr. Capacitative calcium entry revisited. Cell Calcium (1990) 11:611–24. doi: 10.1016/0143-4160(90)90016-N
2. Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol (2009) 10:21–7. doi:10.1038/ni0209-223c
3. Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol (2010) 28:491–533. doi:10.1146/annurev.immunol.021908.132550
4. Prakriya M. Store-operated Orai channels: structure and function. Curr Top Membr (2013) 71:1–32. doi:10.1016/B978-0-12-407870-3.00001-9
5. Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol (2008) 28:5209–22. doi:10.1128/MCB.00360-08
6. McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol (2009) 124:1311–1318.e7. doi:10.1016/j.jaci.2009.10.007
7. Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med (2009) 360:1971–80. doi:10.1056/NEJMoa0900082
8. Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity (2011) 34:703–14. doi:10.1016/j.immuni.2011.03.016
9. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol (2015) 33:291–353. doi:10.1146/annurev-immunol-032414-112212
10. Lo WL, Donermeyer DL, Allen PM. A voltage-gated sodium channel is essential for the positive selection of CD4(+) T cells. Nat Immunol (2012) 13:880–7. doi:10.1038/ni.2379
11. Kotturi MF, Carlow DA, Lee JC, Ziltener HJ, Jefferies WA. Identification and functional characterization of voltage-dependent calcium channels in T lymphocytes. J Biol Chem (2003) 278:46949–60. doi:10.1074/jbc.M309268200
12. Kotturi MF, Hunt SV, Jefferies WA. Roles of CRAC and Cav-like channels in T cells: more than one gatekeeper? Trends Pharmacol Sci (2006) 27:360–7. doi:10.1016/j.tips.2006.05.007
13. Badou A, Jha MK, Matza D, Flavell RA. Emerging roles of L-type voltage-gated and other calcium channels in T lymphocytes. Front Immunol (2013) 4:243. doi:10.3389/fimmu.2013.00243
14. Omilusik KD, Nohara LL, Stanwood S, Jefferies WA. Weft, warp, and weave: the intricate tapestry of calcium channels regulating T lymphocyte function. Front Immunol (2013) 4:164. doi:10.3389/fimmu.2013.00164
15. Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature (1985) 316:440–3. doi:10.1038/316440a0
16. Hofmann F, Flockerzi V, Kahl S, Wegener JW. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev (2014) 94:303–26. doi:10.1152/physrev.00016.2013
17. Grafton G, Stokes L, Toellner KM, Gordon J. A non-voltage-gated calcium channel with L-type characteristics activated by B cell receptor ligation. Biochem Pharmacol (2003) 66:2001–9. doi:10.1016/j.bcp.2003.07.005
18. Mieth A, Revermann M, Babelova A, Weigert A, Schermuly RT, Brandes RP. L-type calcium channel inhibitor diltiazem prevents aneurysm formation by blood pressure-independent anti-inflammatory effects. Hypertension (2013) 62:1098–104. doi:10.1161/HYPERTENSIONAHA.113.01986
19. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International union of pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev (2005) 57:411–25. doi:10.1124/pr.57.4.5
20. Lipscombe D, Pan JQ, Gray AC. Functional diversity in neuronal voltage-gated calcium channels by alternative splicing of Ca(v)alpha1. Mol Neurobiol (2002) 26:21–44. doi:10.1385/MN:26:1:021
21. Kotturi MF, Jefferies WA. Molecular characterization of L-type calcium channel splice variants expressed in human T lymphocytes. Mol Immunol (2005) 42:1461–74. doi:10.1016/j.molimm.2005.01.014
22. Brereton HM, Harland ML, Froscio M, Petronijevic T, Barritt GJ. Novel variants of voltage-operated calcium channel alpha 1-subunit transcripts in a rat liver-derived cell line: deletion in the IVS4 voltage sensing region. Cell Calcium (1997) 22:39–52. doi:10.1016/S0143-4160(97)90088-9
23. Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez-Reyes E, et al. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac alpha 1 subunit. J Biol Chem (1994) 269:1635–40.
24. Gerhardstein BL, Gao T, Bunemann M, Puri TS, Adair A, Ma H, et al. Proteolytic processing of the C terminus of the alpha(1C) subunit of L-type calcium channels and the role of a proline-rich domain in membrane tethering of proteolytic fragments. J Biol Chem (2000) 275:8556–63. doi:10.1074/jbc.275.12.8556
25. Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol (2006) 576:87–102. doi:10.1113/jphysiol.2006.111799
26. Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell (2006) 127:591–606. doi:10.1016/j.cell.2006.10.017
27. Schroder E, Byse M, Satin J. L-type calcium channel C terminus autoregulates transcription. Circ Res (2009) 104:1373–81. doi:10.1161/CIRCRESAHA.108.191387
28. Kobayashi T, Yamada Y, Fukao M, Tsutsuura M, Tohse N. Regulation of Ca(V)1.2 current: interaction with intracellular molecules. J Pharmacol Sci (2007) 103(4):347–53. doi:10.1254/jphs.CR0070012
29. Ben-Johny M, Yue DT. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol (2014) 143:679–92. doi:10.1085/jgp.201311153
30. Harraz OF, Altier C. STIM1-mediated bidirectional regulation of Ca(2+) entry through voltage-gated calcium channels (VGCC) and calcium-release activated channels (CRAC). Front Cell Neurosci (2014) 8:43. doi:10.3389/fncel.2014.00043
31. Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science (2010) 330:101–5. doi:10.1126/science.1191027
32. Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, et al. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science (2010) 330:105–9. doi:10.1126/science.1191086
33. Nguyen N, Biet M, Simard E, Beliveau E, Francoeur N, Guillemette G, et al. STIM1 participates in the contractile rhythmicity of HL-1 cells by moderating T-type Ca(2+) channel activity. Biochim Biophys Acta (2013) 1833:1294–303. doi:10.1016/j.bbamcr.2013.02.027
34. Thakur P, Dadsetan S, Fomina AF. Bidirectional coupling between ryanodine receptors and Ca2+ release-activated Ca2+ (CRAC) channel machinery sustains store-operated Ca2+ entry in human T lymphocytes. J Biol Chem (2012) 287:37233–44. doi:10.1074/jbc.M112.398974
35. Alonso MT, Manjarres IM, Garcia-Sancho J. Privileged coupling between Ca(2+) entry through plasma membrane store-operated Ca(2+) channels and the endoplasmic reticulum Ca(2+) pump. Mol Cell Endocrinol (2012) 353:37–44. doi:10.1016/j.mce.2011.08.021
36. Kelly JG, O’Malley K. Clinical pharmacokinetics of calcium antagonists. An update. Clin Pharmacokinet (1992) 22:416–33. doi:10.2165/00003088-199222060-00002
37. Kunzendorf U, Walz G, Brockmoeller J, Neumayer HH, Jochimsen F, Roots I, et al. Effects of diltiazem upon metabolism and immunosuppressive action of cyclosporine in kidney graft recipients. Transplantation (1991) 52:280–4. doi:10.1097/00007890-199108000-00018
38. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A (2005) 102:8089–96. doi:10.1073/pnas.0502506102
39. Liao P, Soong TW. CaV1.2 channelopathies: from arrhythmias to autism, bipolar disorder, and immunodeficiency. Pflugers Arch (2010) 460:353–9. doi:10.1007/s00424-009-0753-0
40. Pelletier L, Savignac M. Ca(2+) signaling in T-cell subsets with a focus on the role of cav1 channels: possible implications in therapeutics. Front Immunol (2013) 4:150. doi:10.3389/fimmu.2013.00150
41. Badou A, Jha MK, Matza D, Mehal WZ, Freichel M, Flockerzi V, et al. Critical role for the beta regulatory subunits of Cav channels in T lymphocyte function. Proc Natl Acad Sci U S A (2006) 103:15529–34. doi:10.1073/pnas.0607262103
42. Jha MK, Badou A, Meissner M, McRory JE, Freichel M, Flockerzi V, et al. Defective survival of naive CD8+ T lymphocytes in the absence of the beta3 regulatory subunit of voltage-gated calcium channels. Nat Immunol (2009) 10:1275–82. doi:10.1038/ni.1793
43. Omilusik K, Priatel JJ, Chen X, Wang YT, Xu H, Choi KB, et al. The Ca(v)1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity (2011) 35:349–60. doi:10.1016/j.immuni.2011.07.011
44. Dung HC. Relationship between the adrenal cortex and thymic involution in “lethargic” mutant mice. Am J Anat (1976) 147:255–64. doi:10.1002/aja.1001470209
45. Campiglio M, Flucher BE. The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J Cell Physiol (2015) 230:2019–31. doi:10.1002/jcp.24998
46. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27:670–84. doi:10.1016/j.immuni.2007.09.006
47. Agnellini P, Wolint P, Rehr M, Cahenzli J, Karrer U, Oxenius A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc Natl Acad Sci U S A (2007) 104:4565–70. doi:10.1073/pnas.0610335104
48. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity (2015) 42:265–78. doi:10.1016/j.immuni.2015.01.006
49. Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell (2002) 109:719–31. doi:10.1016/S0092-8674(02)00767-5
50. Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med (2010) 16:1147–51. doi:10.1038/nm.2232
51. Badou A, Savignac M, Moreau M, Leclerc C, Pasquier R, Druet P, et al. HgCl2-induced interleukin-4 gene expression in T cells involves a protein kinase C-dependent calcium influx through L-type calcium channels. J Biol Chem (1997) 272:32411–8. doi:10.1074/jbc.272.51.32411
52. Savignac M, Badou A, Moreau M, Leclerc C, Guery JC, Paulet P, et al. Protein kinase C-mediated calcium entry dependent upon dihydropyridine sensitive channels: a T cell receptor-coupled signaling pathway involved in IL-4 synthesis. FASEB J (2001) 15(9):1577–9. doi:10.1096/fj.00-0733fje
53. Cabral MD, Paulet PE, Robert V, Gomes B, Renoud ML, Savignac M, et al. Knocking down Cav1 calcium channels implicated in Th2 cell activation prevents experimental asthma. Am J Respir Crit Care Med (2010) 181:1310–7. doi:10.1164/rccm.200907-1166OC
54. Robert V, Triffaux E, Paulet PE, Guery JC, Pelletier L, Savignac M. Protein kinase C-dependent activation of CaV1.2 channels selectively controls human TH2-lymphocyte functions. J Allergy Clin Immunol (2013) 133:1175–83. doi:10.1016/j.jaci.2013.10.038
55. Baba TW, Giroir BP, Humphries EH. Cell lines derived from avian lymphomas exhibit two distinct phenotypes. Virology (1985) 144:139–51. doi:10.1016/0042-6822(85)90312-5
56. Sadighi Akha AA, Willmott NJ, Brickley K, Dolphin AC, Galione A, Hunt SV. Anti-Ig-induced calcium influx in rat B lymphocytes mediated by cGMP through a dihydropyridine-sensitive channel. J Biol Chem (1996) 271:7297–300. doi:10.1074/jbc.271.13.7297
57. Ma Y, Kobrinsky E, Marks AR. Cloning and expression of a novel truncated calcium channel from non-excitable cells. J Biol Chem (1995) 270:483–93. doi:10.1074/jbc.270.1.483
58. Hoek KL, Antony P, Lowe J, Shinners N, Sarmah B, Wente SR, et al. Transitional B cell fate is associated with developmental stage-specific regulation of diacylglycerol and calcium signaling upon B cell receptor engagement. J Immunol (2006) 177:5405–13. doi:10.4049/jimmunol.177.8.5405
59. Buerstedde JM, Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell (1991) 67:179–88. doi:10.1016/0092-8674(91)90581-I
60. Kurosaki T. Regulation of BCR signaling. Mol Immunol (2011) 48:1287–91. doi:10.1016/j.molimm.2010.12.007
61. Verrou C, Zhang Y, Zurn C, Schamel WW, Reth M. Comparison of the tamoxifen regulated chimeric Cre recombinases MerCreMer and CreMer. Biol Chem (1999) 380:1435–8. doi:10.1515/BC.1999.184
62. Matsumori A, Nishio R, Nose Y. Calcium channel blockers differentially modulate cytokine production by peripheral blood mononuclear cells. Circ J (2010) 74:567–71. doi:10.1253/circj.CJ-09-0467
63. Harfi I, Corazza F, D’hondt S, Sariban E. Differential calcium regulation of proinflammatory activities in human neutrophils exposed to the neuropeptide pituitary adenylate cyclase-activating protein. J Immunol (2005) 175:4091–102. doi:10.4049/jimmunol.175.6.4091
64. Kouoh F, Gressier B, Dine T, Luyckx M, Brunet C, Ballester L, et al. Antioxidant effects and anti-elastase activity of the calcium antagonist nicardipine on activated human and rabbit neutrophils – a potential antiatherosclerotic property of calcium antagonists? Cardiovasc Drugs Ther (2002) 16:515–20. doi:10.1023/A:1022986331231
65. Allanore Y, Borderie D, Perianin A, Lemarechal H, Ekindjian OG, Kahan A. Nifedipine protects against overproduction of superoxide anion by monocytes from patients with systemic sclerosis. Arthritis Res Ther (2005) 7:R93–100. doi:10.1186/ar1614
66. Das R, Burke T, Van Wagoner DR, Plow EF. L-type calcium channel blockers exert an antiinflammatory effect by suppressing expression of plasminogen receptors on macrophages. Circ Res (2009) 105:167–75. doi:10.1161/CIRCRESAHA.109.200311
67. Das R, Plow EF. A new function for old drugs. Cell Cycle (2010) 9:638–9. doi:10.4161/cc.9.4.11016
68. Poggi A, Rubartelli A, Zocchi MR. Involvement of dihydropyridine-sensitive calcium channels in human dendritic cell function. Competition by HIV-1 Tat. J Biol Chem (1998) 273:7205–9. doi:10.1074/jbc.273.13.7205
69. Vukcevic M, Spagnoli GC, Iezzi G, Zorzato F, Treves S. Ryanodine receptor activation by Ca v 1.2 is involved in dendritic cell major histocompatibility complex class II surface expression. J Biol Chem (2008) 283:34913–22. doi:10.1074/jbc.M804472200
70. Vukcevic M, Zorzato F, Keck S, Tsakiris DA, Keiser J, Maizels RM, et al. Gain of function in the immune system caused by a ryanodine receptor 1 mutation. J Cell Sci (2013) 126:3485–92. doi:10.1242/jcs.130310
Keywords: calcium signaling, lymphocytes, myeloid lineage, CaV ion channels, L-type channels, DT40 B-cells, dihydropyridines
Citation: Davenport B, Li Y, Heizer JW, Schmitz C and Perraud A-L (2015) Signature channels of excitability no more: L-type channels in immune cells. Front. Immunol. 6:375. doi: 10.3389/fimmu.2015.00375
Received: 24 March 2015; Accepted: 09 July 2015;
Published: 23 July 2015
Edited by:
Amanda MacKenzie, University of Bath, UKCopyright: © 2015 Davenport, Li, Heizer, Schmitz and Perraud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne-Laure Perraud, Departments of Biomedical Research and Immunology and Microbiology, National Jewish Health, University of Colorado Denver, 1400 Jackson Street, Denver, CO 80206, USA,cGVycmF1ZGFAbmpoZWFsdGgub3Jn
†Present address: Bennett Davenport, Diabetes, Obesity and Metabolism Institute, Mount Sinai Icahn School of Medicine, New York, NY, USA