 Simon Ville
Simon Ville Nicolas Poirier
Nicolas Poirier Gilles Blancho
Gilles Blancho Bernard Vanhove
Bernard Vanhove- 1Unité Mixte de Recherche, UMR_S 1064, Institut National de la Santé et de la Recherche Médicale, Nantes, France
- 2Institut de Transplantation Urologie Néphrologie (ITUN), Université de Nantes, Nantes, France
- 3Effimune SAS, Nantes, France
CD28 and CTLA-4 are prototypal co-stimulatory and co-inhibitory cell surface signaling molecules interacting with CD80/86, known to be critical for immune response initiation and regulation, respectively. Initial “bench-to-beside” translation, two decades ago, resulted in the development of CTLA4-Ig, a biologic that targets CD80/86 and prevents T-cell costimulation. In spite of its proven effectiveness in inhibiting allo-immune responses, particularly in murine models, clinical experience in kidney transplantation with belatacept (high-affinity CTLA4-Ig molecule) reveals a high incidence of acute, cell-mediated rejection. Originally, the etiology of belatacept-resistant graft rejection was thought to be heterologous immunity, i.e., the cross-reactivity of the pool of memory T cells from pathogen-specific immune responses with alloantigens. Recently, the standard view that memory T cells arise from effector cells after clonal contraction has been challenged by a “developmental” model, in which less differentiated memory T cells generate effector cells. This review delineates how this shift in paradigm, given the differences in co-stimulatory and co-inhibitory signal depending on the maturation stage, could profoundly affect our understanding of the CD28/CD80-86/CTLA-4 blockade and highlights the potential advantages of selectively targeting CD28, instead of CD80/86, to control post-transplant immune responses.
Introduction
The importance of costimulation to allo-immune response has been widely demonstrated. A comparative study of xeno- and allo-immune response by Lafferty et al. in the late 60s was at the origin of this concept (1, 2). To their surprise, they found that “as the genetic relationship between donor and recipient becomes more distinct, the degree of reactivity falls to an undetectable level.” They proposed that something more than antigens, with species compatibility, was required to stimulate an allograft response. They called this second signal the allograft stimulus. This became the costimulation signal, when extended to the entire T-cell response, within the second signal theory (3).
Later, CTLA-4, an inhibitory cell surface molecule with the same ligand on antigen-presenting cell (APC) as CD28, namely CD80/86, was discovered, defining the CD28/CD80/86/CTLA-4 balance. This pathway became an attractive focus in the transplantation field and has been the target of much research over the past few decades, leading to the development of CTLA4-Ig (4). This fusion protein binds CD80 and CD86, preventing ligation of CD28 and also of CTLA-4. In spite of its proven effectiveness in inhibiting allo-immune responses, particularly in murine models, clinical experiences in kidney transplantation with belatacept (a high-affinity CTLA4-Ig molecule) have exhibited a high incidence of acute, cell-mediated rejection (5). The etiology of this belatacept-resistant rejection has been ascribed to heterologous immunity, i.e., the cross-reactivity of the pool of memory T cells from pathogen-specific immune responses, with alloantigens (6).
From the beginning, Lafferty et al. found that, once generated, activated cytotoxic T lymphocytes are able to kill any cell that expresses foreign antigens, that is, once activated the requirement for allogenic stimulus is lost (2). Based on a small number of studies, the idea that CD28 costimulation is unnecessary for CD4 + and CD8 + T-cell memory responses has become a generally accepted paradigm in immunology (7). This is consistent with the classic view that most T cells die after reacting to pathogens, but some of them, cells that are capable of destroying the pathogen, give rise to memory cells. Thus, the loss of the costimulation requirement is considered as a selective advantage to the memory T cells, which increases the efficiency of recall responses.
However, lines are shifting: in the memory field a new model, known as “developmental,” where naïve cells directly develop into memory cells without transitioning through an effector stage, is emerging. At the same time, data from experimental models, which are increasingly relevant to anti-infectious immune response, challenge the current paradigm of dispensable CD28 costimulation by memory T cells. Furthermore, advances in the field of cancer immunotherapy provide indication on the impact of CTLA-4 blockade, including on a preexisting immune response. This review delineates how this shift in paradigm could profoundly affect our understanding of the CD28/CD80/86/CTLA-4 blockade and highlights the potential advantages of selectively targeting CD28, instead of CD80/86, to control post-transplant immune responses.
What We Can Learn from CD28-Negative T Cells
A way to investigate the requirement of CD28 for antigen-experienced T cells is to focus on CD28-negative T lymphocytes, for which there is little doubt that activation is CD28 independent.
As noted above, usually the loss of CD28, and consequently of the costimulation requirement, is regarded as a special state, achieved following an immune response by the most efficient clones and generating the best protective anamnestic response due to memory cells (8).
CD28-negative T cells are absent from umbilical cord blood, then emerge over time and finally a majority of peripheral blood T cells become CD28 negative (9). The loss of CD28 is an immunological feature primarily observed in humans and primates. Substantial progress has been made in understanding the molecular, cellular, and functional features of CD28-negative T cells since their initial identification in the early 90s (9, 10). On the one hand, they gain cytolytic activities supported by elevated expression of key molecules including perforin and granzyme, they have a low activation threshold, and in selected cases, render cell activation independent of the recognition of the appropriate antigenic peptide. On the other hand, they have a reduced capacity to proliferate and survive after TCR activation, displaying signs of lymphocyte exhaustion with dominant inhibitory receptors (8–10). Thus, their overall impact is negative, since their accumulation comes at the expense of an appropriate immune response and gives rise to the risk of autoimmunity.
CD28-negative T cells do not appear to be memory cells, whose function would be to improve a recall response, but terminally differentiated cells arising as a consequence of immune-senescence. This contradicts mainstream thinking, where loss of the costimulation requirement is considered as an advantage for memory cells.
The Developmental Model and Possible Prediction of Costimulation Requirement
A new model for the linage relationship of T-cell subsets suggests that less differentiated memory T cells give rise to effector cells, and not vice versa, so memory cells are derived directly from activated naïve cells that have never experienced an effector state (11–13).
This model is called developmental because it proposes that T-cell differentiation is largely a linear and unidirectional process, whose driving force is the cumulative history of antigenic stimulation, going from naïve cell to terminally differenced effector T cell via a memory stage, with progressive chromatin change. Cell maturation has been likened to a ball rolling down a hill, with cells progressively losing potential energy, i.e., “stemness” and proliferative capacities, but gaining effector and homing capacities (Figure 1). This process is associated with progressive characteristic changes in cell surface molecule expression that allow us to classify cells into various subsets. The loss of CD28 is one of the last events occurring during maturation (13). This fits with the features of CD28-negative T cells described above (9, 10).
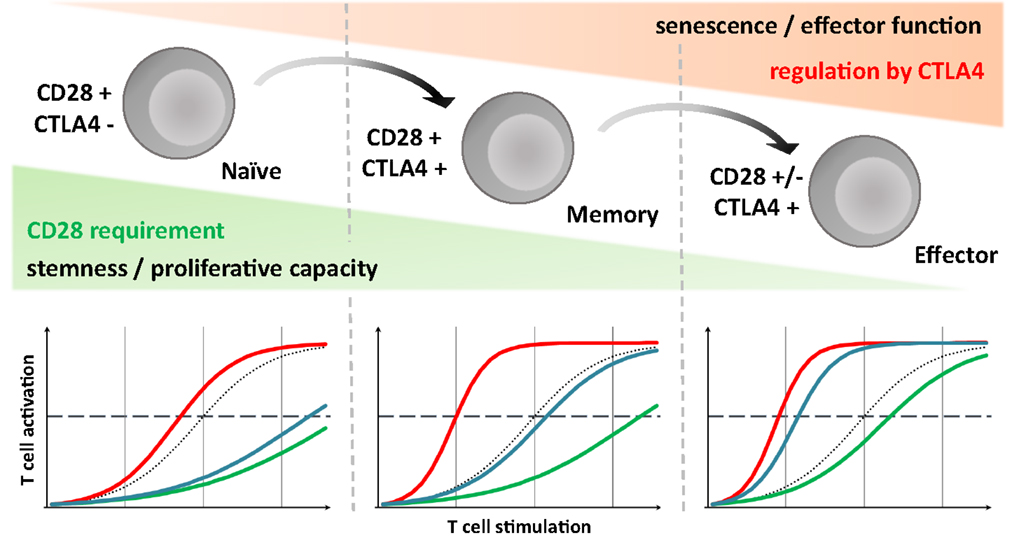
Figure 1. CD28 requirement and CTLA-4 mediated inhibition evolve through T cell run, highlighting consequence of different strategies targeting the CD28/CD80/86/CTLA-4 axis. Upper panel: according to the developmental model, during immune response, T cells differentiate progressively ranging from naive to effector via the memory stage. Throughout this process, like a ball rolling down a hill, they lose their proliferative potential but gain effector and homing competences. We assume that simultaneously their activation threshold, and so CD28 dependency, decreases but that conversely the importance of CTLA-4 intrinsic inhibitory signaling gradually increased. Lower panel: dotted line, control condition; red line, CTLA-4 blockade; green line, CD28 selective blockade; blue line, CD80/86 blockade; broken line represent sufficient level for T-cell activation and mounting an efficient response. For naive T cells, due to the lack of CTLA-4 signaling, selective and non-selective CD28 blockade would be equally efficient in controlling their activation. In the case of terminally differentiated T cells, preserving CTLA-4 mediated signals could be essential, especially in the absence of a CD28 requirement, suggesting a relevant advantage of the CD28 selective blockade compared to CD80/86 antagonist. Memory T cells might represent a middle path in which the intensity of the TCR stimulation, more important in allo-immune context especially with direct presentation, is probably critical.
CD28 loss, and from a broader perspective, loss of the costimulation requirement, would not then reflect an advantage inherent to memory acquired following an immune response, incidentally with a risk of immunopathology by inappropriate reactivation, but a feature of cells reaching the end of a progressive maturation process with limited potential but with effector capacities, and restricted to peripheral tissues.
Origins of the Notion That Memory T Cells are Costimulation-Independent
The costimulation field has become much more complex since the publication of Lafferty’s allogenic stimulus hypothesis. Numerous activators and inhibitors of ligand/receptor interactions have been described on both sides of the immunological synapse and given the appellation cell surface signaling molecules. Their distribution is extremely variable according to their developmental stage, the localization of the different lymphocyte subsets and their propensity to impact on each other through feedback loops. The fate of each cell thus depends on the integration of signals derived from a large complex of stimulatory and inhibitory interactions (14). This framework, much more complex than the standard view of the second signal model, is required to interpret the results of the numerous studies that have been conducted over recent years on the CD28/CD80/86/CTLA-4 triad. Some conclusions, sometimes considered as “ground rules”, have to be tempered by the inevitable limitations of the particular experimental methodologies and model systems. One of these ground rules is the paradigm that memory T-cell activation is CD28 independent; an idea based on a small number of in vitro studies and ones on CD28-deficient mice.
CD28 signaling requirements in memory CD4 + and CD8 + T-cell responses have been much less well studied than those on primary response generation. A first experimental model used by Steinman 30 years ago was the mixed lymphocyte reaction (MLR) (15, 16). “Memory cells” resulting from primary MLRs were actually not true memory cells as defined today, but rather lymphoblasts. Unlike naïve T cells that proliferate only after stimulation with allogenic dendritic cells (DCs), these lymphoblasts proliferate regardless of the APC subset, including macrophage or B cell. The conclusion was that once activated, lymphocytes become independent of second signals.
These data were confirmed by Croft (17, 18). Adoptive transfer of TCR transgenic T cells, previously activated specifically in vitro, allowed exploration of antigen-specific memory responses. Indeed, after homeostatic proliferation in the host, they become memory-like, and, once harvested from spleen, they could be specifically re-activated ex vivo with specific peptides exogenously loaded onto various cultured APCs. Then using APC from CD80/86-deficient mice or CTLA4-Ig, the CD28-independence of these memory T cells was demonstrated (19, 20).
We should stress that all the previously discussed in vitro studies have examined CD28 costimulation requirements under conditions where the T-cell stimulus was not equivalent to the stimulus received in physiological conditions. Peptide was exogenously loaded onto cultured APCs, and thus the requirement for costimulation may have been overcome due to the strength of TCR signaling (21). Indeed, even for a primary response, the costimulation requirement can be overcome if sufficiently high levels of TCR stimulation are obtained. Viola et al., showed in vitro that, independent of the nature of the TCR stimuli, if TCR stimulation exceeds a minimum threshold, complete activation is achieved and in the presence of CD28 costimulation, that threshold is significantly lower (22), especially in memory T cells (23). Thus, the costimulation requirement is a quantitative phenomenon and has to be investigated in the light of the strength of TCR stimulation.
However, evidence in vivo was provided in a report by Suresh et al. showing that, in lymphocytic choriomeningitis virus (LCMV) infected CD28-deficient mice, memory LCMV-specific CD8 + T-cell response seems to be normally reactivated. Indeed when they were re-challenged with a lethal dose of LCMV, all the mice survived while all naive controls died (24).
At first sight, the use of CD28-deficient mice to investigate a memory response in vivo may seem questionable, since the primary response, and consequently the establishment of memory cells in these animals, is greatly reduced (25). But initial studies using LCMV-infected mice revealed that, unlike for principle viruses, an efficient primary CD8 + T-cell response can be generated in the absence of CD28 costimulation (25). The reason for this discrepancy was ascribed to higher levels of TCR stimulation, which could overcome the need for costimulation. Therefore, using this model to explore the recall responses actually makes little sense. In addition, more detailed studies suggest a number of deficiencies in terms of the primary LCMV-specific T-cell response in CD28-deficient mice. In particular, the expansion of virus-specific CD4 T cells was reduced by about a factor of 10 (26) and results with B7-deficient mice indicate that B7 costimulation is required for induction and maintenance of LCMV-specific CD8 + T-cell memory (27). Finally, although CD28-deficient mice have normal levels of B- and T-cell populations, given the importance of CD28 costimulation in thymic T-cell development (28, 29), lack of CD28 induces a defect in regulatory T cells and could lead to defective mature T cells. Taken together, this complicates using these mice to study memory responses.
In the early 2000s, based on in vitro studies and models of LCMV infection in CD28-deficient mice, despite their methodological and technical limitation, the prevailing belief was that CD28 costimulation was not required for memory T-cell activation.
Revisiting the Concept
The accepted hypothesis began to be questioned with studies (30, 31) in which in vivo depletion systems were used to analyze the role of DCs in reactivating CD8 memory T cells during recall response to three different microbial infections. Without DCs during recall response, a profound decrease in the number of CD8 memory T cells was observed, suggesting that costimulation through DCs was required for optimal memory response.
More evidence against the proposition that costimulation is dispensable for memory T cells comes from successes in the treatment of autoimmune diseases, which by definition involve pre-existing auto-reactive T cells. CTLA4-Ig has proved effective both in experimental models (32, 33) and in the clinic with psoriasis (34) and rheumatoid arthritis (35).
Furthermore, except for the specific case of LCMV infection, a lack of costimulation by CD28 impaired secondary response in numerous other infectious models has been found (36–42). Whether these observations indicate requirements for CD28 costimulation during the initial priming or during the recall response is not clear and has not been investigated in detail.
In several more recent works (27, 43–50), reactivation of specific memory T-cell populations in immunized WT mice has been investigated using specific tetramers, or by adoptive transfer of labeled memory T cells. Assessment of a CD28 requirement was made through either adoptive transfer in CD80/86-deficient hosts or through costimulation blockade at the moment of recall, using CTLA4-Ig or anti-CD28 antagonist antibodies.
The essential function of CD28 for conferring host protection during secondary infection has been confirmed using the cre-lox system allowing a CD28-inducible KO in a model of infection by N. Brasiliensis (51). Mice were infected a first time, then a second one after treatment with tamoxifen allowing an efficient CD28 deletion. Compared with WT, these mice had a delayed expulsion of adult worms in the small intestine.
Finally, a more recent study highlighted the critical importance of the CD28 pathway to memory T-cells homeostasis (52). Again in a context of LCMV infection, Kalia et al. demonstrated that without Tregs, memory T cells in a quiescent state proliferated and engaged terminal differentiation. CTLA-Ig by blocking CD80/86 interaction with CD28 rescued memory defects (maintaining a quiescent state) by mimicking Treg known to modulate the level of ligand available for CD28 through CD80/86 trans-endocytosis on APC mediated by CTLA-4 (53).
Thus, currently, extensive research using more relevant experimental models has demonstrated that the optimal elaboration of secondary T-cell immunity, as well as memory T-cell homeostasis, is dependent on productive CD28/CD80/86 interactions, in the setting of anti-infectious immune response.
Allo-Immune Response
As a starting point, we have to distinguish two dramatically different scenarios for the involvement of immune memory response in transplantation. First, recipients who are sensitized to HLA antigens, which occurs mainly through blood transfusions, pregnancy, or previous organ transplantation (54–56). To date, very little research has been done on use of T-cell-specific costimulation blockade strategies in HLA-sensitized recipients and as such it will not be addressed in this review. Second, there are recipients without HLA-specific immunization. In such case, memory T-cell involvement is not, at first sight, obvious.
In the early 90s, shortly after its discovery, the CD28/CD80/86 interaction blockade, later associated with CD40–CD40L blockade, raised great hopes in the transplantation field. In murine models, numerous studies demonstrated that blockade of these co-stimulatory pathways during transplantation was highly effective at tolerizing naive donor-reactive T cells and prolonging graft survival. This occurred irrespective of the blockade modality: CTLA4-Ig (57–59) or anti-CD80/86 antibodies (4). While treatment with CTLA4-Ig in rodents demonstrated high efficacy, experiments in non-human primates demonstrated much more modest prolongation of allograft survival (60–62).
Initially, a weak affinity of the first CTLA4-Ig for CD86, compared with CD80, was hypothesized as the source of this lack of effectiveness (4). Thus, a second generation of CTLA4-Ig, LEA29Y, with a better affinity for CD86, was developed. Translation of LEA29Y into non-human primate models of renal transplantation showed superior prolongation in graft survival compared to CTLA4-Ig as a monotherapy, and dramatically improved survival when used as part of a combined immunosuppressive regimen including either mycophenolate mofetil (MMF) and steroids or anti-IL-2R (basiliximab) (63). Based on these encouraging results, LEA29Y (belatacept) was moved into clinical trials as the principal component of an immunosuppressive regimen consisting of basiliximab, steroids, and MMF (5). As expected, this study showed improvement in renal function compared with cyclosporine-treated recipients owing to reduced CNI-related renal toxicities (5, 64). However, the incidence of biopsy-proven acute rejection was higher in belatacept-treated recipient, giving rise to a new concept: the “belatacept-resistant rejection,” its counterpart being resistance to tolerance induction in rodent experimental models.
As detailed above, based on studies in vitro and in CD28-deficient mice, the perception that memory cells did not require costimulation signaling by CD28 was deeply ingrained. Consequently, memory T cells were presumed to be the guilty party in belatacept-resistant rejection via heterologous immunity, the concept that without bystander activation, virus-specific memory T cells can become activated by unrelated viruses, through molecular mimicry (65). On the top of this, unexpected cross-reactivity between virus-specific CTL clones and uninfected allogenic targets has been demonstrated (66). This activity could be attributed to dual recognition of pathogen-peptide/self-CMH complexes as well as peptide/allo-CMH complexes (6, 67). The most famous example is in seminal studies by Burrows et al. demonstrating that CD8 + T cells specific to EBV-EBNA3A restricted by HLA-B8 were cross-reactive with HLA-B44 presenting a self-peptide. Recently, the molecular understanding of this phenomenon has improved (68, 69) and its magnitude in the transplantation context has been clarified (70).
Heterologous immunity was suspected of playing a major role in mediating costimulation blockade-resistant allograft rejection, observed in situations where transplant recipients have an immune history. Several studies argue for this hypothesis, showing that naive recipients that had previously been infected with different pathogens became refractory to the tolerance-inducing effects of costimulation blockade (71, 72). This resistance is transmitted by adoptive transfer of CD8 and/or CD4 from an immunized to a naive animal (73). Furthermore, in a more relevant model of kidney transplantation in NHP, where tolerance was induced by costimulation blockade combined with donor-specific transfusion (DST), it was revealed that the higher frequency of alloreactive memory cells (when measured by ELISPOST assay) correlated with the occurrence of acute rejection (74).
Collectively, these studies concluded that resistance to the tolerance-inducing effects of costimulation blockade in experimental models and belatacept-resistant rejection in the clinic were caused by heterologous immunity (75, 76). How can this conclusion be reconciled with the recent data showing that effective memory T-cell recall response actually requires CD28 costimulation? One explanation could be that in the non-physiologic context of transplantation, the strength of the antigenic challenge overcomes the costimulation threshold, particularly in Ag-experienced cells.
An Early and Only Cellular Rejection?
Heterologous immunity occurs through the interaction of a recipient Ag-experienced T cell with a donor APC, in transplant immunology this is called the direct recognition pathway. If we assume that the strength of the antigenic challenge during an allo-immune response overcomes the CD28 requirement threshold, it should again be through the direct recognition pathway. Yet the main immunological issue in kidney transplantation concerns the late onset of kidney dysfunction caused by chronic rejection mainly driven by the indirect pathway (i.e., the interaction of a recipient T cell with a recipient APC exposing donor allogenic MHC peptides) (77), which presumably has a higher physiological CD28 requirement threshold. In addition, the onset of de novo DSA can explain a large proportion of chronic rejection. Its onset is dependent on allogenic B-cell response that receives help from a highly specialized subset of CD4 T cells in the germinal center (GC), the follicular helper T cells (Tfh) (78). A recent study revealed that help for a GC alloantibody response could only be provided by CD4 T cells by the indirect pathway (79). The fact that CD28 costimulation is greatly required for primary Tfh response probably explains the lack of DSA in experimental models and belatacept-treated recipients exhibiting remarkably low levels of DSA (64).
The above points suggest that costimulation blockade-resistant rejection should occur early, driven by the direct pathway and consequently without the development of specific alloantibodies, except, obviously, in the case of prior specific immunization.
Are Experienced-T-Cell Subsets on Equal Terms with Costimulation Blockade Resistance?
Even in cases involving the direct recognition pathway, it is likely that all Ag-experienced T cells are not equal in terms of CD28 requirement. Recent studies on tolerance induction by costimulation blockade (80–85) substantiate the view mentioned above that CD28 requirement loss would not reflect an inherent advantage to any memory response acquired following an immune response, but would be a feature confined to cells reaching the end of a progressive process of maturation.
When allo-specific CD8 T Central Memory (TCM) and T Effector Memory (TEM) cells were transferred into wild-type recipients, they were found equally effective at rejecting allografts. When transferred into aly-deficient recipient (aly-deficiency leads to an absence of secondary lymphoid organs), TEM cells were significantly better than TCM at rejecting allografts (86). This suggests that TCM, but not TEM, reactivation requires the presence of APC with costimulation molecules to proliferate and gain effector and homing capacities.
In line with this, in a model of heterologous immunity generated by a latent γHV68 infection of WT mice, effector T cells (CD44highCD127lowCD62Llow) and TEM (CD44highCD127highCD62Llow-int) were found to be responsible for resistance to tolerance induction by costimulation blockade, in contrast to TCM (80).
In a murine model, decreasing the amount/duration of antigen exposure during priming impacted the ability of donor-specific experienced T cells to mediate costimulation blockade-resistant rejection (81). Interestingly, only donor-specific T cells that were generated under conditions of reduced Ag exposure failed to mediate costimulation blockade (referring to as CD80/86 blockade) resistant rejection. Overall antigenic stimulation undergone by T cells during priming is proposed as predicting cell fate, ranging from unpolarized cells to terminally differentiated cells (87). Thus in the case of poor antigenic stimulation, the accumulation of unpolarized cells could explain the success of the costimulation blockade.
The differential effects of belatacept on cell proliferation in response to either viral peptide processed on self APC or allogenic stimulation seem to confirm this proposition (82). Xu et al. showed that a large percentage of the repertoire proliferated in response to alloantigen, but contained few polyfunctional cells (advanced in their maturation and expressing IFNγ, TNFα, and IL-2). By contrast, the proportion of cells responding to a viral peptide was low and consisted predominantly of mature polyfunctional TEM. When belatacept was added to the cell culture medium, only the more mature cells escaped the costimulation blockade. This again demonstrates that only T cells that have reached a late maturation stage are independent of CD28.
Furthermore, this could explain the relative success of the association of belatacept and alefacept, a CD2 antagonist, in an experimental model of kidney transplantation in NHP (83). Indeed, CD8+CD2+ were the most differentiated in terms of cytotoxic molecule expression and polyspecificity.
Hence among experienced T cells, those liable for costimulation blockade resistance are mature cells, having completed the progressive process of differentiation, including the loss of the CD28 costimulation requirement for their activation.
Interestingly, recent data have revealed that end-stage renal disease patients, compared to healthy controls, have a significant greater number of memory T cells showing progressive terminal differentiation, similar to what is observed in old people with immune-senescence (88). Likewise, anti-thymocyte globulins (ATG)-treated recipient exhibit more late stage differentiated T cells, including CD28 negative (89). Hence, kidney transplant candidates, by definition with impaired renal function, could be especially affected by belatacept-resistant rejection.
Beyond having a CD28 requirement, CTLA-4 might also play a role in belatacept-resistant rejection. Halloran et al. have recently demonstrated that CTLA-4 transcripts dominate the molecular landscape of T-cell-mediated rejection (TCMR) (90), highlighting the possibility that an active negative regulation of T cells in tissue could explain the occurrence of robust TCMR in belatacept-treated recipients.
CD28 Selective Blockade
Up to now, “CD28 blockade” referred to inhibiting B7, either with a CTLA4-Ig or anti-CD80/86 antibody. Obviously, concomitant inhibition of the CTLA-4 pathway is the main drawback of this strategy. As suggested above, the selective blockade of CD28 signaling (i.e., blocking only CD28/CD80/86 interactions) should present the advantage of respecting the immune-modulatory signals mediated by CTLA-4. The recent development of monovalent antagonist anti-CD28 binders makes this strategy feasible and safe (91–95), clearly differentiating them from agonist or superagonist anti-CD28 antibodies (96–100).
CD28 antagonists prevent acute allograft rejection in mice (101) and primate (92). The potential benefit of preserving CTLA-4 pathways would be due to its extrinsic action, mainly through regulatory T cells. Indeed, use of CD28 antagonist is associated with Treg accumulation in the graft, where they most likely modulate pathogenic T cells and promote prolonged allograft survival (92).
But CTLA-4 has also intrinsic, cell-autonomous roles (102). For experienced T cells, we would expect the advantages of a selective CD28 blockade compared with CTLA4-Ig, if two conditions are met: (i) cells independent of CD28 costimulation for their activation are at play in the context of allo-immune responses and (ii) that activation of these same cells is regulated by CTLA-4. We have seen above that CD28-independent alloreactive cells do exist even though this concerns probably only a few singular cell subsets. Whether experienced T cells are regulated by CTLA-4 is the focus of the following paragraph.
Are Experienced T Cells Regulated by CTLA-4?
Targeting CTLA-4 with ipilimumab for melanoma immunotherapy was the first clinical demonstration of the physiological role of CTLA-4 acting as an immune checkpoint that controls T-cell reactivity (103, 104). Initial work indicated that the maximal activity of anti-CTLA-4 treatment required the targeting of CTLA-4 on both effectors and Tregs (105). It has also been suggested that anti-CTLA-4 antibodies lead to the depletion of Tregs within the tumor microenvironment in a Fcγ receptor-dependent manner (106–108), concomitant with an increase in the number of activated T cells in peripheral blood (109–112) as well as the tumor site (113–115). Two non-mutually exclusive scenarios can explain this second observation. First, anti-CTLA-4 treatment could improve the priming, then expansion of tumor-specific naive T cells. Second, it could increase the magnitude of the preexisting memory/effector tumor reactive T cells by turning off inhibitory mechanisms (116). Recent advances argue for the latter. Cha et al. measured the frequency of individual rearranged TCRβ genes after anti-CTLA-4 treatment in cancer patients. Clinical outcome was associated with maintenance of high-frequency clones present at the start of the treatment. The bulk of the change in clone frequency occurred in the effector/memory T-cell compartment rather than in the naive T-cell pool, suggesting that treatment boosted meaningful preexisting T-cell responses (117). More recently, it has been evidenced in mice that preexisting anti-tumor T-cell responses are amplified by checkpoint blockade therapy. Anti CTLA-4 and anti PD-1 in a sarcoma model regulated a subset of genes in CD8 tumor-specific infiltrating lymphocyte (TIL) (especially Granzyme B, IFN-γ, and TNF-α that are known to cause acute rejection), whose enhanced expression is similar to that observed in CD8 T cells from mice during acute secondary viral infection. The depressed genes were similar to those of exhausted CD8 T cells in chronic viral infection (particularly LAG-3 and TIM-3) (118). In a melanoma model, anti-CTLA-4 predominantly inhibits Treg cells in TIL but also reinvigorates exhausted PD-1 + Eomes + CD8 T cells (119).
In the context of rejection prophylaxis by CTLA4-Ig, CTLA-4 is also blocked (at least it cannot interact with CD80/86 anymore). It is tempting to speculate that, similar to that which is observed in tumors, some preexisting transplant infiltrating lymphocytes in an advanced stage of differentiation, which are supposed to be costimulation independent, could be reinvigorated by the CTLA-4 blockade with belatacept. Indeed CTLA-4 might inhibit T cells even in the absence of CD28 (120) and data from clinical trials provide indirect evidence for such an “immune checkpoint inhibitory” effect of CTLA4-Ig. In inflammatory bowel disease (IBD), patients treated with CTLA4-Ig demonstrate minimal improvement and disease exacerbation was seen in some treatment groups (121). The development of IBD has also been reported in a patient treated with CTLA-4 Ig for rheumatoid arthritis (122).
Which Cells are Responsible for Belatacept-Resistant Rejection?
Regardless of maturation stage, we can assume that both the threshold of CD28 requirement and the intrinsic regulation by CTLA-4 pathway differ between various T-cell subsets.
Polarized Th17 could be responsible for Belatacept-resistant rejection, since an elevated level of Th17 memory cells has been associated with acute rejection with belatacept (85), and as mentioned above, in IBD, which is a Th17-mediated disease, CTLA4-Ig treatment has exhibited minimal efficacy and even, in a few cases, disease exacerbation (121). In addition, Candida albicans immunization of mice conferred resistance to costimulation blockade following transplantation. C. albicans polarizes the response toward Th17 cells and enhances expression of CTLA-4 on Th17 cells. Mycobacterium tuberculosis, which polarizes the response toward Th1 cells, does not confer such resistance (84). These data were verified using mice genetically deficient for hallmark T-cell transcription factors such as B6.RORγt KO and B6.T-bet KO (123). Thus, Th17 cells might be particularly sensitive to regulation by CTLA-4, and CTLA4-Ig might hamper this regulation.
Turning to Tfh, the initial priming instigating a Tfh response is CD28 dependent, including in the allo-immune response context (124). By contrast, primed Tfh lose their CD28 requirement when they secondarily interact with B cells (125). Furthermore, at that stage, CTLA-4 also regulates Tfh function in a cell-intrinsic manner (126). Again, like Th17, Tfh accumulates with immune history and has the features required to prompt resistance to CTLA4-Ig.
Potential Advantage of Targeting CD28 Instead of CD80/86
Recently (127), we performed a direct assessment of FR104 (93), a selective CD28 Fab antagonist, versus CTLA4-Ig (LEA26Y) in kidney allograft in baboon. The biologics were used de novo together with an initial 1-month treatment with a low dose of tacrolimus, weaned between months 1 and 2, after which the recipients were under monotherapy with the biologics. Biopsy-proven acute rejection animals were treated with boluses of steroids. In the CTLA4-Ig group (n = 5), four out of the five recipients developed severe acute cellular rejection before, during or just after tacrolimus weaning and this proved to be corticoresistant. In the FR104 group (n = 5), only two animals developed an acute rejection episode, just after tacrolimus weaning, and this could be reversed by steroids. A transcriptional analysis of 1-month biopsies did not reveal any significant differences except the remarkable exception of IL-21, stronger in CTLA-4 treated animals, whose main source is Tfh cells. Immunohistochemistry revealed some CD4 T cells expressing PD-1, the main marker used to identify Tfh and IL-21. We then assessed in vitro proliferation of stimulated Tfh (CXCR5 + ICOS + PD-1 +) using human tonsil tissue and found that inhibition was more effective with FR104 than with CTLA4-Ig. This was confirmed in an experimental model of immunization with KLH in mise where, as expected, primary Tfh response was equally inhibited with both CD28 selective blockade and CTLA4-Ig, unlike the recall response in which the CD28 selective blockade was more efficient in controlling Tfh response. Of interest in a model of islet transplantation, mIL21R.Fc rescues CTLA-Ig-treated mice, resulting in tolerance in 100% of the mice versus 55% in a CTLA4-Ig monotherapy group, and it was demonstrated that IL-21 acted as an antitolerogenic cytokine by preventing Treg generation and inhibiting Treg function (128).
Summary
In the field of transplantation, the initial great hopes for CD28/CD80/86/CTLA-4 blocking strategy have been dashed in the confrontation with clinical reality. The presence of a complex repertoire of preexisting experienced T cells either free of a CD28 costimulation requirement and/or controlled by the CTLA-4 immune checkpoint is a likely explanation.
However, the picture might not be so dark. First, because primary Tfh response is strictly under the control of CD28, explaining why a costimulation blockade with belatacept prevents the induction of alloantibodies. Second, a loss of a CD28 requirement might not be exhibited by memory cells, but rather confined to terminally matured cells, to some extent exhausted. Although in the context of allo-immune response, these cells could cause severe rejection, the risk of a rescuing inhibitory signal mediated by CTLA-4 and of eliciting belatacept-resistant cellular rejection could be alleviated by the use of CD28-specific antagonists, which are currently in clinical development that will block CD28-mediated signals, without preventing CTLA-4 signals. This novel approach might have the potential advantage of controlling post-transplant immune responses more effectively.
Conflict of Interest Statement
Nicolas Poirier and Bernard Vanhove are shareholders and employees in Effimune, a company developing CD28 antagonists. Simon Ville and Gilles Blancho have no conflict of interest to declare.
References
1. Lafferty KJ, Jones MA. Reactions of the graft versus host (GVH) type. Aust J Exp Biol Med Sci (1969) 47:17–54. doi:10.1038/icb.1969.3
2. Lafferty KJ, Misko IS, Cooley MA. Allogeneic stimulation modulates the in vitro response of T cells to transplantation antigen. Nature (1974) 249:275–6. doi:10.1038/249275a0
3. Baxter AG, Hodgkin PD. Activation rules: the two-signal theories of immune activation. Nat Rev Immunol (2002) 2:439–46. doi:10.1038/nri823
4. Ford ML, Larsen CP. Translating costimulation blockade to the clinic: lessons learned from three pathways. Immunol Rev (2009) 229:294–306. doi:10.1111/j.1600-065X.2009.00776.x
5. Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, et al. Costimulation blockade with belatacept in renal transplantation. N Engl J Med (2005) 353:770–81. doi:10.1056/NEJMoa050085
6. Selin LK, Brehm MA. Frontiers in nephrology: heterologous immunity, T cell cross-reactivity, and alloreactivity. J Am Soc Nephrol (2007) 18:2268–77. doi:10.1681/ASN.2007030295
7. Sprent J, Surh CD. T cell memory. Annu Rev Immunol (2002) 20:551–79. doi:10.1146/annurev.immunol.20.100101.151926
8. Mou D, Espinosa J, Lo DJ, Kirk AD. CD28 negative T cells: is their loss our gain?: CD28 negative T cells. Am J Transplant (2014) 14:2460–6. doi:10.1111/ajt.12937
9. Weng N, Akbar AN, Goronzy J. CD28 − T cells: their role in the age-associated decline of immune function. Trends Immunol (2009) 30:306–12. doi:10.1016/j.it.2009.03.013
10. Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev (2005) 205:158–69. doi:10.1111/j.0105-2896.2005.00256.x
11. Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol (2013) 25:556–63. doi:10.1016/j.coi.2013.09.003
12. Restifo NP. Big bang theory of stem-like T cells confirmed. Blood (2014) 124:476–7. doi:10.1182/blood-2014-06-578989
13. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol (2013) 43:2797–809. doi:10.1002/eji.201343751
14. Zhu Y, Yao S, Chen L. Cell surface signaling molecules in the control of immune responses: a tide model. Immunity (2011) 34:466–78. doi:10.1016/j.immuni.2011.04.008
15. Inaba K, Steinman RM. Resting and sensitized T lymphocytes exhibit distinct stimulatory (antigen-presenting cell) requirements for growth and lymphokine release. J Exp Med (1984) 160:1717–35. doi:10.1084/jem.160.6.1717
16. Metlay JP, Puré E, Steinman RM. Distinct features of dendritic cells and anti-Ig activated B cells as stimulators of the primary mixed leukocyte reaction. J Exp Med (1989) 169:239–54. doi:10.1084/jem.169.1.239
17. Croft M, Bradley LM, Swain SL. Naive versus memory CD4 T cell response to antigen. Memory cells are less dependent on accessory cell costimulation and can respond to many antigen-presenting cell types including resting B cells. J Immunol (1994) 152:2675–85.
18. Croft M. Activation of naive, memory and effector T cells. Curr Opin Immunol (1994) 6:431–7. doi:10.1016/0952-7915(94)90123-6
19. London CA, Lodge MP, Abbas AK. Functional responses and costimulator dependence of memory CD4+ T cells. J Immunol (2000) 164:265–72. doi:10.4049/jimmunol.164.1.265
20. Bachmann MF, Gallimore A, Linkert S, Cerundolo V, Lanzavecchia A, Kopf M, et al. Developmental regulation of Lck targeting to the CD8 coreceptor controls signaling in naive and memory T cells. J Exp Med (1999) 189:1521–30. doi:10.1084/jem.189.10.1521
21. Boesteanu AC, Katsikis PD. Memory T cells need CD28 costimulation to remember. Semin Immunol (2009) 21:69–77. doi:10.1016/j.smim.2009.02.005
22. Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science (1996) 273:104–6. doi:10.1126/science.273.5271.104
23. Kündig TM, Shahinian A, Kawai K, Mittrücker H-W, Sebzda E, Bachmann MF, et al. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity (1996) 5:41–52. doi:10.1016/S1074-7613(00)80308-8
24. Suresh M, Whitmire JK, Harrington LE, Larsen CP, Pearson TC, Altman JD, et al. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J Immunol (2001) 167:5565–73. doi:10.4049/jimmunol.167.10.5565
25. Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science (1993) 261:609–12. doi:10.1126/science.7688139
26. Christensen JE, Christensen JP, Kristensen NN, Hansen NJV, Stryhn A, Thomsen AR. Role of CD28 co-stimulation in generation and maintenance of virus-specific T cells. Int Immunol (2002) 14:701–11. doi:10.1093/intimm/dxf037
27. Grujic M, Bartholdy C, Remy M, Pinschewer DD, Christensen JP, Thomsen AR. The role of CD80/CD86 in generation and maintenance of functional virus-specific CD8+ T cells in mice infected with lymphocytic choriomeningitis virus. J Immunol (2010) 185:1730–43. doi:10.4049/jimmunol.0903894
28. Vacchio MS, Williams JA, Hodes RJ. A novel role for CD28 in thymic selection: elimination of CD28/B7 interactions increases positive selection. Eur J Immunol (2005) 35:418–27. doi:10.1002/eji.200424918
29. Zheng X, Gao J-X, Chang X, Wang Y, Liu Y, Wen J, et al. B7-CD28 interaction promotes proliferation and survival but suppresses differentiation of CD4-CD8- T cells in the thymus. J Immunol (2004) 173:2253–61. doi:10.4049/jimmunol.173.4.2253
30. Zammit DJ, Cauley LS, Pham Q-M, Lefrançois L. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity (2005) 22:561–70. doi:10.1016/j.immuni.2005.03.005
31. Belz GT, Wilson NS, Smith CM, Mount AM, Carbone FR, Heath WR. Bone marrow-derived cells expand memory CD8+ T cells in response to viral infections of the lung and skin. Eur J Immunol (2006) 36:327–35. doi:10.1002/eji.200535432
32. Bluestone JA, St. Clair EW, Turka LA. CTLA4Ig: bridging the basic immunology with clinical application. Immunity (2006) 24:233–8. doi:10.1016/j.immuni.2006.03.001
33. Khoury SJ, Akalin E, Chandraker A, Turka LA, Linsley PS, Sayegh MH, et al. CD28-B7 costimulatory blockade by CTLA4Ig prevents actively induced experimental autoimmune encephalomyelitis and inhibits Th1 but spares Th2 cytokines in the central nervous system. J Immunol (1995) 155:4521–4.
34. Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest (1999) 103:1243–52. doi:10.1172/JCI5857
35. Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med (2003) 349:1907–15. doi:10.1056/NEJMoa035075
36. Bertram EM, Tafuri A, Shahinian A, Chan VS, Hunziker L, Recher M, et al. Role of ICOS versus CD28 in antiviral immunity. Eur J Immunol (2002) 32:3376–85. doi:10.1002/1521-4141(200212)32:12<3376::AID-IMMU3376>3.0.CO;2-Y
37. Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol (2002) 168:3777–85. doi:10.4049/jimmunol.168.8.3777
38. Bertram EM, Dawicki W, Sedgmen B, Bramson JL, Lynch DH, Watts TH. A switch in costimulation from CD28 to 4-1BB during primary versus secondary CD8 T cell response to influenza in vivo. J Immunol (2004) 172:981–8. doi:10.4049/jimmunol.172.2.981
39. Mittrucker H-W, Kursar M, Kohler A, Hurwitz R, Kaufmann SHE. Role of CD28 for the generation and expansion of antigen-specific CD8+ T lymphocytes during infection with listeria monocytogenes. J Immunol (2001) 167:5620–7. doi:10.4049/jimmunol.167.10.5620
40. Mittrücker H-W, Köhler A, Mak TW, Kaufmann SH. Critical role of CD28 in protective immunity against Salmonella typhimurium. J Immunol (1999) 163:6769–76.
41. Fuse S, Obar JJ, Bellfy S, Leung EK, Zhang W, Usherwood EJ. CD80 and CD86 control antiviral CD8+ T-cell function and immune surveillance of murine gammaherpesvirus 68. J Virol (2006) 80:9159–70. doi:10.1128/JVI.00422-06
42. Villegas EN, Elloso MM, Reichmann G, Peach R, Hunter CA. Role of CD28 in the generation of effector and memory responses required for resistance to Toxoplasma gondii. J Immunol (1999) 163:3344–53.
43. Ndejembi MP, Teijaro JR, Patke DS, Bingaman AW, Chandok MR, Azimzadeh A, et al. Control of memory CD4 T cell recall by the CD28/B7 costimulatory pathway. J Immunol (2006) 177:7698–706. doi:10.4049/jimmunol.177.11.7698
44. Fuse S, Zhang W, Usherwood EJ. Control of memory CD8+ T cell differentiation by CD80/CD86-CD28 costimulation and restoration by IL-2 during the recall response. J Immunol (2008) 180:1148–57. doi:10.4049/jimmunol.180.2.1148
45. Fuse S, Tsai C-Y, Rommereim LM, Zhang W, Usherwood EJ. Differential requirements for CD80/86-CD28 costimulation in primary and memory CD4 T cell responses to vaccinia virus. Cell Immunol (2011) 266:130–4. doi:10.1016/j.cellimm.2010.09.008
46. Teijaro JR, Njau MN, Verhoeven D, Chandran S, Nadler SG, Hasday J, et al. Costimulation modulation uncouples protection from immunopathology in memory T cell responses to influenza virus. J Immunol (2009) 182:6834–43. doi:10.4049/jimmunol.0803860
47. Prilliman KR, Lemmens EE, Palioungas G, Wolfe TG, Allison JP, Sharpe AH, et al. Cutting edge: a crucial role for B7-CD28 in transmitting T help from APC to CTL. J Immunol (2002) 169:4094–7. doi:10.4049/jimmunol.169.8.4094
48. Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, et al. Memory CD8+ T cells require CD28 costimulation. J Immunol (2007) 179:6494–503. doi:10.4049/jimmunol.179.10.6494
49. Eberlein J, Davenport B, Nguyen TT, Victorino F, Sparwasser T, Homann D. Multiple layers of CD80/86-dependent costimulatory activity regulate primary, memory, and secondary lymphocytic choriomeningitis virus-specific T cell immunity. J Virol (2012) 86:1955–70. doi:10.1128/JVI.05949-11
50. Linterman MA, Denton AE, Divekar DP, Zvetkova I, Kane L, Ferreira C, et al. CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. Elife (2014) 3:e03180. doi:10.7554/eLife.03180
51. Ndlovu H, Darby M, Froelich M, Horsnell W, Lühder F, Hünig T, et al. Inducible deletion of CD28 prior to secondary nippostrongylus brasiliensis infection impairs worm expulsion and recall of protective memory CD4+ T cell responses. PLoS Pathog (2014) 10:e1003906. doi:10.1371/journal.ppat.1003906
52. Kalia V, Penny LA, Yuzefpolskiy Y, Baumann FM, Sarkar S. Quiescence of memory CD8+ T cells is mediated by regulatory T cells through inhibitory receptor CTLA-4. Immunity (2015) 42:1116–29. doi:10.1016/j.immuni.2015.05.023
53. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi:10.1126/science.1202947
54. Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol (2005) 5:807–17. doi:10.1038/nri1702
55. Smith RN, Kawai T, Boskovic S, Nadazdin O, Sachs DH, Cosimi AB, et al. Four stages and lack of stable accommodation in chronic alloantibody-mediated renal allograft rejection in cynomolgus monkeys. Am J Transplant (2008) 8:1662–72. doi:10.1111/j.1600-6143.2008.02303.x
56. Haas M, Sis B, Racusen LC, Solez K, Glotz D, Colvin RB, et al. Banff 2013 meeting report: inclusion of C4d-negative antibody-mediated rejection and antibody-associated arterial lesions: Banff 2013 meeting report. Am J Transplant (2014) 14:272–83. doi:10.1111/ajt.12590
57. Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, et al. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science (1992) 257:792–5. doi:10.1126/science.1496399
58. Lenschow DJ, Zeng Y, Thistlethwaite JR, Montag A, Brady W, Gibson MG, et al. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science (1992) 257:789–92. doi:10.1126/science.1323143
59. Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei R-Q, et al. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc Natl Acad Sci U S A (1992) 89:11102–5. doi:10.1073/pnas.89.22.11102
60. Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci U S A (1997) 94:8789–94. doi:10.1073/pnas.94.16.8789
61. Kirk AD, Tadaki DK, Celniker A, Batty DS, Berning JD, Colonna JO, et al. Induction therapy with monoclonal antibodies specific for cd80 and cd86 delays the onset of acute renal allograft rejection in non-human primates1. Transplantation (2001) 72:377–84. doi:10.1097/00007890-200108150-00005
62. Levisetti MG, Padrid PA, Szot GL, Mittal N, Meehan SM, Wardrip CL, et al. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol (1997) 159:5187–91.
63. Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant (2005) 5:443–53. doi:10.1111/j.1600-6143.2005.00749.x
64. Vincenti F, Charpentier B, Vanrenterghem Y, Rostaing L, Bresnahan B, Darji P, et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am J Transplant (2010) 10:535–46. doi:10.1111/j.1600-6143.2009.03005.x
65. Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol (2002) 2:417–26. doi:10.1038/nri820
66. Burrows SR, Khanna R, Burrows JM, Moss DJ. An alloresponse in humans is dominated by cytotoxic T lymphocytes (CTL) cross-reactive with a single Epstein-Barr virus CTL epitope: implications for graft-versus-host disease. J Exp Med (1994) 179:1155–61. doi:10.1084/jem.179.4.1155
67. Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsh RM. Direct visualization of cross-reactive effector and memory allo-specific CD8 T cells generated in response to viral infections. J Immunol (2003) 170:4077–86. doi:10.4049/jimmunol.170.8.4077
68. Felix NJ, Donermeyer DL, Horvath S, Walters JJ, Gross ML, Suri A, et al. Alloreactive T cells respond specifically to multiple distinct peptide-MHC complexes. Nat Immunol (2007) 8:388–97. doi:10.1038/ni1446
69. Macdonald WA, Chen Z, Gras S, Archbold JK, Tynan FE, Clements CS, et al. T cell allorecognition via molecular mimicry. Immunity (2009) 31:897–908. doi:10.1016/j.immuni.2009.09.025
70. Amir AL, D’Orsogna LJA, Roelen DL, van Loenen MM, Hagedoorn RS, de Boer R, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood (2010) 115:3146–57. doi:10.1182/blood-2009-07-234906
71. Pantenburg B, Heinzel F, Das L, Heeger PS, Valujskikh A. T cells primed by Leishmania major infection cross-react with alloantigens and alter the course of allograft rejection. J Immunol (2002) 169:3686–93. doi:10.4049/jimmunol.169.7.3686
72. Valujskikh A, Pantenburg B, Heeger PS. Primed allospecific T cells prevent the effects of costimulatory blockade on prolonged cardiac allograft survival in mice. Am J Transplant (2002) 2:501–9. doi:10.1034/j.1600-6143.2002.20603.x
73. Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest (2003) 111:1887–95. doi:10.1172/JCI17477
74. Nadazdin O, Boskovic S, Murakami T, Tocco G, Smith R-N, Colvin RB, et al. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci Transl Med (2011) 3:86ra51. doi:10.1126/scitranslmed.3002093
75. Adams AB, Pearson TC, Larsen CP. Heterologous immunity: an overlooked barrier to tolerance. Immunol Rev (2003) 196:147–60. doi:10.1046/j.1600-065X.2003.00082.x
76. Lakkis FG. Memory T cells: a hurdle to immunologic tolerance. J Am Soc Nephrol (2003) 14:2402–10. doi:10.1097/01.ASN.0000085020.78117.70
77. Ali JM, Bolton EM, Bradley JA, Pettigrew GJ. Allorecognition pathways in transplant rejection and tolerance. Transplantation (2013) 96:681–8. doi:10.1097/TP.0b013e31829853ce
78. Crotty S. Follicular helper CD4 T cells (T FH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
79. Conlon TM, Saeb-Parsy K, Cole JL, Motallebzadeh R, Qureshi MS, Rehakova S, et al. Germinal center alloantibody responses are mediated exclusively by indirect-pathway CD4 T follicular helper cells. J Immunol (2012) 188:2643–52. doi:10.4049/jimmunol.1102830
80. Stapler D, Lee ED, Selvaraj SA, Evans AG, Kean LS, Speck SH, et al. Expansion of effector memory TCR V 4+CD8+ T cells is associated with latent infection-mediated resistance to transplantation tolerance. J Immunol (2008) 180:3190–200. doi:10.4049/jimmunol.180.5.3190
81. Floyd TL, Koehn BH, Kitchens WH, Robertson JM, Cheeseman JA, Stempora L, et al. Limiting the amount and duration of antigen exposure during priming increases memory T cell requirement for costimulation during recall. J Immunol (2011) 186:2033–41. doi:10.4049/jimmunol.1003015
82. Xu H, Perez SD, Cheeseman J, Mehta AK, Kirk AD. The allo- and viral-specific immunosuppressive effect of belatacept, but not tacrolimus, attenuates with progressive T cell maturation: belatacept efficacy and T cell maturation. Am J Transplant (2014) 14:319–32. doi:10.1111/ajt.12574
83. Weaver TA, Charafeddine AH, Agarwal A, Turner AP, Russell M, Leopardi FV, et al. Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates. Nat Med (2009) 15:746–9. doi:10.1038/nm.1993
84. Krummey SM, Floyd TL, Liu D, Wagener ME, Song M, Ford ML. Candida-elicited murine Th17 cells express high CTLA-4 compared with Th1 cells and are resistant to costimulation blockade. J Immunol (2014) 192:2495–504. doi:10.4049/jimmunol.1301332
85. Krummey SM, Cheeseman JA, Conger JA, Jang PS, Mehta AK, Kirk AD, et al. High CTLA-4 expression on Th17 cells results in increased sensitivity to CTLA-4 coinhibition and resistance to belatacept: Th17 cells have high CTLA-4 expression. Am J Transplant (2014) 14:607–14. doi:10.1111/ajt.12600
86. Oberbarnscheidt MH, Ng Y-H, Chalasani G. The roles of CD8 central and effector memory T-cell subsets in allograft rejection. Am J Transplant (2008) 8:1809–18. doi:10.1111/j.1600-6143.2008.02335.x
87. Kalia V, Sarkar S, Ahmed R. Fine-tuning CD4+ central memory T cell heterogeneity by strength of stimulation. Eur J Immunol (2008) 38:15–9. doi:10.1002/eji.200738044
88. Betjes MG, Langerak AW, van der Spek A, de Wit EA, Litjens NH. Premature aging of circulating T cells in patients with end-stage renal disease. Kidney Int (2011) 80:208–17. doi:10.1038/ki.2011.110
89. Crepin T, Carron C, Roubiou C, Gaugler B, Gaiffe E, Simula-Faivre D, et al. ATG-induced accelerated immune senescence: clinical implications in renal transplant recipients: ATG and immune senescence in transplanted patients. Am J Transplant (2015) 15:1028–38. doi:10.1111/ajt.13092
90. Venner JM, Famulski KS, Badr D, Hidalgo LG, Chang J, Halloran PF. Molecular landscape of T cell-mediated rejection in human kidney transplants: prominence of CTLA4 and PD ligands: molecular phenotype of TCMR. Am J Transplant (2014) 14:2565–76. doi:10.1111/ajt.12946
91. Haspot F, Seveno C, Dugast A-S, Coulon F, Renaudin K, Usal C, et al. Anti-CD28 antibody-induced kidney allograft tolerance related to tryptophan degradation and TCR- class II- B7+ regulatory cells. Am J Transplant (2005) 5:2339–48. doi:10.1111/j.1600-6143.2005.01018.x
92. Poirier N, Azimzadeh AM, Zhang T, Dilek N, Mary C, Nguyen B, et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci Transl Med (2010) 2:17ra10. doi:10.1126/scitranslmed.3000116
93. Poirier N, Mary C, Dilek N, Hervouet J, Minault D, Blancho G, et al. Preclinical efficacy and immunological safety of FR104, an antagonist anti-CD28 monovalent fab′ antibody: preclinical efficacy and safety of FR104. Am J Transplant (2012) 12:2630–40. doi:10.1111/j.1600-6143.2012.04164.x
94. Poirier N, Dilek N, Mary C, Ville S, Coulon F, Branchereau J, et al. FR104, an antagonist anti-CD28 monovalent fab’ antibody, prevents alloimmunization and allows calcineurin inhibitor minimization in nonhuman primate renal allograft: FR104 prevents allograft rejection. Am J Transplant (2015) 15:88–100. doi:10.1111/ajt.12964
95. Suchard SJ, Davis PM, Kansal S, Stetsko DK, Brosius R, Tamura J, et al. A monovalent anti-human CD28 domain antibody antagonist: preclinical efficacy and safety. J Immunol (2013) 191:4599–610. doi:10.4049/jimmunol.1300470
96. Shiao SL, McNiff JM, Masunaga T, Tamura K, Kubo K, Pober JS. Immunomodulatory properties of FK734, a humanized anti-CD28 monoclonal antibody with agonistic and antagonistic activities. Transplantation (2007) 83:304–13. doi:10.1097/01.tp.0000251426.46312.d5
97. Yu X-Z, Albert MH, Martin PJ, Anasetti C. CD28 ligation induces transplantation tolerance by IFN-γ-dependent depletion of T cells that recognize alloantigens. J Clin Invest (2004) 113:1624–30. doi:10.1172/JCI20940
98. Yu X-Z, Bidwell SJ, Martin PJ, Anasetti C. CD28-specific antibody prevents graft-versus-host disease in mice. J Immunol (2000) 164:4564–8. doi:10.4049/jimmunol.164.9.4564
99. Yu X-Z, Martin PJ, Anasetti C. CD28 signal enhances apoptosis of CD8 T cells after strong TCR ligation. J Immunol (2003) 170:3002–6. doi:10.4049/jimmunol.170.6.3002
100. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med (2006) 355:1018–28. doi:10.1056/NEJMoa063842
101. Zhang T, Fresnay S, Welty E, Sangrampurkar N, Rybak E, Zhou H, et al. Selective CD28 blockade attenuates acute and chronic rejection of murine cardiac allografts in a CTLA-4-dependent manner: selective CD28 blockade requires CTLA-4. Am J Transplant (2011) 11:1599–609. doi:10.1111/j.1600-6143.2011.03624.x
102. Wing K, Yamaguchi T, Sakaguchi S. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol (2011) 32:428–33. doi:10.1016/j.it.2011.06.002
103. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
104. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364:2517–26. doi:10.1056/NEJMoa1104621
105. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206:1717–25. doi:10.1084/jem.20082492
106. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. doi:10.1084/jem.20130579
107. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med (2013) 210:1685–93. doi:10.1084/jem.20130573
108. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res (2013) 1:32–42. doi:10.1158/2326-6066.CIR-13-0013
109. Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J Immunol (2005) 175:7746–54. doi:10.4049/jimmunol.175.11.7746
110. Comin-Anduix B, Lee Y, Jalil J, Algazi A, de la Rocha P, Camacho LH, et al. Detailed analysis of immunologic effects of the cytotoxic T lymphocyte-associated antigen 4-blocking monoclonal antibody tremelimumab in peripheral blood of patients with melanoma. J Transl Med (2008) 6:22. doi:10.1186/1479-5876-6-22
111. Ku GY, Yuan J, Page DB, Schroeder SEA, Panageas KS, Carvajal RD, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer (2010) 116:1767–75. doi:10.1002/cncr.24951
112. Wang W, Yu D, Sarnaik AA, Yu B, Hall M, Morelli D, et al. Biomarkers on melanoma patient T cells associated with ipilimumab treatment. J Transl Med (2012) 10:146–146. doi:10.1186/1479-5876-10-146
113. Huang RR, Jalil J, Economou JS, Chmielowski B, Koya RC, Mok S, et al. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin Cancer Res (2011) 17:4101–9. doi:10.1158/1078-0432.CCR-11-0407
114. Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med (2011) 9:204. doi:10.1186/1479-5876-9-204
115. Ji R-R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother (2012) 61:1019–31. doi:10.1007/s00262-011-1172-6
116. Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A (2011) 108:16723–8. doi:10.1073/pnas.1110814108
117. Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med (2014) 6:238ra70. doi:10.1126/scitranslmed.3008211
118. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515:577–81. doi:10.1038/nature13988
119. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520:373–7. doi:10.1038/nature14292
120. Fallarino F, Fields PE, Gajewski TF. B7-1 engagement of cytotoxic T lymphocyte antigen 4 inhibits T cell activation in the absence of CD28. J Exp Med (1998) 188:205–10. doi:10.1084/jem.188.1.205
121. Sandborn WJ, Colombel J, Sands BE, Rutgeerts P, Targan SR, Panaccione R, et al. Abatacept for Crohn’s disease and ulcerative colitis. Gastroenterology (2012) 143:62–69. doi:10.1053/j.gastro.2012.04.010
122. Amezcua-Guerra LM, Hernández-Martínez B, Pineda C, Bojalil R. Ulcerative colitis during CTLA-4Ig therapy in a patient with rheumatoid arthritis. Gut (2006) 55:1059–60. doi:10.1136/gut.2006.095539
123. Sabet-Baktach M, Eggenhofer E, Renner P, Lantow M, Schlitt H, Geissler E, et al. Eomes-expressing CD8+ T cells and Th17 cells mediate costimulatory blockade-resistant allograft rejection in mice. Am J Transplant (2015) 15(Suppl 3):S1.
124. Kim EJ, Kwun J, Gibby AC, Hong JJ, Farris AB, Iwakoshi NN, et al. Costimulation blockade alters germinal center responses and prevents antibody-mediated rejection: costimulation blockade alters GC response. Am J Transplant (2014) 14:59–69. doi:10.1111/ajt.12526
125. Linterman MA, Rigby RJ, Wong R, Silva D, Withers D, Anderson G, et al. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes Cd28and ICOS. Immunity (2009) 30:228–41. doi:10.1016/j.immuni.2008.12.015
126. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity (2014) 41:1026–39. doi:10.1016/j.immuni.2014.12.005
127. Ville S, Poirier N, Vanhove B, Blancho G. Selective blockade of the CD28/B7/CTLA4 pathway with monovalent anti-CD28 antibodies versus targeting of B7 With CTLA4-Ig, in non-human primate kidney allograft. Am J Transplant (2015) 15(Suppl 3):S1.
Keywords: CD28, CTLA-4, costimulation blockade, memory T cell, effector T cell, transplantation immunology, heterologous immunity, CTLA4-Ig
Citation: Ville S, Poirier N, Blancho G and Vanhove B (2015) Co-stimulatory blockade of the CD28/CD80-86/CTLA-4 balance in transplantation: impact on memory T cells? Front. Immunol. 6:411. doi: 10.3389/fimmu.2015.00411
Received: 01 June 2015; Accepted: 27 July 2015;
Published: 10 August 2015
Edited by:
Zhenhua Dai, Guangdong Provincial Academy of Chinese Medical Sciences, ChinaReviewed by:
Philippe Saas, Etablissement Français du Sang BFC, FranceZheng Jenny Zhang, Northwestern University, USA
Copyright: © 2015 Ville, Poirier, Blancho and Vanhove. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernard Vanhove, INSERM UMR1064, 30 Bd Jean Monnet, Nantes Cedex 01 44093, France,YmVybmFyZC52YW5ob3ZlQHVuaXYtbmFudGVzLmZy