 Elizabeth Ann L. Enninga
Elizabeth Ann L. Enninga Wendy K. Nevala
Wendy K. Nevala Shernan G. Holtan3
Shernan G. Holtan3- 1Mayo Graduate School, Mayo Clinic, Rochester, MN, USA
- 2Department of Hematology, Mayo Clinic, Rochester, MN, USA
- 3Department of Transplantation, University of Minnesota, Minneapolis, MN, USA
- 4Department of Oncology, Mayo Clinic, Rochester, MN, USA
The role of the immune system in cancer progression has become increasingly evident over the past decade. Chronic inflammation in the promotion of tumorigenesis is well established, and cancer-associated tolerance/immune evasion has long been appreciated. Recent developments of immunotherapies targeting cancer-associated inflammation and immune tolerance, such as cancer vaccines, cell therapies, neutralizing antibodies, and immune checkpoint inhibitors, have shown promising clinical results. However, despite significant therapeutic advances, most patients diagnosed with metastatic cancer still succumb to their malignancy. Treatments are often toxic, and the financial burden of novel therapies is significant. Thus, new methods for utilizing similar biological systems to compare complex biological processes can give us new hypotheses for combating cancer. One such approach is comparing trophoblastic growth and regulation to tumor invasion and immune escape. Novel concepts regarding immune activation in pregnancy, especially reactivation of the immune system at labor through toll like receptor engagement by fetal derived DNA, may be applicable to cancer immunotherapy. This review summarizes mechanisms of inflammation in cancer, current immunotherapies used in the clinic, and suggestions for looking beyond oncology for novel methods to reverse cancer-associated tolerance and immunologic exhaustion utilizing mechanisms encountered in normal human pregnancy.
Introduction
Cancer is the complex orchestration of tumor and supporting stroma, immune mediators, and angiogenic factors that result in growth and metastasis of tumor cells, which lead to organ dysfunction and death if not successfully treated. What begins as a single cellular mutation in an oncogene and/or a tumor suppressor undergoing repeated insult culminates into a large, heterogeneous population of tumor cells that can migrate to distant sites, invade healthy tissues, and evade the host immune defenses. This last concept – the evasion of the host immune system – has become an increasingly recognized topic and therapeutic target in cancer biology. If the cellular machinery inside the cell cannot fix the mutations or cause apoptosis, the immune system must work to eradicate the developing cancer. However, tumor cells are “self,” making it more challenging for natural killer (NK) cells, cytotoxic T lymphocytes (CTLs), and other cellular effectors involved in immunosurveillance to target tumor cells compared to cells infected with a virus. Matzinger’s danger model states that the immune system is more concerned with damage (acute inflammation) signals than foreign antigens when eliciting a response (1). Since immune cells become tolerized by the tumor, they might not respond to danger signals the way a healthy immune system would. This interesting immunologic paradox also exists in the setting of pregnancy, where the mother is continually exposed to haploidentical “foreign” cells from the fetus but does not mount an immunologic attack that would be deleterious to the pregnancy. Tumor cells may mimic trophoblastic cells of the placenta in that they downregulate danger signals while increasing expression of immunosuppressive mediators (2, 3).
Determining new methods to elicit strong, specific, and durable anti-tumor immune responses is the focus of many laboratories. However, a number of barriers to successful immunotherapy exist. The first barrier is the impaired baseline immunologic function of cancer patients, even before they receive any therapy. For example, patients with advanced melanoma exist in a state of systemic chronic inflammation, which is driven, in part, by high levels of vascular endothelial growth factor (VEGF)-A secreted by the tumor to suppress the cancer targeting activity of immune cells (4, 5). The second barrier to successful immunotherapy is to determine which patients benefit from treatment. Analyzing blood from 21 patients on multiple vaccine trials who responded versus those that did not, researchers observed that melanoma patients who had a complete response were producing Th1-associated cytokines such as tumor necrosis factor (TNF) and interferon gamma (IFN-γ), unlike patients who had no response to the vaccine (6). It is well established that patients who have tumor-infiltrating lymphocytes (TILs) at the site of their malignancy tend to have better prognosis (7–10), and those who lack an organized immune response to melanoma have an extremely poor prognosis (11). The third barrier to successful immunotherapy is understanding when and how to couple immune-based therapies with standard cytotoxic chemotherapies or molecularly targeted treatments. Clinical and experimental data from our group and others have thus provided ample evidence that enriching our understanding of the host immune system’s interaction with malignancy is paramount to improving outcomes, regardless of mutational status and availability of targeted agents (12). The positive synergy accomplished by combining targeted therapy (i.e., BRAF or MEK mutations) with immunotherapy could also provide promising results and is currently being tested in a phase I clinical trial (NCT01767454) (13, 14). In this review, we will discuss the significance of immunity in many different cancers, including melanoma, and current methods to modulate it. Then, we transition to immunologic mechanisms in pregnancy exploited by tumors, and conclude with emerging data regarding the potential benefit of cell-free (fetal) nucleic acids in the reconstitution and prolongation of anti-tumor immunity.
Current Knowledge and Methods in Cancer Biology
Biology of Chronic Inflammation in Cancer
Clinical and experimental data have revealed that patient outcomes in advanced cancers are strongly influenced by the type of immune response that is established prior to initial treatment. There are two important types of responses in cancer: acute (anti-tumor) and chronic (protumor) inflammation. Dr. William Coley was a pioneer in immunotherapy who utilized heat killed bacteria named “Coley’s toxins” to induce an acute inflammatory response in sarcoma patients. Coley’s toxins resulted in 5–10+ years of survival for ~50% of patients (15). This was the first clinical evidence that an acute inflammatory response will destroy tumor cells. After exposure to foreign antigen, innate cells such as macrophages and dendritic cells (DCs) travel to the site of infection and begin processing and presenting antigens to adaptive immune cells. Adaptive immune cells, specifically T and B lymphocytes, with specificity against the antigen then undergo expansion until that antigen is eliminated. In the setting of cancer, an acute inflammatory response causes destruction of the tumor through the activation of a Th1 T-helper cell response driven by IFN-γ and tumor killing by CTLs and NK cells. The expansion of M1, or classically activated macrophages, continues to activate other lymphocytes against the tumor through secretion of IFN-γ and presentation of tumor antigens (16, 17). Thus, immunosurveillance in an immune competent host can eliminate a majority of transformed cells before they induce malignancy (18).
If there is a prolonged (or suboptimal) exposure to the foreign antigen, a chronic inflammatory response is generated, which is supportive of tumor development and growth (19). In chronic inflammation, Th2 T-helper cells promote anergy, a loss of T-cell mediated cytotoxicity and B-cell activation through the secretion of interleukin (IL)-4, IL-5, IL-6, IL-10, and IL-13 (20). Regulatory T-cells (Tregs) expand and migrate to the site of the tumor and suppress DC, CTL, and NK cell anti-tumor effects (21). High numbers of FOXP3+ Tregs in the tumor were found to correlate with disease stage and poor overall survival (22). Tumor-associated macrophages (TAMs), or M2 alternatively activated macrophages, promote tumor progression through their ability to regulate VEGF and angiogenesis (23, 24). M2 polarization is triggered by chronically activated B-cell secretion of granulocyte-macrophage colony stimulating factor (GM-CSF), IL-6, and IL-10 (25). Myeloid derived suppressor cells (MDSCs) bind directly to CTLs and inhibit their anti-tumor effects through nitric oxide (NO) secretion (26). MDSCs also contribute to tumor blood vessel formation and metastasis through the production of matrix metalloproteinase (MMP)-9, which releases high levels of VEGF into the bloodstream (27, 28). These series of events work in concert to create a positive feedback loop that supports, rather than inhibits, tumor growth.
The hypothesis that tumors arise from sites of chronic inflammation was initiated by Virchow in 1863 and increasing amounts of evidence have supported his claim (29–32). Disease progression, metastatic spread to lymph nodes, tumor size, and patient survival correlate with high levels of CD4+ T-helper cells and low levels of CD8+ CTLs at the primary tumor in many types of cancers (33–37). Interestingly, transcription factor NF-κB was determined to be crucial for inflammation, and more recently, tumorigenesis as numerous cancer types show constitutive activation of NF-κB (38–41). Moreover, Helicobacter pylori infection is one of the main risk factors for gastric cancer and is believed to promote tumorigenesis through NF-κB activated transcription of IL-1, IL-6, IL-8, TNF-α, and cyclooxygenase-2 (COX2), which are all mediators of chronic inflammation (42, 43). Finally, chronic viral infections such as human papillomavirus (HPV) and hepatitis (both B and C) have been directly linked to cervical cancer, head and neck cancer, and liver cancer, respectively (44, 45). A case-control study conducted in the United States found that long-term use of non-steroidal anti-inflammatory drugs, as means to dampen chronic inflammation, decreased a person’s risk of developing melanoma by almost 50% (46). Altogether, mediators of chronic inflammation support the tumor’s ability to proliferate, invade, and migrate within the host promoting tumor cell survival.
Therapeutics Designed to Enhance Immunity Against Cancer
Many strategies exist to treat patients with various types of cancer. Targeting and destroying tumors using the host’s immune system is the basic principle of modern cancer immunotherapy. However, many patients do not respond to immunotherapy, the drugs are costly, and patients may suffer immunologic adverse events (AEs) that can be severe or life threatening. Table 1 summarizes results from clinical trials and the toxicities associated with therapy. Checkpoint inhibitors have revolutionized immunotherapy and are considered one of the most effective therapies for utilizing the immune system against tumors. Examples include anti-cytotoxic T lymphocyte antigen 4 (CTLA-4), anti-program death 1 (PD-1), and anti-program death ligand 1 (PD-L1) reviewed by Topalian et al. (47). The use of antibodies to block proteins known to promote tumor growth is of significant current interest in cancer therapy. Many of these drugs have shown to induce a response as a single agent or in combination with chemotherapy. Anti-VEGF)-A, anti-human epidermal growth factor receptor 2 (HER2/neu), and anti-CD20 are a few monoclonal antibodies used in oncology, but there are many more being studied (48). Immune-stimulating vaccines have also been developed for cancer patients. Therapeutic vaccines require a tumor specific antigen and an activation signal (immune adjuvant), such as a toll like receptor (TLR) agonist, in order to stimulate an immune response against an already established tumor. Common tumor antigens include melan-A, NY-ESO-1, B7C, and MAGE-1 (49–52). However, the challenge with many of these peptides is that they can be easily cleared without activating DCs. In addition, tumor antigen heterogeneity and changing expression of these antigens makes targeting ineffective. The most successful cancer vaccines include Provenge and Gardasil. Yet, the challenges with developing therapeutic vaccines include the many differences that are documented between trials, including vaccine strategy, antigen dose, tumor and patient heterogeneity, severity of disease, and vaccine adjuvants, which can all confound the results. These variables must be considered when developing therapeutic vaccines and testing their efficacy in clinical trials. Adoptive cell transfer (ACT) is another modality of cancer immunotherapy where cells, which can be unmanipulated, antigen-specific, or stimulated, are utilized to kill cancer cells in lymphodepleted patients. ACT has been successful at breaking tolerance in many cancers. Chimeric antigen receptor (CAR) therapy utilizes both targeting antibodies and cytotoxic CD8 T-cells for destroying cancer cells in a similar manner as ACT. For CAR therapy, T-cells are collected from cancer patients, expanded in vitro and their receptors are modified to more specifically target the tumor when given back to the patient (53). Despite some of these incredible response rates, ACT is expensive, requires that the patient have adequate lymphocytes for collection, needs specialized manufacturing facilities, regulatory hurdles, and is time prohibitive (54).
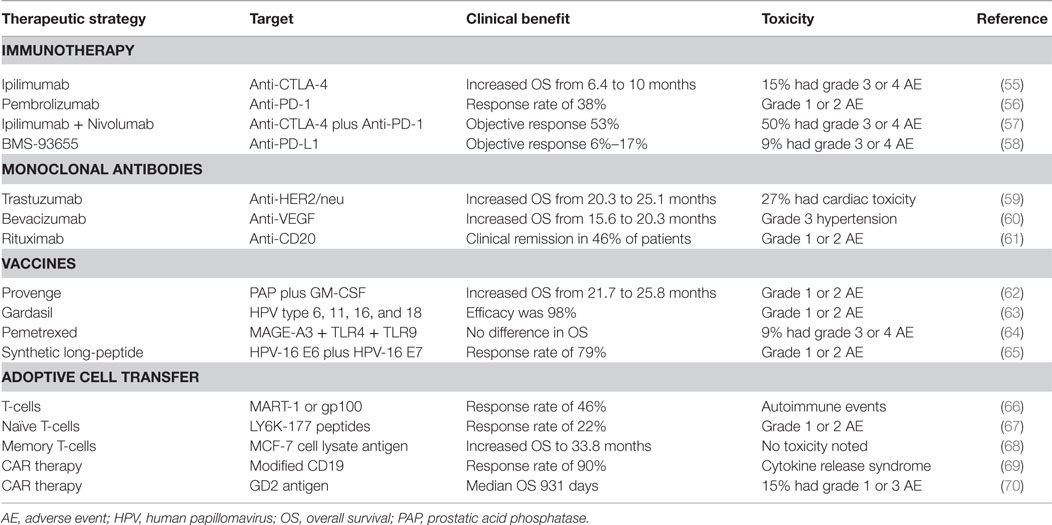
Table 1. Therapeutic efficacy and related toxicities of drugs developed for cancer treatment.
Novel Insights of Immune Tolerance from Pregnancy Biology
Immunology of Pregnancy: Breaking Tolerance Prior to Labor
Exploring similar systems, such as immune tolerance of a haploidentical fetus, takes another approach to understanding complex immune regulation in tumorigenesis. The maternal immune system must effectively balance tolerance to paternal antigens while continuing to protect the mother from infection; failure to do so can result in negative pregnancy outcomes (71). Thus, a T-helper 2, or Th2, immune response has been defined as the dominant phenotype during pregnancy, and this phenotype is also evident in metastatic cancer (4, 72). The most profound similarity is that both must evade immune recognition and destruction by the host/maternal system while expressing foreign antigens (tumor versus paternal). Yet, how a fetal allograft is not rejected although the maternal immune system continues to be capable of responding to other foreign antigens remains of significant scientific interest today. Many have made the observation that tumors mimic the tolerant immune state required by a trophoblast for successful implantation (early pregnancy) (3, 73, 74). However, a modification occurs in late pregnancy leading to an acute inflammatory response driven by proinflammatory cytokines, hormones, and chemokines, which traffic effector leukocytes to the myometrium and initiate labor (75). The exact mechanism as to how labor is initiated in human beings remains unclear. Placental tissue derived from women with recurrent pregnancy loss showed signs of inflammation, such as elevated NK cells, thromboembolism, insufficient trophoblast invasion, and lesions compared to placental tissue from healthy pregnancies (76). In a murine model, complement activation decreased VEGF-A, which leads to miscarriage or growth restriction, and blocking complement activation reversed this affect (77). This demonstrates the necessity of careful immune regulation at the fetal–maternal interface in order to establish a viable pregnancy. Others have studied pregnant women and found that tumor-associated antigens (TAAs) like MUC1, HER2/neu, WT-1, and PRAME, which are highly expressed in placental tissue, elicit the strongest immune response during the first and second trimester, declining after delivery and the completion of nursing; however, history of delivery was not correlated with increasing immune responses (78). This suggests that although the fetus is being tolerated by the mother’s immune response, maternal immunity is still fully capable of reactivating when given a strong enough stimulus. Through better understanding of the mechanisms that drive immune reactivation (labor), we can gain valuable insight into possible tools to use for promoting tumor destruction in cancer patients.
Fetal Derived Nucleic Acids for Immune Activation
Cell-free fetal DNA (cff-DNA) and RNA strands are shed into the maternal system during pregnancy. The concentration of these nucleic acids increases in the circulation with the length of gestation (79, 80). During early gestation, the concentration of cff-DNA in plasma ranges between 0.022 and 0.46 ng/mL, which increases to 5.08ng/mL by late pregnancy (81). Clinically, these sequences have been used to determine chromosomal abnormalities or genetic mutations that a child might have inherited (82). A new hypothesis recently arose suggesting that the increase in cff-DNA at term could activate TLRs on maternal cells, which leads to the breakage of immune tolerance, the activation of innate immune cells, and finally the onset of labor (83). It has been well documented that foreign DNA is recognized by TLR9, and RNA is recognized by TLR3 on immune cells, which activate inflammatory processes (84, 85). TLR9 is found in the endosome of peripheral blood mononuclear cells, such as monocytes, macrophages, T, B, and NK cells, whereas TLR3 is expressed only in the endosomes of myeloid derived cells such as DCs and monocytes (86). Fetal RNA is derived from the placenta and was shown to be surprisingly stabile in plasma, although lower levels are detected compared to cff-DNA (87). Apoptosis of placenta and trophoblastic cells are believed to be the major source of cff-DNA in the maternal bloodstream (88, 89).
Studying pregnancy disorders may also provide important mechanisms to reactivating inflammation for cancer patients. Three weeks before onset of symptoms, cff-DNA was increased between two- and five-fold in plasma of women suffering from preeclampsia compared to women with healthy pregnancies (90). Timing of the measurement and maternal health (BMI) are known confounders of cff-DNA measurement, thus another group found no difference between cff-DNA levels in preeclamptic women (91–93). From these discrepancies, a new idea was proposed: cff-DNA may be proinflammatory and high levels may send a danger signal to the maternal immune system (83). Using a mouse model of pre-term birth and preeclampsia, high levels of cff-DNA stimulated TLR9 to initiate acute inflammation, which caused fetal reabsorption at days 10–14, and knocking out TLR9 diminished this result (94). In general, TLRs and proinflammatory cytokines are overexpressed in women suffering from preeclampsia versus those with healthy pregnancies (95). In women suffering from hyperemesis gravidarum, cff-DNA levels were found to be 2.5-fold higher than healthy controls, and this is believed to be due to the increased activation of the maternal immune system, specifically the increased levels of cytotoxic T-cells and NK cells in the decidua and blood of the mother which target the haploidentical fetus (96, 97). Figure 1 shows a schematic of this possible process RNA and/or DNA could use to activate the labor process in pregnancy and how this mechanism could be manipulated for tumor rejection in cancer patients. Fetal DNA or RNA, at a critical concentration in maternal plasma, could be recognized by TLRs and processed by myeloid cells such as DCs or NK cells. These cells could then present this fetal antigen to lymphocytes resulting in an acute inflammatory response that leads to labor. This mechanism might also be applicable to reactivating anti-tumor immunity in cancer patients. Taken together, understanding of cff-DNA as possible proinflammatory mediator and predictive marker for disease onset during pregnancy could become a powerful resource for obstetrics research with translational value to oncology.
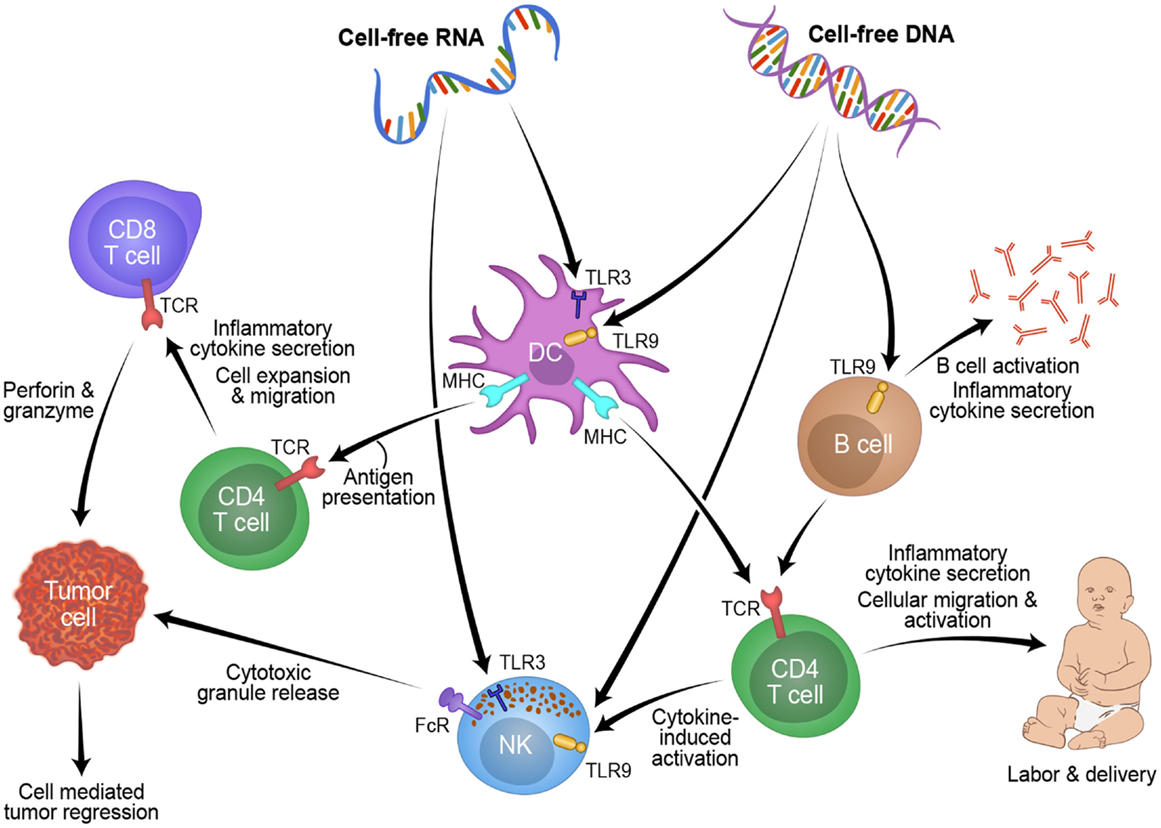
Figure 1. Overview of how cell-free fetal derived RNA or DNA from pregnancy can activate an inflammatory immune response through toll like receptors, which could be applied to novel cancer treatments.
Sequence length, epigenetic modification, and/or confirmation of DNA may also be a critical factor when it comes to the ability of soluble DNA to activate immune cells. CpG motifs derived from Escherichia coli were the first sequences characterized, which bind and activate TLR9 in B-cells and plasmacytoid dendritic cells (pDCs) (84, 98). These unmethylated, often palindromic CpG motifs are more frequently found in bacterial DNA compared to DNA from human beings and short oligodeoxynuleotide (ODN) have been synthesized showing the same ability to activate TLR9 as bacterial DNA, suggesting sequence, not length, is imperative to achieve TLR9 activation (84). Interestingly, cff-DNA is comprised of mostly short sequences of 0.3 kb compared to maternal DNA (99). It is possible that this shorter length of cff-DNA may contain similar palindromic CpG sequences to the ODN sequences, which can effectively activate TLR9 signaling. Methylation typically occurs in CpG-rich regions (or promotor sites) in the mammalian genome and results in the suppression of transcription (100). Cff-DNA also has distinct methylation patterns based on differentiation during embryogenesis that are unlike the maternal genome (101). Poon et al. utilized the IGF2-H19 locus to distinguish chimeric fetal DNA from maternal since this region is only methylated on the paternal allele (102). Another example is the maspin gene promotor, which was found to be hypomethylated in placental tissue compared to hypermethylated in maternal blood cells (103). Since cff-DNA is likely derived from the placenta, utilizing methylation status in plasma could be a more effective way to distinguish fetal derived DNA irrespective of the sex of the fetus or polymorphisms. This finding is similar to unmethylated ODN sequences used to activate TLR9 signaling. When the ODN sequence was methylated, researchers found no immune stimulatory effects compared to the unmethylated sequence, demonstrating the importance of this epigenetic marker (84). Further research into whether these epigenetic markers make a difference in affinity for TLR binding and activation of innate immunity needs to be completed. Additionally, the folding pattern of cff-DNA may also be important for effective receptor triggering and signaling. DNA can self-assemble into many different structures based on nucleic acid abundance and/or intrinsic atom properties (104). Thus, specific folding patterns of nucleic acids could be better at activating innate immune receptors compared to other patterns. Taken together, understanding the multiple factors that could be pivotal to circulating DNA may help us better understand haploidentical cell-free DNA or RNA and how it could be utilized in cancer patients to activate anti-tumor effects.
Circulating Nucleic Acids and Toll like Receptor Signaling in Cancer
Interestingly, there are cell-free nucleic acids found in healthy individuals and these levels were found to be elevated in blood of patients with many different cancers (105–109). It is unclear how this DNA gets into the bloodstream, but it is believed to be the by-product of macrophage engulfment of necrotic and/or apoptotic cells (110). Since tumors have areas of high necrosis, this hypothesis would explain the increase in circulating tumor (ct)-DNA fragments found in cancer patients. In addition, particle associated RNA was also found at increased levels in cancer patients compared to healthy individuals, although much less studied (111). The size of these DNA fragments is also important to note, varying from small fragments (70–200 bp) to large fragments around 21 kb (112). Serum from cancer patients has an average of 180 ng/mL of cell-free DNA compared to healthy subjects having an average of only 30 ng/mL (107, 113). Determining which DNA sequences have tumor origin versus background circulating DNA fragments is difficult and the use of these fragments for diagnostic or prognostic value remains controversial. In breast cancer, high levels of ct-DNA correlated with tumor size, grade, staging, lymph node status, and metastasis (105, 109). Survival was also reported to correlate with ct-DNA levels: breast cancer patients with high levels of ct-DNA in their blood had a lower OS than those with low levels of ct-DNA (114). Yet, others find no correlation between level of ct-DNA and survival in lung cancer or colorectal cancer (115, 116). Some of these differences could be due to the challenges and methods of isolating these short fragments including blood collection and processing methods, time elapsed between draw and isolation, and the isolation technique (117). Epigenetics of ct-DNA was also found to be important in the development of carcinogenesis, and new methods are being developed for measuring differentially methylated tumor DNA. Methylation studies have demonstrated transcriptional repression at CpG islands of tumor suppressor genes that lead to cancer progression (118). Thus, many new technologies are being developed to find hypermethylated promotors, especially proto-oncogenes regions, which are not present in healthy persons. CDKN2A, PARP-1, and GSTP1 are just a few genes that were found to be hypermethylated in ct-DNA and tumor tissue, and are being studied for biomarker use (119–121). In many instances, hypermethylation has been found to correlate with worse survival; therefore, many groups are working on development of epigenetic therapy, which has been reviewed by Jones and Baylin (122). Although the results of ct-DNA studies remain inconclusive, the promise of using a minimally invasive method to diagnosis and treat cancer patients makes it worth continually pursuing.
As suggested above, TLR signaling could be significant for the reactivation of immunity in pregnancy and possibly cancer. However, biology tells us that a patient’s cell-free DNA does not activate TLR signaling on their immune cells; if it did, autoimmunity would occur. Table 2 compares and contrasts cff-DNA to ct-DNA. Major known differences include methylation status and size; however, sequence and structure have yet to be considered as possible differences between the two. Overall, a better understanding of TLR adjuvants is critical to improve patient care. Clinically, there are several TLR agonists which have been tested for the treatment of cancer. TLR9 is expressed on chronic lymphocytic leukemia cells and will undergo apoptosis when given CpG ODN 2006 (TLR9 agonist) in vitro (123). TLR3 agonist bacillus Calmette–Guerin (BCG) given to mice prior to tumor injection, were less likely to develop tumors than their untreated littermates and this was due to the increase in TNF (124). Clinically, TLR agonists have not been as impressive as their pre-clinical results. In phase II studies of CpG ODN 2006, anti-tumor effects were modest in T-cell lymphoma and melanoma as a single agent (125, 126). BCG was approved for early stage bladder cancer after randomized studies showed that 88% of patients had a complete response and a reduction in tumor recurrence (127, 128). The challenge with development of TLR agonists includes understanding which TLRs are involved in protumor versus anti-tumor effects and how to target these agonists to the site of the tumor more effectively. Although the effects have been moderate, there is hope that combination with other drugs along with specifically targeting the anti-tumor T-cells will improve clinical efficacy (129). Immune responses are tightly regulated; just as TLR stimulation will activate responses it will also suppress them as to prevent autoimmunity. TLR7/8 and TLR4 agonists promote expression of negative co-stimulatory PD-L1 on DCs, which inhibits anti-tumor effects (130). However, a combined PD-1/PD-L1 blockade with a TLR3 agonist resulted in an increase of CD8 T-cell effectors and anti-tumor responses in a murine melanoma model (131). Maternal microenvironment may also play a key role in regulating responses to nucleic acids. During pregnancy, IL-27, a Th1 promoting cytokine, increases with length of gestation and decreases just after delivery in a very similar fashion to fDNA in maternal plasma (132). Thus, along with a critical concentration of DNA, other factors, such as known proinflammatory cytokines, are likely required to further drive activation of lymphoid and myeloid cells against a haploidentical fetus and an altered-self tumor.

Table 2. Differences and similarities between fetal-derived and tumor-derived circulating DNA.
Basic in vitro Observations of cff-DNA and TLR Activation
To begin understanding if DNA might reactivate immunity at parturition, we isolated fDNA from the plasma of four pregnant women between 36 and 40 weeks of gestation using the Akonni Circulating DNA TruTip method (133). CD14+ monocytes isolated from healthy donors (males 36–44 years) were treated with 0.5 μg/mL of cff-DNA for 6 h before they were lysed and RNA was isolated per Qiagen’s RNeasy protocol. We added a much higher concentration of cff-DNA to the monocytes compared to what is biologically seen, to address the fact that our DNA population is not pure (fetal and maternal DNA likely included). Samples were then processed to cDNA and run on a human TLR qPCR array (SABioscience, Valencia, CA). When three different monocyte populations were treated with the same cff-DNA sample, we found TLR3, TIRAP, IL-2, and Table 1 to have a fold change greater than two in all three populations compared to untreated controls (Figure 2A). TLR9, the known receptor for DNA binding, was downregulated in two out of the three samples suggesting endosomal degradation possibly due to incubating the cells with DNA too long. TLR3 is known to bind double stranded RNA, but was consistently impacted by our cff-DNA fraction, suggesting that either our fraction contains RNA or cff-DNA is similar enough to viral RNA that it can bind and activate TLR3 dependent inflammation. We repeated the same experiment using the same donor cells (44–year-old male) and tested three different cff-DNA samples isolated from different women near term. Although there were some differences based on the cff-DNA sample, we found similar genes to be changed twofold with treatment of cff-DNA (Figure 2B). This data can only illustrate the TLR effect of cff-DNA; further research is necessary to determine the role of fetal nucleic acids in pregnancy and the possibility that these short fragments could be used as a potential therapy for reactivating a proinflammatory response in immune tolerant cancer patients. Yet, the differential expression of genes involved in TLR signaling between cells treated with and without cff-DNA suggests that a mechanism may be worth pursuing.
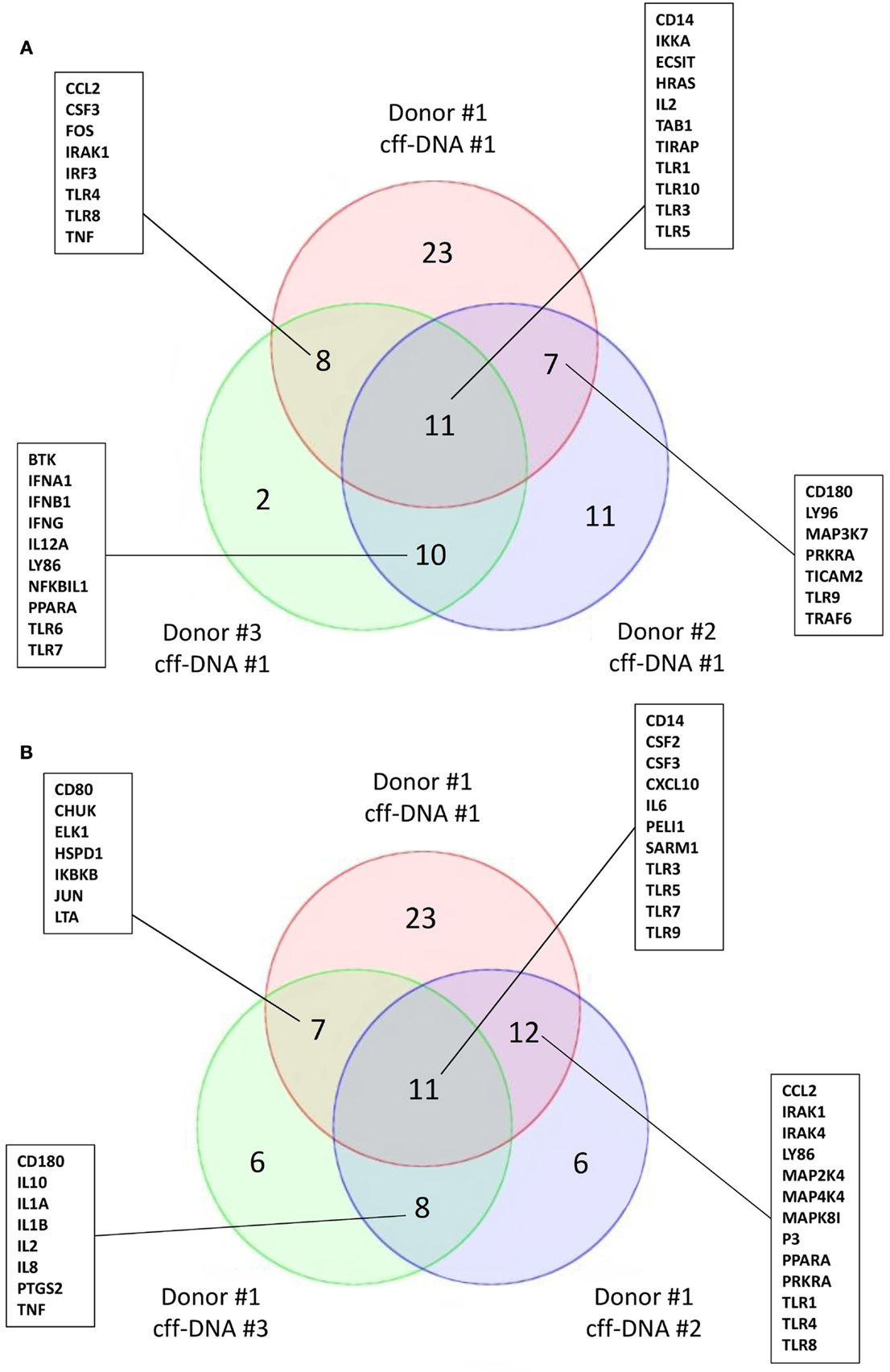
Figure 2. Activation of TLR signaling cascade on CD14+ monocytes with addition of cff-DNA. Venn diagrams showing similar genes involved in the TLR signaling pathway having a fold change cut off of 2. (A) Three different healthy CD14+ monocyte populations treated with the same cff-DNA. (B) One healthy CD14+ monocyte population treated with three different cff-DNAs.
Conclusion
The role of the immune system in cancer progression and response to therapy has become increasingly appreciated in the past decade. Research into the development of chronic inflammation and the promotion of tumor growth has shed light on the need for immunologic intervention in order to cure cancer. Advancements in immunotherapies, including vaccines, cell therapy, and checkpoint inhibitors, have had exciting success; however, a majority of patients see little effect but still run the risk debilitating side effects. We have seen promise in using concepts from pregnancy for reactivating the proinflammatory immune response necessary for anti-tumor effects in melanoma patients. Specifically, a better understanding of the role of cff-DNA in the breakage of tolerance during late pregnancy may offer insight into predicting time of delivery in obstetrics and provide new ideas along with methods that may be applicable to cancer immunology. Further research into cell-free nucleic acids and TLR signaling could be the key to more effectively disrupting chronic inflammation in cancer patients and improving outcomes.
Author Contributions
EE, WN, SH, and SM contributed to the ideas, concepts, and interpretations of this work and critically drafted/revised it for scientific integrity and accountability. Final draft was approved by all the authors. EE prepared the manuscript.
Conflict of Interest Statement
The authors declare that this research was conducted without any commercial/financial relationships which could cause a conflict of interest.
Acknowledgments
This publication was supported by CTSA grant number TL1 TR000137 (EE) from the National Center for Advancing Translational Science (NCATS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
1. Matzinger P. The danger model: a renewed sense of self. Science (2002) 296:301–5. doi:10.1126/science.1071059
2. Enninga EA, Holtan SG, Creedon DJ, Dronca RS, Nevala WK, Ognjanovic S, et al. Immunomodulatory effects of sex hormones: requirements for pregnancy and relevance in melanoma. Mayo Clin Proc (2014) 89:520–35. doi:10.1016/j.mayocp.2014.01.006
3. Holtan SG, Creedon DJ, Haluska P, Markovic SN. Cancer and pregnancy: parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin Proc (2009) 84:985–1000. doi:10.1016/S0025-6196(11)60669-1
4. Nevala WK, Vachon CM, Leontovich AA, Scott CG, Thompson MA, Markovic SN. Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin Cancer Res (2009) 15:1931–9. doi:10.1158/1078-0432.CCR-08-1980
5. Stewart TJ, Smyth MJ. Improving cancer immunotherapy by targeting tumor-induced immune suppression. Cancer Mets Rev (2011) 30:125–40. doi:10.1007/s10555-011-9280-5
6. Marshall JA, Forster TH, Purdie DM, Lanagan CM, O’Connor LE, O’Rourke MG, et al. Immunological characteristics correlating with clinical response to immunotherapy in patients with advanced metastatic melanoma. Immunol Cell Biol (2006) 84:295–302. doi:10.1111/j.1440-1711.2006.01445.x
7. Kawata A, Une Y, Hosokawa M, Uchino J, Kobayashi H. Tumor-infiltrating lymphocytes and prognosis of hepatocellular carcinoma. Jpn J Clin Oncol (1992) 22:256–63.
8. Lyle S, Salhany KE, Elder DE. TIA-1 positive tumor-infiltrating lymphocytes in nevi and melanomas. Mod Pathol (2000) 13:52–5. doi:10.1038/modpathol.3880009
9. Prall F, Duhrkop T, Weirich V, Ostwald C, Lenz P, Nizze H, et al. Prognostic role of CD8+ tumor-infiltrating lymphocytes in stage III colorectal cancer with and without microsatellite instability. Hum Pathol (2004) 35:808–16. doi:10.1016/j.humpath.2004.01.022
10. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. New Engl J Med (2003) 348:203–13. doi:10.1056/NEJMoa020177
11. Holtan SG, Mansfield AS, Creedon DJ, Nevala WK, Haluska P, Leontovich AA, et al. An organ system based approach to prognosis in advanced melanoma. Front Biosci (2012) 4:2823–33. doi:10.2741/E586
12. Lowe DB, Storkus WJ. Chronic inflammation and immunologic-based constraints in malignant disease. Immunotherapy (2011) 3:1265–74. doi:10.2217/imt.11.113
13. Kim T, Amaria RN, Spencer C, Reuben A, Cooper ZA, Wargo JA. Combining targeted therapy and immune checkpoint inhibitors in the treatment of metastatic melanoma. Cancer Biol Med (2014) 11:237–46. doi:10.7497/j.issn.2095-3941.2014.04.002
14. Puzanov I, CM, Linette GP, Patel SP, Luke JJ, Sosman JA, et al. Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation-positive unresectable or metastatic melanoma (MM). J Clin Oncol (2014) 32:(5s Suppl) abstr 2511.
16. Romagnani S. The Th1/Th2 paradigm. Immunol Today (1997) 18:263–6. doi:10.1016/S0167-5699(97)80019-9
17. Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol (2001) 13:459–63. doi:10.1093/intimm/13.4.459
18. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410:1107–11. doi:10.1038/35074122
19. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res (2014) 2014:149185. doi:10.1155/2014/149185
20. Parker DC. T cell-dependent B cell activation. Annu Rev Immunol (1993) 11:331–60. doi:10.1146/annurev.iy.11.040193.001555
21. Yokokawa J, Cereda V, Remondo C, Gulley JL, Arlen PM, Schlom J, et al. Enhanced functionality of CD4+CD25(high)FoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clin Cancer Res (2008) 14:1032–40. doi:10.1158/1078-0432.CCR-07-2056
22. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol (2006) 24:5373–80. doi:10.1200/JCO.2006.05.9584
23. Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res (2002) 62:7042–9.
24. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66:11238–46. doi:10.1158/0008-5472.CAN-06-1278
25. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi:10.1016/S1471-4906(02)02302-5
26. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol (2001) 166:5398–406. doi:10.4049/jimmunol.166.9.5398
27. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol (2000) 2:737–44. doi:10.1038/35036374
28. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004) 6:409–21. doi:10.1016/j.ccr.2004.08.031
29. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell (2005) 7:211–7. doi:10.1016/j.ccr.2005.02.013
30. David H. Rudolf Virchow and modern aspects of tumor pathology. Pathol Res Pract (1988) 183:356–64. doi:10.1016/S0344-0338(88)80138-9
31. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
32. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
33. Chin Y, Janseens J, Vandepitte J, Vandenbrande J, Opdebeek L, Raus J. Phenotypic analysis of tumor-infiltrating lymphocytes from human breast cancer. Anticancer Res (1992) 12:1463–6.
34. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi:10.1126/science.1129139
35. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer (2013) 108:914–23. doi:10.1038/bjc.2013.32
36. Knol AC, Nguyen JM, Pandolfino MC, Quereux G, Brocard A, Peuvrel L, et al. Tissue biomarkers in melanoma patients treated with TIL. PLoS One (2012) 7:e48729. doi:10.1371/journal.pone.0048729
37. Kohrt HE, Nouri N, Nowels K, Johnson D, Holmes S, Lee PP. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med (2005) 2:e284. doi:10.1371/journal.pmed.0020284
38. Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS Jr. Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene (2000) 19:1123–31. doi:10.1038/sj.onc.1203412
39. Houldsworth J, Mathew S, Rao PH, Dyomina K, Louie DC, Parsa N, et al. REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood (1996) 87:25–9.
40. Lu D, Thompson JD, Gorski GK, Rice NR, Mayer MG, Yunis JJ. Alterations at the rel locus in human lymphoma. Oncogene (1991) 6:1235–41.
41. Uffort DG, Grimm EA, Ellerhorst JA. NF-kappaB mediates mitogen-activated protein kinase pathway-dependent iNOS expression in human melanoma. J Invest Dermatol (2009) 129:148–54. doi:10.1038/jid.2008.205
42. Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology (1997) 113:1099–109. doi:10.1053/gast.1997.v113.pm9322504
43. Kim H, Lim JW, Kim KH. Helicobacter pylori-induced expression of interleukin-8 and cyclooxygenase-2 in AGS gastric epithelial cells: mediation by nuclear factor-kappaB. Scand J Gastroenterol (2001) 36:706–16. doi:10.1080/003655201300191969
44. Bosch FX, Ribes J, Borras J. Epidemiology of primary liver cancer. Semin Liver Dis (1999) 19:271–85. doi:10.1055/s-2007-1007117
45. Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. New Engl J Med (2003) 348:518–27. doi:10.1056/NEJMoa021641
46. Curiel-Lewandrowski C, Nijsten T, Gomez ML, Hollestein LM, Atkins MB, Stern RS. Long-term use of nonsteroidal anti-inflammatory drugs decreases the risk of cutaneous melanoma: results of a United States case-control study. J Invest Dermatol (2011) 131:1460–8. doi:10.1038/jid.2011.58
47. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi:10.1016/j.ccell.2015.03.001
48. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer (2012) 12:278–87. doi:10.1038/nrc3236
49. Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A (1997) 94:1914–8. doi:10.1073/pnas.94.5.1914
50. Coulie PG, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med (1994) 180:35–42. doi:10.1084/jem.180.1.35
51. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci U S A (1994) 91:6458–62. doi:10.1073/pnas.91.14.6458
52. Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res (2008) 68:889–92. doi:10.1158/0008-5472.CAN-07-3095
53. Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med (2014) 65:333–47. doi:10.1146/annurev-med-060512-150254
54. Trainor N, Pietak A, Smith T. Rethinking clinical delivery of adult stem cell therapies. Nat Biotechnol (2014) 32:729–35. doi:10.1038/nbt.2970
55. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
56. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. New Engl J Med (2013) 369:134–44. doi:10.1056/NEJMoa1305133
57. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. New Engl J Med (2013) 369:122–33. doi:10.1056/NEJMoa1302369
58. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. New Engl J Med (2012) 366:2455–65. doi:10.1056/NEJMoa1200694
59. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. New Engl J Med (2001) 344:783–92. doi:10.1056/NEJM200103153441101
60. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. New Engl J Med (2004) 350:2335–42. doi:10.1056/NEJMoa032691
61. Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood (1997) 90:2188–95.
62. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Engl J Med (2010) 363:411–22. doi:10.1056/NEJMoa1001294
63. Garland SM, Hernandez-Avila M, Wheeler CM, Perez G, Harper DM, Leodolter S, et al. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. New Engl J Med (2007) 356:1928–43. doi:10.1056/NEJMoa061760
64. Paz-Ares LG, Biesma B, Heigener D, von Pawel J, Eisen T, Bennouna J, et al. Phase III, randomized, double-blind, placebo-controlled trial of gemcitabine/cisplatin alone or with sorafenib for the first-line treatment of advanced, nonsquamous non-small-cell lung cancer. J Clin Oncol (2012) 30:3084–92. doi:10.1200/JCO.2011.39.7646
65. Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. New Engl J Med (2009) 361:1838–47. doi:10.1056/NEJMoa0810097
66. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science (2002) 298:850–4. doi:10.1126/science.1076514
67. Ishikawa H, Imano M, Shiraishi O, Yasuda A, Peng YF, Shinkai M, et al. Phase I clinical trial of vaccination with LY6K-derived peptide in patients with advanced gastric cancer. Gastric Cancer (2014) 17:173–80. doi:10.1007/s10120-013-0258-6
68. Domschke C, Ge Y, Bernhardt I, Schott S, Keim S, Juenger S, et al. Long-term survival after adoptive bone marrow T cell therapy of advanced metastasized breast cancer: follow-up analysis of a clinical pilot trial. Cancer Immunol Immunother (2013) 62:1053–60. doi:10.1007/s00262-013-1414-x
69. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med (2014) 371:1507–17. doi:10.1056/NEJMoa1407222
70. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi:10.1182/blood-2011-05-354449
71. Raghupathy R. Th1-type immunity is incompatible with successful pregnancy. Immunol Today (1997) 18:478–82. doi:10.1016/S0167-5699(97)01127-4
72. Lin H, Mosmann TR, Guilbert L, Tuntipopipat S, Wegmann TG. Synthesis of T helper 2-type cytokines at the maternal-fetal interface. J Immunol (1993) 151:4562–73.
73. Enninga EA, Holtan SG, Creedon DJ, Dronca RS, Nevala WK, Ognjanovic S, et al. Immunomodulatory effects of sex hormones: requirements for pregnancy and relevance in melanoma. Mayo Clin Proc (2014) 89:520–35. doi:10.1016/j.mayocp.2014.01.006
74. Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update (2007) 13:121–41. doi:10.1093/humupd/dml048
75. Peltier MR. Immunology of term and preterm labor. Reprod Biol Endocrinol (2003) 1:122. doi:10.1186/1477-7827-1-122
76. Kwak JY, Beer AE, Kim SH, Mantouvalos HP. Immunopathology of the implantation site utilizing monoclonal antibodies to natural killer cells in women with recurrent pregnancy losses. Am J Reprod Immunol (1999) 41:91–8. doi:10.1111/j.1600-0897.1999.tb00080.x
77. Girardi G, Yarilin D, Thurman JM, Holers VM, Salmon JE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med (2006) 203:2165–75. doi:10.1084/jem.20061022
78. Lutz M, Worschech A, Alb M, Gahn S, Bernhard L, Schwab M, et al. Boost and loss of immune responses against tumor-associated antigens in the course of pregnancy as a model for allogeneic immunotherapy. Blood (2014) 125:261–72. doi:10.1182/blood-2014-09-601302
79. Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. Lancet (1997) 350:485–7. doi:10.1016/S0140-6736(97)02174-0
80. Poon LL, Leung TN, Lau TK, Lo YM. Presence of fetal RNA in maternal plasma. Clin Chem (2000) 46:1832–4.
81. Pertl B, Bianchi DW. Fetal DNA in maternal plasma: emerging clinical applications. Obstet Gynecol (2001) 98:483–90. doi:10.1016/S0029-7844(01)01195-4
82. Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W, Leung TY, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci U S A (2008) 105:20458–63. doi:10.1073/pnas.0810641105
83. Phillippe M. Cell-free fetal DNA – a trigger for parturition. New Engl J Med (2014) 370:2534–6. doi:10.1056/NEJMcibr1404324
84. Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature (1995) 374:546–9. doi:10.1038/374546a0
85. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science (2007) 317:1522–7. doi:10.1126/science.1139522
86. O’Neill LA, Golenbock D, Bowie AG. The history of toll-like receptors – redefining innate immunity. Nat Rev Immunol (2013) 13:453–60. doi:10.1038/nri3446
87. Ng EK, Tsui NB, Lau TK, Leung TN, Chiu RW, Panesar NS, et al. mRNA of placental origin is readily detectable in maternal plasma. Proc Natl Acad Sci U S A (2003) 100:4748–53. doi:10.1073/pnas.0637450100
88. Litton C, Stone J, Eddleman K, Lee MJ. Noninvasive prenatal diagnosis: past, present, and future. Mt Sinai J Med (2009) 76:521–8. doi:10.1002/msj.20153
89. Masuzaki H, Miura K, Yoshiura KI, Yoshimura S, Niikawa N, Ishimaru T. Detection of cell free placental DNA in maternal plasma: direct evidence from three cases of confined placental mosaicism. J Med Genet (2004) 41:289–92. doi:10.1136/jmg.2003.015784
90. Levine RJ, Qian C, Leshane ES, Yu KF, England LJ, Schisterman EF, et al. Two-stage elevation of cell-free fetal DNA in maternal sera before onset of preeclampsia. Am J Obstet Gynecol (2004) 190:707–13. doi:10.1016/j.ajog.2003.12.019
91. Crowley A, Martin C, Fitzpatrick P, Sheils O, O’Herlihy C, O’Leary JJ, et al. Free fetal DNA is not increased before 20 weeks in intrauterine growth restriction or pre-eclampsia. Prenat Diagn (2007) 27:174–9. doi:10.1002/pd.1645
92. Sifakis S, Zaravinos A, Maiz N, Spandidos DA, Nicolaides KH. First-trimester maternal plasma cell-free fetal DNA and preeclampsia. Am J Obstet Gynecol (2009) 201(472):e471–7. doi:10.1016/j.ajog.2009.05.025
93. Vora NL, Johnson KL, Basu S, Catalano PM, Hauguel-De Mouzon S, Bianchi DW. A multifactorial relationship exists between total circulating cell-free DNA levels and maternal BMI. Prenat Diagn (2012) 32:912–4. doi:10.1002/pd.3919
94. Scharfe-Nugent A, Corr SC, Carpenter SB, Keogh L, Doyle B, Martin C, et al. TLR9 provokes inflammation in response to fetal DNA: mechanism for fetal loss in preterm birth and preeclampsia. J Immunol (2012) 188:5706–12. doi:10.4049/jimmunol.1103454
95. Dabagh-Gorjani F, Anvari F, Zolghadri J, Kamali-Sarvestani E, Gharesi-Fard B. Differences in the expression of TLRs and inflammatory cytokines in pre-eclamptic compared with healthy pregnant women. Iran J Immunol (2014) 11:233–45. doi:lJlv11i4A2
96. Minagawa M, Narita J, Tada T, Maruyama S, Shimizu T, Bannai M, et al. Mechanisms underlying immunologic states during pregnancy: possible association of the sympathetic nervous system. Cell Immunol (1999) 196:1–13. doi:10.1006/cimm.1999.1541
97. Sugito Y, Sekizawa A, Farina A, Yukimoto Y, Saito H, Iwasaki M, et al. Relationship between severity of hyperemesis gravidarum and fetal DNA concentration in maternal plasma. Clin Chem (2003) 49:1667–9. doi:10.1373/49.10.1667
98. Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol (2001) 31:3026–37. doi:10.1002/1521-4141(2001010)31:10<3026::AID-IMMU3026>3.0.CO;2-H
99. Chan KC, Zhang J, Hui AB, Wong N, Lau TK, Leung TN, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem (2004) 50:88–92. doi:10.1373/clinchem.2003.024893
100. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet (2003) 33(Suppl):245–54. doi:10.1038/ng1089
101. Oligny LL. Human molecular embryogenesis: an overview. Pediatr Dev Pathol (2001) 4:324–43. doi:10.1007/s10024001-0033-2
102. Poon LL, Leung TN, Lau TK, Chow KC, Lo YM. Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin Chem (2002) 48:35–41.
103. Chim SS, Tong YK, Chiu RW, Lau TK, Leung TN, Chan LY, et al. Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc Natl Acad Sci U S A (2005) 102:14753–8. doi:10.1073/pnas.0503335102
104. Rothemund PW. Folding DNA to create nanoscale shapes and patterns. Nature (2006) 440:297–302. doi:10.1038/nature04586
105. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res (1977) 37:646–50.
106. Mandel P, MP. Les acides nucleiques du plasma sanguine chez l’homme. C R Seances Soc Biol Fil (1948) 142:241–3.
107. Shapiro B, Chakrabarty M, Cohn EM, Leon SA. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer (1983) 51:2116–20. doi:10.1002/1097-0142(19830601)51:11<2116::AID-CNCR2820511127>3.0.CO;2-S
108. Sozzi G, Conte D, Leon M, Ciricione R, Roz L, Ratcliffe C, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol (2003) 21:3902–8. doi:10.1200/JCO.2003.02.006
109. Zhong XY, Ladewig A, Schmid S, Wight E, Hahn S, Holzgreve W. Elevated level of cell-free plasma DNA is associated with breast cancer. Arch Gynecol Obstet (2007) 276:327–31. doi:10.1007/s00404-007-0345-1
110. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A (2005) 102:16368–73. doi:10.1073/pnas.0507904102
111. Ng EK, Tsui NB, Lam NY, Chiu RW, Yu SC, Wong SC, et al. Presence of filterable and nonfilterable mRNA in the plasma of cancer patients and healthy individuals. Clin Chem (2002) 48:1212–7.
112. Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res (2001) 61:1659–65.
114. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. New Engl J Med (2013) 368:1199–209. doi:10.1056/NEJMoa1213261
115. Lecomte T, Berger A, Zinzindohoue F, Micard S, Landi B, Blons H, et al. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer (2002) 100:542–8. doi:10.1002/ijc.10526
116. Sozzi G, Conte D, Mariani L, Lo Vullo S, Roz L, Lombardo C, et al. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res (2001) 61:4675–8.
117. Chan KC, Yeung SW, Lui WB, Rainer TH, Lo YM. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem (2005) 51:781–4. doi:10.1373/clinchem.2004.046219
118. Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet (2007) 39:157–8. doi:10.1038/ng1941
119. Goessl C, Krause H, Muller M, Heicappell R, Schrader M, Sachsinger J, et al. Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res (2000) 60:5941–5.
120. Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res (2000) 60:892–5.
121. Zhang L, Wang M, Wang W, Mo J. Incidence and prognostic value of multiple gene promoter methylations in gliomas. J Neurooncol (2014) 116:349–56. doi:10.1007/s11060-013-1301-5
122. Jones PA, Baylin SB. The epigenomics of cancer. Cell (2007) 128:683–92. doi:10.1016/j.cell.2007.01.029
123. Jahrsdorfer B, Wooldridge JE, Blackwell SE, Taylor CM, Griffith TS, Link BK, et al. Immunostimulatory oligodeoxynucleotides induce apoptosis of B cell chronic lymphocytic leukemia cells. J Leukoc Biol (2005) 77:378–87. doi:10.1189/jlb.0604373
124. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A (1975) 72:3666–70. doi:10.1073/pnas.72.9.3666
125. Pashenkov M, Goess G, Wagner C, Hormann M, Jandl T, Moser A, et al. Phase II trial of a toll-like receptor 9-activating oligonucleotide in patients with metastatic melanoma. J Clin Oncol (2006) 24:5716–24. doi:10.1200/JCO.2006.07.9129
126. Wysocka M, Benoit BM, Newton S, Azzoni L, Montaner LJ, Rook AH. Enhancement of the host immune responses in cutaneous T-cell lymphoma by CpG oligodeoxynucleotides and IL-15. Blood (2004) 104:4142–9. doi:10.1182/blood-2004-03-1190
127. Lamm DL, Thor DE, Harris SC, Reyna JA, Stogdill VD, Radwin HM. Bacillus Calmette-Guerin immunotherapy of superficial bladder cancer. J Urol (1980) 124:38–40.
128. Merz VW, Marth D, Kraft R, Ackermann DK, Zingg EJ, Studer UE. Analysis of early failures after intravesical instillation therapy with bacille Calmette-Guerin for carcinoma in situ of the bladder. Br J Urol (1995) 75:180–4. doi:10.1111/j.1464-410X.1995.tb07307.x
129. Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol (2013) 93:847–63. doi:10.1189/jlb.1012501
130. Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol (2011) 41:413–24. doi:10.1002/eji.201040979
131. Pulko V, Liu X, Krco CJ, Harris KJ, Frigola X, Kwon ED, et al. TLR3-stimulated dendritic cells up-regulate B7-H1 expression and influence the magnitude of CD8 T cell responses to tumor vaccination. J Immunol (2009) 183:3634–41. doi:10.4049/jimmunol.0900974
132. Enninga EA, Nevala WK, Creedon DJ, Markovic SN, Holtan SG. Fetal sex-based differences in maternal hormones, angiogenic factors, and immune mediators during pregnancy and the postpartum period. Am J Reprod Immunol (2014) 73:251–62. doi:10.1111/aji.12303
Keywords: cell-free fetal DNA, circulating tumor DNA, immunotherapy, inflammation, toll like receptors
Citation: Enninga EAL, Nevala WK, Holtan SG and Markovic SN (2015) Immune reactivation by cell-free fetal DNA in healthy pregnancies re-purposed to target tumors: novel checkpoint inhibition in cancer therapeutics. Front. Immunol. 6:424. doi: 10.3389/fimmu.2015.00424
Received: 22 May 2015; Accepted: 03 August 2015;
Published: 26 August 2015
Edited by:
Anahid Jewett, University of California, Los Angeles, USAReviewed by:
Luis De La Cruz-Merino, Hospital Universitario Virgen Macarena, SpainNicholas Cacalano, University of California, Los Angeles, USA
Copyright: © 2015 Enninga, Nevala, Holtan and Markovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svetomir N. Markovic, Department of Hematology/Oncology, 200 First Street SW, Rochester, MN 55905, USA,bWFya292aWMuc3ZldG9taXJAbWF5by5lZHU=