 Toshiaki Kawakami
Toshiaki Kawakami Tomoaki Ando
Tomoaki Ando Yuko Kawakami1
Yuko Kawakami1
- 1Division of Cell Biology, La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA
- 2Laboratory for Allergic Disease, RIKEN Center for Integrative Medical Sciences (IMS-RCAI), Yokohama, Japan
Atopic dermatitis (AD) is a chronic inflammatory skin disease. Myeloproliferative neoplasms (MPNs) are hematopoietic malignancies caused by uncontrolled proliferation of hematopoietic stem/progenitor cells. Recent studies have described several mutant mice exhibiting both AD-like skin inflammation and MPN. Common pathways for skin inflammation encompass overexpression of thymic stromal lymphopoietin and reduced signaling of epidermal growth factor receptor in the epidermis, while overproduction of granulocyte-colony-stimulating factor by keratinocytes and constitutive activation of Stat5 in hematopoietic stem cells are important for the development of MPN. The murine studies suggest the existence of a similar human disease tentatively termed as the atopic dermatitis-myeloproliferative neoplasm syndrome.
Atopic dermatitis (AD) is a chronic pruritic inflammatory skin disease. Although the etiology of AD is not completely understood, clinical and basic studies suggest that impaired skin barrier function and immune dysregulation underlie the disease (1). For example, loss-of-function mutations in the filaggrin gene (FLG) are the most well-characterized risk factor for the development of AD (2, 3). Filaggrin plays a critical role in skin barrier function (4). Tight junctions below the stratum corneum also contribute to skin barrier. The expression of the tight junction proteins, claudin-1 and claudin-23, is reduced in patients with AD (5). In immunological terms, AD has been characterized by excessive Th2 cell and eosinophilic infiltration in acute lesions and a mixed Th1 and Th2 pattern in chronic lesions (6). Recent studies also suggest the involvement of Th17 cells in the acute but not the chronic phase (7, 8). Additionally, the expression of genes encoding neutrophil chemoattractants was revealed in AD lesions, consistent with increased neutrophil infiltration in the lesions (9).
Mechanistic studies of AD have been mostly conducted by mouse experiments (10, 11) despite perceived differences in human and mouse biology (12–14). Importantly, several mutant mice develop both AD-like skin inflammation and myeloproliferative neoplasm (MPN), suggesting the existence of a disease tentatively termed as the atopic dermatitis-myeloproliferative neoplasm (AD-MPN) syndrome. A majority of AD models do not develop MPN, as AD is a common disease but MPN is a rare disease. MPNs are hematopoietic malignancies characterized by an overproduction of any combination of white cells, red cells, and platelets. Human MPNs include polycythemia vera, essential thrombocythemia, primary myelofibrosis, and chronic myelogenous leukemia (15). Interestingly, pruritus is a common symptom in Philadelphia chromosome-negative MPNs. Suggestive of the role of mast cells in both disease phenotypes, mast cells from MPNs release greater amounts of pruritogenic factors, such as histamine, leukotrienes, and IL-31 (16). The mutant mice with epidermal deletion [by K5 (or K14)-Cre-mediated deletion of a floxed gene] of JunB (transcription factor) (17), a disintegrin and metalloproteinase 17 (ADAM17) (18, 19) or Notch receptors/their signal transducers (20) exhibited both AD-like skin inflammation and MPN. We recently described AD-like dermatitis and MPN in phospholipase C (PLC)-β3-deficient mice (21, 22). Similar skin inflammation accompanied by increased serum IgE and IgG1 was also described with mice deficient in SOCS7, a negative regulator of cytokine action by inhibiting transcription factors Stat5 and Stat3 (23). Therefore, in these mutant mice, AD and MPN pathologies likely develop by the shared or overlapping molecular and cellular mechanisms.
Myeloproliferative neoplasms are caused by uncontrolled proliferation and/or survival of hematopoietic stem/progenitor cells (24). Mechanistically, myeloproliferation could be caused by granulocyte-colony-stimulating factor (G-CSF) produced by JunB-deficient keratinocytes in JunBfl/fl;K5-Cre mice (17) and ADAM17-deficient keratinocytes in Adam17fl/fl;K14-Cre mice (Figure 1) (18). Consistent with the crucial role for keratinocyte-derived G-CSF, the loss of G-CSF in vivo prevented MPN and reduced skin inflammation in JunBfl/fl;K5-Cre mice. Transplantation of bone marrow cells derived from JunBfl/fl;K5-Cre mice failed to transfer of the MPN phenotype to WT mice. JunB is a direct repressor of Csf3 (gene encoding G-CSF) transcription (17). ADAM17 cleaves membrane proteins, working as the principal sheddase for tumor necrosis factor and several ligands of the epidermal growth factor receptor (EGFR). ADAM17 also activates Notch in adult epidermis. In Adam17−/− mice, impaired Notch signaling (i.e., decreased Notch intracellular domain) resulted in increased c-Fos recruitment to the Csf3 promoter leading to increased Csf3 transcription, as Notch signaling antagonized c-Fos recruitment to the Csf3 promoter (18). G-CSF might also be involved in skin inflammation by promoting keratinocyte proliferation (25). Consistent with the role of reduced EGFR signaling in AD-like skin inflammation in Adam17fl/fl;K14-Cre mice, an ADAM17-targeted EGFR ligand transforming growth factor-α, whose expression was strongly reduced in Adam17−/−keratinocytes, could reconstitute the skin barrier function in Adam17−/− mice, and mice with epidermal deletion of EGFR exhibited AD-like dermatitis just like Adam17fl/fl;K14-Cre mice (19). Another common pathway for skin inflammation might be overexpression of thymic stromal lymphopoietin (TSLP) (Figure 1), an IL-7-like cytokine produced by keratinocytes (26, 27), in lesional epidermis (18, 22, 28). Notch signaling also antagonized c-Fos recruitment to the Tslp promoter (18). Skin-specific overexpression of TSLP in transgenic mice resulted in the development of eczematous lesions and elevated serum levels of IgE (29, 30). Both spontaneous and allergen-induced dermatitis in Plcb3−/−mice required the receptor for TSLP (31). Expression of TSLP was extremely high in lesional epidermis of Plcb3−/− mice. TSLP could directly activate cutaneous sensory neurons, triggering itch (32), a major symptom that impairs the quality of life in AD patients. Mast cells, which were required for spontaneous and allergen-induced AD models (22, 31), were increased in lesional skin of Plcb3−/−mice due to the increased Stat5 and reduced SHP-1 (protein tyrosine phosphatase) activities (Figure 1) (22). PLC-β3 functions as an adaptor by directly binding to SHP-1 and Stat5, and Stat5 activity is inactivated by dephosphorylation of Stat5–Tyr694 by SHP-1 (21, 33). Consistent with the role of Stat5 in AD and MPN, itchy dermatitis with increased mast cells developed in transgenic mice expressing Jak2 V617F, an activating mutation found in many human MPN patients that also caused a polycythemia vera-like disease in the transgenic mice (34). The STAT5-regulatory mechanism for increased mast cells seemed important for AD pathogenesis in human patients as well (22). Breakdown of the same Stat5-regulatory mechanism was also responsible for the uncontrolled growth of hematopoietic stem cells (HSCs) in MPN in Plcb3−/−mice (21).
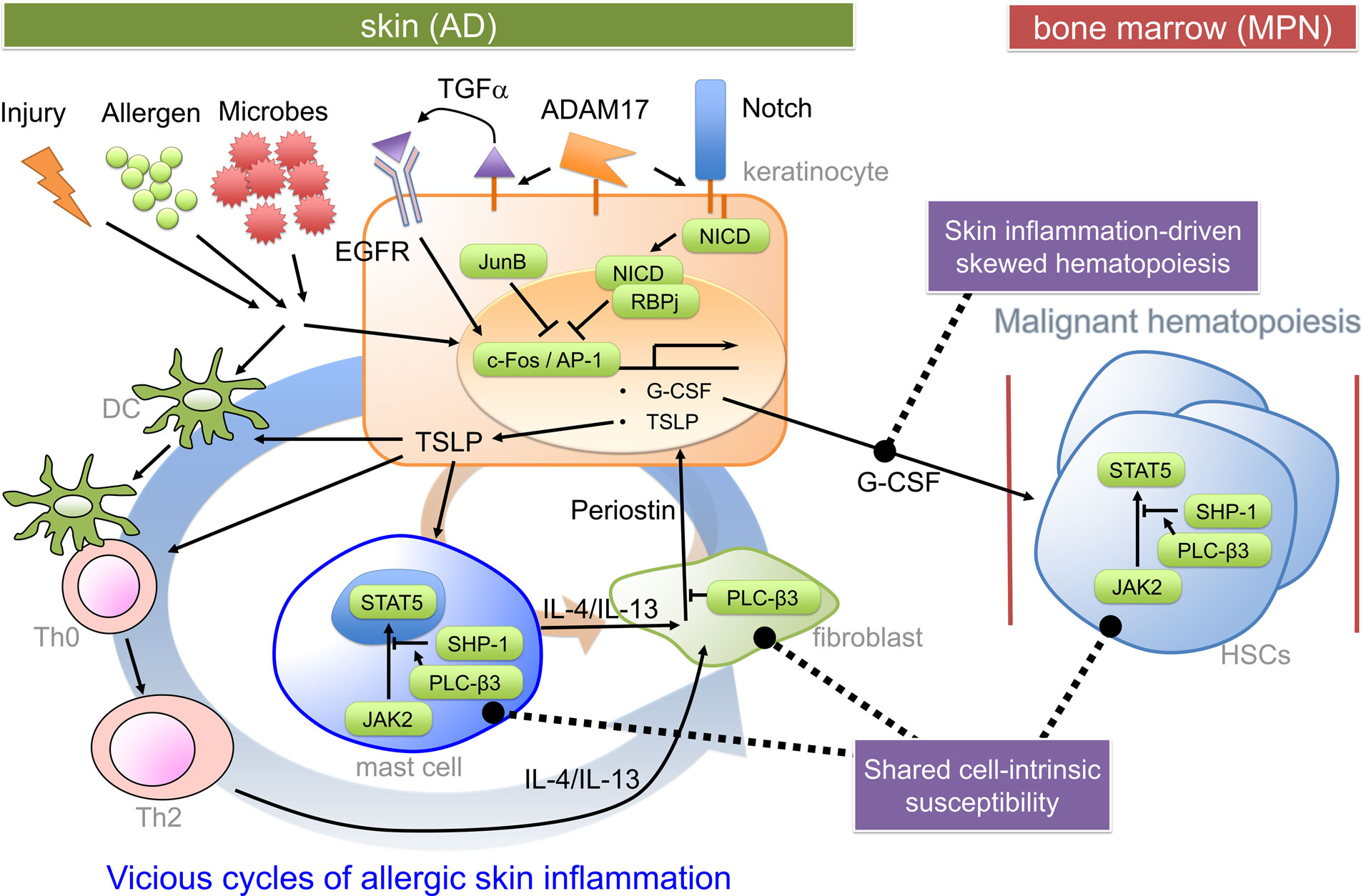
Figure 1. Pathogenic mechanisms for the hypothetical AD-MPN syndrome. Keratinocyte-derived TSLP (and impaired skin barrier) seems to play a critical role in dendritic cell-dependent Th2 responses to offending allergens or injury. Th2 cytokines predominantly cause an acute inflammation and activation of several types of cells, including dermal fibroblasts. Th2 cytokine-stimulated fibroblasts secrete periostin, an αv integrin-interacting matricellular protein, which in turn stimulates keratinocytes to secrete TSLP (35). TSLP also stimulates mast cells to secrete Th2 and other cytokines. Periostin secretion from dermal fibroblasts depends on Th2 cytokines and mast cells, forming at least two positive feedback loops (indicated thick and thin circular loops). Keratinocyte-derived G-CSF is important for MPN development in certain mutant mice. Expression of genes coding for G-CSF and TSLP is under control of c-Fos/AP-1, which is positively regulated by EGFR and negatively regulated by Notch signaling and JunB. The proliferative and survival properties of hematopoietic stem cells (HSCs) are positively regulated by Stat5, whose constitutive activation can lead to MPN. PLC-β3 negatively regulates TSLP production in keratinocytes, periostin production in fibroblasts, and Stat5 activity in HSCs, mast cells, and Th2 cells. DC, dendritic cell; NICD, Notch intracellular domain; RBPj, recombination signal binding protein for immunoglobulin kappa region.
Are allergic diseases such as AD protective or causative for the hematopoietic malignancies? Behind this question, there are two hypotheses to explain positive versus negative associations of the atopic diseases to malignancy: the “immunosurveillance” theory proposes that the atopic inflammatory state stimulates the immune system to eliminate the transformed cells. On the contrary, in the “antigenic stimulation” theory, the atopic or immune-stimulating condition leads to randomly occurring pro-oncogenic mutations in actively proliferating cells (36). Some prospective cohort studies support the hypothesis that the allergic diseases could be the risk of hematopoietic malignancies (leukemia, lymphoma, and myeloma), prostate cancer, and breast cancer (36–43), suggesting that specific types of allergies might increase risks for certain malignancies. But ovarian cancer risk was decreased in allergic patients (38). However, a meta-analysis of several case–control studies reported no increased risk of leukemia (44).
The hypothetical AD-MPN syndrome might have similarities with malignancy-associated Sweet’s syndrome or acute febrile neutrophilic dermatosis (45). Sweet’s syndrome occurs most often associated with acute myelogenous leukemia and is characterized by fever, neutrophilia, erythematous skin lesions, and a diffuse infiltrate consisting predominantly of mature neutrophils (i.e., these skin lesions are different from typical AD lesions). Thus, acute myelogenous leukemia-associated Sweet’s syndrome might be an acute evolution of an insidious MPN. Moreover, some Sweet’s syndrome patients have mutations in the PTPN6 gene encoding SHP-1 (46) (Figure 1). Interestingly, TSLP receptor (also known as CRLF2) overexpression has been associated with certain subtypes of pediatric leukemia (47, 48). Therefore, it will be interesting to investigate whether humans have an AD-MPN syndrome.
Author Contributions
TK, TA, and YK all contributed to write the manuscript. TA prepared the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was funded in part by grants from the MPN Foundation, NIH/NIAMS (1R01 AR064418-01A1), and the Ministry of Education, Culture, Sports, Science and Technology, Japan (25253071 and 12345678).
Abbreviations
AD, atopic dermatitis; EGFR, epidermal growth factor receptor; G-CSF, granulocyte-colony-stimulating factor; MPN, myeloproliferative neoplasm; PLC, phospholipase C; TSLP, thymic stromal lymphopoietin.
References
2. Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med (2011) 365(14):1315–27. doi:10.1056/NEJMra1011040
3. Margolis DJ, Apter AJ, Gupta J, Hoffstad O, Papadopoulos M, Campbell LE, et al. The persistence of atopic dermatitis and filaggrin (FLG) mutations in a US longitudinal cohort. J Allergy Clin Immunol (2012) 130(4):912–7. doi:10.1016/j.jaci.2012.07.008
4. McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol (2013) 131(2):280–91. doi:10.1016/j.jaci.2012.12.668
5. De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol (2011) 127(3):e1–7. doi:10.1016/j.jaci.2010.10.018
6. Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest (2004) 113(5):651–7. doi:10.1172/JCI21060
7. Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol (2008) 128(11):2625–30. doi:10.1038/jid.2008.111
8. Toda M, Leung DY, Molet S, Boguniewicz M, Taha R, Christodoulopoulos P, et al. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J Allergy Clin Immunol (2003) 111(4):875–81. doi:10.1067/mai.2003.1414
9. Choy DF, Hsu DK, Seshasayee D, Fung MA, Modrusan Z, Martin F, et al. Comparative transcriptomic analyses of atopic dermatitis and psoriasis reveal shared neutrophilic inflammation. J Allergy Clin Immunol (2012) 130(6):1335.e–43.e. doi:10.1016/j.jaci.2012.06.044
10. Kawakami T, Ando T, Kimura M, Wilson BS, Kawakami Y. Mast cells in atopic dermatitis. Curr Opin Immunol (2009) 21(6):666–78. doi:10.1016/j.coi.2009.09.006
11. Jin H, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol (2009) 129(1):31–40. doi:10.1038/jid.2008.106
12. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (2004) 172(5):2731–8. doi:10.4049/jimmunol.172.5.2731
13. Shay T, Jojic V, Zuk O, Rothamel K, Puyraimond-Zemmour D, Feng T, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci U S A (2013) 110(8):2946–51. doi:10.1073/pnas.1222738110
14. Emes RD, Goodstadt L, Winter EE, Ponting CP. Comparison of the genomes of human and mouse lays the foundation of genome zoology. Hum Mol Genet (2003) 12(7):701–9. doi:10.1093/hmg/ddg078
15. Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol (2011) 29(5):573–82. doi:10.1200/JCO.2010.29.8711
16. Ishii T, Wang J, Zhang W, Mascarenhas J, Hoffman R, Dai Y, et al. Pivotal role of mast cells in pruritogenesis in patients with myeloproliferative disorders. Blood (2009) 113(23):5942–50. doi:10.1182/blood-2008-09-179416
17. Meixner A, Zenz R, Schonthaler HB, Kenner L, Scheuch H, Penninger JM, et al. Epidermal JunB represses G-CSF transcription and affects haematopoiesis and bone formation. Nat Cell Biol (2008) 10(8):1003–11. doi:10.1038/ncb1761
18. Murthy A, Shao YW, Narala SR, Molyneux SD, Zuniga-Pflucker JC, Khokha R. Notch activation by the metalloproteinase ADAM17 regulates myeloproliferation and atopic barrier immunity by suppressing epithelial cytokine synthesis. Immunity (2012) 36(1):105–19. doi:10.1016/j.immuni.2012.01.005
19. Franzke CW, Cobzaru C, Triantafyllopoulou A, Loffek S, Horiuchi K, Threadgill DW, et al. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J Exp Med (2012) 209(6):1105–19. doi:10.1084/jem.20112258
20. Dumortier A, Durham AD, Di Piazza M, Vauclair S, Koch U, Ferrand G, et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PLoS One (2010) 5(2):e9258. doi:10.1371/journal.pone.0009258
21. Xiao W, Hong H, Kawakami Y, Kato Y, Wu D, Yasudo H, et al. Tumor suppression by phospholipase C-beta3 via SHP-1-mediated dephosphorylation of Stat5. Cancer Cell (2009) 16(2):161–71. doi:10.1016/j.ccr.2009.05.018
22. Ando T, Xiao W, Gao P, Namiranian S, Matsumoto K, Tomimori Y, et al. Critical role for mast cell Stat5 activity in skin inflammation. Cell Rep (2014) 6(2):366–76. doi:10.1016/j.celrep.2013.12.029
23. Knisz J, Banks A, McKeag L, Metcalfe DD, Rothman PB, Brown JM. Loss of SOCS7 in mice results in severe cutaneous disease and increased mast cell activation. Clin Immunol (2009) 132(2):277–84. doi:10.1016/j.clim.2009.04.003
24. Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood (2002) 100(1):238–45. doi:10.1182/blood.V100.1.238
25. Kawada A, Hiruma M, Noguchi H, Ishibashi A, Motoyoshi K, Kawada I. Granulocyte and macrophage colony-stimulating factors stimulate proliferation of human keratinocytes. Arch Dermatol Res (1997) 289(10):600–2. doi:10.1007/s004030050246
26. Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med (2006) 203(2):269–73. doi:10.1084/jem.20051745
27. Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol (2012) 130(4):845–52. doi:10.1016/j.jaci.2012.07.010
28. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol (2002) 3(7):673–80. doi:10.1038/ni805
29. Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med (2005) 202(4):541–9. doi:10.1084/jem.20041503
30. Li M, Messaddeq N, Teletin M, Pasquali JL, Metzger D, Chambon P. Retinoid X receptor ablation in adult mouse keratinocytes generates an atopic dermatitis triggered by thymic stromal lymphopoietin. Proc Natl Acad Sci U S A (2005) 102(41):14795–800. doi:10.1073/pnas.0507385102
31. Ando T, Matsumoto K, Namiranian S, Yamashita H, Glatthorn H, Kimura M, et al. Mast cells are required for full expression of allergen/SEB-induced skin inflammation. J Invest Dermatol (2013) 133(12):2695–705. doi:10.1038/jid.2013.250
32. Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell (2013) 155(2):285–95. doi:10.1016/j.cell.2013.08.057
33. Yasudo H, Ando T, Xiao W, Kawakami Y, Kawakami T. Short Stat5-interacting peptide derived from phospholipase C-beta3 inhibits hematopoietic cell proliferation and myeloid differentiation. PLoS One (2011) 6(9):e24995. doi:10.1371/journal.pone.0024995
34. Jin X, Zhao W, Kirabo A, Park SO, Ho WT, Sayeski PP, et al. Elevated levels of mast cells are involved in pruritus associated with polycythemia vera in JAK2V617F transgenic mice. J Immunol (2014) 193(2):477–84. doi:10.4049/jimmunol.1301946
35. Masuoka M, Shiraishi H, Ohta S, Suzuki S, Arima K, Aoki S, et al. Periostin promotes chronic allergic inflammation in response to Th2 cytokines. J Clin Invest (2012) 122(7):2590–600. doi:10.1172/JCI58978
36. Soderberg KC, Hagmar L, Schwartzbaum J, Feychting M. Allergic conditions and risk of hematological malignancies in adults: a cohort study. BMC Public Health (2004) 4:51. doi:10.1186/1471-2458-4-51
37. Talbot-Smith A, Fritschi L, Divitini ML, Mallon DF, Knuiman MW. Allergy, atopy, and cancer: a prospective study of the 1981 Busselton cohort. Am J Epidemiol (2003) 157(7):606–12. doi:10.1093/aje/kwg020
38. Mills PK, Beeson WL, Fraser GE, Phillips RL. Allergy and cancer: organ site-specific results from the adventist health study. Am J Epidemiol (1992) 136(3):287–95.
39. Eriksson NE, Holmen A, Hogstedt B, Mikoczy Z, Hagmar L. A prospective study of cancer incidence in a cohort examined for allergy. Allergy (1995) 50(9):718–22. doi:10.1111/j.1398-9995.1995.tb01212.x
40. McWhorter WP. Allergy and risk of cancer. A prospective study using NHANESI followup data. Cancer (1988) 62(2):451–5. doi:10.1002/1097-0142(19880715)62:2<451::AID-CNCR2820620234>3.0.CO;2-D
41. Musolino C, Allegra A, Minciullo PL, Gangemi S. Allergy and risk of hematologic malignancies: associations and mechanisms. Leuk Res (2014) 38(10):1137–44. doi:10.1016/j.leukres.2014.08.004
42. Koshiol J, Lam TK, Gridley G, Check D, Brown LM, Landgren O. Racial differences in chronic immune stimulatory conditions and risk of non-Hodgkin’s lymphoma in veterans from the United States. J Clin Oncol (2011) 29(4):378–85. doi:10.1200/JCO.2010.30.1515
43. Nunez-Enriquez JC, Fajardo-Gutierrez A, Buchan-Duran EP, Bernaldez-Rios R, Medina-Sanson A, Jimenez-Hernandez E, et al. Allergy and acute leukaemia in children with Down syndrome: a population study. Report from the Mexican inter-institutional group for the identification of the causes of childhood leukaemia. Br J Cancer (2013) 108(11):2334–8. doi:10.1038/bjc.2013.237
44. Linabery AM, Jurek AM, Duval S, Ross JA. The association between atopy and childhood/adolescent leukemia: a meta-analysis. Am J Epidemiol (2010) 171(7):749–64. doi:10.1093/aje/kwq004
45. Cohen PR. Sweet’s syndrome – a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis (2007) 2:34. doi:10.1186/1750-1172-2-34
46. Nesterovitch AB, Gyorfy Z, Hoffman MD, Moore EC, Elbuluk N, Tryniszewska B, et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol (2011) 178(4):1434–41. doi:10.1016/j.ajpath.2010.12.035
47. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet (2009) 41(11):1243–6. doi:10.1038/ng.469
Keywords: atopic dermatitis, myeloproliferative neoplasm, hematopoietic stem cell, mast cell, mouse models
Citation: Kawakami T, Ando T and Kawakami Y (2015) Hypothetical atopic dermatitis-myeloproliferative neoplasm syndrome. Front. Immunol. 6:434. doi: 10.3389/fimmu.2015.00434
Received: 19 June 2015; Accepted: 10 August 2015;
Published: 24 August 2015
Edited by:
Masaaki Murakami, Hokkaido University, JapanReviewed by:
Daisuke Kamimura, Hokkaido University, JapanYasunobu Arima, Osaka University, Japan
Hideyuki Ujiie, Hokkaido University, Japan
Copyright: © 2015 Kawakami, Ando and Kawakami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshiaki Kawakami, Division of Cell Biology, La Jolla Institute for Allergy and Immunology, 9420 Athena Circle, La Jolla, CA 92037, USA,dG9zaGlAbGppLm9yZw==