 Sachiko Hirosue
Sachiko Hirosue Juan Dubrot
Juan Dubrot- 1Institute of Bioengineering, École Polytechnique Fédéral de Lausanne, Lausanne, Switzerland
- 2Department of Pathology and Immunology, Université de Genève, Geneva, Switzerland
Antigen presentation is no longer the exclusive domain of cells of hematopoietic origin. Recent works have demonstrated that lymph node stromal cell (LNSC) populations, such as fibroblastic reticular cells, lymphatic and blood endothelial cells, not only provide a scaffold for lymphocyte interactions but also exhibit active immunomodulatory roles that are critical to mounting and resolving effective immune responses. Importantly, LNSCs possess the ability to present antigens and establish antigen-specific interactions with T cells. One example is the expression of peripheral tissue antigens, which are presented on major histocompatibility complex (MHC)-I molecules with tolerogenic consequences on T cells. Additionally, exogenous antigens, including self and tumor antigens, can be processed and presented on MHC-I complexes, which result in dysfunctional activation of antigen-specific CD8+ T cells. While MHC-I is widely expressed on cells of both hematopoietic and non-hematopoietic origins, antigen presentation via MHC-II is more precisely regulated. Nevertheless, LNSCs are capable of endogenously expressing, or alternatively, acquiring MHC-II molecules. Transfer of antigen between LNSC and dendritic cells in both directions has been recently suggested to promote tolerogenic roles of LNSCs on the CD4+ T cell compartment. Thus, antigen presentation by LNSCs is thought to be a mechanism that promotes the maintenance of peripheral tolerance as well as generates a pool of diverse antigen-experienced T cells for protective immunity. This review aims to integrate the current and emerging literature to highlight the importance of LNSCs in immune responses, and emphasize their role in antigen trafficking, retention, and presentation.
Introduction
One of the hallmarks of adaptive immunity is the T cell-antigen-presenting cell (APC) crosstalk that takes place in lymph nodes (LNs). As secondary lymphoid organs, LNs are essential in maintaining tolerance as well as initiating and resolving immune responses (1). LNs constitute particular locations where peripheral tissue environments are sampled in the form of endogenous and exogenous antigens. These processes occur in specific LN regions and are finely controlled by resident stromal cells that promote lymphocyte trafficking and maintain lymphocyte homeostasis.
Lymph node stromal cells (LNSCs) constitute a heterogeneous mixture of non-hematopoietic cells (CD45−) of mesenchymal and endothelial origins (2), and have long been appreciated to provide the scaffold on which immune cells encounter antigen (3, 4). Despite their low frequency (<1% of the LN cell count), recent studies have elucidated that LNSCs have active and important roles in shaping the immune response.
The main CD45− LNSC populations can be defined based on their expression of podoplanin (gp38) and PECAM-1 (CD31) by flow cytometry (5–7). They include fibroblastic reticular cells (FRCs: CD31− gp38+), lymphatic endothelial cells (LECs: CD31+ gp38+), blood endothelial cells (BECs: CD31+ gp38−) (5, 6, 8) and a less studied double negative population (DN: CD31− gp38−). Follicular dendritic cells (FDC: CD21+, CD35+, FDC-M1+) constitute an additional subset derived from non-hematopoietic origins (9), which can also express gp38 (5, 10, 11). These LNSC populations help define and organize the structure of the LN as a whole, and because of their distinct anatomic localizations, they can each shape immune responses in complementary ways.
Within LNs, FRCs form an intricate and highly organized reticular network that not only contributes to lymphocyte trafficking but also organizes the lymphocyte populations into different functional zones (12). As the FRC network defines the T cell zone in the cortex, FRCs have been associated with T cell biology. They are a major source of IL7, which is essential for T cell homeostasis (5), and additionally are known to be a source of the chemokines CCL19 and together with LECs, CCL21 (13, 14), which both act on the homing receptor CCR7 on T cells. Moreover, FRCs contribute to the regulation of T cell activation by providing a reticular network that facilitates dendritic cell (DC)–T cell interactions, and subsequent T cell priming (12, 15–17). At the same time, local T cell-derived interferon (IFN)-γ and tumor necrosis factor (TNF)-α act synergistically to induce nitric oxide production by FRCs through the activation of inducible nitric oxide synthase (iNOS) (18–20), thus inhibiting T cell proliferation. More recently, FRCs have been shown to support B cell survival (21).
The lymphatic vascular network, formed by LECs, ensures the transport of antigens from peripheral tissues to local LNs, and then to downstream LNs (22). Within LNs, LECs define the floor and the ceiling of the subcapsular sinus (SCS) and the cortical and medullary sinuses. As lymphatic vessels are the primary route of soluble and APC-carried antigen to the LN, LECs are well-situated to interact directly with lymph-borne antigens, as well as LN-resident and migratory DCs (23), via production of IL7 and CCL21 (24). Under inflammatory conditions, LECs can directly attenuate DC activation through contact-dependent mechanisms (25). Moreover, LECs directly influence T cell activation, in part by dampening activated T cell proliferation through the production of nitric oxide (18, 19). LECs also help direct T cell egress from LNs through secretion of S1P, which is augmented by tenascin-C interactions with α9-integrin on activated LECs (26–28).
BECs line blood vessels that irrigate LNs. The majority of naïve lymphocytes enter LNs from the blood and reach the specific T-cell zones through the specialized high endothelial venules (HEVs) in the cortex (29). HEVs are known to actively promote T cell recruitment and ingress into the LN parenchyma by producing CCL21 (30) and transcytosing CCL19 (31) from the parenchyma.
Specifically located within B cell follicles, FDCs are essential in defining and maintaining the B cell follicular structure, thus shaping humoral immunity (32–34). FDCs have been recently shown to differentiate from marginal reticular cells, which share surface markers with FRCs, yet represent a distinct LNSC subtype located primarily near the SCS and B cell follicles (10). FDCs have a specialized role in maintaining and coordinating B cell responses, including self-reactive B cell deletion (35).
Together, LNSCs create the framework of the LN, with key functional consequences, including guiding peripheral antigens and cells into the LN, and regulating the residence time and interactions of lymphocytes and APCs in the LN (36–38). Immunological roles of LECs and FRCs have been recently reviewed elsewhere (24, 39). Here, we focus on how peripheral antigen sampling and presentation by LNSC may contribute to the overall immune response.
LNSCs Coordinate Antigen Availability in the LN
Adaptive immunity is a feature of evolved immune systems and relies on the ability of lymphocytes to recognize cognate antigens in the context of a major histocompatibility complex (MHC) (40).
In this regard, there are at least two levels of LNSC participation in antigen presentation. One is the traffic control of antigen distribution, involving the spatial and temporal delivery of LN-infiltrating antigens to specific LN compartments (41). The other resides in the ability of LNSCs themselves to process and present antigens directly to T cells (36).
Antigen Traffic Control in the LN
The lymphatic system represents an efficient and sophisticated mode of communication between peripheral tissues and their draining LNs, with the lymph as its medium. The lymph is composed of a mixture of biomolecules, including proteins, peptides, and exogenously derived antigens that reflect the state of the peripheral tissue. Under normal, physiological conditions, lymph has been shown to be significantly enriched, relative to plasma, in intracellular and extracellular matrix proteins and peptides derived from tissue homeostatic turn-over (42). Human lymph has also been shown to carry a more diverse self-peptidome relative to plasma (43). Hence, the lymph fluid appears to represent a comprehensive status report of the peripheral tissues. Following an infection or an inflammatory challenge, exogenous antigens and self-peptides from damaged cells, together with homeostatic components of lymph, enter tissue-draining LNs. This mixture of antigens, including soluble, particulate, and APC-borne forms, is ushered to different anatomic regions of the LN, notably through the interaction with resident LNSCs (Figure 1).
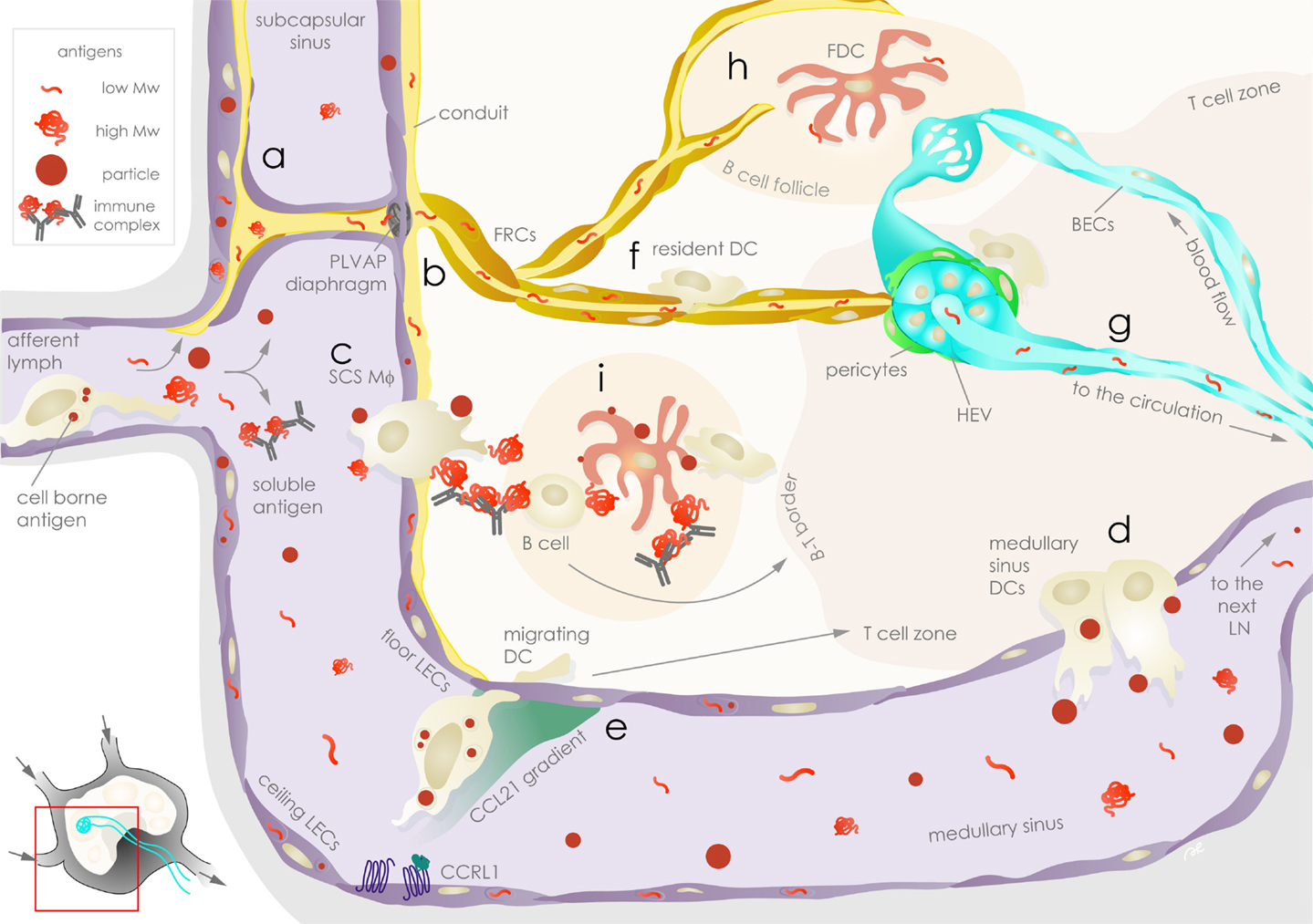
Figure 1. Exogenous antigen distribution in the lymph node is choreographed by stromal cells. Lymph-borne peripheral exogenous antigens arrive in the lymph node (LN) via the afferent lymphatic vessels as fast-draining soluble or slow arriving cell-associated forms. Floor and ceiling lymphatic endothelial cells (LECs, violet) are the first stromal cells encountered by the antigens in the subcapsular sinus (SCS). Soluble antigens can be taken up and sequestered by LECs (a), while the remaining fraction of antigens leave the SCS: lower molecular weight antigens are channeled to conduits surrounded by fibroblastic reticular cells (FRCs, ochre) and are filtered through plasmalemma vesicle-associated protein (PLVAP) diaphragms on floor LECs (b); immune complexes and particles are ferried across the SCS by SCS macrophages (Mϕ) (c). The bulk of soluble antigens not retained or channeled in the above compartments pass through to the medullary sinus, where they are sampled by medullary sinus dendritic cells (DCs) prior to draining to the downstream LN via efferent lymphatic vessels (d). Floor and ceiling LECs establish chemokine gradients that guide antigen-bearing migrating DCs arriving from the periphery by differential expression of atypical chemokine receptors, such as CCRL1, that scavenge CCL21 (e). In the parenchyma, soluble antigens channeled in the conduits are sampled by FRCs as well as LN-resident DCs (f) before they reach the high endothelial venules (HEVs, aqua), which are surrounded by FRCs or α7+ pericytes (green, part of the double negative DN stromal population), and enter the circulation where they can be sampled by blood endothelial cells (BECs, aqua) (g). Conduits also deliver lower molecular weight antigens to follicular dendritic cells (FDCs, rose) in the B cell follicle (h), while SCS Mϕ relay larger antigens to FDCs or to B cells (i). Antigens on FDCs are available for sampling by DCs and antigen-specific B cells for extended durations. Stromal cells as well as APCs communicate with T cells, contributing to antigen-specific immunological outcomes.
LN Entry of Soluble Antigens
Soluble molecules in the lymph enter LNs and are immediately subjected to size-based sorting. Much of our understanding of soluble antigen entry into LNs comes from intravital microscopy studies focused on understanding the generation of humoral immunity (44–47). Upon entry into the SCS, lower molecular weight antigens (≲70 kDa) are immediately channeled to FRC-lined conduits entering the B cell follicles (17), allowing for direct cognate B cell (48) as well as FDC (49) sampling in the follicles, which are important steps toward a humoral response. SCS macrophages (50) and DCs surrounding HEVs (51) also sample antigens. In addition, low molecular weight proteins can also be rapidly sampled by DCs in the LN to elicit T cell responses (52). Meanwhile, a bulk of the remaining, unsampled low molecular weight antigens will end up exiting through HEVs to reach the circulation, or alternatively, through efferent lymphatic vessels, which feed into the next LN (53, 54).
Larger molecules (≳70 kDa) lag behind the small molecular weight antigens by several minutes following LN entry and may take hours to reach cognate B cells (48). This size-dependent distribution and retention time in the LN is in part due to sieve-like diaphragms, composed of plasmalemma vesicle-associated protein (PLVAP) in the SCS LECs (55). PLVAP was originally identified as a BEC-specific molecule that regulates vessel permeability and was recently shown to be important for the formation of fenestrae in liver sinusoidal endothelial cells (LSECs), enabling the regulation of macromolecular transport in the liver (56). In LNs, PLVAP co-localized with the subcapsular and medullary sinus-lining LYVE-1+ LECs as well as HEVs (55). PLVAP-deficient mice exhibit an increased permeability of 180 kDa proteins and 500 kDa dextran into the LN parenchyma through the FRC conduits. These observations emphasize the importance of PLVAP in size-based mechanical filtration of antigens in the LN sinus. As a result, larger molecules are generally excluded from the cortex and are forced to pass through the LEC-lined medullary sinuses to drain to the next LN. As a result, these larger molecules pass through an environment rich in LN-resident DCs (sinusoidal DCs) that extend protrusions into the medullary sinus to actively sample the lymph (57). The size-exclusion properties of the LNSC architecture seems to be maintained during inflammatory LN remodeling, as was observed in the case of intradermally administered vaccinia virus (53).
LN Trafficking of Particles
Aside from soluble antigens, cell-free antigens can come in the form of particulates, including exosomes, microvesicles, apoptotic bodies, and other membrane-bound particles arising from steady-state and inflammatory conditions (58). Viruses and bacteria, lipid macrostructures, such as alpha-GalCer, and synthetic particles generally occur within a similar size range (59), and are promptly intercepted by the SCS macrophages upon LN entry (57, 60–67), although this capture does not always translate to the development of humoral immunity (63).
Immune complexes, which consist of antigens coated with natural or antigen-specific antibodies and complement proteins, may also behave like “particles” (68). The captured immune complexes are then relayed either directly or alternatively via complement receptor-expressing naïve B cells to FDCs (69, 70).
The remaining unsampled particulates bypass the LN cortex and flow directly to the LN medullary sinus, where they may be captured by LN-resident sinusoidal DCs in a similar fashion as described earlier for the large soluble antigens. Particulate antigens, ranging from 40 nm up to at least 1 μm in diameter were shown to be captured by the sinusoidal DCs (57). This pattern of particles captured in the medullary sinus is consistent with what has been observed for other particulate antigens (63, 71). Furthermore, it has been shown to be essential for the timely generation of both CD4+ and CD8+ T cell responses to particle-conjugated OVA, while immune responses against soluble antigen developed independently of this mechanism (57).
Thus, while lymphatic transport of larger particulates from the periphery may not be as efficient as that of soluble antigens (72), LNSCs indirectly improve the timeliness of T cell responses by contributing avenues that promote rapid routing of particles in the size range of pathogens to LN-resident sinusoidal DCs.
Cell-Associated Antigens in the LN
Antigens may also be carried to the draining LN by peripheral APCs. The trafficking of APCs from the periphery to the LN has been extensively studied (22, 73, 74). In this process, LECs play an important role by promoting APC entry into lymphatic vessels (75–77) and regulating their intra-lymphatic motility (76–78) even when only partial dermal lymph drainage is available (79). In areas of inflammation, inflammatory signals and changes in interstitial fluid drainage into local lymphatic vessels promote DC recruitment and transmigration in several ways. Following binding of immune complexes, DCs become more responsive to recruitment via gradients of CCL19 (80). Inflammatory signals can also increase LEC expression of the adhesion molecules, ICAM-1, VCAM-1, and E-selectin, upregulate CD137 (4-1BB) and CCL21 secretion, and promote CX3CL1 (fractalkine) shedding (81–85). Altogether, these phenotypic changes in LECs and DCs coordinate increased DC transmigration into lymphatic vessels and into draining LNs, in which direct interactions between LECs and DCs are required for efficient DC migration into the LN (86).
While the effects of peripheral lymphatics on DC motility are well documented, the influence of LN-resident LNSC on DC motility is less well known. DC entry into the LN is driven by a chemokine gradient generated by LECs (14). In the subcapsular space, LECs on the afferent side (ceiling LECs) but not on the floor of the capsule (floor LECs) were shown to reduce CCL21 levels due to the expression of atypical chemokine receptor, CCRL1. Such atypical chemokine receptors bind their ligands but fail to signal (87, 88). More importantly, the scavenger function of CCRL1 led to the generation of a localized CCL21 gradient that prompted the entry of DC into the LN parenchyma. In addition to CCRL1, CCL21 has other binding partners, such as podoplanin (89), which may also contribute to the modulation of DC-mediated antigen transport. LNSCs have been shown to partake in intra-LN control of chemokine distribution by other atypical chemokine receptors, such as D6 on afferent LECs and HEVs (90, 91), and Duffy antigen receptor for chemokine transcellular transport (DARC) on HEVs (92). In the LN, binding of CLEC-2 on DCs with gp38 on FRCs, but not LECs, has been demonstrated as a key axis for DC movement (77, 93). Inflammation seems to coordinate CLEC-2 and gp38 expression, which provides an explanation for enhanced interaction between DCs and FRCs (11, 77, 94). By modulating the recruitment of antigen-loaded DCs from peripheral tissues into and within the LN parenchyma, LNSCs help specify the location and duration of cell-borne antigen availability in the LN. Further studies to understand these DC-stromal cell interactions at a molecular level will likely reveal additional insights on the stromal functions in the LN.
LN Antigen Retention
Antigen retention is a way of modulating antigen availability during an immune response. Indeed, LNSCs have been known to be safe havens for some pathogens (4, 33). LNSCs are infection targets in some experimental settings, as described for FRCs in a mouse model of persistent lymphocytic choriomeningitis virus (LCMV clone 13) infections (95, 96). The chronicity of the viral burden is sustained in part by upregulation of PD-L1 in infected FRCs that counteract the antiviral CD8+ T cell response.
In addition, during acute infections or after vaccination, LECs have been described as antigen reservoirs. Antigen archiving required the induction of LEC proliferation, which, in turn, was an effect of non-cognate T cell proliferation. These observations suggest that T cells can indirectly prolong the persistence of virally derived antigens after the clearance of the virus by promoting antigen archiving function in LECs, which translated to protective memory in the CD8+ T cell compartment (97).
Antigens derived from apoptotic cells as well as pathogens are known to be retained in LNs by FDCs (33). FDCs retain antigens on FcγRIIb inhibitory Fc receptors and CD21/CD35 (complement receptors Cr2/Cr1) for presentation to cognate B cells (32, 98, 99). Binding to any of these receptors results in rapid internalization of the immune complexes (100, 101). However, rather than continuing onto a degradation pathway, the engaged complement receptor 2 (Cr2) undergoes periodic recycling and the untampered antigen is therefore made available on the FDC surface over an extended period (100). Immune complexes on FDCs can then be sampled by LN-resident DCs to be presented on MHC-I and II with demonstrated deletion of antigen-specific CD8+ T cells (102). Whether other LNSC utilize a similar, recycling, non-degradative compartment remains to be seen. Neither CD21 nor CD35 have been reported to be expressed on other LNSCs (Figure S1 in Supplementary Material), making FDCs unique in this mode of prolonged, intact antigen presentation.
It must be remarked that many studies that discuss antigen uptake and persistence often overlook LNSCs. For example, studies demonstrating particulate antigen capture by CD169+ macrophages do not routinely counter-stain for LECs with LYVE-1. However, intradermally injected vesicular stomatitis virus (VSV) results in significant internalization by LYVE-1+ cells, regardless of macrophage depletion by clodronate liposomes (61). Similarly, few dynamic imaging studies investigating antigen-specific responses in the LN marked the stroma (12), precluding the opportunity to study the participation of LNSCs. On these grounds, there is a need to clearly define and place LNSC contributions within the current understanding of antigen availability in the LN.
Structure–Function Relationships in LNSCs Govern Antigen Availability and Influence Immune Responses
The crucial role of LNSCs in coordinating antigen availability and presentation kinetics of APCs in the LN (3) has been illustrated in a number of inducible knockout models (Table S1 in Supplementary Material). Removing specific LNSC subsets has been achieved in different experimental conditions by expressing the diphtheria toxin (DT) receptor in FDCs (Cd21-Cre) (32), FRCs (Ccl19-Cre, FAP) (103, 104), and LECs (Lyve1-Cre) (105). The absence of any of these LNSC subsets was expectedly sufficient to severely disrupt LN compartmentalization and lymphocyte numbers, leading to an impaired immune response. FDC-depleted LNs cannot retain immune complexes and exhibit deficiencies in germinal center responses (32, 106). Selective depletion of FRCs resulted not only in a deficient T cell response but also correlated with a dramatic reduction of the B cell chemoattractant molecule CXCL12 and the pro-survival factor BAFF, accompanied by follicle destabilization and impaired germinal center formation (103). FRCs appear to regulate the trafficking of mainly naïve lymphocytes as FRC-depleted mice responded to inflammatory challenge with similar numbers of effector T and B cells to normal controls (104). The DT depletion approach was less informative when applied to LECs. In a mouse model where DTR expression is induced in LYVE1+ cells, systemic DT administration depleted LYVE1+ cells in the intestinal–blood barrier, leading to sepsis (105).
In these depletion studies, lymph and thus antigen flow from the periphery and in the LN is partially maintained. Diminished lymphatic drainage is known to perturb LN B cell organization and humoral immunity (107, 108). However, even when lymphatic drainage is retained, the absence of LNSCs has consequences. Despite the remaining flow of lymph through the ghost FRC conduits, it becomes clear that not only FDCs but also FRCs are indispensable for organizing the boundaries of B cell follicles and the humoral response. This is reflected in the fact that Notch signaling in FRCs has been shown to be important for T follicular helper cell differentiation and germinal center formation (109). This example highlights the synergistic effects of multiple LNSC subtypes and different pathways toward the humoral response, and is likely responsible for regulating the number of B-cell follicles per LN, which otherwise generally scales with the LN size under homeostatic conditions (110).
Another example of synergy between LNSC subsets has been described, where FRCs support the expansion of LECs and BECs by expressing vascular endothelial growth factor (VEGF) and other molecules (8, 111), aided by LN-resident CD11c+ cells (112). In addition to supporting the expansion of other LNSC subsets, under inflammatory conditions, the gp38-CLEC-2 signaling axis between FRCs and DCs results in LN structural relaxation by decreasing the contractility of FRCs, ultimately creating space to accommodate increased cell numbers in the LN (113, 114). This pathway is also important in the maintenance of HEV structural integrity as CLEC-2 on platelets can signal through gp38 on FRCs that surround HEVs (115).
In this regard, it is of notable importance that antigen capture, MHC processing, and T cell activation by LN-resident DC are all spatially linked processes (57). While peripheral DC migration to the LN is necessary for a robust immune response, in part by prolonging the time frame of antigen availability within the LN (52, 116), the lymphatic sinus DCs were found to be sufficient to elicit both antigen-specific CD4+ and CD8+ T cell responses as well as T follicular helper responses, leading to humoral immunity (57). This suggests that antigen drainage and capture occur immediately, triggering an early adaptive immune response and migratory DCs act to amplify the response later upon their arrival to the T cell zone (23). Thus, APC distribution in the LN and their spatial signature (23) affect the kinetics of the immune response. LNSCs are essentially upstream of this process, and in turn, are significantly involved in channeling antigens into the correct subanatomic locations so as to modulate antigen availability to various APC subpopulations.
Taken together, functional lymph flow, antigen sorting, and LN structural integrity maintained by LNSCs are important for the generation of adaptive immune responses. These are manifested in the dynamic LN architecture, which in turn affects the kinetics and time scale of B and T cell activation, proliferation, and polarization as well as feeds back to alter the LN structure and prolong antigen availability.
Direct Antigen Presentation by LNSCs
There is a growing appreciation that LNSCs not only guide the antigens to APCs but also that they themselves present antigen to educate T cells.
Brief Introduction to Antigen Presentation
Typically, antigen-presentation pathways are introduced in such a way that intracellular antigens are presented to CD8+ T cells as peptide:MHC-I complexes, while extracellular antigens are presented to CD4+ T cells as peptide:MHC-II complexes. Presentation of self-antigens by MHC-I on all nucleated cells prevents attack by effector CD8+ T cells (117). At the same time, MHC-I presentation allows the presentation of intracellular pathogen-associated epitopes for elimination of infected cells. On the other hand, the processing of extracellular antigens for MHC-II presentation to activate CD4+ T cells had been conventionally thought to be restricted to professional APCs. Upon internalization, the extracellular antigens in the trafficking vesicle remain topologically outside while they are processed to generate peptides for MHC-II loading (118, 119). However, neither topological segregation of antigens on the level of the APC nor the exclusivity of certain antigen-presentation pathways to professional APCs captures the full story of MHC-I and MHC-II presentation.
With regards to antigen presentation on MHC-I, it is clear that intracellular antigens from cytoplasmic or intranuclear sources are processed by the proteasome and other cytosolic proteases in the cytoplasm, imported into the endoplasmic reticulum by the transporter associated with antigen-processing (TAP) complex, trimmed, and loaded onto MHC-I. The MHC-I model must also account for exogenous antigens, such as self-antigens draining to the LN under homeostatic conditions, which are presented on MHC-I, the well-known phenomenon of cross-presentation (120). In this process, exogenous antigens leave endocytic vesicles to access the cytoplasmic space for processing and loading onto the MHC-I presenting machinery in a TAP-dependent manner in the endoplasmic reticulum (121–123) or in phagolysosomes (124, 125). Alternatively, exogenous antigens can be loaded onto MHC-I molecules within recycling vesicles in a TAP-independent manner (126, 127). The importance of the cross-presentation pathway can be appreciated in that tumors (aberrant self), and intracellular pathogens (non-self) either lose, or have evolved mechanisms to prevent MHC-I antigen presentation, to evade cytotoxic T lymphocyte (CTL) destruction of tumors, or pathogen-infected host cells, respectively (128, 129). This antigen-processing pathway, previously thought to be the domain of professional APCs, has now been described for other cell types (130).
With regards to MHC-II presentation, phagocytosis and processing of extracellular antigens eventually leads to peptides that are loaded onto MHC-II mainly in MHC-II-enriched multivescicular compartments (131, 132). For epitope peptides to bind to MHC-II, the MHC-II chaperone invariant chain (Ii) must first be cleaved to Class II-associated invariant chain peptide (CLIP). CLIP is then released by the enzyme H2-M to bind the MHC-II epitope peptides for the peptide:MHC-II complex to finally be available on the APC cell surface (133). Interestingly, these proteins involved in MHC-II presentation are not completely absent from LNSCs on the transcription level (8, 134, 135) (Figure S2 in Supplementary Material).
In addition, MHC-II can be loaded with intracellular antigens as suggested by the analysis of MHC-binding peptide repertoire in the thymus (136, 137). Autophagy and endocytosis of apoptotic material can introduce intracellular antigens to the MHC-II pathway (136). Again, existence of viral evasion mechanisms suggests the importance of this pathway (138).
Other intracellular pathways are shared for both MHC-I and -II loading, such as the gamma-interferon-inducible lysosomal thiolreductase (GILT) enzymes in DCs (139), which are thought to aid in antigen translocation into the cytosol (MHC-I) as well as exposure of buried epitopes (MHC-II) (140).
These observations demonstrate that antigen presentation is not limited by the topological origins of the antigen and that antigen presentation pathways previously thought to be restricted to professional APCs can be active in other cell types. As such, both self- and non-self-antigens can be presented on MHC-I and MHC-II molecules. The antigenic self-peptide repertoire is highly plastic (141), and may be changed under inflammatory conditions with the induction of the immunoproteasome (142, 143) or altered MHC trafficking in activated DCs (133). Finally, antigen-presentation capacity is variable in efficiency and duration as well as environmentally modulated in different DC subsets (144). This opens the possibility for antigen presentation by LNSCs to partake in the modulation of the immune response.
MHC-I Presentation by LNSCs
MHC-I Expression in LNSCs
Nearly all nucleated cells constitutively express MHC-I, LNSCs being no exception. The MHC-I complex at the cell surface consists of a polymorphic MHC-I together with a non-polymorphic β2 microglobulin (β2M), both of which are regulated by a specific transactivator NLRC5 (145). MHC-I expression can be induced in response to IFN-γ through the IFN-stimulated response element. This upregulation of MHC-I was observed when LNSCs were in the presence of toll-like receptor (TLR) agonists, or engaged in an antigen-specific interaction with T cells (146–148). Along with MHC-I expression, NLRC5 induces TAP1 and LMP2 (proteasome subunit β type 9), coordinating the expression of some main proteins involved in the cytoplasmic antigen-processing and MHC-I-presentation pathways.
Self-Antigen Expression in LNSCs
Peripheral tissue antigens (PTAs) can be promiscuously expressed in the thymus in medullary thymic epithelial cells for the purpose of negative selection of T cells in central tolerance (149). However, some self-reactive T cells remain in the periphery (150, 151). Tissue draining, but also non-draining, LN FRCs, LECs, and BECs have been described to express PTA repertoires, including both endogenous PTAs as well as transgenically introduced PTAs (146, 152–155). Interestingly, these repertoires are not overlapping between the LNSC subsets (146, 156), likely attributable to differential expression of transcription factors driving PTA expression, such as Autoimmune regulator (Aire) (146, 154) and deformed epidermal autoregulatory factor 1 (Deaf-1) characterized in the pancreatic LN (157). Within a LNSC subset, PTA expression can further be localized subanatomically (156). For example, in the LN, the PTA Tyr369 is presented only by the medullary LECs as evidenced by antigen-specific CD8+ T cell proliferation, when LN LECs were separated into subcapsular, medullary, and cortical LECs based on the differential expression of MAdCAM-1 and PD-L1 (156). The intracellular mechanisms of PTA MHC-I presentation have not been addressed in any of these studies, though it is assumed to be the same as with any endogenously expressed intracellular antigen.
The PTA expression pattern in the mouse suggests that the LN is particular in the presentation of antigens by stromal cells. In the spleen, another important secondary lymphoid organ, stromal cells did not express detectable PTAs compared to LNSCs (152, 153). Moreover, the same stromal cell types in the periphery express fewer PTAs than those in the LN. For example, unlike their LN counterparts, LECs isolated from the diaphragm and colon cannot present the PTA Tyrosinase to antigen-specific T cells (156).
Whether there exist overarching drivers or triggers for the LN-restricted expression of PTA by LNSCs, and how this is subdivided into specific cell types in subanatomic locations, has not yet been explored.
Exogenous Antigen Cross-Presentation in LNSCs
As polarized cells with active endocytotic and transcytotic pathways, LNSCs actively take up exogenous molecules and some subsets process antigens for cross-presentation and cross-priming of antigen-specific CD8+ T cells. This was confirmed in several models using OVA as a model exogenous antigen, as detected by the 25d1.16 antibody, which recognizes the MHC-I epitope of OVA complexed with MHC-I, SIINFEKL:MHC-I, on APCs. OVA derived from OVA-expressing B16F10 tumors was found to be displayed on MHC-I in LECs in the tumor and the draining LN (158). Similarly, SIINFEKL:MHC-I complexes were confirmed on LECs incubated with synthetic nanoparticles bearing OVA-derived long peptides in vitro (148, 158). In these studies, OVA-loaded primary LN LECs were shown to be capable of cross-priming OT-I CD8+ T cells in a TAP1-dependent manner (148).
For LN FRCs, FDCs and BECs, on the other hand, no intracellular antigen-processing pathways have been described. However, cross-presentation by BECs has been described in other organs. Primary cultures of murine aortic BECs have been shown to cross-present exogenous male antigen to a T cell hybridoma cell line MHH, specific for the MHC-I (Db) restricted male antigen HYUty (159). LSECs also present exogenous antigen in a TAP1-dependent manner (130, 160, 161). This suggests that LN BECs may also cross-present exogenous antigens on MHC-I.
Active exogenous antigen (protein and particle) uptake and degradation, a necessary upstream process for exogenous antigen presentation including cross-presentation, has been documented in LNSCs. In addition to protein and particle uptake by LECs in vitro (147, 148), fluorescently labeled OVA has been detected in LYVE-1+ LECs within minutes of intradermal injection in vivo (148). In a similar study, intracellular antigen degradation and processing was visualized by increased DQ-OVA fluorescence within 90 min of subcutaneous injection (17). A recent report suggested that LECs can retain antigen over extended time periods, with detectable OVA fluorescence in LECs even at 1–3 weeks after injection. However, the same report also described that DQ-OVA fluorescence, delivered with TLR agonists and anti-CD40, was no longer detectable in LNSC populations by flow cytometry a week after injection (97). This suggests that more than one pathway of OVA uptake or intracellular trafficking is active in LECs such that ingested antigens may be trafficked and processed differently when antigen reaches the cells together with inflammatory signals, such as TLR agonists and anti-CD40 (162–164).
Such feedback mechanisms where the composition of an ingested antigen influences antigen trafficking are known to exist on professional APCs. Scavenging receptors can bind to antigens associated with heat shock proteins and other chaperone proteins, with resulting cross-presentation of the antigen (165, 166). They can also interact with TLRs, and affect the immunological phenotypes of APCs, such as the polarization of macrophages (167). Engagement of one such receptor, the mannose receptor, has been shown to route the binding antigen to a cross-presentation pathway (168). Further facilitating MHC-I processing, subsequent ubiquitination of mannose receptor can lead to cytoplasmic escape of the mannose receptor bound antigen, enabling easier access to MHC-I loading machinery (169, 170). Although cross-presentation, involving these mechanisms, has been shown thus far in model cell lines and in DCs, it cannot be ignored that the majority of the scavenger receptor superfamily (167), other known cross-presentation receptors, and C-type lectins are abundantly expressed by skin draining and mesenteric LNSCs (Figure S1 in Supplementary Material). Together with their expression of other proteins important in cross-presentation and a number of TLRs (146) (Figure S1 in Supplementary Material), the existence of these scavenging pattern recognition receptors (PRRs) on LNSCs may play a role in the cross-presentation of exogenous antigens, thus shaping immunological outcomes.
While the importance of scavenging receptors on LNSC immunobiology is only beginning to be appreciated, their gene expression pattern may help explain the preferential accumulation of the intradermally administered OVA in LECs versus in other LNSCs (97, 148). This LNSC distribution is somewhat unexpected because OVA is a low molecular weight protein (45 kDa), and therefore FRC and BEC association would follow the expected drainage pattern (Figure 1). However, because OVA is mannosylated (171), this may preferentially drive internalization by mannose receptor CD206 (172), which amongst the LNSCs, is present only on LECs (Figure S1 in Supplementary Material). This phenomenon may also be mirrored in the uptake and persistence of Herpes Simplex Virus glycoprotein in LECs (97), as baculovirus production of antigens can result in N-glycans with high-mannose glycosylation patterns (173).
There are also a number of other major cross-presentation pathways that have yet to be investigated, and appear to play minor roles in LNSC. Although it has been shown that DCs efficiently cross-present immune complexes upon rapid internalization by Fcγ receptors (174–176), LNSCs do not seem to express high levels of activating nor inhibitory Fcγ receptors (Figure S1 in Supplementary Material). The one exception is FcRn, which is expressed across all LNSC. Better known for its IgG recycling and transport functions in neonatal (177, 178) and drug delivery contexts (179), FcRn binds to IgG Fc and albumin in acidic environments and regulates their homeostatic levels in the circulation (180). FcRn also binds antibody-coated antigens, which has implications in antigen recycling, antigen archiving, MHC-II presentation as well as MHC-I presentation (175, 181). Another example involves the complement receptors, which are constitutively active in professional APCs, but are largely absent in resting LNSCs. However, C1q receptor for phagocytosis (C1qrp) stands out as an exception (Figure S1 in Supplementary Material). In professional APCs, C1q has been suggested to favor cross-presentation as seen by priming of antigen-specific T cells, but the direct involvement of C1q receptor in this process was not demonstrated (182). Given that multiple C1q-binding partners are known (183, 184), the functional utility of C1q receptor on LNSCs remains to be clarified.
Whether any of these transcriptional findings of antigen scavenging receptor genes on resting state LNSCs have productive functions in antigen presentation is an open question. In addition, the efficacy of cross-presentation mainly studied in professional APCs depends on physical properties of the antigen (162, 185, 186). Furthermore, uptake mechanisms can determine the fate of the internalized antigen in one of the several trafficking pathways depending on the APC type and the concurrent inflammatory signals (163). These mechanisms are only beginning to become elucidated in LNSCs. Nevertheless, it is clear that more work needs to be done to understand the cross-presentation pathways in LNSCs.
Consequences of Direct MHC-I Presentation by LNSC
One of the most important hypotheses to describe the activation of CD8+ T cells involves a three-signal mechanism, which stipulates that naïve cells will need to (1) recognize their cognate antigen presented in the context of an MHC-I molecule, (2) receive a co-stimulatory signal through their co-stimulatory receptors, and (3) be primed by cytokines (187). The impact of the first signal, direct MHC-I presentation with LNSCs playing the role of APCs, has been extensively reviewed in the recent years (1, 24, 36, 155, 188–196). To summarize, peptide:MHC-I complexes on LNSC surfaces resulted in initial antigen-specific CD8+ T cell proliferation in all of the models used. Following the hypothesis, antigen presentation alone without the second signals – signaling through co-stimulatory receptors – will not lead to fully functional T cell activation. Notably, these second signals are lacking in LNSCs (Figure S3 in Supplementary Material), which have a negligible level of expression of co-stimulatory molecules compared to DCs. Furthermore, LNSCs do not upregulate the co-stimulatory molecules CD40, CD80, and CD86 following stimulation with inflammatory cues, for example, following TLR engagement (most studies have used TLR3 agonist polyI:C), or in presence of IFN-γ or TNF-α (18, 146–148, 197). While LECs upregulate the immunostimulatory molecules HVEM, CD48, and MHC-II under such conditions (197), they also concomitantly upregulate PD-L1, as do FRCs, and BECs under the same inflammatory conditions (146, 148, 197, 198). Such observations explain findings, describing the early generation apoptosis of antigen-specific CD8+ T cells following co-culture with antigen-loaded LEC in vitro (148, 197). Furthermore, LEC-educated CD8+ T cells express less IFN-γ or IL2 than DC-educated counterparts, and cannot be reactivated by the addition of IL2. In vivo LNSC antigen-presentation studies have also reported poor recovery of transferred antigen-specific T cells (135, 146, 152–155, 158), due to inhibitory LNSC-CD8+ T cell crosstalk via the PD-L1:PD-1 and MHC-II:LAG3 (135) signaling axes. Thus, with the exception of one report using a model of PTA expression (134), most studies have suggested that direct antigen presentation by LNSCs, specifically by LECs and FRCs, appears to promote a dysfunctionally activated phenotype in interacting CD8+ T cells, which can result in deletional tolerance.
MHC-II Presentation by LNSC
The hallmark of professional APCs, namely DCs, macrophages, and B cells, is the constitutive cell surface presence of MHC-II and their proven ability for antigen processing and presentation (133). Constitutive MHC-II expression is restricted to a small number of cells of the immune system. Nonetheless, as reviewed recently (199, 200), there are now many different cell types from both hematopoietic and non-hematopoietic origins that can indeed express MHC-II and interact with CD4+ T cells in the periphery.
MHC-II Expression by LNSCs
LNSCs constitute such non-professional APC cell types that express MHC-II. Unlike MHC-I molecules, however, MHC-II expression is highly regulated. The class II transactivator (CIITA) is the master regulator for the expression of MHC-II. This transcription factor forms a DNA-binding multiprotein complex that controls MHC-II expression in all MHC-II expressing cell types (201). CIITA expression itself is regulated by cell-specific promoters, of which three have been identified, namely pI, pIII, and pIV. Amongst them, the promoter pIV is essential for driving constitutive CIITA expression in thymic epithelial cells and for mediating IFN-γ-dependent MHC-II expression in cells of non-hematopoietic origin, such as endothelial cells, epithelial cells, fibroblasts, and astrocytes (202). Although MHC-II expression in human umbilical vascular and foreskin endothelial cells can be induced by lymphocyte adhesion in a CIITA-independent fashion (203–205), recent evidence demonstrates the major role of pIV-driven CIITA for MHC-II regulation in LNSCs.
The first observation of MHC-II expression on endothelial cells was reported more than 30 years ago (206). The capacity of human blood and lymphatic endothelia to express MHC-II was believed to be linked to cytokine stimulation via IFN-γ or TNF-α (207–209). For example, MHC-II expression on an immortalized murine FRC cell line also depended on IFN-γ (210). In recent studies, it was demonstrated that BECs, but not LECs from skin explants, constitutively express MHC-II, revealing functional differences prompted by environmental factors (211). Even more recently, steady-state MHC-II expression in LN LECs, BECs, and FRCs has been reported at both transcriptional and protein expression levels (8, 91, 134, 147). By using transgenic mouse models lacking the different CIITA promoters, steady-state levels of MHC-II molecules on the surface of LNSCs were demonstrated to be a combination of IFN-γ-inducible, pIV-driven CIITA basal activity, and acquired MHC-II complexes from DCs (198). In vivo, LNSCs from CIITA pIV−/− mice and IFN-γ receptor-deficient mice exhibit an identical MHC-II profile that was above background levels, pointing to other contributions. This suggests that differences in MHC-II expression levels observed in published studies may arise from environmental factors, for example, from differences in animal facilities. In addition to endogenous pIV-mediated MHC-II expression, LNSCs are capable of acquiring MHC-II from DCs as demonstrated in irradiated MHC-II-deficient mice re-constituted with MHC-II-expressing bone marrow (BM) precursors. In these studies, radioresistant MHC-II-deficient LNSCs displayed significant levels of surface MHC-II molecules compared to LNSCs in mice re-constituted with MHC-II-deficient BM. This is supported in vitro by observations that following removal of hematopoietic cells, LEC and FRC cultures lack any expression of MHC-II unless co-cultured with DCs. In vivo, manipulation of DC numbers in LNs significantly affects levels of MHC-II expressed by LECs, BECs, and FRCs, demonstrating the existence of intercellular MHC-II transfer from DCs to LNSCs in vivo.
Antigen Transfer and LNSCs in Antigen-Specific CD4+ T Cell Responses
Antigen transfer can occur as a peptide exchange on cell surfaces as well as the transfer of peptide:MHC complexes through membrane exchange between cells.
In peptide exchange, peptide epitopes can bind directly on cell surface or early endosomal MHC molecules (212–216), where both MHC-I and II are receptive to binding lymph-borne peptides. This is particularly relevant as recent analysis of the human lymph peptidome showed a predominantly self-peptide repertoire, including products deriving from extracellular processing of proteins, rather than that of the traditional repertoire derived from intracellular MHC-II processing (43, 217–220). While these peptides have not been shown to actually bind to LNSCs, these cells are ideally localized to fully sample these low molecular weight peptides.
Membrane exchange between cells is not an uncommon occurrence in biology and immunology (221). In the LN, for example, blebs of CD169+ macrophages are acquired by innate-like T cells and NK cells under steady-state conditions (222). Peptide:MHC-I and MHC-II complexes have been shown to be transferred between DC and tumor cells (223) as well as between DCs (224). In another example, MHC-II has been demonstrated to be present on exosomes secreted by DCs (225, 226).
Between hematopoietic APCs and LNSCs, transfer of antigens and peptide:MHC-II complexes seems to play an important role in MHC-II restricted antigen presentation. Exosomal transfer appears to represent a major route of MHC-II transfer from professional APCs to LNSCs. While FDCs are not able to synthesize MHC-II themselves (227) nor phagocytose antigens (228), human tonsillar FDCs have been reported to retain exogenous MHC-II vesicles (229). By immunoelectron microscopy, they were thought to be exosomes due to their protein immunoreactivity, and were possibly peptide loaded. While the source of the exosomes was not clarified (B cells or DCs), such MHC-II peptide complexes on FDC cell surface can interact with CD4+ T cells. Exosomes were also implicated in the transfer of peptide:MHC-II complexes from DCs to LNSCs (198). The transfer of peptide:MHC-II complexes from DCs, but not from B cells or macrophages, maintained the steady-state levels of MHC-II on FRCs, LECs, and BECs (198). Consistent with known surface marker expression patterns on LNSCs, if the transfer of DC co-stimulatory molecules occurred, it did not result in detectable forms.
Antigen transfer between LECs and DCs is however, not restricted to one direction. Indeed, the transfer of transgenic LEC-specific PTA to hematopoietic cells has been described (135). Neither membrane-bound nor cytoplasmically expressed PTAs were presented by LECs to prime antigen-specific CD4+ T cell responses (135). This was attributed to the orders of magnitude lower expression of H2-M in LECs compared to professional APCs, which is required to free the MHC-II groove for peptide binding. Instead, the model PTA β-gal expressed by LECs was shown to be loaded onto MHC-II in hematopoietic cells (135). While the exchange mechanism is still open to examination, it was reported not to be dependent on recognition of apoptotic cells or DC phagocytosis. On the other hand, FRCs and BECs express the adaptor molecule H2-DM under steady-state and upon inflammation (8, 134) (Figure S2 in Supplementary Material). Despite this key difference, it remains unclear whether FRCs or BECs may act solely as PTA reservoirs or, in addition, can directly present antigen and impact CD4+ T cells. Supporting direct CD4+ T cell priming by FRCs, the aforementioned immortalized FRC cell line was shown to induce CD4+ T cell proliferation in vitro (210).
These complementary bidirectional observations highlight the close relationship and communication between professional APCs and LNSCs to enable MHC-II presentation by LNSCs.
As the nature and regulation of MHC-II expression in LNSC has only recently begun to be elucidated, the functional contributions of LNSC antigen presentation to CD4+ T cells remain unclear in part due to its complexity. Lack of measurable productive T cell responses has been one of the major difficulties preventing the clarification of the impact of antigen presentation by LNSCs on CD4+ T cell outcome. As for CD8+ T cell responses, the absence of co-stimulatory signals, such as CD80 or CD86 and the constitutive expression of PD-L1 by LNSCs (197, 198), preclude the possibility of functional effector CD4+ T cell priming. In other words, MHC-II expression is mandatory but not sufficient to elicit a measurable CD4+ T cell response. In this regard, it has been shown that human LN-derived LECs fail to induce allogeneic CD4+ T cell proliferation even after IFN-γ stimulation (147). In these particular in vitro settings, LECs were unable to induce proliferation of either naïve or memory CD4+ T cells. Likewise, DC-acquired peptide:MHC-II complexes presented by murine LNSCs to cognate CD4+ T cells not only failed to promote T cell proliferation but also, in the case of LECs, induced increased cell death in an antigen-dependent manner. Moreover, this process led to CD4+ T cell unresponsiveness to further restimulation with anti-CD3 and anti-CD28 antibodies (198). These challenges present a major obstacle in understanding the relative contributions of acquired peptide:MHC-II complexes and endogenous MHC-II molecules to CD4+ T cell responses in vivo.
A recent study described the role of LNSC MHC-II in maintaining antigen-specific regulatory T cells (Tregs) (134). An elegant model of LN transplantation was used, where MHC-II-deficient LN is transplanted into wild-type recipients (Table S1 in Supplementary Material). In this model, the LNSCs remain MHC-II deficient, while MHC-II-sufficient hematopoietic cells repopulate the transplanted LN. This local absence of MHC-II expression in LNSCs was directly correlated with a reduction in Treg numbers. Importantly, only local Treg numbers were affected, and neither wild-type transplant control nor endogenous distal LNs exhibited the same effects.
However, the specific contribution of each LNSC subset remains to be determined. Indeed, with regards to LEC-restricted features, such as high levels of PD-L1 expression (156) as well as non-overlapping PTA expression in LECs, BECs, and FRCs (146, 152–155), it is likely that each LNSC subtype is capable of impacting CD4+ T cell outcomes, including Treg development and differentiation. Moreover, antigen-processing and MHC-II-mediated antigen presentation by LNSCs still needs to be carefully dissected, especially in a context where antigen exchange between neighboring cells has been demonstrated.
In summary, LNSCs possess important roles under physiological conditions, and the function of these cells as APCs also appears to be crucial during ongoing immune responses and inflammatory conditions. The fact that the main stromal cell subsets indeed upregulate MHC-II expression in a IFN-γ-dependent manner implies a greater ability of these cells to influence CD4+ T cell responses in inflamed LNs. In fact, LNSCs have been suggested to constrain CD4+ T cell expansion after viral challenge (230). The nature of such effects, however, has not been deeply investigated and the conclusions remain to be clarified.
Shared Responsibilities
It is striking that many of the mechanisms and functional consequences of antigen presentation are shared across the LNSC subsets, with the exception of FDCs. The described MHC-I and II presentation are much the same between FRCs and LECs, and although less studied, some aspects are also similar in BECs. These three LNSC subtypes are IFN-γ responsive through expression of IFN-γ receptor (8). In FRCs and LECs, PD-L1 expression and lack of co-stimulation seem to be important in their education of CD8+ T cells. Furthermore, key chemokine and cytokine expression supporting T cell education is distributed amongst FRCs, LECs, and BECs, for example, CCL21 expression for attracting CCR7+ DC and T cells (FRCs, LECs, and BECs) (231), and IL7 for supporting naive T cells (FRCs and LECs) (5, 232–234), and IL15 for the development and homeostasis for naïve and memory T cells and other cells (FRCs, BECs, and DN) (235) in the LN. As the understanding of LNSC antigen presentation deepens, the shared characteristics between LNSCs must be reconstructed with the LN microarchitecture and antigen flow in mind.
Challenges in Studying LNSC Antigen Presentation
Lack of suitable in vivo models presents a major difficulty in demonstrating the relative importance of antigen presentation by LNSC subsets. Directly studying the role of antigen presentation in each LNSC subset, including specific targeting, capture, and processing of exogenous antigens is not easily accomplished in vivo (Table S1 in Supplementary Material). While depletion of hematopoietic cell subtypes is routine and well-established, ablation of any of the stromal subsets results in a major disruption of the LN microenvironment and organization, and any immunological effects of LNSC antigen presentation become difficult to interpret due to experimental artifacts. In this regard, while care must be taken to exclude the contribution of radioresistant APCs (134, 236), BM chimeras and LN transplants represent less drastic alternatives that have begun to enable the deconvolution of hematopoietic from stromal contributions to antigen presentation. Concerning MHC-II, the CIITA pIV−/− mice can be used as a model for MHC-II abrogation in all non-hematopoietic cells, including LNSCs (237, 238).
Supposing the antigen presentation can be limited in one LNSC subtype, another challenge in studying antigen presentation by LNSCs is the reliance on T cell proliferation as a practical read-out. In addition to the balance favoring coinhibitory to co-stimulatory molecules, LNSCs can suppress T cell activation in a contact-dependent manner (18–20). As T cell proliferation integrates several outcomes deriving from LNSC and T cell contact, not limited to antigen peptide:MHC and T cell receptor recognition, other reporting systems of direct antigen presentation need to be developed.
What have been rewarding are the models of LNSC subset-restricted PTA expression (Table S1 in Supplementary Material). Direct presentation of PTAs by LNSCs has been established largely by using two animal disease models: inflammatory bowel disease (146, 152, 239) and autoimmune vitiligo (153, 154, 156, 197, 240), paired with T cells harvested from respective antigen-specific TCR transgenic mice.
Useful in addressing the challenge of LNSC-restricted antigen presentation and PTA expression, a number of mouse models are available in which antigen-presentation machinery can be knocked out or altered in a stromal cell-specific manner. This selectivity can be accomplished using the Cre-lox recombination system, in which target genes marked by loxP recombination sites are excised by a Cre recombinase enzyme expressed under a cell-specific promoter (241). Especially in conditionally inducible form, these transgenic mouse systems present a useful means to study the immunological roles restricted to each of the stromal subsets. Examples of this, some of which have already been mentioned above, include the CCL19-Cre system for FRC-specific knockouts (242), and the LYVE-1-Cre and inducible Prox1-CreERT2 systems for LEC-targeted knockouts (27, 243, 244). As with any genetic model, there are caveats. For example, using LYVE-1-Cre, deletion of floxed genes has been observed in some hematopoietic cells (27). While sparing the major APCs of the LNs (135), PROX-1, which is essential for lymphatic development and maintenance (245), have been shown to be expressed by several other cell types in multiple organs (246). Depending on the experimental goals and objectives of a study, both Cre lines may need to be used as complementary models to evaluate the immunological outcomes of LEC-specific PTA expression (135). Podoplanin-Cre has been described to target both LECs and FRCs, and has contributed to the discovery of a new stromal subset found in spleen (247). For BECs, Tie2-Cre (248) and Flk1-Cre (249) have been described (244). It must be noted that both Tie2 and Flk1 are expressed in cells of hematopoietic origin (250, 251), requiring BM chimeras to limit the effect of Cre recombination to stromal cells. FDCs have been targeted by using Cd21-Cre (252), and their developmental origins have been explored by crossing Cd21-Cre with the Ubow mice in fate-mapping studies (10). Again, it is notable that Cd21-Cre-mediated deletion was also observed in other tissues, including the forebrain (253).
In a few cases, these LNSC subset-targeting Cre mice have been crossed with available mouse models carrying floxed MHC-I and II genes. In fact, MHC-II flox has been successfully used with Tie2-Cre to eliminate MHC-II expression in both the hematopoietic and endothelial lineages, while MHC-II expression remains unaffected in the thymic epithelial cells (250). The naïve CD4+ T cell numbers were comparable with Cre-negative littermates, while numbers of Tregs and antigen-experienced cells were significantly decreased in the knockouts. As for generating MHC-I conditional knockouts to be used with Cre systems, mice with β2M floxed allele can be generated in two steps (254).
Aside from genetically modified animal models, other mechanisms of specifically targeting molecules responsible for antigen uptake and processing in LNSC subsets have not been actively explored (255). Once targeting can be achieved, relevant methods exist to modulate MHC-I antigen presentation, for example, to inhibit MHC-I loading of peptides using viral inhibitors (128).
Another limitation of drawing any general conclusions for antigen presentation by LNSC derives from the fact that often, LNSC are defined as bulk populations, based on CD45, gp38 and CD31 expression. Recent papers have delved deeper into nuances of the LNSC subtypes beyond those defined by these surface markers. The findings demonstrate the need to further subdivide LNSC based on their anatomical location, molecular phenotype, and functional differences (2, 4, 38). For instance, the grossly defined FRC population contains marginal reticular cells (256), and the BEC population can be functionally distinguished as being composed of HEVs and capillary endothelial cells (91). Finally, aside from the recently identified integrin-α7-expressing pericytes (8), and gp38− FDC (2, 11), the DN fraction (gp38− CD31−) has yet to be fully defined. Thus far, no animal models address these subdivisions. More transgenic mouse models precisely engineered (257, 258) to study antigen presentation by LNSC subsets may become available in the near future.
Alternatively, in vitro models may be used to investigate antigen presentation in LNSC subtypes. These models present technical difficulties of their own, especially when using sorted primary LNSCs. LNSC subtypes are not only defined by surface markers and cytokine production profiles but also by their biochemical and biophysical communication with other cells. This interdependence can be indirectly appreciated from transcriptome studies comparing freshly isolated versus cultured human LECs and BECs (211), which demonstrated that some key characteristic gene expression signatures of LECs and BECs were lost following in vitro culture. Another study compared freshly isolated human LN FRCs to LN FRCs propagated over 60 days in culture, and arrived at similar conclusions (259).
Such loss of phenotype may be potentially averted in several ways. For instance, physiological complexity can be introduced to existing in vitro LNSC co-culture systems by engineering an environment that recapitulates physiologically inferred mechanical tension and fluid flow (260). For primary FRCs, CCL21 expression, which is otherwise lost following in vitro culture, was rescued by growing the cells under conditions mimicking interstitial flow rates on a 3D composite matrix capable of mechanically supporting cell tension (261). Moreover, FRC co-culture with lymphocytes in a semi-3D nylon mesh promoted the generation of a robust reticular network structure (15). This further highlights that LNSC differentiation states and likely antigen-presentation properties are interdependent on biophysical and biochemical cues between the neighboring cells and the microenvironment.
Inescapably, the LN architecture must be reconstructed to grasp the relevant cell-cell interdependencies in the context of their LN locations. Established slice cultures (115), development of organoid cultures and complex in vitro systems (262) improves on conventional 2D co-culture systems to interrogate LNSC antigen presentation and immune cell education. Furthermore, dissecting the microanatomical differences of the LNSC subsets using techniques, such as histo-cytometry (23, 263), which yields multiparameter cell marker analysis coupled with anatomical location, may prove to be informative in understanding the role of LNSC antigen presentation within the complex coordination of antigen, DC and T cell interactions (264).
Nevertheless, in vivo models will remain a cornerstone in understanding LNSC antigen presentation as the biological complexity remains irreproducible in vitro. This includes the dynamics of the cellularity of the LNSCs, where each LNSC population differentially expands and contracts during an immune response upon viral infection and vaccination (94, 230). Two-photon intravital microscopy has greatly contributed to our understanding of the kinetics of antigen–cell and cell–cell interactions in the initiation of immune responses in the LN (265). These studies make use of genetically engineered mouse models, which express fluorescent proteins in specific stromal cells subsets: for LECs (246, 266–269), vascular endothelial cells (270, 271), and for LECs and BECs simultaneously (78, 272) [for review, see Ref. (244)]. So far, these mice have generally been used for real-time observations in organs other than the LN. However, the same mouse models may greatly contribute to studying the dynamic interactions of antigens, DCs and T cells with stromal cells (273, 274). By capturing early T cell interactions with antigen-presenting LNSCs, the issues of using T cell proliferation as a read-out may be avoided.
Much remains to be explored to understand the role of LNSCs as APCs in both pathological and physiological processes, such as wound healing, cancer, transplantation, and autoimmunity. Last but not least, most of the observations of LNSC antigen presentation have been made in mice, and need to be addressed in human LNs and diseases. In summary, phenotypic differences inherent to various LNSC subtypes, their anatomic locations, and their interactions with surrounding hematopoietic and stromal cells collectively determine their ability to coordinate antigen presentation and T cell education. Grasping the contributions of individual LNSC subtypes within this complex system remains elusive due to significant technical hurdles, thus emerging tools, models, and technologies are likely to be crucial to the elucidation of key mechanisms.
Relative Contributions of LNSC Antigen Presentation on Immunological Outcomes
While there is accumulating evidence that LNSCs modulate peripheral tolerance and the initiation and resolution of immune responses through both direct and indirect mechanisms, the relative importance of LNSC versus DC antigen presentation has not been integrated into a larger model. Several mechanisms of direct interactions between LNSCs and APCs have been described, including active antigen transfer and direct sampling of antigens on LNSCs by APCs. Depending on the model system, hematopoietic APCs are highly important in LNSC-harbored antigen. For example, in BM chimera studies, there was no antigen-specific T cell proliferation if LECs are, but the hematopoietic APCs are not capable of presenting antigen (135). It is certain that DCs are the dominant actors in antigen presentation and tolerance induction against peripheral self-antigens (275). In addition to CD11c+ DCs, extrathymic Aire-expressing cells (eTACs) also appear to play an important role, namely in peripheral tolerance. eTACs are a radioresistant CD45lo CD11clo CD11blo MHC-IIhi PTA-expressing APC of BM origin, which reside in the inter-follicular region in the LNs (276). eTACs share many features with antigen-presenting LNSCs, such as similarly low expression of CD80 and CD86, and high expression of PD-L1. This extends to their ability to initiate CD8+ T cell deletion (276). They further deactivate CD4+ T cell responses independently of the enriched antigen-specific Treg population (277).
Nevertheless, it is clear that the tipping point to initiate an immune response or to tolerate an antigen is dependent on a fine balance between antigen presentation by LNSCs or by APCs of hematopoietic origin.
Initiation and Maintenance of the Immune Response
The importance of LNSC antigen presentation can be appreciated in studies of infections in BM chimeric mice. It was shown that during pathogen infection, non-hematopoietic cells have a noticeable role in initiating antigen-specific CD4+ T cell responses (230) and CD8+ T cell proliferation (278, 279). When LNSCs could not present antigen, the overall clonal expansion was diminished in several viral infection models, suggesting a role of LNSC antigen presentation for early clonal CD4+ and CD8+ T cell expansion. This initial clonal expansion in the CD4+ T cell populations were 10× less than when the antigen is sterile, i.e., non-replicating (230), which points to the importance of the amount of available antigen. On the other hand, to sustain the CD8+ T cell expansion, hematopoietic APCs were reported to be necessary (279).
In these settings, even when focusing on the levels of LNSC antigen retention and presentation alone, the interpretation is difficult and does not necessarily correlate with the T cell proliferation response. When compared side by side, the magnitude of LNSC-primed CD8+ T cell responses differs with the infectious agent (279). The least T cell proliferation upon LNSC antigen presentation was seen in Listeria monocytogenes infections (279), which is thought to mainly target macrophages, DCs, and hepatocytes (280) and not be harbored in LECs (97). However, the greatest effect of LNSC antigen presentation on T cell proliferation was shown for the LCMV Armstrong strain, which unlike clone 13 of the same virus, does not extensively infect FRCs (95). Rather, like in an infection with strain WE, the Armstrong infection results in FRC network destruction and decreased long-term antigen presence in the LN (95, 96). Intermediate contributions of LNSC antigen presentation were supported by T cell responses to vaccinia virus, which is archived in LECs (97), and in VSV infections, both of which infect sessile cells just below the SCS (281). Thus, systematic analysis must be conducted to understand how LNSC antigen retention and presentation aid the initiation of the antigen-specific T cell response.
Distinct T Cell Differentiation States and Immunological Protection
In light of existing data, an attractive hypothesis is that LNSC antigen presentation contributes to the generation of diverse T cell phenotypes in the face of antigen-specific challenge. This hypothesis is inspired by observations that CD8+ T cells primed by cross-presenting LSECs, which were previously thought to be tolerized (282), then were shown to be reactivated under viral challenge (283). LSECs occupy a large surface area exposed to blood that carries external food and commensal bacterial antigens and are known to cross-present exogenous antigens (130, 160, 161). Similar to MHC-I restricted antigen presentation by LNSCs, antigen cross-presentation by LSECs resulted in tolerized CD8+ T cells, where LSEC PD-L1 expression was important (284, 285). The surviving LSEC-educated T cells had an antigen-experienced central memory-like phenotype in secondary lymphoid organs (283). Furthermore, LSEC-educated T cells could be reactivated in vitro and in vivo in an antigen-specific manner in the presence of CD28 and IL12, and they could participate in an antigen-specific viral challenge with recombinant adenovirus (283).
Given the similarities between LSECs and LNSCs, the observation that LSEC-educated T cells are not terminally tolerized, but are primed for reactivation, leads to the question of whether or not LNSC-educated T cells behave comparably. Earlier, we described how LNSCs appear to induce a dysfunctionally activated phenotype in interacting T cells, through their ability to present antigens but not the co-stimulatory signals required to induce T cell activation. Like LSEC-educated CD8+ T cells, LNSC-educated and tolerized CD8+ T cells in the iFABP-tOVA PTA model were reactivated when challenged with intravenously injected VSV-OVA (286) (Table S1 in Supplementary Material). The iFABP-tOVA mouse line with lower PTA expression levels resolved the infection and retained a cognate CD8+ T cell population after a VSV-OVA challenge, while the line with higher antigen expression levels suffered death by T cell mediated destruction of the PTA-expressing intestinal epithelial cells (286). These T cells were capable of target lysis, yet unable to secrete IFN-γ and TNF-α, and resided in inter-epithelial layers (286). For lack of an existing phenotypic category, the authors called this phenotype of the LNSC-primed T cell, split-anergy.
Concluding Remarks
The immunological roles of LNSCs cannot be dissociated from the structural microarchitecture of the LN, albeit in a much more nuanced manner than previously appreciated. LNSCs can act at different levels to promote but also regulate antigen presentation both directly and indirectly by interacting with APCs and T cells. Moreover, the interactions and relationships between the different stromal cells together with DCs (11, 112, 287) are crucial in framing the immunological roles of LNSCs, which result in the physical and biochemical modulation of the LN microenvironment.
The immune system relies heavily on suppression under resting conditions and during resolution of immune responses. It is not surprising, then, that more layered and likely redundant cellular actors and intercellular interactions are involved in suppression of undesirable immune activation. The LNSC-educated T cells find a place in the recently articulated adaptation model of immunity that goes beyond the self/non-self recognition and danger theory to explain tolerance during an ongoing immune response: (1) recognition of self/non-self/mimic of self by T cells, (2) activation of the immune response, and (3) efficacy of the immune response (288). Within this framework, the efficacy of the immune response is programed by the expression and engagement of the sets of adaptation receptor–ligand pairs on the T cells and their interacting APCs/LNSCs (for example, PD-1/PD-L1, other co-stimulatory and coinhibitory molecules, adhesion molecules, and pattern recognition molecules) (288). At least in mouse models, LNSCs fulfill the first two requirements: antigen presentation to T cells, and the initiation of antigen-specific T cell responses. Furthermore, LNSCs are capable of interacting with and signaling through distinct sets of adaptation receptors on T cells from those on hematopoietic APCs (147). Interdependence of co-stimulatory and coinhibitory pathways generates another layer of complexity (197).
Antigen presentation in the LN takes place in a wide spectrum of differently timed and equipped antigen-presenting LNSCs and hematopoietic APCs, not to mention that the encounter with lymphocytes is highly influenced by the inflammation responsive LN microarchitecture. This results in ever-expanding and plastic phenotypes of antigen-experienced T cells. As more becomes known about the roles of LNSCs in antigen-specific responses, their contributions to adaptive immune responses must be considered especially when applying this knowledge to the engineering of vaccines and immunotherapies.
Author Contributions
SH has developed the concept, wrote the manuscript, prepared the figure, supplemental figures and the table, and critically read, revised, and approved the manuscript. JD has co-developed the concept, co-wrote the manuscript and the supplemental table, critically read, revised, and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank E. Vokali, S. S. Yu, C. M. Card, V. Triacca, E. Güç, L. Jeanbart, C. F. Buchanan, M. A. Swartz, and S. Hugues for insightful discussions and their critical reading of the manuscript. The authors are grateful to Prix Leenaards awarded to M. A. Swartz and S. Hughes (SH, JD), and the Swiss National Science Foundation grant CR2312-143754 awarded to M. A. Swartz and S. Kunz (SH). This review has benefited from the data assembled by the ImmGen Consortium (139).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2015.00446
Abbreviations
APC(s), antigen-presenting cell(s); BEC(s), blood endothelial cell(s); BM, bone marrow; CIITA, class II transactivator; DC(s), dendritic cell(s); DN, double negative; DT, diptheria toxin; eTAC(s), extrathymic Aire-expressing cell(s); FDC(s), follicular dendritic cell(s); FRC(s), fibroblastic reticular cell(s); gp38, podoplanin; HEV(s), high endothelial venule(s); IFN-γ, interferon-γ; LEC(s), lymphatic endothelial cell(s); LN(s), lymph node(s); LNSC(s), lymph node stromal cell(s); LSEC(s), liver sinusoidal endothelial cell(s); MHC(s), major histocompatibility complex(es); OVA, chicken egg ovalbumin; PLVAP, plasmalemma vesicle-associated protein; PTA(s), peripheral tissue antigen(s); SCS(s), subcapsular sinus(es); TAP, transporter associated with antigen processing; TLR(s), toll-like receptor(s); TNF-α, tumor necrosis factor-α; Treg(s), regulatory T cell(s); VEGF, vascular endothelial growth factor; VSV, vesicular stomatitis virus.
References
1. Malhotra D, Fletcher AL, Turley SJ. Stromal and hematopoietic cells in secondary lymphoid organs: partners in immunity. Immunol Rev (2013) 251:160–76. doi:10.1111/imr.12023
2. Roozendaal R, Mebius RE. Stromal cell-immune cell interactions. Annu Rev Immunol (2011) 29:23–43. doi:10.1146/annurev-immunol-031210-101357
3. Junt T, Scandella E, Ludewig B. Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat Rev Immunol (2008) 8:764–75. doi:10.1038/nri2414
4. Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol (2009) 9:618–29. doi:10.1038/nri2588
5. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol (2007) 8:1255–65. doi:10.1038/ni1513
6. Fletcher AL, Malhotra D, Acton SE, Lukacs-Kornek V, Bellemare-Pelletier A, Curry M, et al. Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front Immunol (2011) 2:35. doi:10.3389/fimmu.2011.00035
7. Broggi MA, Schmaler M, Lagarde N, Rossi SW. Isolation of murine lymph node stromal cells. J Vis Exp (2014):e51803. doi:10.3791/51803
8. Malhotra D, Fletcher AL, Astarita J, Lukacs-Kornek V, Tayalia P, Gonzalez SF, et al. Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol (2012) 13:499–510. doi:10.1038/ni.2262
9. Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell (2012) 150:194–206. doi:10.1016/j.cell.2012.05.032
10. Jarjour M, Jorquera A, Mondor I, Wienert S, Narang P, Coles MC, et al. Fate mapping reveals origin and dynamics of lymph node follicular dendritic cells. J Exp Med (2014) 211:1109–22. doi:10.1084/jem.20132409
11. Kumar V, Dasoveanu DC, Chyou S, Tzeng TC, Rozo C, Liang Y, et al. A dendritic-cell-stromal axis maintains immune responses in lymph nodes. Immunity (2015) 42:719–30. doi:10.1016/j.immuni.2015.03.015
12. Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity (2006) 25:989–1001. doi:10.1016/j.immuni.2006.10.011
13. Alvarenga HG, Marti L. Multifunctional roles of reticular fibroblastic cells: more than meets the eye? J Immunol Res (2014) 2014:402038. doi:10.1155/2014/402038
14. Ulvmar MH, Werth K, Braun A, Kelay P, Hub E, Eller K, et al. The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat Immunol (2014) 15:623–30. doi:10.1038/ni.2889
15. Katakai T, Hara T, Lee JH, Gonda H, Sugai M, Shimizu A. A novel reticular stromal structure in lymph node cortex: an immuno-platform for interactions among dendritic cells, T cells and B cells. Int Immunol (2004) 16:1133–42. doi:10.1093/intimm/dxh113
16. Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, et al. Visualizing dendritic cell networks in vivo. Nat Immunol (2004) 5:1243–50. doi:10.1038/ni1139
17. Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, et al. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity (2005) 22:19–29. doi:10.1016/j.immuni.2004.11.013
18. Khan O, Headley M, Gerard A, Wei W, Liu L, Krummel MF. Regulation of T cell priming by lymphoid stroma. PLoS One (2011) 6:e26138. doi:10.1371/journal.pone.0026138
19. Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, et al. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol (2011) 12:1096–104. doi:10.1038/ni.2112
20. Siegert S, Huang HY, Yang CY, Scarpellino L, Carrie L, Essex S, et al. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PLoS One (2011) 6:e27618. doi:10.1371/journal.pone.0027618
21. Mionnet C, Mondor I, Jorquera A, Loosveld M, Maurizio J, Arcangeli ML, et al. Identification of a new stromal cell type involved in the regulation of inflamed B cell follicles. PLoS Biol (2013) 11:e1001672. doi:10.1371/journal.pbio.1001672
22. Forster R, Braun A, Worbs T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol (2012) 33:271–80. doi:10.1016/j.it.2012.02.007
23. Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity (2012) 37:364–76. doi:10.1016/j.immuni.2012.07.011
24. Card CM, Yu SS, Swartz MA. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Invest (2014) 124:943–52. doi:10.1172/JCI73316
25. Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, et al. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J Immunol (2009) 183:1767–79. doi:10.4049/jimmunol.0802167
26. Grigorova IL, Schwab SR, Phan TG, Pham TH, Okada T, Cyster JG. Cortical sinus probing, S1P1-dependent entry and flow-based capture of egressing T cells. Nat Immunol (2009) 10:58–65. doi:10.1038/ni.1682
27. Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med (2010) 207:17–27. doi:10.1084/jem.20091619
28. Ito K, Morimoto J, Kihara A, Matsui Y, Kurotaki D, Kanayama M, et al. Integrin alpha9 on lymphatic endothelial cells regulates lymphocyte egress. Proc Natl Acad Sci U S A (2014) 111:3080–5. doi:10.1073/pnas.1311022111
29. Miyasaka M, Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol (2004) 4:360–70. doi:10.1038/nri1354
30. Stein JV, Rot A, Luo Y, Narasimhaswamy M, Nakano H, Gunn MD, et al. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J Exp Med (2000) 191:61–76. doi:10.1084/jem.191.1.61
31. Baekkevold ES, Yamanaka T, Palframan RT, Carlsen HS, Reinholt FP, Von Andrian UH, et al. The CCR7 ligand elc (CCL19) is transcytosed in high endothelial venules and mediates T cell recruitment. J Exp Med (2001) 193:1105–12. doi:10.1084/jem.193.9.1105
32. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med (2011) 208:2497–510. doi:10.1084/jem.20111449
33. El Shikh ME, Pitzalis C. Follicular dendritic cells in health and disease. Front Immunol (2012) 3:292. doi:10.3389/fimmu.2012.00292
34. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: origin, phenotype, and function in health and disease. Trends Immunol (2014) 35:105–13. doi:10.1016/j.it.2013.11.001
35. Yau IW, Cato MH, Jellusova J, De Mendoza T, Brink R, Rickert RC. Censoring of self-reactive B cells by follicular dendritic cell-displayed self-antigen. J Immunol (2013) 191:1082–90. doi:10.4049/jimmunol.1201569
36. Turley SJ, Fletcher AL, Elpek KG. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat Rev Immunol (2010) 10:813–25. doi:10.1038/nri2886
37. Qi H, Kastenmuller W, Germain RN. Spatiotemporal basis of innate and adaptive immunity in secondary lymphoid tissue. Annu Rev Cell Dev Biol (2014) 30:141–67. doi:10.1146/annurev-cellbio-100913-013254
38. Chang JE, Turley SJ. Stromal infrastructure of the lymph node and coordination of immunity. Trends Immunol (2015) 36:30–9. doi:10.1016/j.it.2014.11.003
39. Fletcher AL, Acton SE, Knoblich K. Lymph node fibroblastic reticular cells in health and disease. Nat Rev Immunol (2015) 15:350–61. doi:10.1038/nri3846
40. Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol (2003) 3:984–93. doi:10.1038/nri1246
41. Bajenoff M. Stromal cells control soluble material and cellular transport in lymph nodes. Front Immunol (2012) 3:304. doi:10.3389/fimmu.2012.00304
42. Hansen K, D’alessandro A, Clement CC, Santambrogio L. Lymph formation, composition and circulation: a proteomics perspective. Int Immunol (2015) 27:219–27. doi:10.1093/intimm/dxv012
43. Clement CC, Cannizzo ES, Nastke MD, Sahu R, Olszewski W, Miller NE, et al. An expanded self-antigen peptidome is carried by the human lymph as compared to the plasma. PLoS One (2010) 5:e9863. doi:10.1371/journal.pone.0009863
44. Gonzalez SF, Pitcher LA, Mempel T, Schuerpf F, Carroll MC. B cell acquisition of antigen in vivo. Curr Opin Immunol (2009) 21:251–7. doi:10.1016/j.coi.2009.05.013
45. Harwood NE, Batista FD. The antigen expressway: follicular conduits carry antigen to B cells. Immunity (2009) 30:177–9. doi:10.1016/j.immuni.2009.01.004
46. Cyster JG. B cell follicles and antigen encounters of the third kind. Nat Immunol (2010) 11:989–96. doi:10.1038/ni.1946
47. Gonzalez SF, Degn SE, Pitcher LA, Woodruff M, Heesters BA, Carroll MC. Trafficking of B cell antigen in lymph nodes. Annu Rev Immunol (2011) 29:215–33. doi:10.1146/annurev-immunol-031210-101255
48. Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, et al. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity (2009) 30:264–76. doi:10.1016/j.immuni.2008.12.014
49. Bajenoff M, Germain RN. B-cell follicle development remodels the conduit system and allows soluble antigen delivery to follicular dendritic cells. Blood (2009) 114:4989–97. doi:10.1182/blood-2009-06-229567
50. Pape KA, Catron DM, Itano AA, Jenkins MK. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity (2007) 26:491–502. doi:10.1016/j.immuni.2007.02.011
51. Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science (2006) 312:1672–6. doi:10.1126/science.1125703
52. Itano AA, Mcsorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity (2003) 19:47–57. doi:10.1016/S1074-7613(03)00175-4
53. Gretz JE, Norbury CC, Anderson AO, Proudfoot AE, Shaw S. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med (2000) 192:1425–40. doi:10.1084/jem.192.10.1425
54. Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, et al. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med (2001) 194:1361–73. doi:10.1084/jem.194.9.1361
55. Rantakari P, Auvinen K, Jappinen N, Kapraali M, Valtonen J, Karikoski M, et al. The endothelial protein PLVAP in lymphatics controls the entry of lymphocytes and antigens into lymph nodes. Nat Immunol (2015) 16:386–96. doi:10.1038/ni.3101
56. Herrnberger L, Hennig R, Kremer W, Hellerbrand C, Goepferich A, Kalbitzer HR, et al. Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage. PLoS One (2014) 9:e115005. doi:10.1371/journal.pone.0115005
57. Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity (2015) 42:172–85. doi:10.1016/j.immuni.2014.12.024
58. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol (2013) 200:373–83. doi:10.1083/jcb.201211138
59. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol (2010) 10:787–96. doi:10.1038/nri2868
60. Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity (2007) 27:160–71. doi:10.1016/j.immuni.2007.06.007
61. Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature (2007) 450:110–4. doi:10.1038/nature06287
62. Barral P, Polzella P, Bruckbauer A, Van Rooijen N, Besra GS, Cerundolo V, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol (2010) 11:303–12. doi:10.1038/ni.1853
63. Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Kim YA, et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol (2010) 11:427–34. doi:10.1038/ni.1856
64. Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, et al. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature (2010) 465:1079–83. doi:10.1038/nature09118
65. Coombes JL, Han SJ, Van Rooijen N, Raulet DH, Robey EA. Infection-induced regulation of natural killer cells by macrophages and collagen at the lymph node subcapsular sinus. Cell Rep (2012) 2:124–35. doi:10.1016/j.celrep.2012.06.001
66. Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature (2014) 507:519–22. doi:10.1038/nature12978
67. Saunderson SC, Dunn AC, Crocker PR, Mclellan AD. CD169 mediates the capture of exosomes in spleen and lymph node. Blood (2014) 123:208–16. doi:10.1182/blood-2013-03-489732
68. Link A, Zabel F, Schnetzler Y, Titz A, Brombacher F, Bachmann MF. Innate immunity mediates follicular transport of particulate but not soluble protein antigen. J Immunol (2012) 188:3724–33. doi:10.4049/jimmunol.1103312
69. Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol (2007) 8:992–1000. doi:10.1038/ni1494
70. Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol (2009) 10:786–93. doi:10.1038/ni.1745
71. Kourtis IC, Hirosue S, De Titta A, Kontos S, Stegmann T, Hubbell JA, et al. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS One (2013) 8:e61646. doi:10.1371/journal.pone.0061646
72. Reddy ST, Swartz MA, Hubbell JA. Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol (2006) 27:573–9. doi:10.1016/j.it.2006.10.005
73. Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol (2005) 5:617–28. doi:10.1038/nri1670
74. Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012) 12:762–73. doi:10.1038/nri3298
75. Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, Friedel RH, et al. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nat Immunol (2010) 11:594–600. doi:10.1038/ni.1885
76. Tal O, Lim HY, Gurevich I, Milo I, Shipony Z, Ng LG, et al. DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J Exp Med (2011) 208:2141–53. doi:10.1084/jem.20102392
77. Acton SE, Astarita JL, Malhotra D, Lukacs-Kornek V, Franz B, Hess PR, et al. Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity (2012) 37:276–89. doi:10.1016/j.immuni.2012.05.022
78. Nitschke M, Aebischer D, Abadier M, Haener S, Lucic M, Vigl B, et al. Differential requirement for ROCK in dendritic cell migration within lymphatic capillaries in steady-state and inflammation. Blood (2012) 120:2249–58. doi:10.1182/blood-2012-03-417923
79. Platt AM, Rutkowski JM, Martel C, Kuan EL, Ivanov S, Swartz MA, et al. Normal dendritic cell mobilization to lymph nodes under conditions of severe lymphatic hypoplasia. J Immunol (2013) 190:4608–20. doi:10.4049/jimmunol.1202600
80. Clatworthy MR, Aronin CE, Mathews RJ, Morgan NY, Smith KG, Germain RN. Immune complexes stimulate CCR7-dependent dendritic cell migration to lymph nodes. Nat Med (2014) 20:1458–63. doi:10.1038/nm.3709
81. Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med (2006) 203:2763–77. doi:10.1084/jem.20051759
82. Miteva DO, Rutkowski JM, Dixon JB, Kilarski W, Shields JD, Swartz MA. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ Res (2010) 106:920–31. doi:10.1161/CIRCRESAHA.109.207274
83. Teijeira A, Palazon A, Garasa S, Marre D, Auba C, Rogel A, et al. CD137 on inflamed lymphatic endothelial cells enhances CCL21-guided migration of dendritic cells. FASEB J (2012) 26:3380–92. doi:10.1096/fj.11-201061
84. Johnson LA, Jackson DG. The chemokine CX3CL1 promotes trafficking of dendritic cells through inflamed lymphatics. J Cell Sci (2013) 126:5259–70. doi:10.1242/jcs.135343
85. Johnson LA, Jackson DG. Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis (2014) 17:335–45. doi:10.1007/s10456-013-9407-0
86. Russo E, Nitschke M, Halin C. Dendritic cell interactions with lymphatic endothelium. Lymphat Res Biol (2013) 11:172–82. doi:10.1089/lrb.2013.0008
87. Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol (2006) 6:907–18. doi:10.1038/nri1964
88. Bachelerie F, Graham GJ, Locati M, Mantovani A, Murphy PM, Nibbs R, et al. New nomenclature for atypical chemokine receptors. Nat Immunol (2014) 15:207–8. doi:10.1038/ni.2812
89. Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol (2004) 15:603–12. doi:10.1097/01.ASN.0000113316.52371.2E
90. Fra AM, Locati M, Otero K, Sironi M, Signorelli P, Massardi ML, et al. Cutting edge: scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J Immunol (2003) 170:2279–82. doi:10.4049/jimmunol.170.5.2279
91. Lee M, Kiefel H, Lajevic MD, Macauley MS, Kawashima H, O’hara E, et al. Transcriptional programs of lymphoid tissue capillary and high endothelium reveal control mechanisms for lymphocyte homing. Nat Immunol (2014) 15:982–95. doi:10.1038/ni.2983
92. Kashiwazaki M, Tanaka T, Kanda H, Ebisuno Y, Izawa D, Fukuma N, et al. A high endothelial venule-expressing promiscuous chemokine receptor DARC can bind inflammatory, but not lymphoid, chemokines and is dispensable for lymphocyte homing under physiological conditions. Int Immunol (2003) 15:1219–27. doi:10.1093/intimm/dxg121
93. Astarita JL, Acton SE, Turley SJ. Podoplanin: emerging functions in development, the immune system, and cancer. Front Immunol (2012) 3:283. doi:10.3389/fimmu.2012.00283
94. Yang CY, Vogt TK, Favre S, Scarpellino L, Huang HY, Tacchini-Cottier F, et al. Trapping of naive lymphocytes triggers rapid growth and remodeling of the fibroblast network in reactive murine lymph nodes. Proc Natl Acad Sci U S A (2014) 111:E109–18. doi:10.1073/pnas.1312585111
95. Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, et al. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci U S A (2007) 104:15430–5. doi:10.1073/pnas.0702579104
96. Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat Immunol (2008) 9:667–75. doi:10.1038/ni.1605
97. Tamburini BA, Burchill MA, Kedl RM. Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat Commun (2014) 5:3989. doi:10.1038/ncomms4989
98. Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med (2009) 206:1485–93. doi:10.1084/jem.20090209
99. Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity (2012) 37:199–207. doi:10.1016/j.immuni.2012.08.002
100. Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, et al. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity (2013) 38:1164–75. doi:10.1016/j.immuni.2013.02.023
101. Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol (2014) 14:495–504. doi:10.1038/nri3689
102. McCloskey ML, De Lafaille MA, Carroll MC, Erlebacher A. Acquisition and presentation of follicular dendritic cell-bound antigen by lymph node-resident dendritic cells. J Exp Med (2011) 208:135–48. doi:10.1084/jem.20100354
103. Cremasco V, Woodruff MC, Onder L, Cupovic J, Nieves-Bonilla JM, Schildberg FA, et al. B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nat Immunol (2014) 15:973–81. doi:10.1038/ni.2965
104. Denton AE, Roberts EW, Linterman MA, Fearon DT. Fibroblastic reticular cells of the lymph node are required for retention of resting but not activated CD8+ T cells. Proc Natl Acad Sci U S A (2014) 111:12139–44. doi:10.1073/pnas.1412910111
105. Jang JY, Koh YJ, Lee SH, Lee J, Kim KH, Kim D, et al. Conditional ablation of LYVE-1+ cells unveils defensive roles of lymphatic vessels in intestine and lymph nodes. Blood (2013) 122:2151–61. doi:10.1182/blood-2013-01-478941
106. Mackay F, Browning JL. Turning off follicular dendritic cells. Nature (1998) 395:26–7. doi:10.1038/25630
107. Mebius RE, Streeter PR, Breve J, Duijvestijn AM, Kraal G. The influence of afferent lymphatic vessel interruption on vascular addressin expression. J Cell Biol (1991) 115:85–95. doi:10.1083/jcb.115.1.85
108. Thomas SN, Rutkowski JM, Pasquier M, Kuan EL, Alitalo K, Randolph GJ, et al. Impaired humoral immunity and tolerance in K14-VEGFR-3-Ig mice that lack dermal lymphatic drainage. J Immunol (2012) 189:2181–90. doi:10.4049/jimmunol.1103545
109. Fasnacht N, Huang HY, Koch U, Favre S, Auderset F, Chai Q, et al. Specific fibroblastic niches in secondary lymphoid organs orchestrate distinct Notch-regulated immune responses. J Exp Med (2014) 211:2265–79. doi:10.1084/jem.20132528
110. Kumar V, Scandella E, Danuser R, Onder L, Nitschke M, Fukui Y, et al. Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell-lymphotoxin-dependent pathway. Blood (2010) 115:4725–33. doi:10.1182/blood-2009-10-250118
111. Chyou S, Ekland EH, Carpenter AC, Tzeng TC, Tian S, Michaud M, et al. Fibroblast-type reticular stromal cells regulate the lymph node vasculature. J Immunol (2008) 181:3887–96. doi:10.4049/jimmunol.181.6.3887
112. Chyou S, Benahmed F, Chen J, Kumar V, Tian S, Lipp M, et al. Coordinated regulation of lymph node vascular-stromal growth first by CD11c+ cells and then by T and B cells. J Immunol (2011) 187:5558–67. doi:10.4049/jimmunol.1101724
113. Acton SE, Farrugia AJ, Astarita JL, Mourao-Sa D, Jenkins RP, Nye E, et al. Dendritic cells control fibroblastic reticular network tension and lymph node expansion. Nature (2014) 514:498–502. doi:10.1038/nature13814
114. Astarita JL, Cremasco V, Fu J, Darnell MC, Peck JR, Nieves-Bonilla JM, et al. The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat Immunol (2015) 16:75–84. doi:10.1038/ni.3035
115. Herzog BH, Fu J, Wilson SJ, Hess PR, Sen A, Mcdaniel JM, et al. Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature (2013) 502:105–9. doi:10.1038/nature12501
116. Misslitz AC, Bonhagen K, Harbecke D, Lippuner C, Kamradt T, Aebischer T. Two waves of antigen-containing dendritic cells in vivo in experimental Leishmania major infection. Eur J Immunol (2004) 34:715–25. doi:10.1002/eji.200324391
117. Koopmann JO, Hammerling GJ, Momburg F. Generation, intracellular transport and loading of peptides associated with MHC class I molecules. Curr Opin Immunol (1997) 9:80–8. doi:10.1016/S0952-7915(97)80163-X
118. Cresswell P. Antigen presentation. Getting peptides into MHC class II molecules. Curr Biol (1994) 4:541–3.
119. Kasai M, Kominami E, Mizuochi T. The antigen presentation pathway in medullary thymic epithelial cells, but not that in cortical thymic epithelial cells, conforms to the endocytic pathway. Eur J Immunol (1998) 28:1867–76.
120. Nair-Gupta P, Blander JM. An updated view of the intracellular mechanisms regulating cross-presentation. Front Immunol (2013) 4:401. doi:10.3389/fimmu.2013.00401
121. Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol (2002) 3:265–71. doi:10.1038/ni762
122. Norbury CC, Basta S, Donohue KB, Tscharke DC, Princiotta MF, Berglund P, et al. CD8+ T cell cross-priming via transfer of proteasome substrates. Science (2004) 304:1318–21. doi:10.1126/science.1096378
123. Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev (2005) 207:166–83. doi:10.1111/j.0105-2896.2005.00301.x
124. Ackerman AL, Kyritsis C, Tampe R, Cresswell P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci U S A (2003) 100:12889–94. doi:10.1073/pnas.1735556100
125. Harriff MJ, Burgdorf S, Kurts C, Wiertz EJ, Lewinsohn DA, Lewinsohn DM. TAP mediates import of Mycobacterium tuberculosis-derived peptides into phagosomes and facilitates loading onto HLA-I. PLoS One (2013) 8:e79571. doi:10.1371/journal.pone.0079571
126. Gromme M, Uytdehaag FG, Janssen H, Calafat J, Van Binnendijk RS, Kenter MJ, et al. Recycling MHC class I molecules and endosomal peptide loading. Proc Natl Acad Sci U S A (1999) 96:10326–31. doi:10.1073/pnas.96.18.10326
127. Kurotaki T, Tamura Y, Ueda G, Oura J, Kutomi G, Hirohashi Y, et al. Efficient cross-presentation by heat shock protein 90-peptide complex-loaded dendritic cells via an endosomal pathway. J Immunol (2007) 179:1803–13. doi:10.4049/jimmunol.179.3.1803
128. Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol (2009) 9:503–13. doi:10.1038/nri2575
129. Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst (2013) 105:1172–87. doi:10.1093/jnci/djt184
130. Limmer A, Ohl J, Wingender G, Berg M, Jungerkes F, Schumak B, et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur J Immunol (2005) 35:2970–81. doi:10.1002/eji.200526034
131. Neefjes J. CIIV, MIIC and other compartments for MHC class II loading. Eur J Immunol (1999) 29:1421–5.
132. Stern LJ, Potolicchio I, Santambrogio L. MHC class II compartment subtypes: structure and function. Curr Opin Immunol (2006) 18:64–9. doi:10.1016/j.coi.2005.11.005
133. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol (2015) 15:203–16. doi:10.1038/nri3818
134. Baptista AP, Roozendaal R, Reijmers RM, Koning JJ, Unger WW, Greuter M, et al. Lymph node stromal cells constrain immunity via MHC class II self-antigen presentation. Elife (2014) 3. doi:10.7554/eLife.04433
135. Rouhani SJ, Eccles JD, Riccardi P, Peske JD, Tewalt EF, Cohen JN, et al. Roles of lymphatic endothelial cells expressing peripheral tissue antigens in CD4 T-cell tolerance induction. Nat Commun (2015) 6:6771. doi:10.1038/ncomms7771
136. Adamopoulou E, Tenzer S, Hillen N, Klug P, Rota IA, Tietz S, et al. Exploring the MHC-peptide matrix of central tolerance in the human thymus. Nat Commun (2013) 4:2039. doi:10.1038/ncomms3039
137. Aichinger M, Wu C, Nedjic J, Klein L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J Exp Med (2013) 210:287–300. doi:10.1084/jem.20122149
138. Gannage M, Munz C. MHC presentation via autophagy and how viruses escape from it. Semin Immunopathol (2010) 32:373–81. doi:10.1007/s00281-010-0227-7
139. Heng TS, Painter MW, Project C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol (2008) 9:1091–4. doi:10.1038/ni1008-1091
140. Hastings KT, Cresswell P. Disulfide reduction in the endocytic pathway: immunological functions of gamma-interferon-inducible lysosomal thiol reductase. Antioxid Redox Signal (2011) 15:657–68. doi:10.1089/ars.2010.3684
141. de Verteuil D, Granados DP, Thibault P, Perreault C. Origin and plasticity of MHC I-associated self peptides. Autoimmun Rev (2012) 11:627–35. doi:10.1016/j.autrev.2011.11.003
142. Vigneron N, Van den Eynde BJ. Proteasome subtypes and the processing of tumor antigens: increasing antigenic diversity. Curr Opin Immunol (2012) 24:84–91. doi:10.1016/j.coi.2011.12.002
143. Mishto M, Liepe J, Textoris-Taube K, Keller C, Henklein P, Weberruss M, et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur J Immunol (2014) 44:3508–21. doi:10.1002/eji.201444902
144. Nierkens S, Tel J, Janssen E, Adema GJ. Antigen cross-presentation by dendritic cell subsets: one general or all sergeants? Trends Immunol (2013) 34:361–70. doi:10.1016/j.it.2013.02.007
145. Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol (2012) 12:813–20. doi:10.1038/nri3339
146. Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, et al. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med (2010) 207:689–97. doi:10.1084/jem.20092642
147. Norder M, Gutierrez MG, Zicari S, Cervi E, Caruso A, Guzman CA. Lymph node-derived lymphatic endothelial cells express functional costimulatory molecules and impair dendritic cell-induced allogenic T-cell proliferation. FASEB J (2012) 26:2835–46. doi:10.1096/fj.12-205278
148. Hirosue S, Vokali E, Raghavan VR, Rincon-Restrepo M, Lund AW, Corthesy-Henrioud P, et al. Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: a new role for lymphatic endothelial cells. J Immunol (2014) 192:5002–11. doi:10.4049/jimmunol.1302492
149. Kyewski B, Derbinski J, Gotter J, Klein L. Promiscuous gene expression and central T-cell tolerance: more than meets the eye. Trends Immunol (2002) 23:364–71. doi:10.1016/S1471-4906(02)02248-2
150. Anderson AC, Nicholson LB, Legge KL, Turchin V, Zaghouani H, Kuchroo VK. High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: mechanisms of selection of the self-reactive repertoire. J Exp Med (2000) 191:761–70. doi:10.1084/jem.191.5.761
151. Enouz S, Carrie L, Merkler D, Bevan MJ, Zehn D. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J Exp Med (2012) 209:1769–79. doi:10.1084/jem.20120905
152. Lee JW, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura AR, et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol (2007) 8:181–90. doi:10.1038/ni1427
153. Nichols LA, Chen Y, Colella TA, Bennett CL, Clausen BE, Engelhard VH. Deletional self-tolerance to a melanocyte/melanoma antigen derived from tyrosinase is mediated by a radio-resistant cell in peripheral and mesenteric lymph nodes. J Immunol (2007) 179:993–1003. doi:10.4049/jimmunol.179.2.993
154. Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A, et al. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med (2010) 207:681–8. doi:10.1084/jem.20092465
155. Fletcher AL, Malhotra D, Turley SJ. Lymph node stroma broaden the peripheral tolerance paradigm. Trends Immunol (2011) 32:12–8. doi:10.1016/j.it.2010.11.002
156. Cohen JN, Tewalt EF, Rouhani SJ, Buonomo EL, Bruce AN, Xu X, et al. Tolerogenic properties of lymphatic endothelial cells are controlled by the lymph node microenvironment. PLoS One (2014) 9:e87740. doi:10.1371/journal.pone.0087740
157. Yip L, Su L, Sheng D, Chang P, Atkinson M, Czesak M, et al. Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat Immunol (2009) 10:1026–33. doi:10.1038/ni.1773
158. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep (2012) 1:191–9. doi:10.1016/j.celrep.2012.01.005
159. Bagai R, Valujskikh A, Canaday DH, Bailey E, Lalli PN, Harding CV, et al. Mouse endothelial cells cross-present lymphocyte-derived antigen on class I MHC via a TAP1- and proteasome-dependent pathway. J Immunol (2005) 174:7711–5. doi:10.4049/jimmunol.174.12.7711
160. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med (2000) 6:1348–54. doi:10.1038/82161
161. Berg M, Wingender G, Djandji D, Hegenbarth S, Momburg F, Hammerling G, et al. Cross-presentation of antigens from apoptotic tumor cells by liver sinusoidal endothelial cells leads to tumor-specific CD8+ T cell tolerance. Eur J Immunol (2006) 36:2960–70. doi:10.1002/eji.200636033
162. Mant A, Chinnery F, Elliott T, Williams AP. The pathway of cross-presentation is influenced by the particle size of phagocytosed antigen. Immunology (2012) 136:163–75. doi:10.1111/j.1365-2567.2012.03558.x
163. Mantegazza AR, Magalhaes JG, Amigorena S, Marks MS. Presentation of phagocytosed antigens by MHC class I and II. Traffic (2013) 14:135–52. doi:10.1111/tra.12026
164. Oliveira CC, van Hall T. Importance of TAP-independent processing pathways. Mol Immunol (2013) 55:113–6. doi:10.1016/j.molimm.2012.10.005
165. Murshid A, Gong J, Calderwood SK. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J Immunol (2010) 185:2903–17. doi:10.4049/jimmunol.0903635
166. Murshid A, Gong J, Calderwood SK. The role of heat shock proteins in antigen cross presentation. Front Immunol (2012) 3:63. doi:10.3389/fimmu.2012.00063
167. Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol (2013) 13:621–34. doi:10.1038/nri3515
168. Rauen J, Kreer C, Paillard A, Van Duikeren S, Benckhuijsen WE, Camps MG, et al. Enhanced cross-presentation and improved CD8+ T cell responses after mannosylation of synthetic long peptides in mice. PLoS One (2014) 9:e103755. doi:10.1371/journal.pone.0103755
169. Zehner M, Chasan AI, Schuette V, Embgenbroich M, Quast T, Kolanus W, et al. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross-presentation. Proc Natl Acad Sci U S A (2011) 108:9933–8. doi:10.1073/pnas.1102397108
170. Zehner M, Burgdorf S. Regulation of antigen transport into the cytosol for cross-presentation by ubiquitination of the mannose receptor. Mol Immunol (2013) 55:146–8. doi:10.1016/j.molimm.2012.10.010
171. Harvey DJ, Wing DR, Kuster B, Wilson IB. Composition of N-linked carbohydrates from ovalbumin and co-purified glycoproteins. J Am Soc Mass Spectrom (2000) 11:564–71. doi:10.1016/S1044-0305(00)00122-7
172. Burgdorf S, Lukacs-Kornek V, Kurts C. The mannose receptor mediates uptake of soluble but not of cell-associated antigen for cross-presentation. J Immunol (2006) 176:6770–6. doi:10.4049/jimmunol.176.11.6770
173. Harrison RL, Jarvis DL. Protein N-glycosylation in the baculovirus-insect cell expression system and engineering of insect cells to produce “mammalianized” recombinant glycoproteins. Adv Virus Res (2006) 68:159–91. doi:10.1016/S0065-3527(06)68005-6
174. den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(-) dendritic cells in vivo. J Exp Med (2002) 196:817–27. doi:10.1084/jem.20020295
175. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcgamma receptors in dendritic cells and macrophages. Nat Rev Immunol (2014) 14:94–108. doi:10.1038/nri3582
176. Platzer B, Stout M, Fiebiger E. Antigen cross-presentation of immune complexes. Front Immunol (2014) 5:140. doi:10.3389/fimmu.2014.00140
177. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7:715–25. doi:10.1038/nri2155
178. Rath T, Baker K, Pyzik M, Blumberg RS. Regulation of immune responses by the neonatal fc receptor and its therapeutic implications. Front Immunol (2014) 5:664. doi:10.3389/fimmu.2014.00664
179. Sockolosky JT, Szoka FC. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv Drug Deliv Rev (2015). doi:10.1016/j.addr.2015.02.005
180. Montoyo HP, Vaccaro C, Hafner M, Ober RJ, Mueller W, Ward ES. Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proc Natl Acad Sci U S A (2009) 106:2788–93. doi:10.1073/pnas.0810796106
181. Baker K, Rath T, Pyzik M, Blumberg RS. The role of FcRn in antigen presentation. Front Immunol (2014) 5:408. doi:10.3389/fimmu.2014.00408
182. van Montfoort N, De Jong JM, Schuurhuis DH, Van Der Voort EI, Camps MG, Huizinga TW, et al. A novel role of complement factor C1q in augmenting the presentation of antigen captured in immune complexes to CD8+ T lymphocytes. J Immunol (2007) 178:7581–6. doi:10.4049/jimmunol.178.12.7581
183. Nicholson-Weller A, Klickstein LB. C1q-binding proteins and C1q receptors. Curr Opin Immunol (1999) 11:42–6. doi:10.1016/S0952-7915(99)80008-9
184. Ghebrehiwet B, Hosszu KK, Valentino A, Peerschke EI. The C1q family of proteins: insights into the emerging non-traditional functions. Front Immunol (2012) 3:52. doi:10.3389/fimmu.2012.00052
185. Tran KK, Shen H. The role of phagosomal pH on the size-dependent efficiency of cross-presentation by dendritic cells. Biomaterials (2009) 30:1356–62. doi:10.1016/j.biomaterials.2008.11.034
186. Chen C, Li HQ, Liu YJ, Guo ZF, Wu HJ, Li X, et al. A novel size-based sorting mechanism of pinocytic luminal cargoes in microglia. J Neurosci (2015) 35:2674–88. doi:10.1523/JNEUROSCI.4389-14.2015
187. Mescher MF, Agarwal P, Casey KA, Hammerbeck CD, Xiao Z, Curtsinger JM. Molecular basis for checkpoints in the CD8 T cell response: tolerance versus activation. Semin Immunol (2007) 19:153–61. doi:10.1016/j.smim.2007.02.007
188. Collier AY, Lee JW, Turley SJ. Self-encounters of the third kind: lymph node stroma promotes tolerance to peripheral tissue antigens. Mucosal Immunol (2008) 1:248–51. doi:10.1038/mi.2008.19
189. Mueller SN, Ahmed R. Lymphoid stroma in the initiation and control of immune responses. Immunol Rev (2008) 224:284–94. doi:10.1111/j.1600-065X.2008.00657.x
190. Firner S, Onder L, Nindl V, Ludewig B. Tight control – decision-making during T cell-vascular endothelial cell interaction. Front Immunol (2012) 3:279. doi:10.3389/fimmu.2012.00279
191. Siegert S, Luther SA. Positive and negative regulation of T cell responses by fibroblastic reticular cells within paracortical regions of lymph nodes. Front Immunol (2012) 3:285. doi:10.3389/fimmu.2012.00285
192. Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer (2012) 12:210–9. doi:10.1038/nrc3186
193. Tewalt EF, Cohen JN, Rouhani SJ, Engelhard VH. Lymphatic endothelial cells – key players in regulation of tolerance and immunity. Front Immunol (2012) 3:305. doi:10.3389/fimmu.2012.00305
194. Mueller CG, Coles MC. Emerging immune functions of non-hematopoietic stromal cells. Front Immunol (2014) 5:437. doi:10.3389/fimmu.2014.00437
195. Rouhani SJ, Eccles JD, Tewalt EF, Engelhard VH. Regulation of T-cell tolerance by lymphatic endothelial cells. J Clin Cell Immunol (2014) 5:242. doi:10.4172/2155-9899.1000242
196. Brown FD, Turley SJ. Fibroblastic reticular cells: organization and regulation of the T lymphocyte life cycle. J Immunol (2015) 194:1389–94. doi:10.4049/jimmunol.1402520
197. Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qiao H, Fahl SP, et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood (2012) 120:4772–82. doi:10.1182/blood-2012-04-427013
198. Dubrot J, Duraes FV, Potin L, Capotosti F, Brighouse D, Suter T, et al. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4(+) T cell tolerance. J Exp Med (2014) 211:1153–66. doi:10.1084/jem.20132000
199. Duraes FV, Thelemann C, Sarter K, Acha-Orbea H, Hugues S, Reith W. Role of major histocompatibility complex class II expression by non-hematopoietic cells in autoimmune and inflammatory disorders: facts and fiction. Tissue Antigens (2013) 82:1–15. doi:10.1111/tan.12136
200. Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol (2014) 14:719–30. doi:10.1038/nri3754
201. Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol (2001) 19:331–73. doi:10.1146/annurev.immunol.19.1.331
202. Reith W, Leibundgut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol (2005) 5:793–806. doi:10.1038/nri1708
203. Pober JS, Gimbrone MA Jr. Expression of Ia-like antigens by human vascular endothelial cells is inducible in vitro: demonstration by monoclonal antibody binding and immunoprecipitation. Proc Natl Acad Sci U S A (1982) 79:6641–5. doi:10.1073/pnas.79.21.6641
204. Pardi R, Bender JR, Engleman EG. Lymphocyte subsets differentially induce class II human leukocyte antigens on allogeneic microvascular endothelial cells. J Immunol (1987) 139:2585–92.
205. Collinge M, Pardi R, Bender JR. Class II transactivator-independent endothelial cell MHC class II gene activation induced by lymphocyte adhesion. J Immunol (1998) 161:1589–93.
206. Pober JS, Collins T, Gimbrone MA Jr, Cotran RS, Gitlin JD, Fiers W, et al. Lymphocytes recognize human vascular endothelial and dermal fibroblast Ia antigens induced by recombinant immune interferon. Nature (1983) 305:726–9. doi:10.1038/305726a0
207. Wedgwood JF, Hatam L, Bonagura VR. Effect of interferon-gamma and tumor necrosis factor on the expression of class I and class II major histocompatibility molecules by cultured human umbilical vein endothelial cells. Cell Immunol (1988) 111:1–9. doi:10.1016/0008-8749(88)90046-9
208. Detmar M, Imcke E, Ruszczak Z, Orfanos CE. Effects of recombinant tumor necrosis factor-alpha on cultured microvascular endothelial cells derived from human dermis. J Invest Dermatol (1990) 95:219S–22S.
209. Swerlick RA, Garcia-Gonzalez E, Kubota Y, Xu YL, Lawley TJ. Studies of the modulation of MHC antigen and cell adhesion molecule expression on human dermal microvascular endothelial cells. J Invest Dermatol (1991) 97:190–6. doi:10.1111/1523-1747.ep12479643
210. Ng CT, Nayak BP, Schmedt C, Oldstone MB. Immortalized clones of fibroblastic reticular cells activate virus-specific T cells during virus infection. Proc Natl Acad Sci U S A (2012) 109:7823–8. doi:10.1073/pnas.1205850109
211. Amatschek S, Kriehuber E, Bauer W, Reininger B, Meraner P, Wolpl A, et al. Blood and lymphatic endothelial cell-specific differentiation programs are stringently controlled by the tissue environment. Blood (2007) 109:4777–85. doi:10.1182/blood-2006-10-053280
212. Pinet V, Malnati MS, Long EO. Two processing pathways for the MHC class II-restricted presentation of exogenous influenza virus antigen. J Immunol (1994) 152:4852–60.
213. Griffin JP, Chu R, Harding CV. Early endosomes and a late endocytic compartment generate different peptide-class II MHC complexes via distinct processing mechanisms. J Immunol (1997) 158:1523–32.
214. Haque A, Hajiaghamohseni LM, Li P, Toomy K, Blum JS. Invariant chain modulates HLA class II protein recycling and peptide presentation in nonprofessional antigen presenting cells. Cell Immunol (2007) 249:20–9. doi:10.1016/j.cellimm.2007.10.005
215. Mukherjee R, Chaturvedi P, Lee-Chan E, Singh B. Exogenous CLIP localizes into endocytic compartment of cells upon internalization: implications for antigen presentation by MHC class II molecules. Mol Immunol (2008) 45:2166–76. doi:10.1016/j.molimm.2007.12.016
216. Mahmutefendic H, Zagorac GB, Tomas MI, Groettrup M, Momburg F, Lucin P. Endosomal trafficking of open Major Histocompatibility Class I conformers – implications for presentation of endocytosed antigens. Mol Immunol (2013) 55:149–52. doi:10.1016/j.molimm.2012.10.008
217. Clement CC, Rotzschke O, Santambrogio L. The lymph as a pool of self-antigens. Trends Immunol (2011) 32:6–11. doi:10.1016/j.it.2010.10.004
218. Clement CC, Aphkhazava D, Nieves E, Callaway M, Olszewski W, Rotzschke O, et al. Protein expression profiles of human lymph and plasma mapped by 2D-DIGE and 1D SDS-PAGE coupled with nanoLC-ESI-MS/MS bottom-up proteomics. J Proteomics (2013) 78:172–87. doi:10.1016/j.jprot.2012.11.013
219. Clement CC, Santambrogio L. The lymph self-antigen repertoire. Front Immunol (2013) 4:424. doi:10.3389/fimmu.2013.00424
220. Santambrogio L, Stern LJ. Carrying yourself: self antigen composition of the lymphatic fluid. Lymphat Res Biol (2013) 11:149–54. doi:10.1089/lrb.2013.0009
221. Davis DM. Intercellular transfer of cell-surface proteins is common and can affect many stages of an immune response. Nat Rev Immunol (2007) 7:238–43. doi:10.1038/nri2020
222. Gray EE, Friend S, Suzuki K, Phan TG, Cyster JG. Subcapsular sinus macrophage fragmentation and CD169+ bleb acquisition by closely associated IL-17-committed innate-like lymphocytes. PLoS One (2012) 7:e38258. doi:10.1371/journal.pone.0038258
223. Zhang QJ, Li XL, Wang D, Huang XC, Mathis JM, Duan WM, et al. Trogocytosis of MHC-I/peptide complexes derived from tumors and infected cells enhances dendritic cell cross-priming and promotes adaptive T cell responses. PLoS One (2008) 3:e3097. doi:10.1371/journal.pone.0003097
224. de Heusch M, Blocklet D, Egrise D, Hauquier B, Vermeersch M, Goldman S, et al. Bidirectional MHC molecule exchange between migratory and resident dendritic cells. J Leukoc Biol (2007) 82:861–8. doi:10.1189/jlb.0307167
225. Buschow SI, Nolte-’T Hoen EN, Van Niel G, Pols MS, Ten Broeke T, Lauwen M, et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic (2009) 10:1528–42. doi:10.1111/j.1600-0854.2009.00963.x
226. Gauvreau ME, Cote MH, Bourgeois-Daigneault MC, Rivard LD, Xiu F, Brunet A, et al. Sorting of MHC class II molecules into exosomes through a ubiquitin-independent pathway. Traffic (2009) 10:1518–27. doi:10.1111/j.1600-0854.2009.00948.x
227. Gray D, Kosco M, Stockinger B. Novel pathways of antigen presentation for the maintenance of memory. Int Immunol (1991) 3:141–8. doi:10.1093/intimm/3.2.141
228. Schnizlein CT, Kosco MH, Szakal AK, Tew JG. Follicular dendritic cells in suspension: identification, enrichment, and initial characterization indicating immune complex trapping and lack of adherence and phagocytic activity. J Immunol (1985) 134:1360–8.
229. Denzer K, Van Eijk M, Kleijmeer MJ, Jakobson E, De Groot C, Geuze HJ. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol (2000) 165:1259–65. doi:10.4049/jimmunol.165.3.1259
230. Abe J, Shichino S, Ueha S, Hashimoto S, Tomura M, Inagaki Y, et al. Lymph node stromal cells negatively regulate antigen-specific CD4+ T cell responses. J Immunol (2014) 193:1636–44. doi:10.4049/jimmunol.1302946
231. Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, Mccoll SR. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev (2013) 24:269–83. doi:10.1016/j.cytogfr.2013.03.001
232. Hara T, Shitara S, Imai K, Miyachi H, Kitano S, Yao H, et al. Identification of IL-7-producing cells in primary and secondary lymphoid organs using IL-7-GFP knock-in mice. J Immunol (2012) 189:1577–84. doi:10.4049/jimmunol.1200586
233. Onder L, Narang P, Scandella E, Chai Q, Iolyeva M, Hoorweg K, et al. IL-7-producing stromal cells are critical for lymph node remodeling. Blood (2012) 120:4675–83. doi:10.1182/blood-2012-03-416859
234. Miller CN, Hartigan-O’connor DJ, Lee MS, Laidlaw G, Cornelissen IP, Matloubian M, et al. IL-7 production in murine lymphatic endothelial cells and induction in the setting of peripheral lymphopenia. Int Immunol (2013) 25:471–83. doi:10.1093/intimm/dxt012
235. Cui G, Hara T, Simmons S, Wagatsuma K, Abe A, Miyachi H, et al. Characterization of the IL-15 niche in primary and secondary lymphoid organs in vivo. Proc Natl Acad Sci U S A (2014) 111:1915–20. doi:10.1073/pnas.1318281111
236. Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol (2008) 8:935–47. doi:10.1038/nri2455
237. Waldburger JM, Suter T, Fontana A, Acha-Orbea H, Reith W. Selective abrogation of major histocompatibility complex class II expression on extrahematopoietic cells in mice lacking promoter IV of the class II transactivator gene. J Exp Med (2001) 194:393–406. doi:10.1084/jem.194.4.393
238. Waldburger JM, Rossi S, Hollander GA, Rodewald HR, Reith W, Acha-Orbea H. Promoter IV of the class II transactivator gene is essential for positive selection of CD4+ T cells. Blood (2003) 101:3550–9. doi:10.1182/blood-2002-06-1855
239. Magnusson FC, Liblau RS, Von Boehmer H, Pittet MJ, Lee JW, Turley SJ, et al. Direct presentation of antigen by lymph node stromal cells protects against CD8 T-cell-mediated intestinal autoimmunity. Gastroenterology (2008) 134:1028–37. doi:10.1053/j.gastro.2008.01.070
240. Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J Immunol (2010) 184:1909–17. doi:10.4049/jimmunol.0902778
241. Wang X. Cre transgenic mouse lines. Methods Mol Biol (2009) 561:265–73. doi:10.1007/978-1-60327-019-9_17
242. Chai Q, Onder L, Scandella E, Gil-Cruz C, Perez-Shibayama C, Cupovic J, et al. Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity (2013) 38:1013–24. doi:10.1016/j.immuni.2013.03.012
243. Srinivasan RS, Dillard ME, Lagutin OV, Lin FJ, Tsai S, Tsai MJ, et al. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev (2007) 21:2422–32. doi:10.1101/gad.1588407
244. Eklund L, Bry M, Alitalo K. Mouse models for studying angiogenesis and lymphangiogenesis in cancer. Mol Oncol (2013) 7:259–82. doi:10.1016/j.molonc.2013.02.007
245. Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell (1999) 98:769–78. doi:10.1016/S0092-8674(00)81511-1
246. Truman LA, Bentley KL, Smith EC, Massaro SA, Gonzalez DG, Haberman AM, et al. ProxTom lymphatic vessel reporter mice reveal Prox1 expression in the adrenal medulla, megakaryocytes, and platelets. Am J Pathol (2012) 180:1715–25. doi:10.1016/j.ajpath.2011.12.026
247. Onder L, Scandella E, Chai Q, Firner S, Mayer CT, Sparwasser T, et al. A novel bacterial artificial chromosome-transgenic podoplanin-cre mouse targets lymphoid organ stromal cells in vivo. Front Immunol (2011) 2:50. doi:10.3389/fimmu.2011.00050
248. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol (2001) 230:230–42. doi:10.1006/dbio.2000.0106
249. Licht AH, Raab S, Hofmann U, Breier G. Endothelium-specific Cre recombinase activity in flk-1-Cre transgenic mice. Dev Dyn (2004) 229:312–8. doi:10.1002/dvdy.10416
250. Shimoda M, Mmanywa F, Joshi SK, Li T, Miyake K, Pihkala J, et al. Conditional ablation of MHC-II suggests an indirect role for MHC-II in regulatory CD4 T cell maintenance. J Immunol (2006) 176:6503–11. doi:10.4049/jimmunol.176.11.6503
251. Lugus JJ, Park C, Ma YD, Choi K. Both primitive and definitive blood cells are derived from Flk-1+ mesoderm. Blood (2009) 113:563–6. doi:10.1182/blood-2008-06-162750
252. Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell (2004) 117:787–800. doi:10.1016/j.cell.2004.05.014
253. Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol (2007) 8:665–8. doi:10.1038/ni0707-665
254. Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature (2011) 474:337–42. doi:10.1038/nature10163
255. Laakkonen P, Porkka K, Hoffman JA, Ruoslahti E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med (2002) 8:751–5. doi:10.1038/nm720
256. Katakai T. Marginal reticular cells: a stromal subset directly descended from the lymphoid tissue organizer. Front Immunol (2012) 3:200. doi:10.3389/fimmu.2012.00200
257. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell (2013) 154:1370–9. doi:10.1016/j.cell.2013.08.022
258. Singh P, Schimenti JC, Bolcun-Filas E. A mouse geneticist’s practical guide to CRISPR applications. Genetics (2015) 199:1–15. doi:10.1534/genetics.114.169771
259. Vega F, Coombes KR, Thomazy VA, Patel K, Lang W, Jones D. Tissue-specific function of lymph node fibroblastic reticulum cells. Pathobiology (2006) 73:71–81. doi:10.1159/000094491
260. Pisano M, Triacca V, Barbee KA, Swartz MA. An in vitro model of the tumor-lymphatic microenvironment with simultaneous transendothelial and luminal flows reveals mechanisms of flow enhanced invasion. Integr Biol (Camb) (2015) 7:525–33. doi:10.1039/c5ib00085h
261. Tomei AA, Boschetti F, Gervaso F, Swartz MA. 3D collagen cultures under well-defined dynamic strain: a novel strain device with a porous elastomeric support. Biotechnol Bioeng (2009) 103:217–25. doi:10.1002/bit.22236
262. Cupedo T, Stroock A, Coles M. Application of tissue engineering to the immune system: development of artificial lymph nodes. Front Immunol (2012) 3:343. doi:10.3389/fimmu.2012.00343
263. Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods (2014) 11:417–22. doi:10.1038/nmeth.2869
264. Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol (2006) 24:519–40. doi:10.1146/annurev.immunol.23.021704.115825
265. Germain RN, Robey EA, Cahalan MD. A decade of imaging cellular motility and interaction dynamics in the immune system. Science (2012) 336:1676–81. doi:10.1126/science.1221063
266. Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, et al. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood (2011) 117:362–5. doi:10.1182/blood-2010-07-298562
267. Hagerling R, Pollmann C, Kremer L, Andresen V, Kiefer F. Intravital two-photon microscopy of lymphatic vessel development and function using a transgenic Prox1 promoter-directed mOrange2 reporter mouse. Biochem Soc Trans (2011) 39:1674–81. doi:10.1042/BST20110722
268. Truong TN, Li H, Hong YK, Chen L. Novel characterization and live imaging of Schlemm’s canal expressing Prox-1. PLoS One (2014) 9:e98245. doi:10.1371/journal.pone.0098245
269. Bianchi R, Teijeira A, Proulx ST, Christiansen AJ, Seidel CD, Rulicke T, et al. A transgenic Prox1-Cre-tdTomato reporter mouse for lymphatic vessel research. PLoS One (2015) 10:e0122976. doi:10.1371/journal.pone.0122976
270. Larina IV, Shen W, Kelly OG, Hadjantonakis AK, Baron MH, Dickinson ME. A membrane associated mCherry fluorescent reporter line for studying vascular remodeling and cardiac function during murine embryonic development. Anat Rec (Hoboken) (2009) 292:333–41. doi:10.1002/ar.20821
271. Poche RA, Larina IV, Scott ML, Saik JE, West JL, Dickinson ME. The Flk1-myr:mCherry mouse as a useful reporter to characterize multiple aspects of ocular blood vessel development and disease. Dev Dyn (2009) 238:2318–26. doi:10.1002/dvdy.21886
272. Zhu J, Dugas-Ford J, Chang M, Purta P, Han KY, Hong YK, et al. Simultaneous in vivo imaging of blood and lymphatic vessel growth in Prox1-GFP/Flk1:myr-mCherry mice. FEBS J (2015) 282:1458–67. doi:10.1111/febs.13234
273. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427:154–9. doi:10.1038/nature02238
274. Henrickson SE, Perro M, Loughhead SM, Senman B, Stutte S, Quigley M, et al. Antigen availability determines CD8(+) T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity (2013) 39:496–507. doi:10.1016/j.immuni.2013.08.034
275. Hopp AK, Rupp A, Lukacs-Kornek V. Self-antigen presentation by dendritic cells in autoimmunity. Front Immunol (2014) 5:55. doi:10.3389/fimmu.2014.00055
276. Gardner JM, Devoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science (2008) 321:843–7. doi:10.1126/science.1159407
277. Gardner JM, Metzger TC, Mcmahon EJ, Au-Yeung BB, Krawisz AK, Lu W, et al. Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4(+) T cells. Immunity (2013) 39:560–72. doi:10.1016/j.immuni.2013.08.005
278. Grinshtein N, Yang TC, Parsons R, Millar J, Denisova G, Dissanayake D, et al. Recombinant adenovirus vaccines can successfully elicit CD8+ T cell immunity under conditions of extreme leukopenia. Mol Ther (2006) 13:270–9. doi:10.1016/j.ymthe.2005.09.018
279. Thomas S, Kolumam GA, Murali-Krishna K. Antigen presentation by nonhemopoietic cells amplifies clonal expansion of effector CD8 T cells in a pathogen-specific manner. J Immunol (2007) 178:5802–11. doi:10.4049/jimmunol.178.9.5802
280. Harty JT, Bevan MJ. CD8 T-cell recognition of macrophages and hepatocytes results in immunity to Listeria monocytogenes. Infect Immun (1996) 64:3632–40.
281. Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol (2008) 9:155–65. doi:10.1038/ni1557
282. von Oppen N, Schurich A, Hegenbarth S, Stabenow D, Tolba R, Weiskirchen R, et al. Systemic antigen cross-presented by liver sinusoidal endothelial cells induces liver-specific CD8 T-cell retention and tolerization. Hepatology (2009) 49:1664–72. doi:10.1002/hep.22795
283. Bottcher JP, Schanz O, Wohlleber D, Abdullah Z, Debey-Pascher S, Staratschek-Jox A, et al. Liver-primed memory T cells generated under noninflammatory conditions provide anti-infectious immunity. Cell Rep (2013) 3:779–95. doi:10.1016/j.celrep.2013.02.008
284. Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol (2007) 178:2714–20. doi:10.4049/jimmunol.178.5.2714
285. Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology (2008) 47:296–305. doi:10.1002/hep.21965
286. Vezys V, Olson S, Lefrancois L. Expression of intestine-specific antigen reveals novel pathways of CD8 T cell tolerance induction. Immunity (2000) 12:505–14. doi:10.1016/S1074-7613(00)80202-2
287. Tzeng TC, Chyou S, Tian S, Webster B, Carpenter AC, Guaiquil VH, et al. CD11c(hi) dendritic cells regulate the re-establishment of vascular quiescence and stabilization after immune stimulation of lymph nodes. J Immunol (2010) 184:4247–57. doi:10.4049/jimmunol.0902914
Keywords: lymph node, stromal cells, antigen presentation, cross-presentation, immunomodulation
Citation: Hirosue S and Dubrot J (2015) Modes of antigen presentation by lymph node stromal cells and their immunological implications. Front. Immunol. 6:446. doi: 10.3389/fimmu.2015.00446
Received: 06 May 2015; Accepted: 17 August 2015;
Published: 08 September 2015
Edited by:
Burkhard Ludewig, Kantonal Hospital St. Gallen, SwitzerlandReviewed by:
Theresa T. Lu, Weill Cornell Medical Center, USAVeronika Lukacs-Kornek, University of Saarland, Germany
Copyright: © 2015 Hirosue and Dubrot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sachiko Hirosue, Laboratory of Lymphatic and Cancer Bioengineering, Institute of Bioengineering, École Polytechnique Fédéral de Lausanne, Station 15, 1015 Lausanne, Switzerland,c2FjaGlrby5oaXJvc3VlQGVwZmwuY2g=