 Shafqat Ahrar Jaigirdar
Shafqat Ahrar Jaigirdar Megan K. L. MacLeod
Megan K. L. MacLeod- Centre for Immunobiology, Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow, UK
Immunological memory is one of the defining features of the adaptive immune system. As key orchestrators and mediators of immunity, CD4 T cells are central to the vast majority of adaptive immune responses. Generated following an immune response, memory CD4 T cells retain pertinent information about their activation environment enabling them to make rapid effector responses upon reactivation. These responses can either benefit the host by hastening the control of pathogens or cause damaging immunopathology. Here, we will discuss the diversity of the memory CD4 T cell pool, the signals that influence the transition of activated T cells into that pool, and highlight how activation requirements differ between naïve and memory CD4 T cells. A greater understanding of these factors has the potential to aid the design of more effective vaccines and to improve regulation of pathologic CD4 T cells, such as in the context of autoimmunity and allergy.
Introduction
The ability to remember the past and act on that memory presents a major evolutionary advantage. Learning how best to respond to a situation can save time, resources and, certainly in terms of immunological memory, lives. This is perhaps best illustrated by the dramatic reduction in child mortality, following the development of vaccines for diseases, such as diphtheria, measles, and polio (1).
Immunological memory is generated following an immunization or infection that activates the adaptive immune system. Following infection, T and B cells specific for the pathogen are stimulated, proliferate, and generated an effector response that leads to the control and/or clearance of the infection. The vast majority of these pathogen-specific cells undergo apoptosis, returning the immune system to homeostasis (2). The few thousand cells that survive are memory T and B cells that respond rapidly and effectively to re-infection. Key to immune protection by memory cells is that they remember the differentiation instructions they received during the initial immune response. Effective vaccines must mimic these instructions, triggering the right differentiation environment to ensure the generation of effective memory cells.
Most current successful vaccines provide protection by generating high affinity class switched antibodies that can neutralize or otherwise clear infections (3). However, there are many diseases, including HIV, malaria, and tuberculosis (TB), to which we have no effective vaccines. In these cases antibody is, at best, partially protective. Rather, a diverse memory immune response involving T and B cells that attack the pathogen on multiple levels is likely to be most effective. A better understanding of T cell memory will be key to achieving this. Much of the work on T cell memory focus on CD8 T cells – cells with clear mechanisms for eliminating intracellular pathogens (4, 5). Here, we will concentrate on CD4 memory T cells which, as helper cells that direct and assist many other cell types, have the potential to act like a catalyst, hastening immune protection via multiple different pathways. The superior responses of memory over naive CD4 T cells can present problems when these cells are directed against self or innocuous antigens. A better understanding of how memory CD4 T cells differ from their naïve counterparts will also inform on how best to control pathogenic CD4 T cells delivering new therapeutic approaches for antigen-specific tolerance in autoimmunity and allergy.
Memory CD4 T Cells are Heterogeneous in Terms of Function and Location
CD4 T cells are first primed in secondary lymphoid organs by dendritic cells (DCs) that process and present antigen on major histocompatibility complex (MHC) class II molecules. These peptide-MHC (pMHC) II complexes impart specificity on the ensuing immune response as they bind to T cell receptors (TCRs) on the surface of CD4 T cells. TCRs, in concert with associated CD3 molecules, transmit activation signals via an array of cytoplasmic proteins to alert transcription factors to initiate gene transcription, thereby shaping the ensuing response. TCR signals alone are not sufficient to prime naïve T cells. Activating DCs must also provide additional, or costimulatory, signals. These signals act in co-operation with inflammatory signals to instruct the T cell on the appropriate type of differentiation pathway to control the invading pathogen. TCR activation in the absence of costimulatory signals leads to T cell tolerance or the induction of regulatory rather than memory CD4 T cells (6–8).
There are a myriad of signals that direct and influence CD4 T cell activation and, in turn, memory cell generation. Traditionally, these signals, and the distinct CD4 T cell differentiation pathways they induce, are divided into discrete subsets, most notably T helper (Th)1, Th2, and Th17 (9). More recent data indicate that T cells retain a significant degree of plasticity (10). Both models require that T cells integrate information from the activation environment and use this information to make decisions about proliferation, differentiation and, critically for memory cells, survival.
There has been much debate about whether memory T cells develop from undifferentiated or differentiated activated T cells (11–13). Using elegant fate reporter mice in which the Th1 effector cytokine, interferon (IFN)γ, drives permanent expression of a fluorescent molecule, Weaver and colleagues demonstrated that both these populations of CD4 T cells can become memory cells (14). This dual pathway to memory converges with the description of T central memory cells (Tcm) and T effector memory cells (Tem). Originally described in human peripheral blood by Sallusto and colleagues (15), these populations can be distinguished based on cell surface expression of homing and selectin molecules, effector cytokine production and, more easily assessed in mouse models, location.
T central memory cells express the secondary lymphoid homing molecules, CD62L and CCR7, involved in migration to and within lymph nodes. These cells require further differentiation signals to make effector cytokines but proliferate substantially upon reactivation. In contrast, Tem cells make rapid effector responses, proliferate less and are mainly found in the spleen, blood and peripheral organs [reviewed in Ref. (16, 17)].
More recently, additional memory cell subgroups have been described (Figure 1), suggesting further heterogeneity in the steps between activated and memory T cell. While all memory cells are by definition long lived, memory stem T cells (Tscm) perhaps represent the most stable subset (18–21). The majority of studies on Tscm have been carried out on human peripheral blood cells, where Tscm express makers of naïve (CD45RA+) and memory (CXCR3 and CD95+) cells. Analysis of antigen-specific CD8 Tscm demonstrates that these cells have previously responded to antigen, can self-renew, and rapidly differentiate into cytokine producing effector cells upon reactivation (18–22). Transcriptional analysis of human CD4 T cell populations identified using CD45RO, CCR7, and CD95, has positioned Tscm as a distinct population with a profile somewhat in-between naïve and Tcm cells (23).
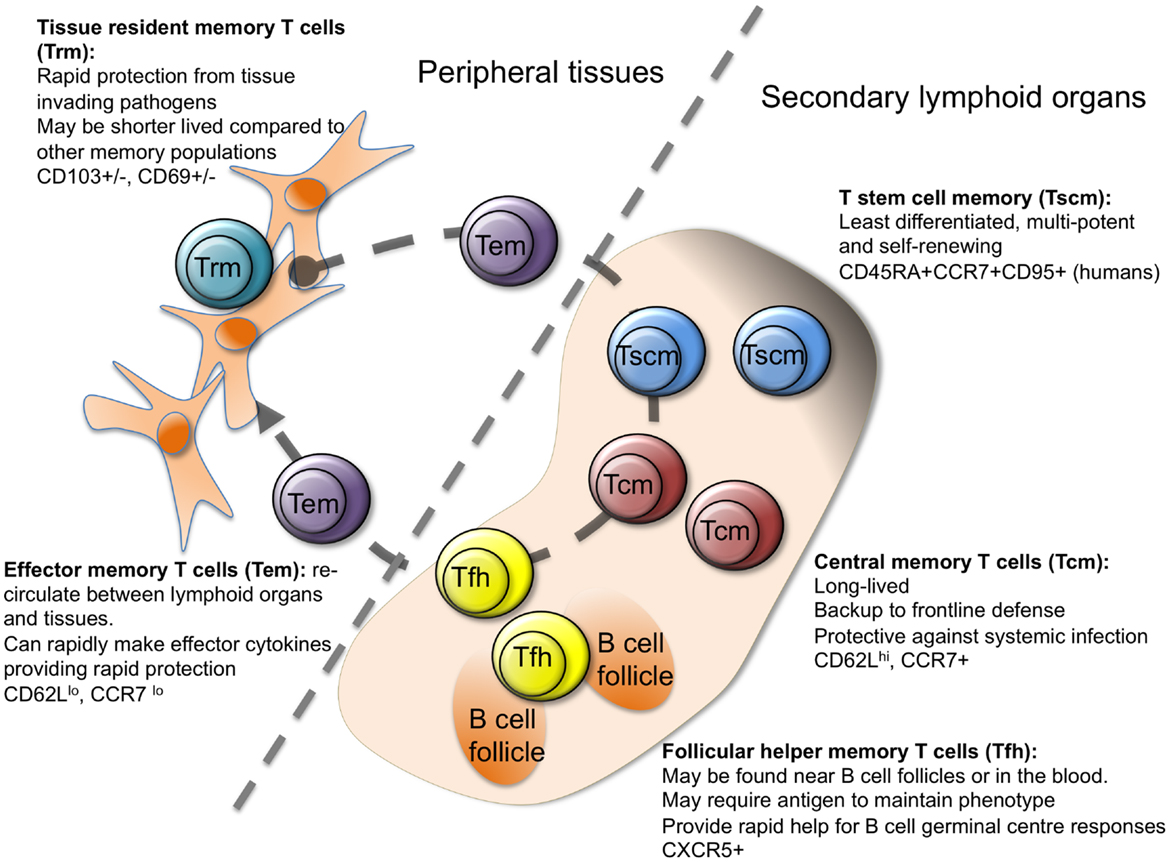
Figure 1. Heterogeneity in memory CD4 T cells. Memory CD4 T cells can be found in lymphoid organs, blood, and at tissue sites. Stem cell memory T cells (Tscm) and central memory T (Tcm) cells are found in lymphoid organs and in the blood. Both populations are relatively undifferentiated compared to other memory CD4 T cell subsets, and are long lived. Follicular helper memory T (Tfh) cells can also be found in the blood and lymphoid organs. They express the B cell follicle homing receptor, CXCR5, which can position them near B cell follicles to provide rapid B cell help upon reactivation. Effector memory (Tem) and tissue resident memory T (Trm) cells can both be found in peripheral tissues and are more differentiated than Tcm and Tscm. Tem are migratory, passing through tissues and the blood, while Trm are restricted to tissues. Both populations can respond rapidly to tissue invading pathogens (15, 17, 23–27).
Memory stem T cells have also been described in non-human primates, where Tscm are found in peripheral blood, secondary lymphoid organs, and the bone marrow (26). Tscm cells in mice have proven harder to track down although cells with a similar profile to human Tscm can be generated in vitro or in vivo in a model of graft versus host disease (28–30). This small subset has, therefore, proven difficult to analyze. While human CD8 Tscm can control tumor growth more effectively than other memory cell subsets in a humanized mouse model (18), demonstrating a physiologically protective role for these cells will be more challenging. This is especially the case as most investigators use challenge infection mouse models that favor rapid effector functions over long-term memory cell stability.
There is much more evidence concerning the protective ability of T cells that reside at infection sites (8, 31, 32). Like Tem, T resident memory (Trm) cells are found in peripheral organs but, as their name implies, they are non-migratory (31). Many of the studies on Trm cells have focused on CD8 T cells; for example, transcriptional evidence that Trm cells are distinct from Tem cells has been performed on CD8, but not CD4 T cells (33). Moreover, as current methods to dissociate memory T cells in peripheral organs underestimate the numbers of Trm cells, our current knowledge on the presence and activities of these cells is still limited (34).
It is clear that the retention of CD8 Trm cells is, at least in some organs, dependent on expression of CD69 and/or the integrin CD103 (31). CD69 acts to maintain Trm cells at tissue sites by antagonizing the SIPR1 receptor, which promotes the exit of T cells from tissue sites (33, 35, 36). CD103, which is induced by TGFβ, promotes interactions between Trm cells and local epithelial cells, thereby supporting tissue retention (36–38).
Both mouse and human lung CD4 Trm cells express CD69 and those found in human epidermis express TGFβ-driven CD103 (39–41). CD4 T cells in human skin dermis, however, are less likely to express CD103, perhaps reflecting differences in the local levels of TGFβ. Like mouse Trm cells, both populations of human skin Trm cells display rapid effector cytokine production when compared with circulating memory CD4 T cells. There are differences between human and mouse skin Trm cells, however. Both populations of human skin Trm are resident, as confirmed by their survival following treatment of T cell lymphoma patients with the leukocyte depleting monoclonal antibody alemtuzumab (CD154) (41, 42). In contrast, mouse CD4 T cells raised against herpes simplex virus (HSV) are primarily found in the dermis and are more likely to display a migratory phenotype (43).
These differences between human and mouse skin CD4 Trm cells may be species specific or due to differences in the antigens that triggered the T cell responses. It is vital that we have a better understanding of human memory T cell subsets if we are to exploit findings from animal models to improve human vaccine design. An extensive study by Farber and colleagues examined both CD4 and CD8 memory T cells in various internal organs from human donors (40). One of the major findings from this study is that the TCRs from memory CD4 T cells are more likely to contain sequences unique to individual organs than those from CD8 memory T cells. This finding suggests that CD4 T cells are either more compartmentalized than CD8 memory T cells or that they are less cross-reactive. Interestingly, mouse CD8 T cells are more promiscuous in the expression of tissue homing molecules than CD4 T cells with the latter homing specifically to the original site of infection, while CD8 T cells have a more wide spread tissue distribution (34, 43).
Neither local tissue inflammation nor antigen may be required for the recruitment of CD4 T cells and their subsequent differentiation into Trm cells. A recent study from von Andrian and colleagues examined the development of uterine CD4 Trm cells, following immunization by subcutaneous, intranasal, or intrauterine routes (8). Immunization by either mucosal route, but not via the skin, led to the development of Trm cells in the uterus. The recruitment of effector T cells did require alpha 4 integrin expression by the T cells, likely driven by activation in a mucosal environment (8, 44). This comprehensive study highlighted that CD4 Trm cells provide significantly more protection than circulating memory CD4 T cells to a challenge infection with Chlamydia trachomatis.
This heterogeneity in memory cell populations raises two key questions, the answers to which will provide critical knowledge for improved vaccine design. First, which cells provide the most effective protection to pathogen challenge and, second, what activation environments drive their development?
How do Memory CD4 T Cells Provide Immune Protection?
The requirement for rapid pathogen control at the site of infection suggests that Tem or Trm cells should, in theory, offer the most effective form of immune protection. Many mouse infection models backup this supposition. For example, lung CD4 Trm cells offer superior protection to influenza virus infection than those from lymphoid organs (39). They achieve this via IFNγ dependent and independent mechanisms that induce a rapid innate cytokine and chemokine response which accelerates the clearance of influenza virus (45). Similarly, IFNγ production by HSV2 specific memory CD4 T cells in the vaginal cavity leads to a local chemokine response that attracts viral specific CD8 T cells that subsequently control a challenge infection (46).
High effector cytokine production is not necessarily a mark of a protective memory T cell. In mice infected with Mycobacterium tuberculosis, two populations of antigen-specific cells can be found in the lung. These populations can be differentiated on the basis of expression of the cell surface inhibitory molecules, KLRG1 and PD1. KLRG1hiPD1− cells are mainly located in the vasculature and produce high levels of IFNγ. KLRGloPD1+, on the other hand, make lower levels of IFNγ and are situated in the lung parenchyma (47, 48). In adoptive transfer studies, the high cytokine producing KLRG1hiPD1− cells survive poorly upon transfer and provide only limited protection to challenge infection. In contrast, the KLRG1loPD1+ cells respond well in the adoptive host and reduce bacteria burden to over a log less compared to controls (48). Similarly, while effector cytokine producing Tem cells confer protection to Leishmania major, these cells fail to survive in the absence of antigen, suggesting these are effector rather than genuine memory cells (49). In contrast to these locally protective CD4 T cells, Tcm and Tem cells from the spleen and mesenteric lymph nodes of drug-cured Trichuris muris-infected mice can transfer equivalent immune protection to naïve recipients (50). Both populations contain cells that have the potential to make interleukin (IL)-4 as indicated by the expression of an mRNA reporter for this type 2 cytokine which is key for the expulsion of the intestinal parasite (51). Whether gut Trm or Tem cells can transfer similar or even enhanced protection in the T. muris model is a key question, which has not yet been tested.
These studies demonstrate that both Th1 and Th2 effector cells transition into the memory pool. Whether IL-17 effector CD4 T cells have this ability is a point of contention and likely complicated by the relative ease with which these cells switch to IFNγ production. This makes these cells difficult to track over time without elegant fate mapping transgenic animals (52–55). The high expression of CD27 by Th17 cells has been linked to their limited survival, but this is not a universal finding; Muranski et al. found that in vitro activated Th17 cells survived long-term in vivo regardless of their expression of CD27 (30, 56).
There is similar discord about the persistence of T follicular helper (Tfh) cells as a distinct memory cell population. During immune responses, Tfh cells migrate to B cell follicles, where they provide help for the germinal center reaction (57). A Tfh cell phenotype can be maintained over time by persistent antigen (24), but whether such cells are genuine memory cells is questionable. We have found that, even in the absence of antigen, some memory CD4 T cells are located at the B/T cell border (25). These cells express high levels of the B cell follicle homing chemokine receptor, CXCR5, and provide accelerated help for B cell class switched antibody responses even when present at naïve precursor frequencies. These data suggest that B cells could directly reactivate memory CD4 T cells. Alternatively, using ex vivo imaging, Suan et al. have recently shown that macrophages can reactivate memory CD4 T cells located in the subcapsular region of the lymph node (27). These reactivated memory T cells rapidly migrate to B cell follicles to generate secondary germinal centers.
CXCR5+ cells with Tfh cell characteristics can also be found in the blood of both humans and mice (58). Upon reactivation, these cells rapidly differentiate into Tfh cells and promote antibody production by B cells. There are few direct demonstrations of Tfh-mediated protective immunity. However, mouse influenza-specific CD4 T cells either primed in vitro or in vivo by vaccination can reduce viral titers at least in part by assisting in the production of anti-viral antibody following intranasal challenge (59, 60).
Taken together these studies indicate that all the memory CD4 T cell subsets defined to date are capable of protective responses. The type and location of the memory CD4 T cells that vaccines should aim to generate are, therefore, likely to be dependent on the location and type of challenge. In support of this, while the recently modified Vaccinia virus Ankara TB vaccine, MVA85A, increased TB specific Th1 cells in the blood, it failed to provide any additional protection beyond standard BCG vaccination (61, 62). Generating lung Trm cells may be key, therefore, to protective immunity against TB. Such cells may, however, have a reduced life span compared to Tcm. Certainly in mouse models of influenza virus infection, the cross viral protective response attributed to lung CD4 and CD8 T cells wanes within months (63). Effective T cell vaccines may have to steer a difficult course between local cytokine producing T cells with a limited life span and potentially longer lived Tscm and Tcm cells.
What Factors Regulate Memory Cell Generation?
A more detailed understanding of the signals that drive the differentiation of memory cell subsets will aid in navigating this course between effector cell differentiation and life span. The type and amount of antigen, inflammatory cytokines, and different antigen presenting cell (APC) populations all influence T cell activation and subsequent memory T cell generation. Small doses of antigen, or presentation for a limited period of time, may be sufficient for initial T cell activation but fail to generate memory cells (64). Too much activation can be detrimental: more differentiated CD4 T cells are less likely to transition into the memory pool than those displaying a less differentiated phenotype; and chronic antigen can lead to exhausted memory T cells that respond poorly upon reactivation (65–67). The type of antigen can also affect memory cell generation with an epitope delivered in the context of a protein, rather than a single peptide, encouraging a broader T cell repertoire to transition into the memory pool (68). Memory cell development is, therefore, not a default outcome of specific T cell:DC interactions, but requires appropriate levels of TCR activation delivered by inflammation-matured APCs.
The strength and timing of the interactions between pMHC II complexes and TCRs clearly influence memory cell differentiation. Strong TCR signals induce greater Tem cell differentiation, while low affinity signals are insufficient for memory cell development (69–71). One prediction of this model is that different TCR clones should be present in distinct memory populations. In support of this, the overall binding strength of the TCR–pMHC II interactions influences the differentiation of Th1 versus Tfh cells (72). In contrast, Sallusto and colleagues recently found the same TCR clones in Th1, Th2, and Th17 differentiated human T cells (73). Similarly, in subcutaneously immunized mice, the same TCRβ clones were found in equivalent abundance in lymph nodes (both draining and distant) and the skin injection site several weeks after immunization (74). Elegant studies tracking individual CD8 T cell clones in vivo have, moreover, clearly demonstrated that the same T cell can differentiate down different memory cell routes (75, 76).
These data suggest that it is not the individual TCR–pMHC II interaction that directs memory cell differentiation. Following the initial priming event, individual daughter cells will experience distinct environments and interact with different APCs that display various levels of pMHC II complexes and other activating and inhibiting cell surface and soluble signals. It is this variety of subsequent activation environments that is likely to lead to a diverse memory cell population. Such signals influence initial T cell expansion and effector cell differentiation, which obviously have a knock on effect on the development of memory T cells (16, 17). Here, we will focus on the molecules and cells that are thought to influence the transition of activated cells into the memory pool.
Common-γ chain cytokines have long been thought to encourage activated T cells to become memory cells (77). IL-7, a key survival factor for all T cells, signals through the common-γ chain and a specific IL-7 receptor, CD127. Typically, activation leads to downregulation of CD127. In CD8 T cells, recently activated T cells that express high levels of CD127 are more prone to develop into memory cells (78). The same is not true, however, for CD4 T cells (65). Although exogenous IL-7 can increase the survival of effector CD4 T cells, limiting endogenous IL-7 does not reduce memory cell generation and supplying IL-7 signals to activated CD4 and CD8 T cells does not increase the total memory T cell pool (79–81). IL-2 may also support memory cell development with CD4 T cells that receive late IL-2 signals more likely to survive into memory (82). Similarly, CD4 T cells that are activated for the first time toward the end of the immune response, as inflammatory signals wane, are more likely to transition into Tcm cells (83).
These data suggest that it will be critical to consider the effect vaccines have not just in the first few days after delivery but over the 2–3 weeks of the primary immune response. Potentially, vaccines that induce antigen depots or a second antigen shot delivered close to the initial immunization may promote memory cell generation.
In particular, targeting antigen to B cells may be key not just to establish a good antibody response but also to generate optimal CD4 T cell activation and subsequent memory T cell generation. In many, but not all cases, CD4 T cell primary responses are diminished in the absence of B cells; the memory cell pool is more consistently reduced (84–88). Presentation of antigen by B cells plays a significant role in this effect as demonstrated by a reduction in CD4 memory T cells in chimeric animals in which B cells do not express MHC II (89, 90). A role for B cells in the transition into, or survival of, memory CD4 T cells has also been demonstrated with B cells required to maintain a stable memory T cell pool, a process that may be independent of soluble antibody (85, 87).
This role for B cells in CD4 memory T cell generation has prompted the hypothesis that Tfh cells may contain a subset of memory precursor cells (88–92). While initial Tfh cell differentiation is triggered by antigen-specific interactions between CD4 T cells and DCs, B cells are required to sustain Tfh cell differentiation (57, 88, 93, 94,). Memory CD4 T cells derived from Tfh phenotype cells have a propensity to re-express markers of Tfh cells, following reactivation (88, 92). Therefore, vaccines that can effectively target antigen to B cells could bias the generation of memory cells that are better equipped to provide B cell help upon reactivation.
In contrast, promoting Trm generation is likely to require immunization at the appropriate tissue site, triggering local inflammation that delivers signals to draining lymph nodes and encourages local recruitment of activated CD4 T cells. It may not be necessary to vaccinate at the potential infection site but stimulate an environment at a distant site that drives the maturation of DCs that instruct appropriate homing molecule expression on the activated T cells (17, 95, 8). Intranasal immunization can, for example, lead to the formation of memory CD4 T cells in the uterus, albeit at slightly lower numbers than a local immunization (8). These signals are likely to be needed during initial T cell activation. The active vitamin D3 metabolite 1,25(OH)2D3 and prostaglandin E2 drive expression of the skin-homing chemokine receptor, CCR8, only following initial T cell activation and alpha4 integrin expressing CD4 T cells migrate to mucosal sites within the first 4 days of the response (8, 96).
Both the initial and sustained T cell activation environment, therefore, play vital roles in influencing commitment to memory and the type(s) of memory cells that are generated. Our knowledge on which of these signals are necessary and sufficient for memory cell commitment is, however, still limited. Clues to the identification of these signals may be found in a better understanding of how memory T cells differ from their naïve counterparts, providing an enhanced clarity on how memory T cells can provide superior protection and what cells vaccines must aim to generate.
What Makes Memory CD4 T Cells Different?
A major tenet of vaccination is that memory T cells are better equipped to control and clear a pathogen compared to their naïve counterparts. “Better” encompasses a range of functions including speed, sensitivity, and the type of response the memory cell can deliver (Figure 2).
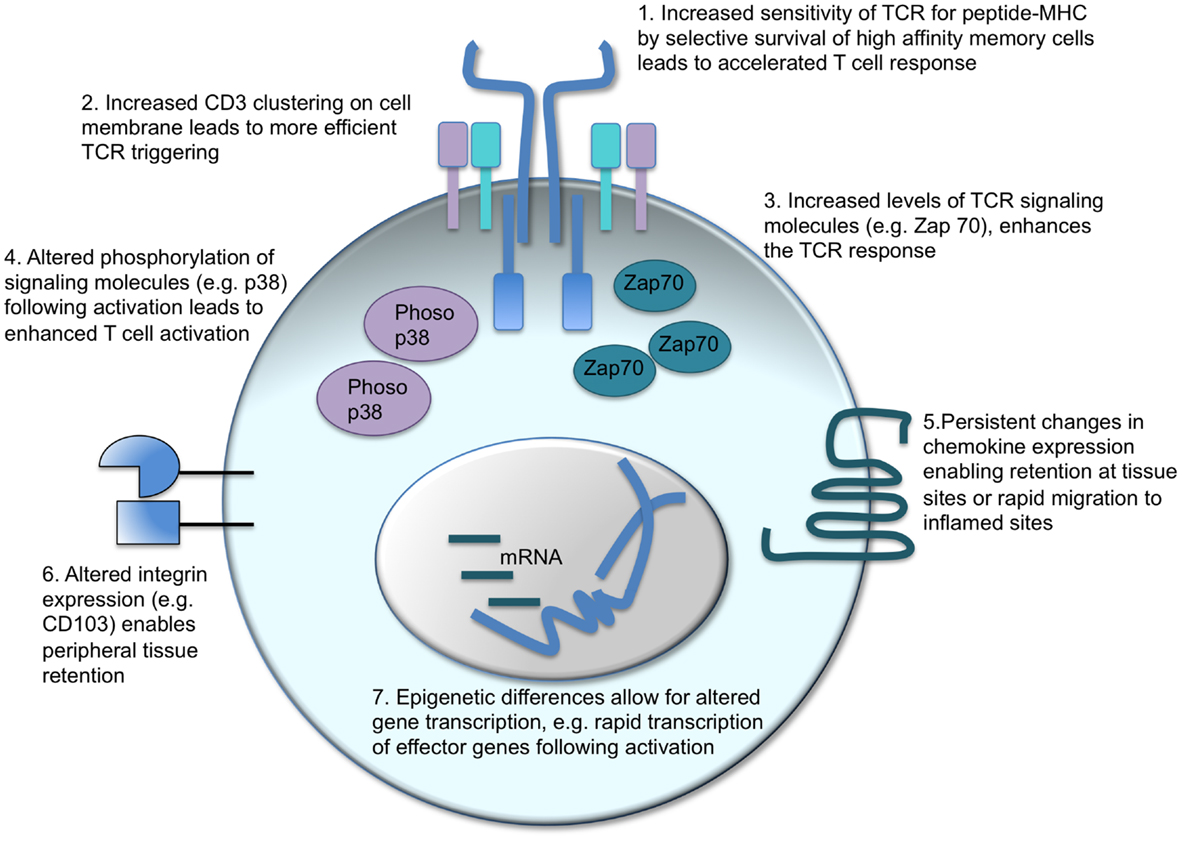
Figure 2. Intrinsic changes in memory CD4 T cells which enhance their response to activation. There are a number of factors that can contribute to the distinct responses of memory CD4 T cells as compared to their naïve counterparts. T cell receptor (TCR) triggering can be enhanced in two ways: in polyclonal populations, the cells with the highest affinity for the antigen can come to dominate the response (1) alternatively, or in addition, the CD3 TCR-signaling complex is clustered more effectively in memory than naïve CD4 T cells (2). Intracellular signaling molecules are also altered: memory CD4 T cells contain more Zap 70 than naïve T cells (3) and TCR activation leads to increased phosphorylation of the MAPK, p38 (4). Changes in cell surface proteins, such as chemokine receptors (5) and integrins (6), affect cell migration and location enabling memory CD4 T cells to migrate rapidly to inflamed sites or reside permanently in pathogen-targeted tissues. Changes in gene transcription before and following T cell reactivation are also evident. Epigenetic differences between naïve and memory CD4 T cells enable more rapid transcription of effector molecules, such as cytokines, thereby accelerating the control and clearance of pathogens (17, 97–101).
While there is a general acceptance that memory T cells respond more quickly to antigen exposure than naive T cells, the data do not necessarily support this conclusion. It is clear, as described above, that both memory CD4 and CD8 T cells make effector cytokine and CTL responses more rapidly than naïve T cells. However, whether memory T cells start to proliferate more quickly than naïve T cells is more contentious. Veiga-Fernandes et al. (CD8 T cells) and Rogers et al. (CD4 T cells) found that memory cells proliferated more quickly than naïve T cells in vitro, others, however, have demonstrated similar entry into cell cycle (102–105).
It is likely that the perception that memory CD4 T cells respond faster is based on their ability to perform a function in vivo more rapidly, e.g., viral clearance or induction of a delayed-type hypersensitivity response. These accelerated responses are likely to be based on a number of differences between naïve and memory T cells, including increased precursor frequency, location, and rapid effector functions, rather than increased speed of antigen recognition and cell proliferation.
The increased sensitivity of memory T cells to antigen is also an area of debate and has been challenged by a recent study that suggests memory CD8 T cells may actually be less sensitive than naive T cells (100, 106). In general, memory T cells are thought to have an increased sensitivity to antigen compared to primary responding cells (107–109). This may be due to alterations in the repertoire with higher affinity TCR clones dominating secondary responses, although this is not always the case (108). Such preferential reactivation of high affinity clones cannot, however, account for the increased sensitivity of memory TCR transgenic T cells indicating that increases in TCR affinity at the population level are not the whole story (108, 110, 111).
The ability of these memory T cells to respond to lower doses of antigen could be due to alterations in the levels of TCR and/or downstream signaling molecules. Kumar et al. found that CD3 molecules were more likely to form distinct clusters on the cell surface of memory as compared to naïve T cells allowing more efficient TCR triggering (99). Downstream signaling molecules are also altered: the key kinase, Zap70, is elevated in memory CD4 T cells, and, while activated memory cells have reduced levels of phosphorylated ERK, p38 is more highly phosphorylated in memory as compared to activated naïve CD4 T cells (97, 98).
Rather than changes in TCR signaling, the more rapid effector cytokine response that memory CD4 T cells mount is likely due to accelerated gene transcription as a consequence of epigenetic changes to the memory cell’s genome (101, 112). Such epigenetic changes can be passed on to dividing daughter cells providing a cellular mechanism for memory in the absence of continued polarizing signals. Epigenetic changes that influence gene transcription either affect the methylation status of the DNA, with demethylation opening up the chromatin for transcription, or altering DNA-associated histone proteins making the DNA more or less accessible to transcription factors and chromatin-modifying enzymes (101, 112).
Given that 95% of gene expression in naïve and memory CD4 T cells is the same (101), it is perhaps not surprising that in a recent large scale DNA methylation analysis of naïve and memory human CD4 T cells, only 132 out of 2100 genes analyzed showed a distinct methylation status (113). Altered genes include Th cell subset signature cytokines. In Th1 cells, the Ifnγ locus is demethylated, opening up the locus for rapid transcription following TCR activation, while the Il4 locus is heavily methylated (114, 115). Similarly, the Il4 locus is progressively demethylated in Th2 cells to ensure rapid IL4 production in effector and memory CD4 T cells (116–118).
Open chromatin is also characterized by hyperacetylation of associated histone proteins (101, 112). Human and mouse naïve CD4 T cells differentiated under Th1 or Th2 conditions in vitro gain acetylation marks at the Ifnγ and Il4 loci, respectively (119, 120). These changes are sustained in Tem cells but Tcm are hypoacetylated at these sites, clearly linking epigenetic changes to cytokine responses (120). In the same study, evidence for epigenetic flexibility was demonstrated: polarized T cells activated under opposing conditions showed evidence of acetylation at the opposing cytokine loci. Similarly, in a genome-wide analysis of methylation changes to histone proteins, Wei et al. found that while in vitro differentiated Th cell subsets had the predicted histone modifications to signature cytokines, the master transcription factors associated with these subsets had both permissive and repressive histone modifications. This pattern of methylation is suggestive of a poised state that could allow for flexibility in subsequent effector response (121). Together these studies demonstrate that activated and memory CD4 T cells have the flexibility to alter their differentiation state to adapt to changing environments, allowing for the possibility to retrain or reinforce a phenotype as required.
Why We Might Need to Restrain or Retrain Memory Cells
The ability of memory cells to respond in a flexible manner is likely to be key to controlling pathogenic CD4 T cell responses. While most memory responses are advantageous to the host, the rapid production of high levels of cytokine can be damaging. Large numbers of influenza virus-specific TCR transgenic cells producing IFNγ can cause lung pathology following intranasal infection of mice (122). Similarly, infection of mice that contain specific memory CD4 T cells with chronic LCMV leads to rapid and extensive weight loss, widespread inflammation and tissue destruction (123). This effect could be mitigated if virus-specific CD8 T cells or antibody was present, suggesting that uncontrolled activation of large numbers of CD4 T cells by continued presentation of the persistent virus antigen was responsible for the pathology. Whether these models are representative of human CD4 memory T cell-driven pathology, which is unlikely to exist in the absence of memory CD8 T or B cells, is unclear. For example, while pandemic influenza virus infection in humans can lead to excessive inflammation, this is likely driven by dysregulated innate responses (124–126). However, it does suggest a cautious approach to any peptide vaccines that only stimulate CD4 T cells (60).
In humans, autoreactive rather than pathogen-specific CD4 T cells cause the most significant damage. It is still unclear whether in T cell driven autoimmune diseases, such as multiple sclerosis and rheumatoid arthritis, autoreactive T cells are constantly activated by self-pMHC II complexes or whether T cell:DC interactions are restricted to periods of active disease. Human autoreactive CD4 T cells can be tracked using MHC II tetramers containing self-peptides. These studies suggest that autoreactive T cells can be found in healthy controls and patients, but that T cells from the latter have a memory cell phenotype and are more likely to make effector cytokines (127–129). While immunosuppressive biologics have significantly improved treatment for chronic inflammatory autoimmune diseases, approaches that tolerize autoreactive CD4 T cells offer an opportunity for a permanent solution. Such targeted immunotherapy has delivered some success in allergic individuals with tolerogenic exposure to allergen causing the deletion of specific CD4 T cells and significantly reducing allergic symptoms (130, 131).
While much is known about tolerizing naïve CD4 T cells, memory CD4 T cells present more of a challenge (132). Classically, tolerance in naïve CD4 T cells involves presentation of antigen in the absence of accompanying costimulatory signals (133). Strategies that aim to tolerize autoreactive CD4 T cells must take into account alterations between naïve and memory CD4 T cells. Memory CD4 T cells are less dependent on CD40–CD40L signals for activation, but do require CD28 signaling to respond fully, suggesting that they may be affected by reactivation in the absence of costimulation (134, 135). Recent studies from ourselves and others suggest that memory CD4 T cells activated in vivo with soluble peptide delivered without adjuvant are not immediately tolerized. Further activation, even in the presence of adjuvant, does lead to cell death (136, 137). These data support the hypothesis that memory CD4 T cells are flexible and can adapt to new polarizing signals. Such changes are likely to affect cells at an epigenetic level. Indeed, activated CD4 T cells that have received a tolerization signal have a demethylated PD-1 promoter leading to increased PD-1 cell surface expression (138).
Concluding Remarks
Teleologically, it makes sense to have a heterogeneous memory cell pool as pathogens are diverse and adept at adaptation to protective immunity. By positioning memory cells with immediate effector functions in pathogen-targeted peripheral tissues and highly proliferative “backup” cells in secondary lymphoid organs, the memory system, in effect, covers all bases. Vaccines that can reproduce such diversity are likely to be the most protective, but may also have the potential to cause adverse effects. For example, local vaccine-induced inflammation may be necessary for the development of Trm cells, but may not be acceptable to the general public. The opportunity to retrain memory CD4 T cells subsequent to their generation opens up additional avenues for manipulation, especially in the setting of autoimmunity and allergy. Only by increasing our knowledge of which signals influence memory cell generation, the underlying mechanisms responsible for their enhanced functions, and the molecular regulation of epigenetic flexibility, will such manipulation be possible.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Robert Benson, Shana Coley, and Vicky Morrison for their helpful comments. This work was supported by an Arthritis Research UK Career Development Award to MM (19905) and an Arthritis Research UK PhD Studentship to SJ (20339).
References
1. Hinman AR, Orenstein WA, Schuchat A. Vaccine-preventable diseases, immunizations, and MMWR – 1961–2011. 7 ed. Morbidity and Mortality Weekly Report (MMWR). (2011). Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/su6004a9.htm
2. Sprent J, Cho JH, Boyman O, Surh CD. T cell homeostasis. Immunol Cell Biol (2008) 86:312–9. doi: 10.1038/icb.2008.12
3. Plotkin SA. Vaccines: correlates of vaccine-induced immunity. Clin Infect Dis (2008) 47:401–9. doi:10.1086/589862
4. Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol (2000) 18:275–308. doi:10.1146/annurev.immunol.18.1.275
5. Vasconcelos JR, Dominguez MR, Araujo AF, Ersching J, Tararam CA, Bruna-Romero O, et al. Relevance of long-lived CD8(+) T effector memory cells for protective immunity elicited by heterologous prime-boost vaccination. Front Immunol (2012) 3:358. doi:10.3389/fimmu.2012.00358
6. Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med (1987) 165:302–19. doi:10.1084/jem.165.2.302
7. Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity (1994) 1:327–39. doi:10.1016/1074-7613(94)90084-1
8. Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, et al. VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science (2015) 348:aaa8205. doi:10.1126/science.aaa8205
9. Schmitt N, Ueno H. Regulation of human helper T cell subset differentiation by cytokines. Curr Opin Immunol (2015) 34:130–6. doi:10.1016/j.coi.2015.03.007
10. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327:1098–102. doi:10.1126/science.1178334
11. Joshi NS, Kaech SM. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol (2008) 180:1309–15. doi:10.4049/jimmunol.180.3.1309
12. Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol (2008) 8:247–58. doi:10.1038/nri2274
13. Opata MM, Stephens R. Early decision: effector and effector memory T cell differentiation in chronic infection. Curr Immunol Rev (2013) 9:190–206. doi:10.2174/1573395509666131126231209
14. Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature (2008) 452:356–60. doi:10.1038/nature06672
15. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi:10.1038/44385
16. Pepper M, Jenkins MK. Origins of CD4(+) effector and central memory T cells. Nat Immunol (2011) 12:467–71. doi:10.1038/ni.2038
17. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol (2013) 31:137–61. doi:10.1146/annurev-immunol-032712-095954
18. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17:1290–7. doi:10.1038/nm.2446
19. Luckey CJ, Weaver CT. Stem-cell-like qualities of immune memory; CD4+ T cells join the party. Cell Stem Cell (2012) 10:107–8. doi:10.1016/j.stem.2012.01.011
20. Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci Transl Med (2015) 7:273ra213. doi:10.1126/scitranslmed.3010314
21. Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Abed Maillard S, et al. Long-lasting stem cell-like memory CD8+ T cells with a naive-like profile upon yellow fever vaccination. Sci Transl Med (2015) 7:282ra248. doi:10.1126/scitranslmed.aaa3700
22. Schmueck-Henneresse M, Sharaf R, Vogt K, Weist BJ, Landwehr-Kenzel S, Fuehrer H, et al. Peripheral blood-derived virus-specific memory stem T cells mature to functional effector memory subsets with self-renewal potency. J Immunol (2015) 194:5559–67. doi:10.4049/jimmunol.1402090
23. Takeshita M, Suzuki K, Kassai Y, Takiguchi M, Nakayama Y, Otomo Y, et al. Polarization diversity of human CD4 stem cell memory T cells. Clin Immunol (2015) 159:107–17. doi:10.1016/j.clim.2015.04.010
24. Fazilleau N, Eisenbraun MD, Malherbe L, Ebright JN, Pogue-Caley RR, Mcheyzer-Williams LJ, et al. Lymphoid reservoirs of antigen-specific memory T helper cells. Nat Immunol (2007) 8:753–61. doi:10.1038/ni1472
25. MacLeod MK, David A, Mckee AS, Crawford F, Kappler JW, Marrack P. Memory CD4 T cells that express CXCR5 provide accelerated help to B cells. J Immunol (2011) 186:2889–96. doi:10.4049/jimmunol.1002955
26. Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest (2013) 123:594–9. doi:10.1172/JCI66327
27. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity (2015) 42:704–18. doi:10.1016/j.immuni.2015.03.002
28. Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med (2005) 11:1299–305. doi:10.1038/nm1326
29. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med (2009) 15:808–13. doi:10.1038/nm.1982
30. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity (2011) 35:972–85. doi:10.1016/j.immuni.2011.09.019
31. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity (2014) 41:886–97. doi:10.1016/j.immuni.2014.12.007
32. Zens KD, Farber DL. Memory CD4 T cells in influenza. Curr Top Microbiol Immunol (2015) 386:399–421. doi:10.1007/82_2014_401
33. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14:1294–301. doi:10.1038/ni.2744
34. Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell (2015) 161:737–49. doi:10.1016/j.cell.2015.03.031
35. Ledgerwood LG, Lal G, Zhang N, Garin A, Esses SJ, Ginhoux F, et al. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat Immunol (2008) 9:42–53. doi:10.1038/ni1534
36. Lee YT, Suarez-Ramirez JE, Wu T, Redman JM, Bouchard K, Hadley GA, et al. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J Virol (2011) 85:4085–94. doi:10.1128/JVI.02493-10
37. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188:4866–75. doi:10.4049/jimmunol.1200402
38. Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi:10.1016/j.immuni.2013.08.019
39. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol (2011) 187:5510–4. doi:10.4049/jimmunol.1102243
40. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell (2014) 159:814–28. doi:10.1016/j.cell.2014.10.026
41. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med (2015) 7:279ra239. doi:10.1126/scitranslmed.3010302
42. Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med (2012) 4:117ra117. doi:10.1126/scitranslmed.3003008
43. Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature (2011) 477:216–9. doi:10.1038/nature10339
44. Davila SJ, Olive AJ, Starnbach MN. Integrin alpha4beta1 is necessary for CD4+ T cell-mediated protection against genital Chlamydia trachomatis infection. J Immunol (2014) 192:4284–93. doi:10.4049/jimmunol.1303238
45. Strutt TM, Mckinstry KK, Dibble JP, Winchell C, Kuang Y, Curtis JD, et al. Memory CD4+ T cells induce innate responses independently of pathogen. Nat Med (2010) 16:558–64. doi:10.1038/nm.2142
46. Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature (2009) 462:510–3. doi:10.1038/nature08511
47. Sakai S, Kauffman KD, Schenkel JM, Mcberry CC, Mayer-Barber KD, Masopust D, et al. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J Immunol (2014) 192:2965–9. doi:10.4049/jimmunol.1400019
48. Moguche AO, Shafiani S, Clemons C, Larson RP, Dinh C, Higdon LE, et al. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J Exp Med (2015) 212:715–28. doi:10.1084/jem.20141518
49. Peters NC, Pagan AJ, Lawyer PG, Hand TW, Henrique Roma E, Stamper LW, et al. Chronic parasitic infection maintains high frequencies of short-lived Ly6C+CD4+ effector T cells that are required for protection against re-infection. PLoS Pathog (2014) 10:e1004538. doi:10.1371/journal.ppat.1004538
50. Zaph C, Rook KA, Goldschmidt M, Mohrs M, Scott P, Artis D. Persistence and function of central and effector memory CD4+ T cells following infection with a gastrointestinal helminth. J Immunol (2006) 177:511–8. doi:10.4049/jimmunol.177.1.511
51. Else KJ, Finkelman FD, Maliszewski CR, Grencis RK. Cytokine-mediated regulation of chronic intestinal helminth infection. J Exp Med (1994) 179:347–51. doi:10.1084/jem.179.1.347
52. Bending D, De La Pena H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, et al. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest (2009) 119:565–72. doi:10.1172/JCI37865
53. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, et al. Late developmental plasticity in the T helper 17 lineage. Immunity (2009) 30:92–107. doi:10.1016/j.immuni.2008.11.005
54. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol (2011) 12:255–63. doi:10.1038/ni.1993
55. McGeachy MJ. Th17 memory cells: live long and proliferate. J Leukoc Biol (2013) 94:921–6. doi:10.1189/jlb.0313113
56. Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, et al. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol (2010) 11:83–9. doi:10.1038/ni.1826
57. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
58. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity (2013) 39:770–81. doi:10.1016/j.immuni.2013.09.007
59. Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol (2006) 177:2888–98. doi:10.4049/jimmunol.177.5.2888
60. Alexander J, Bilsel P, Del Guercio MF, Stewart S, Marinkovic-Petrovic A, Southwood S, et al. Universal influenza DNA vaccine encoding conserved CD4+ T cell epitopes protects against lethal viral challenge in HLA-DR transgenic mice. Vaccine (2010) 28:664–72. doi:10.1016/j.vaccine.2009.10.103
61. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381:1021–8. doi:10.1016/S0140-6736(13)60177-4
62. Tameris M, Geldenhuys H, Luabeya AK, Smit E, Hughes JE, Vermaak S, et al. The candidate TB vaccine, MVA85A, induces highly durable Th1 responses. PLoS One (2014) 9:e87340. doi:10.1371/journal.pone.0087340
63. Liang S, Mozdzanowska K, Palladino G, Gerhard W. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J Immunol (1994) 152:1653–61.
64. Obst R, Van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D. Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation. Proc Natl Acad Sci U S A (2007) 104:15460–5. doi:10.1073/pnas.0707331104
65. Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity (2011) 35:633–46. doi:10.1016/j.immuni.2011.08.016
66. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity (2014) 40:289–302. doi:10.1016/j.immuni.2014.01.005
67. Kahan SM, Wherry EJ, Zajac AJ. T cell exhaustion during persistent viral infections. Virology (2015) 479-480C:180–93. doi:10.1016/j.virol.2014.12.033
68. Baumgartner CK, Yagita H, Malherbe LP. A TCR affinity threshold regulates memory CD4 T cell differentiation following vaccination. J Immunol (2012) 189:2309–17. doi:10.4049/jimmunol.1200453
69. Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol (2003) 4:355–60. doi:10.1038/ni908
70. Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity (2008) 28:533–45. doi:10.1016/j.immuni.2008.02.014
71. Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol (2012) 188:4305–14. doi:10.4049/jimmunol.1103568
72. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell (2013) 153:785–96. doi:10.1016/j.cell.2013.04.007
73. Becattini S, Latorre D, Mele F, Foglierini M, De Gregorio C, Cassotta A, et al. T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science (2015) 347:400–6. doi:10.1126/science.1260668
74. Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med (2015) 21:647–53. doi:10.1038/nm.3860
75. Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science (2013) 340:630–5. doi:10.1126/science.1235454
76. Gerlach C, Rohr JC, Perie L, Van Rooij N, Van Heijst JW, Velds A, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science (2013) 340:635–9. doi:10.1126/science.1235487
77. Schluns KS, Lefrancois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol (2003) 3:269–79. doi:10.1038/nri1052
78. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol (2003) 4:1191–8. doi:10.1038/ni1009
79. Sun JC, Lehar SM, Bevan MJ. Augmented IL-7 signaling during viral infection drives greater expansion of effector T cells but does not enhance memory. J Immunol (2006) 177:4458–63. doi:10.4049/jimmunol.177.7.4458
80. Tripathi P, Mitchell TC, Finkelman F, Hildeman DA. Cutting edge: limiting amounts of IL-7 do not control contraction of CD4+ T cell responses. J Immunol (2007) 178:4027–31. doi:10.4049/jimmunol.178.7.4027
81. Haring JS, Jing X, Bollenbacher-Reilley J, Xue HH, Leonard WJ, Harty JT. Constitutive expression of IL-7 receptor alpha does not support increased expansion or prevent contraction of antigen-specific CD4 or CD8 T cells following Listeria monocytogenes infection. J Immunol (2008) 180:2855–62. doi:10.4049/jimmunol.180.5.2855
82. McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, et al. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat Commun (2014) 5:5377. doi:10.1038/ncomms6377
83. Catron DM, Rusch LK, Hataye J, Itano AA, Jenkins MK. CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. J Exp Med (2006) 203:1045–54. doi:10.1084/jem.20051954
84. Linton PJ, Harbertson J, Bradley LM. A critical role for B cells in the development of memory CD4 cells. J Immunol (2000) 165:5558–65. doi:10.4049/jimmunol.165.10.5558
85. van Essen D, Dullforce P, Brocker T, Gray D. Cellular interactions involved in Th cell memory. J Immunol (2000) 165:3640–6. doi:10.4049/jimmunol.165.7.3640
86. Liu Q, Liu Z, Rozo CT, Hamed HA, Alem F, Urban JF Jr, et al. The role of B cells in the development of CD4 effector T cells during a polarized Th2 immune response. J Immunol (2007) 179:3821–30. doi:10.4049/jimmunol.179.6.3821
87. Whitmire JK, Asano MS, Kaech SM, Sarkar S, Hannum LG, Shlomchik MJ, et al. Requirement of B cells for generating CD4+ T cell memory. J Immunol (2009) 182:1868–76. doi:10.4049/jimmunol.0802501
88. Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity (2011) 35:583–95. doi:10.1016/j.immuni.2011.09.009
89. Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol (2006) 176:3498–506. doi:10.4049/jimmunol.176.6.3498
90. Barr TA, Brown S, Mastroeni P, Gray D. TLR and B cell receptor signals to B cells differentially program primary and memory Th1 responses to Salmonella enterica. J Immunol (2010) 185:2783–9. doi:10.4049/jimmunol.1001431
91. Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol (2013) 190:4014–26. doi:10.4049/jimmunol.1202963
92. Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity (2013) 38:805–17. doi:10.1016/j.immuni.2013.02.020
93. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29:138–49. doi:10.1016/j.immuni.2008.05.009
94. Poholek AC, Hansen K, Hernandez SG, Eto D, Chandele A, Weinstein JS, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol (2010) 185:313–26. doi:10.4049/jimmunol.0904023
95. Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, et al. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature (2003) 424:88–93. doi:10.1038/nature01726
96. McCully ML, Collins PJ, Hughes TR, Thomas CP, Billen J, O’donnell VB, et al. Skin metabolites define a new paradigm in the localization of skin tropic memory T Cells. J Immunol (2015) 195. doi:10.4049/jimmunol.1402961
97. Chandok MR, Okoye FI, Ndejembi MP, Farber DL. A biochemical signature for rapid recall of memory CD4 T cells. J Immunol (2007) 179:3689–98. doi:10.4049/jimmunol.179.6.3689
98. Adachi K, Davis MM. T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T cells. Proc Natl Acad Sci U S A (2011) 108:1549–54. doi:10.1073/pnas.1017340108
99. Kumar R, Ferez M, Swamy M, Arechaga I, Rejas MT, Valpuesta JM, et al. Increased sensitivity of antigen-experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity (2011) 35:375–87. doi:10.1016/j.immuni.2011.08.010
100. von Essen MR, Kongsbak M, Geisler C. Mechanisms behind functional avidity maturation in T cells. Clin Dev Immunol (2012) 2012:163453. doi:10.1155/2012/163453
101. Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol (2012) 12:306–15. doi:10.1038/nri3173
102. Zimmermann C, Prevost-Blondel A, Blaser C, Pircher H. Kinetics of the response of naive and memory CD8 T cells to antigen: similarities and differences. Eur J Immunol (1999) 29:284–90. doi:10.1002/(SICI)1521-4141(199901)29:01<284::AID-IMMU284>3.0.CO;2-C
103. Rogers PR, Dubey C, Swain SL. Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. J Immunol (2000) 164:2338–46. doi:10.4049/jimmunol.164.5.2338
104. Veiga-Fernandes H, Walter U, Bourgeois C, Mclean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol (2000) 1:47–53. doi:10.1038/76907
105. Whitmire JK, Eam B, Whitton JL. Tentative T cells: memory cells are quick to respond, but slow to divide. PLoS Pathog (2008) 4:e1000041. doi:10.1371/journal.ppat.1000041
106. Mehlhop-Williams ER, Bevan MJ. Memory CD8+ T cells exhibit increased antigen threshold requirements for recall proliferation. J Exp Med (2014) 211:345–56. doi:10.1084/jem.20131271
107. Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med (1999) 189:701–10. doi:10.1084/jem.189.4.701
108. Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol (2001) 2:711–7. doi:10.1038/90650
109. Turula H, Smith CJ, Grey F, Zurbach KA, Snyder CM. Competition between T cells maintains clonal dominance during memory inflation induced by MCMV. Eur J Immunol (2013) 43:1252–63. doi:10.1002/eji.201242940
110. Ericsson PO, Orchansky PL, Carlow DA, Teh HS. Differential activation of phospholipase C-gamma 1 and mitogen-activated protein kinase in naive and antigen-primed CD4 T cells by the peptide/MHC ligand. J Immunol (1996) 156:2045–53.
111. Rogers PR, Croft M. CD28, Ox-40, LFA-1, and CD4 modulation of Th1/Th2 differentiation is directly dependent on the dose of antigen. J Immunol (2000) 164:2955–63. doi:10.4049/jimmunol.164.6.2955
112. Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science (2010) 330:612–6. doi:10.1126/science.1191078
113. Komori HK, Hart T, Lamere SA, Chew PV, Salomon DR. Defining CD4 T cell memory by the epigenetic landscape of CpG DNA methylation. J Immunol (2015) 194:1565–79. doi:10.4049/jimmunol.1401162
114. Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, et al. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol (2007) 8:732–42. doi:10.1038/ni1474
115. Dong J, Chang HD, Ivascu C, Qian Y, Rezai S, Okhrimenko A, et al. Loss of methylation at the IFNG promoter and CNS-1 is associated with the development of functional IFN-gamma memory in human CD4(+) T lymphocytes. Eur J Immunol (2013) 43:793–804. doi:10.1002/eji.201242858
116. Agarwal S, Avni O, Rao A. Cell-type-restricted binding of the transcription factor NFAT to a distal IL-4 enhancer in vivo. Immunity (2000) 12:643–52. doi:10.1016/S1074-7613(00)80215-0
117. Lee DU, Agarwal S, Rao A. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity (2002) 16:649–60. doi:10.1016/S1074-7613(02)00314-X
118. Tykocinski LO, Hajkova P, Chang HD, Stamm T, Sozeri O, Lohning M, et al. A critical control element for interleukin-4 memory expression in T helper lymphocytes. J Biol Chem (2005) 280:28177–85. doi:10.1074/jbc.M502038200
119. Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol (2002) 3:643–51. doi:10.1038/ni808
120. Messi M, Giacchetto I, Nagata K, Lanzavecchia A, Natoli G, Sallusto F. Memory and flexibility of cytokine gene expression as separable properties of human T(H)1 and T(H)2 lymphocytes. Nat Immunol (2003) 4:78–86. doi:10.1038/ni872
121. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30:155–67. doi:10.1016/j.immuni.2008.12.009
122. Teijaro JR, Njau MN, Verhoeven D, Chandran S, Nadler SG, Hasday J, et al. Costimulation modulation uncouples protection from immunopathology in memory T cell responses to influenza virus. J Immunol (2009) 182:6834–43. doi:10.4049/jimmunol.0803860
123. Penaloza-MacMaster P, Barber DL, Wherry EJ, Provine NM, Teigler JE, Parenteau L, et al. Vaccine-elicited CD4 T cells induce immunopathology after chronic LCMV infection. Science (2015) 347:278–82. doi:10.1126/science.aaa2148
124. Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog (2008) 4:e1000115. doi:10.1371/journal.ppat.1000115
125. Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature (2012) 484:519–23. doi:10.1038/nature10921
126. Paquette SG, Banner D, Chi Le TB, Leomicronn AJ, Xu L, Ran L, et al. Pandemic H1N1 influenza A directly induces a robust and acute inflammatory gene signature in primary human bronchial epithelial cells downstream of membrane fusion. Virology (2014) 448:91–103. doi:10.1016/j.virol.2013.09.022
127. Law SC, Street S, Yu CH, Capini C, Ramnoruth S, Nel HJ, et al. T-cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA-DRB1 shared epitope alleles. Arthritis Res Ther (2012) 14:R118. doi:10.1186/ar3848
128. James EA, Rieck M, Pieper J, Gebe JA, Yue BB, Tatum M, et al. Citrulline-specific Th1 cells are increased in rheumatoid arthritis and their frequency is influenced by disease duration and therapy. Arthritis Rheumatol (2014) 66:1712–22. doi:10.1002/art.38637
129. Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med (2015) 7:287ra274. doi:10.1126/scitranslmed.aaa8038
130. Wambre E, Delong JH, James EA, Torres-Chinn N, Pfutzner W, Mobs C, et al. Specific immunotherapy modifies allergen-specific CD4(+) T-cell responses in an epitope-dependent manner. J Allergy Clin Immunol (2014) 133(872–879):e877. doi:10.1016/j.jaci.2013.10.054
131. Zaleska A, Eiwegger T, Soyer O, Van De Veen W, Rhyner C, Soyka MB, et al. Immune regulation by intralymphatic immunotherapy with modular allergen translocation MAT vaccine. Allergy (2014) 69:1162–70. doi:10.1111/all.12461
132. MacLeod MK, Anderton SM. Antigen-based immunotherapy (AIT) for autoimmune and allergic disease. Curr Opin Pharmacol (2015) 23:11–6. doi:10.1016/j.coph.2015.05.003
133. Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev (2011) 241:180–205. doi:10.1111/j.1600-065X.2011.01011.x
134. MacLeod M, Kwakkenbos MJ, Crawford A, Brown S, Stockinger B, Schepers K, et al. CD4 memory T cells survive and proliferate but fail to differentiate in the absence of CD40. J Exp Med (2006) 203:897–906. doi:10.1084/jem.20050711
135. Ndlovu H, Darby M, Froelich M, Horsnell W, Luhder F, Hunig T, et al. Inducible deletion of CD28 prior to secondary nippostrongylus brasiliensis infection impairs worm expulsion and recall of protective memory CD4(+) T cell responses. PLoS Pathog (2014) 10:e1003906. doi:10.1371/journal.ppat.1003906
136. David A, Crawford F, Garside P, Kappler JW, Marrack P, Macleod M. Tolerance induction in memory CD4 T cells requires two rounds of antigen-specific activation. Proc Natl Acad Sci U S A (2014) 111:7735–40. doi:10.1073/pnas.1406218111
137. Mackenzie KJ, Nowakowska DJ, Leech MD, Mcfarlane AJ, Wilson C, Fitch PM, et al. Effector and central memory T helper 2 cells respond differently to peptide immunotherapy. Proc Natl Acad Sci U S A (2014) 111:E784–93. doi:10.1073/pnas.1316178111
Keywords: CD4 T cell, memory, autoimmunity, vaccine, infection, epigenetic, differentiation, cytokine
Citation: Jaigirdar SA and MacLeod MKL (2015) Development and function of protective and pathologic memory CD4 T cells. Front. Immunol. 6:456. doi: 10.3389/fimmu.2015.00456
Received: 02 July 2015; Accepted: 24 August 2015;
Published: 08 September 2015
Edited by:
Eric Huseby, University of Massachusetts Medical School, USAReviewed by:
Tara Marlene Strutt, University of Massachusetts Medical School, USAMarion Pepper, University of Washington, USA
Copyright: © 2015 Jaigirdar and MacLeod. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan K. L. MacLeod, Centre for Immunobiology, Institute of Infection, Immunity and Inflammation, University of Glasgow, B528 SGDB, 120 University Place, Glasgow G12 8TA, UK,bWVnYW4ubWFjbGVvZEBnbGFzZ293LmFjLnVr