 Pieter H. C. Leliefeld
Pieter H. C. Leliefeld Leo Koenderman
Leo Koenderman Janesh Pillay
Janesh Pillay- 1Department of Surgery, University Medical Center Utrecht, Utrecht, Netherlands
- 2Laboratory of Translational Immunology, University Medical Center Utrecht, Utrecht, Netherlands
- 3Department of Respiratory Medicine, University Medical Center Utrecht, Utrecht, Netherlands
- 4Department of Anaesthesiology and Critical Care, University Medical Center Utrecht, Utrecht, Netherlands
Neutrophils are classically considered as cells pivotal for the first line of defense against invading pathogens. In recent years, evidence has accumulated that they are also important in the orchestration of adaptive immunity. Neutrophils rapidly migrate in high numbers to sites of inflammation (e.g., infection, tissue damage, and cancer) and are subsequently able to migrate to draining lymph nodes (LNs). Both at the site of inflammation as well as in the LNs, neutrophils can engage with lymphocytes and antigen-presenting cells. This crosstalk occurs either directly via cell–cell contact or via mediators, such as proteases, cytokines, and radical oxygen species. In this review, we will discuss the current knowledge regarding locations and mechanisms of interaction between neutrophils and lymphocytes in the context of homeostasis and various pathological conditions. In addition, we will highlight the complexity of the microenvironment that is involved in the generation of suppressive or stimulatory neutrophil phenotypes.
Introduction
Neutrophils are particularly known for their potent anti-microbial functions (1). This notion is enforced by various congenital neutrophil deficiencies, which show marked clinical phenotypes characterized by enhanced susceptibility to bacterial and fungal infections (2, 3). Infections, sterile inflammation, and other non-chronic challenges to the immune system are characterized by a rapid influx of neutrophils into the affected tissue (4). These neutrophils respond to chemo-attractants and adhesion molecules expressed on endothelial cells, and their main function is to clear infections and/or debris. In addition, they influence inflammatory responses through interactions with various cells of the immune system, such as antigen-presenting cells (APCs) and lymphocytes (5, 6). This has been observed in both murine models and in ex vivo studies with isolated cells from humans. Although neutrophils have long been considered to be composed of a homogenous population, an increasing body of literature supports the presence of multiple neutrophil phenotypes in cancer and inflammation (7–10). This heterogeneity can be induced by specific differentiation programs in the bone marrow or orchestrated by extracellular signals derived from inflammatory tissue (e.g., cytokines, bioactive lipids, or chemokines) (11, 12). The contribution of distinct neutrophil populations to immune suppression has not been resolved. In addition, in murine models and some human studies, clear distinctions were suggested between neutrophils and granulocytic myeloid-derived suppressor cells (G-MDSCs). These issues have been reviewed in detail (13, 14). This review will focus on the location and relevant diseases in which both (suppressive) neutrophils and G-MDSCs modulate adaptive immune responses and the mechanisms behind this process.
Location of the Interaction between Neutrophils and Lymphocytes
Site of Inflammation – Bystander Response
The early phase of infection is characterized by an influx of neutrophils and monocytes, which precedes the development of an antigen-specific response. Simultaneously, small numbers of T-cells are recruited into the infected tissue. Some of these T-cells can be activated and proliferate ‘‘in situ’’ in response to antigen presentation by myeloid cells (15). In addition, inflammatory cytokines cause proliferation and activation of non-specific T-cells in the profoundly pro-inflammatory microenvironment. This process has been coined the ‘‘bystander response’’ and was first seen in viral infections (16). It has recently been suggested that this bystander response contributes to early pathogen control in mice by enabling bystander memory T-cells to recognize and eliminate micro-organisms, such as Listeria monocytogenes infected cells in a NKG2D-dependent manner (17).
It is conceivable that a large and uncontrolled bystander response might predispose for auto-immunity and self-reactivity through the proliferation of self-reactive T-cells (18). It is tempting to speculate that neutrophils are involved to limit and control this bystander T-cell response as the timing of massive neutrophil tissue infiltration and the bystander response coincide.
Lymph Nodes and Primary Lymphatic Tissue
Neutrophils are found both in LNs and spleen particularly under inflammatory conditions (19–24). Dynamic imaging studies have shown that neutrophils are recruited to and form swarms in infected LNs in mice (25, 26). In addition, neutrophil migration to afferent LNs in response to tissue inflammation has been shown in various murine models (19–24).
There are two possible routes for neutrophils to enter LNs, via blood vessels or via afferent lymphatics (Figure 1). The first route requires exiting the circulation via high endothelial venules (HEVs). This mechanism is controversial, as human neutrophils seem to lack the expression of CCR7, a receptor for CCL21 and required for lymphocyte exiting through HEVs (27). Nonetheless, it has been shown in a murine model of ovalbumin-induced inflammation that neutrophil homing to LNs via the HEV takes place and requires integrins αMβ2 (MAC-1), αLβ2 (LFA-1), and l- and P-selectin (19). In LN-draining inflammatory tissue, additional chemokines and cytokines could orchestrate the attraction of neutrophils via HEVs. This has also been shown in tumor-draining LNs, when the tumor was subjected to photodynamic therapy. This treatment induces additional sterile inflammation. In this model, neutrophils are recruited to tumor-draining LNs via the HEV in an IL-17-dependent manner (20).
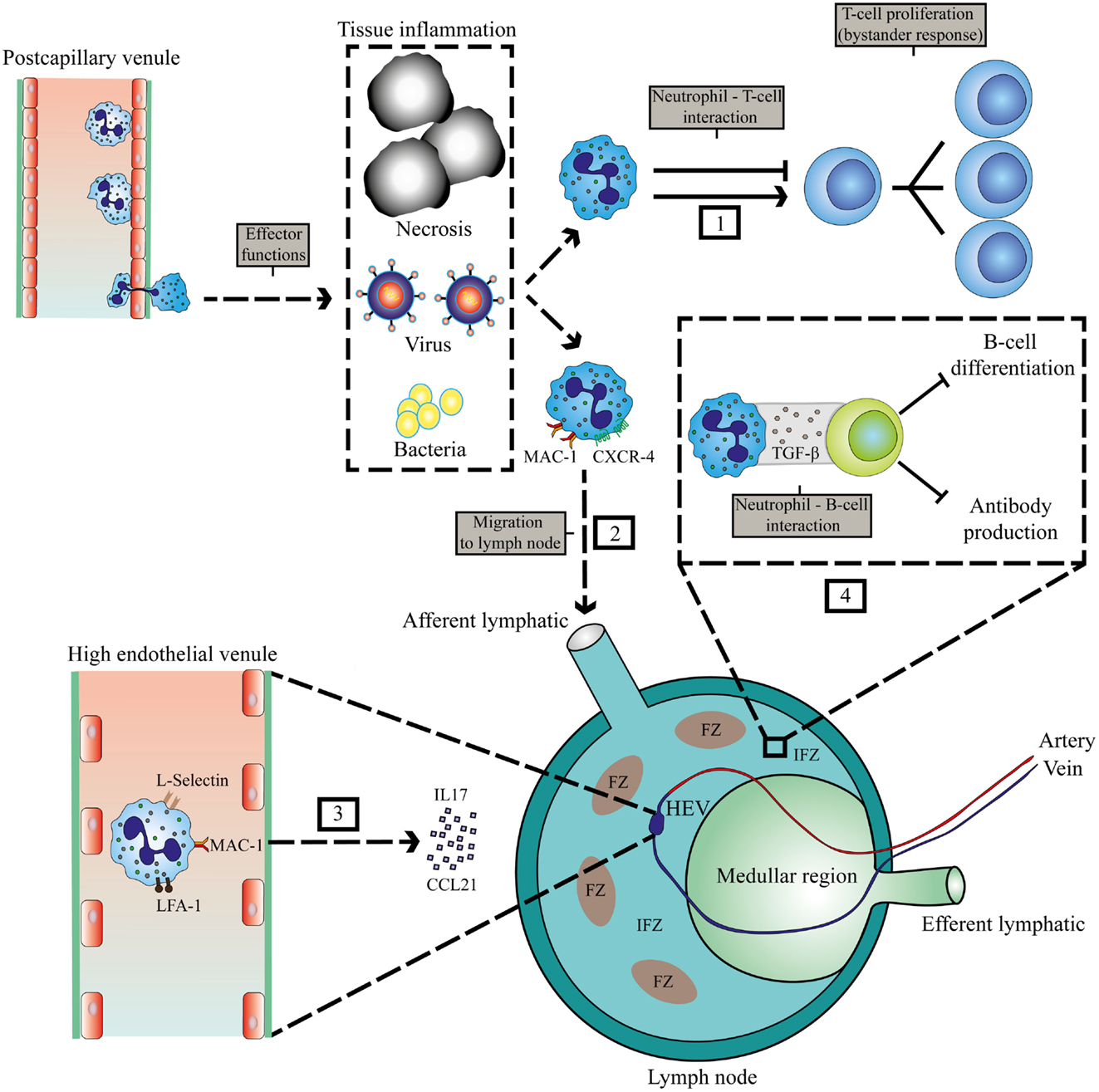
Figure 1. Localization of interaction between neutrophils and lymphocytes at sites of inflammation and in lymphoid tissue. Invasion of pathogens or inflammation due to necrosis and cancer leads to extravasation of neutrophils. (1) Interaction of neutrophils with T-cells in the peripheral tissue. (2) MAC-1 and CXCR4-dependent migration of neutrophils to LNs via afferent lymphatics during inflammation (22). (3) IL17 and CCL21-mediated migration of neutrophils via HEVs to the LN that requires MAC-1, LFA-1, and l-selectin (19, 20). (4) Inhibition of humoral responses by neutrophils in the IFZ and medullar region (23). HEV, high endothelial venule; FZ, follicular zone; IFZ, interfollicular zone.
Neutrophil migration to LNs via afferent lymphatics has been observed in various murine models of infections, vaccinations, and cancer and seems to depend on MAC-1 and CXCR4 expression on neutrophils (19, 22) (see Figure 1). The area in the LN that is occupied by neutrophils will determine which cells they encounter, and how they can influence subsequent immune responses. Neutrophils have been reported to occupy the medullary region and interfollicular zone (23). Neutrophils migrate to these areas in the LN during infection with Staphylococcus aureus. There they exhibit short- and long-term interactions with B-cells, thereby inhibiting production of antibodies, and thus humoral responses (23). Furthermore, neutrophil B-cell interactions have also been observed in primary lymphoid organs in various mouse models. In the marginal zone of the spleen, neutrophils were observed to contribute to antibody production and class switching by activating B-cells by producing BAFF, APRIL, and IL-21 (24). In this study, evidence was provided for the existence of a similar population of neutrophils that modulate B-cell responses in humans. However, this remains controversial as in a subsequent study, no splenic neutrophil–B cell interactions could be observed in humans (28). It seems firmly established, at least in various murine models that neutrophils enter primary and secondary lymphatic sites during the immune response evoked by various inflammatory stimuli. Apart from regulating immunity in inflamed tissue, they may play a role in regulating immune responses at these privileged immune sites (see below).
The Role of Neutrophils in Controlling Immune Responses Evoked by Bacterial and Viral Infections, Sterile Inflammation and Cancer
Viral Infections
Acute viral infections, such as influenza, are ideal models to study cellular kinetics during the immune responses and the putative modulating effects of neutrophils hereon. Influenza and RSV infections are characterized by an early large influx of neutrophils in the lung tissue followed several days later by a virus specific CD8+ T-cell response (29–31). Neutrophils might facilitate the development of this antigen-specific response as they are able to serve as APCs in influenza infection in mice (31, 32). Such antigen presentation by influenza-infected neutrophils has been demonstrated and was found to be mediated by MHC-I and co-stimulatory molecules CD80 and CD86, which leads to induction and activation of anti-viral responses of CD8+ T-cells (32). On the other hand, it is tempting to speculate that neutrophils may also inhibit T-cell responses in viral infections by inhibiting T-cell proliferation and inducing T-cell apoptosis. The mechanisms of this suppression will be discussed below and involve reactive oxygen species (ROS), arginase-I (ARG), and PD-L1. This has also been found in other inflammatory scenarios (33–35). The role of immune suppression by neutrophils in vivo in murine models of viral infections has not been adequately experimentally verified, but may be deduced from the fact that pathology in mice is T-cell dependent and that depleting neutrophils often results in an exaggerated response and pathology (31, 36). In chronic viral infections, such as human hepatitis B virus (HBV), it has recently been shown that recruitment of neutrophils to the liver limits immune pathology through inhibiting bystander and HBV specific T-cells in an arginase-dependent way, thus protecting the host from immune-mediated damage (37). These neutrophils were isolated from the PBMC fraction and were termed G-MDSC (13).
Bacterial Infections
As in viral infections, bacterial infections are associated with a large recruitment of neutrophils. The pivotal difference with viral infections is that phagocytosis and killing of bacterial targets by phagocytes is the principle mechanism of pathogen eradication (1). Ineffective killing of phagocytosed bacteria results in intracellular (phagosomal) survival and can lead to pathogen shuttling to distant sites and LNs (38, 39). Recently, it has been shown that neutrophils from mice infected with S. aureus migrated to the draining LNs and limited humoral responses through direct cellular interactions with B-cells. These direct cellular interactions were also found for neutrophil–T-cell interactions in humans (40). Kamenyeva et al. suggested that suppression of antibody production by neutrophils ex vivo was dependent on TGF-β (23). However, the contribution of the reduced humoral response to pathogen load was not assessed.
As mentioned above, in the early course of infection, large numbers of neutrophils are recruited to the affected tissue where modulation of T-cell responses most likely occurs with early recruited T-cells. These early lymphocytes mainly belong to the family of γδ-T-cells (41). These γδ-T-cells are thought to play a role in early pathogen clearance through production of cytokines and their crosstalk with innate immune cells (41). Neutrophils play an important role in the initiation of these γδ-T-cell responses. Phagocytosis of bacteria enables neutrophils to activate γδ-T-cells and induce their proliferation (42). This is dependent on (1) the microbial metabolite (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMB-PP), which neutrophils release after phagocytosis of bacteria and (2) the presence of monocytes for cellular contact-induced activation (42). However, other human studies have revealed the ability of neutrophils to suppress γδ-T-cells activation, possibly providing a negative-feedback mechanism (43, 44).
The early instruction of γδ-T-cells in humans by neutrophils parallels the role of these innate immune cells in the early instruction of T-cell responses in mice. In a murine model of Legionella pneumophila, neutrophils from pulmonary tissue are pivotal for the development of a TH1 response. In this model which resembles human disease, neutrophils were depleted by neutrophil-specific antibody Ly-6G, and this led to more TH2 skewing and more disease (45).
T-cell instruction, activation, and proliferation mostly require antigen-presenting cells, such as dendritic cells, B-cells, and macrophages. Neutrophils have been shown to both negatively and positively affect antigen presentation by these APCs under different conditions. This has extensively been reviewed previously (46, 47). Neutrophils may simply affect the amount of available antigen by phagocytosis, and thus limit antigen presentation by professional APCs (48). Alternatively, neutrophils might function as APCs themselves (49, 50). This possibility is supported by several studies showing the expression of MHCII and co-stimulatory molecules on neutrophils under different clinical conditions (51–53).
Disseminated Bacterial Infections (Sepsis)
Severe bacterial infections can result in systemic dissemination of bacteria that can lead to severe clinical conditions, such as sepsis and septic shock. These conditions are characterized by severe systemic inflammation, which can result in severe inflammatory damage to the host when not properly controlled. Immune inhibitory mechanisms have evolved in order to prevent this exaggerated inflammatory response (54). The specific role of neutrophils in this immune suppression has not been adequately studied. This is a challenging research question as depletion or inhibition of neutrophil functions with the purpose of studying their anti-inflammatory role has profound impact on bacterial clearance. Identification of suppressive mechanisms that do not influence pathogen clearance and neutrophil-specific murine knockout models may aid in answering this question.
In humans, evidence has accumulated that neutrophils might contribute to the immune suppression seen in sepsis. Neutrophils in septic-shock patients express ARG and suppress T-cell functions, probably through depletion of l-arginine as detailed below (55). Immune suppression in sepsis can be at least in part attributed to the PD-1/PD-L1 axis that is involved in control of apoptosis in T-cells (56). Interestingly, expression of PD-L1 on tissue neutrophils has also been shown during chronic inflammation (57). The expression of PD-L1 on human neutrophils was found to be induced by the TH1 cytokine, interferon-γ, in vitro (35).
Sterile Inflammation/Vaccination
Neutrophils also play a role in the fine tuning of inflammation under sterile conditions. Many studies have been performed in ovalbumine (OVA)-induced immune responses in murine models. The OVA models are used as vaccination and allergy models and are useful to study the development of adaptive immune responses. The role of neutrophils in the OVA model follows the above-described findings in microbial models. The cells seem to effectively cross-prime CD8+ T-cells in an MHCI-dependent manner (58). They function as APCs or influence the capacity to present antigens by professional APCs (59, 60). For instance, dendritic cells have been shown to take up antigens acquired from phagocytosed apoptotic neutrophils (61).
These examples show that neutrophils can increase antigen presentation as APC or by delivering antigen to APCs. On the other hand, there are reports that neutrophils decrease the level of antigen presentation by APCs through an unknown mechanism during brief cellular interactions (48). These findings show that it is difficult to predict in which circumstances neutrophils stimulate or suppress antigen presentation even when very similar and well-controlled models are used.
Cancer
There is a large body of literature, which shows a heterogeneous population of myeloid cells characterized by their potential to inhibit adaptive immunity, and thus anti-tumor immune responses (14). These myeloid-derived suppressor cells consist of mononuclear cells and neutrophils in different stages of maturation. G-MDSCs facilitate tumor growth in various murine models through suppression of CD8+ responses and production of cytokines (62–64). In addition, in human cancer patients, G-MDSCs and suppressive neutrophils are isolated from the peripheral blood (65, 66). Although the distinction between neutrophils and G-MDSCs is not clear, the modulating role of these cells in the immune responses induced by tumors has become an accepted paradigm, and is extensively reviewed elsewhere (13, 67). Neutrophils are involved in both pro- or anti-tumor immune responses. Importantly, they have recently been shown to promote metastasis (7, 68). Their anti-tumor effects are mediated by their direct antibody-dependent cytotoxicity and their production of pro-inflammatory cytokines near and inside the tumor (69). These properties will not be discussed in this short review.
The pro-tumor effects of neutrophils are mediated by different mechanisms. First, neutrophils play an essential role in angiogenesis through expression of matrix metallo-proteases, such as MMP9 (70, 71). Second, in multiple murine models they inhibit anti-tumor CD8+ T-cell responses through mechanisms described below. The suppression of anti-tumor T-cell responses by neutrophils was recently shown to be pivotal in tumor metastasis in a murine model of breast cancer (7). In this study, γδ-T-cells facilitated neutrophil recruitment to the tumor via an IL-17 and G-CSF mediated pathway. The microenvironmental cues for the switch in neutrophil phenotype from pro- to anti-tumor are slowly being unraveled. In a model of lung cancer, TGF-β induces or recruits neutrophils with a pro-tumor phenotype (termed N1), whereas blocking TGF-β induces an anti-tumor neutrophil phenotype (termed N2) (11).
Mechanisms of T-Cell Suppression by Neutrophils and G-MDSCs
Despite the evidence that neutrophils can stimulate T-cell responses, most studies point toward a direct suppressive role of these cells on different T-cell responses in various disease models as described above. The mechanisms of suppression have been reviewed in detail elsewhere and are summarized in Figure 2 (13). Most of the mechanisms that neutrophils employ to suppress T-cell functions are closely related to their anti-microbial functions, i.e., the same or similar mediators are used. Two of the most frequently reported mechanisms are via ARG and ROS.
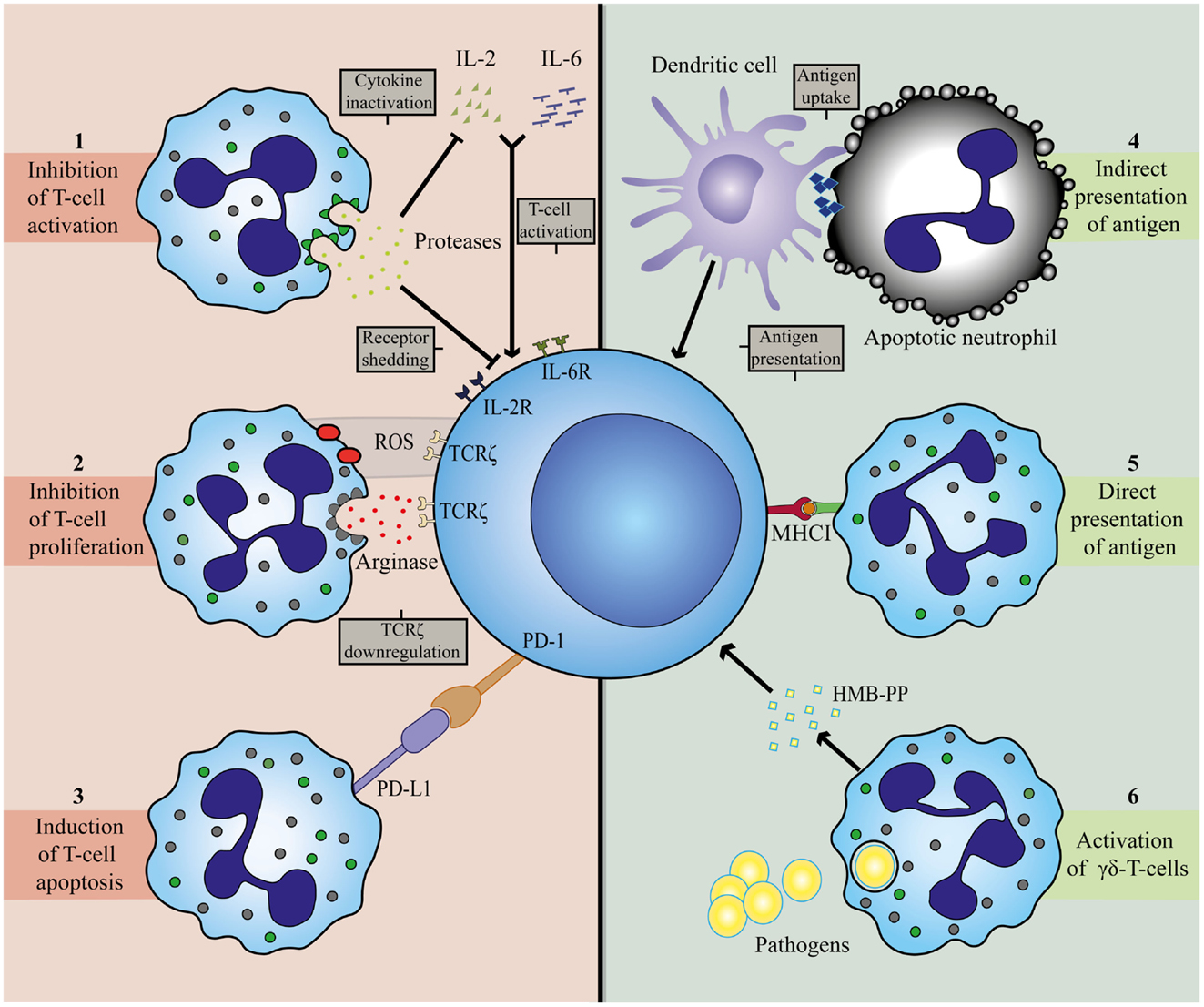
Figure 2. Mechanisms involved in T-cell inhibition (left panel) and activation (right panel) by neutrophils. Neutrophils can establish T-cell inhibition by (1) degranulation of granular constituents. The serine proteases elastase and cathepsin G inactivate T-cell stimulating cytokines, IL-2 and IL-6, and catalyze shedding of cytokine receptors for IL-2 and IL-6 on T-cells (72, 73). (2) Production of ROS and release of arginase. Both agents can result in downregulation of TCRζ on T-cells, thereby arresting the cell in the G0-G1 phase (40, 74–78). (3) Expression of PD-L1. Upregulation of this ligand is associated with interferon-dependent PD1-mediated T-cell apoptosis (35, 56). T-cell activation by neutrophils is attained by (4) indirect antigen presentation. Dendritic cells take up antigens from apoptotic neutrophils and serve as APC for T-cells (61). (5) Direct antigen presentation. Neutrophils posses the capacity to cross-prime CD8+ T-cells directly in a MHCI-dependent manner (32). (6) Release of microbial metabolites (HMB-PP). Neutrophils release bacterial products after ingestion to activate γδ-T-cells (42).
ARG is found in the gelatinase containing granules of neutrophils and is thought to contribute to antifungal immunity trough depletion of l-arginine (79, 80). Depletion of l-arginine also results in a cell cycle arrest in activated T-cells in the G0-G1 phase, which limits T-cell proliferation (74). This is thought to occur through downregulation of TCRζ (75, 76). It seems that the expression of TCRζ requires l-arginine for adequate expression and functionality (81). In addition, l-arginine is required for dephosphorylation of cofilin. Cofilin is pivotal for the stability of the immunological synapse. Therefore, depletion of l-arginine by ARG impairs the formation and stability of an immunological synapse, which is required for T-cell activation and proliferation (82, 83).
ROS are an intricate part of neutrophil anti-microbial defense and the lack of ability to produce ROS as, seen in chronic granulomatous disease, is characterized by severe infections (84–86). One of the products of NADPH oxidase activation is hydrogenperoxide (H2O2). H2O2 can suppress T-cell proliferation and activation through various mechanisms. It induces apoptosis, decreases Nf-κB activation, and downregulates TCRζ (77, 78). In addition, T-cell suppression by ROS is also accompanied by the oxidation of cofilin (77, 87). Cofilin can, therefore, be influenced by both ARG and ROS: both mechanisms being employed by neutrophils. Therefore, the T-cell suppression by disruption of cofilin might prove to be a useful therapeutic target.
Interestingly, regulatory T-cells are resistant to oxidative stress (88). This suggests that regulatory T-cells are less sensitive to suppression than other T-cells, thus enhancing the overall suppressive effect of H2O2.
Suppression of T-cell activation and proliferation requires high concentrations of H2O2 (33, 87). This amount of ROS might only be reached in inflammatory tissue with massive neutrophil influx. A more elegant way of suppression via H2O2 is through cell-cell contact between neutrophils and T-cells. Such a direct mechanism for delivery of ROS in an immunological synapse has been identified (29). Here, neutrophil-T-cell contacts were mediated by MAC-1.
Another important suppressive pathway requires similar cell-cell contacts: inhibition via T-cell PD-1 by PD-L1, a potent inducer of apoptosis in T-cells 1, expressed on neutrophils during sepsis (25). The underlying mechanism of PD-L1 expression is an interferon dependent process (35). The PD-1/PD-L1 axis is thought to be an important mechanism in the immune suppression found in sepsis patients by inducing lymphocyte apoptosis and monocyte dysfunction (56). Blocking this axis after the induction of sepsis by administering a PD-1 blocking antibody improves survival in mice by increasing pathogen clearance (89). This suppressive mechanism might be protective in tissues with severe inflammatory infiltrates, but may be detrimental as immune suppression aggravates sepsis. At this moment, one can only speculate regarding the role of PD-L1 on neutrophils in this immune suppressed state.
Finally, neutrophils can modulate T-cells by degranulating granular constituents, such as neutrophil elastase. These proteases are able to cleave and inactivate essential cytokines, such as IL-2 and receptors, such as the IL-2 and IL-6 receptor on T-cells (72, 73).
Conclusion
The studies mentioned in this review have led to the consensus that neutrophils are capable of modulating adaptive immune responses through interactions with T- and B-cells and possibly APCs. The mechanistic studies in mice have been corroborated with human ex vivo data. These studies show that neutrophils are capable of directly interacting with lymphocytes and modulating their responses at local sites of inflammation as well as in draining LNs. One of the key remaining issues is the question whether human neutrophils show functional plasticity as has been suggested by us and others (13, 90). This plasticity can occur at different levels: (1) the existence of functional subsets, which are intrinsically different or (2) the transdifferentiation into suppressive neutrophils or even into an APC type of hybrid cell (90–92). The microenvironmental cues mediating the switch from classical neutrophils to suppressive neutrophils have barely been studied although TGF-β seems to play an important role in microbial and tumor models (11, 23).
In conclusion, murine and human studies to date show that neutrophils are potent modulators of immunity. The first step of establishing a strategy to target immune modulatory neutrophils without influencing their essential anti-microbial functions is finding relevant human diseases in which this modulation plays a pivotal role (93). The unraveling of microenvironmental cues mediating the recruitment of and/or “switching” into suppressive neutrophils in such diseases is essential in understanding and targeting of this pathway.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev (2007) 219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x
2. Dinauer MC. Inherited neutrophil disorders: molecular basis and new therapies. Hematology (2000) 2000:303–18. doi:10.1182/asheducation-2000.1.303
3. Keszei M, Westerberg LS. Congenital defects in neutrophil dynamics. J Immunol Res (2014) 2014:303782. doi:10.1155/2014/303782
4. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6:173–82. doi:10.1038/nri1785
5. Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood (2014) 124:710–9. doi:10.1182/blood-2014-03-453217
6. Kalyan S, Kabelitz D. When neutrophils meet T cells: beginnings of a tumultuous relationship with underappreciated potential. Eur J Immunol (2014) 44:627–33. doi:10.1002/eji.201344195
7. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522:345–8. doi:10.1038/nature14282
8. Pillay J, Ramakers BP, Kamp VM, Loi ALT, Lam SW, Hietbrink F, et al. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J Leukoc Biol (2010) 88:211–20. doi:10.1189/jlb.1209793
9. Buckley C, Ross E. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol (2006) 79:303–11. doi:10.1189/jlb.0905496.Journal
10. Pillay J, Hietbrink F, Koenderman L, Leenen LPH. The systemic inflammatory response induced by trauma is reflected by multiple phenotypes of blood neutrophils. Injury (2007) 38:1365–72. doi:10.1016/j.injury.2007.09.016
11. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi:10.1016/j.ccr.2009.06.017
12. Christoffersson G, Vågesjö E, Vandooren J, Lidén M, Massena S, Reinert RB, et al. VEGF-Arecruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood (2012) 120:4653–62. doi:10.1182/blood-2012-04-421040
13. Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell Mol Life Sci (2013) 70:3813–27. doi:10.1007/s00018-013-1286-4
14. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
15. McGill J, Legge KL. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J Immunol (2009) 183:4177–81. doi:10.4049/jimmunol.0901109
16. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science (1996) 272:1947–50. doi:10.1126/science.272.5270.1947
17. Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, et al. Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep (2013) 3:701–8. doi:10.1016/j.celrep.2013.02.020
18. Burroughs NJ, Ferreira M, Oliveira BMPM, Pinto AA. Autoimmunity arising from bystander proliferation of T cells in an immune response model. Math Comput Model (2011) 53:1389–93. doi:10.1016/j.mcm.2010.01.020
19. Gorlino CV, Ranocchia RP, Harman MF, García IA, Crespo MI, Morón G, et al. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J Immunol (2014) 193:1966–74. doi:10.4049/jimmunol.1301791
20. Brackett CM, Muhitch JB, Evans SS, Gollnick SO. IL-17 promotes neutrophil entry into tumor-draining lymph nodes following induction of sterile inflammation. J Immunol (2013) 191:4348–57. doi:10.4049/jimmunol.1103621
21. De Veer M, Neeland M, Burke M, Pleasance J, Nathanielsz J, Elhay M, et al. Cell recruitment and antigen trafficking in afferent lymph after injection of antigen and poly(I: C) containing liposomes, in aqueous or oil-based formulations. Vaccine (2013) 31:1012–8. doi:10.1016/j.vaccine.2012.12.049
22. Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat Commun (2015) 6:7139. doi:10.1038/ncomms8139
23. Kamenyeva O, Boularan C, Kabat J, Cheung GYC, Cicala C, Yeh AJ, et al. Neutrophil recruitment to lymph nodes limits local humoral response to Staphylococcus aureus. PLoS Pathog (2015) 11:e1004827. doi:10.1371/journal.ppat.1004827
24. Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell–helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol (2011) 13:170–80. doi:10.1038/ni.2194
25. Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M, Herzmark P, et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity (2008) 29:487–96. doi:10.1016/j.immuni.2008.07.012
26. Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature (2013) 498:371–5. doi:10.1038/nature12175
27. Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol (2008) 8:362–71. doi:10.1038/nri2297
28. Nagelkerke SQ, Aan De Kerk DJ, Jansen MH, Van Den Berg TK, Kuijpers TW. Failure to detect functional neutrophil B helper cells in the human spleen. PLoS One (2014) 9:1–6. doi:10.1371/journal.pone.0088377
29. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol (2011) 179:199–210. doi:10.1016/j.ajpath.2011.03.013
30. Lukens MV, van de Pol AC, Coenjaerts FEJ, Jansen NJG, Kamp VM, Kimpen JLL, et al. A systemic neutrophil response precedes robust CD8(+) T-cell activation during natural respiratory syncytial virus infection in infants. J Virol (2010) 84:2374–83. doi:10.1128/JVI.01807-09
31. Tate MD, Brooks AG, Reading PC, Mintern JD. Neutrophils sustain effective CD8+ T-cell responses in the respiratory tract following influenza infection. Immunol Cell Biol (2012) 90:197–205. doi:10.1038/icb.2011.26
32. Hufford MM, Richardson G, Zhou H, Manicassamy B, García-Sastre A, Enelow RI, et al. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8+ T cells. PLoS One (2012) 7 e46581. doi:10.1371/journal.pone.0046581
33. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res (2001) 61:4756–60.
34. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSC) inhibit graft-versus-host (GVHD) disease via an arginase-1 dependent mechanism that is upregulated by IL-13. Blood (2010) 116:612–26. doi:10.1182/blood-2010-06-287839
35. De Kleijn S, Langereis JD, Leentjens J, Kox M, Netea MG, Koenderman L, et al. IFN-γ-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLoS One (2013) 8 e72249. doi:10.1371/journal.pone.0072249
36. Rygiel TP, Rijkers ESK, de Ruiter T, Stolte EH, van der Valk M, Rimmelzwaan GF, et al. Lack of CD200 enhances pathological T cell responses during influenza infection. J Immunol (2009) 183:1990–6. doi:10.4049/jimmunol.0900252
37. Pallett LJ, Gill US, Quaglia A, Sinclair LV, Jover-Cobos M, Schurich A, et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat Med (2015) 21:591–600. doi:10.1038/nm.3856
38. Thwaites GE, Gant V. Are bloodstream leukocytes Trojan horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol (2011) 9:215–22. doi:10.1038/nrmicro2508
39. Bland DM, Anderson DM. Imaging early pathogenesis of bubonic plague: are neutrophils commandeered for lymphatic transport of bacteria? MBio (2013) 4:e837–813. doi:10.1128/mBio.00837-13
40. Pillay J, Kamp VM, Van Hoffen E, Visser T, Tak T, Lammers J, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest (2012) 122:327–36. doi:10.1172/JCI57990DS1
41. Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol (2010) 10:467–78. doi:10.1038/nri2781
42. Davey MS, Lin C-Y, Roberts GW, Heuston S, Brown AC, Chess JA, et al. Human neutrophil clearance of bacterial pathogens triggers anti-microbial γδ T cell responses in early infection. PLoS Pathog (2011) 7:e1002040. doi:10.1371/journal.ppat.1002040
43. Sabbione F, Gabelloni ML, Ernst G, Gori MS, Salamone G, Oleastro M, et al. Neutrophils suppress γδ T-cell function. Eur J Immunol (2014) 44:819–30. doi:10.1002/eji.201343664
44. Kalyan S, Chandrasekaran V, Quabius ES, Lindhorst TK, Kabelitz D. Neutrophil uptake of nitrogen-bisphosphonates leads to the suppression of human peripheral blood γδ T cells. Cell Mol Life Sci (2014) 71:2335–46. doi:10.1007/s00018-013-1495-x
45. Tateda K, Moore TA, Deng JC, Newstead MW, Zeng X, Matsukawa A, et al. Early recruitment of neutrophils determines subsequent T1/T2 host responses in a murine model of Legionella pneumophila pneumonia. J Immunol (2001) 166:3355–61. doi:10.4049/jimmunol.166.5.3355
46. Soehnlein O, Weber C, Lindbom L. Neutrophil granule proteins tune monocytic cell function. Trends Immunol (2009) 30:538–46. doi:10.1016/j.it.2009.06.006
47. Appelberg R. Neutrophils and intracellular pathogens: beyond phagocytosis and killing. Trends Microbiol (2007) 15:87–92. doi:10.1016/j.tim.2006.11.009
48. Yang C-W, Strong BSI, Miller MJ, Unanue ER. Neutrophils influence the level of antigen presentation during the immune response to protein antigens in adjuvants. J Immunol (2010) 185:2927–34. doi:10.4049/jimmunol.1001289
49. Müller I, Munder M, Kropf P, Hänsch GM. Polymorphonuclear neutrophils and T lymphocytes: strange bedfellows or brothers in arms? Trends Immunol (2009) 30:522–30. doi:10.1016/j.it.2009.07.007
50. Potter NS, Harding CV. Neutrophils process exogenous bacteria via an alternate class I MHC processing pathway for presentation of peptides to T lymphocytes. J Immunol (2001) 167:2538–46. doi:10.4049/jimmunol.167.5.2538
51. Wagner C, Iking-Konert C, Hug F, Stegmaier S, Heppert V, Wentzensen A, et al. Cellular inflammatory response to persistent localized Staphylococcus aureus infection: phenotypical and functional characterization of polymorphonuclear neutrophils (PMN). Clin Exp Immunol (2006) 143:70–7. doi:10.1111/j.1365-2249.2005.02963.x
52. Gosselin EJ, Wardwell K, Rigby WF, Guyre PM. Induction of MHC class II on human polymorphonuclear neutrophils by granulocyte/macrophage colony-stimulating factor, IFN-gamma, and IL-3. J Immunol (1993) 151:1482–90.
53. Radsak M, Iking-Konert C, Stegmaier S, Andrassy K, Hänsch GM. Polymorphonuclear neutrophils as accessory cells for T-cell activation: major histocompatibility complex class II restricted antigen-dependent induction of T-cell proliferation. Immunology (2000) 101:521–30. doi:10.1046/j.1365-2567.2000.00140.x
54. Boomer JS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA (2011) 306:2594. doi:10.1001/jama.2011.1829
55. Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y, et al. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care (2014) 18:R163. doi:10.1186/cc14003
56. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol (2013) 13:862–74. doi:10.1038/nri3552
57. Tak T, Hilvering B, Tesselaar K, Koenderman L. Similar activation state of neutrophils in sputum of asthma patients irrespective of sputum eosinophilia. Clin Exp Immunol (2015). doi:10.1111/cei.12676
58. Beauvillain C, Delneste Y, Scotet M, Peres A, Gascan H, Guermonprez P, et al. Neutrophils efficiently cross-prime naive T cells in vivo. Blood (2007) 110:2965–73. doi:10.1182/blood-2006-12-063826
59. Maletto BA, Ropolo AS, Alignani DO, Liscovsky MV, Ranocchia RP, Moron VG, et al. Presence of neutrophil-bearing antigen in lymphoid organs of immune mice. Blood (2006) 108:3094–102. doi:10.1182/blood-2006-04-016659
60. Abdallah DSA, Egan CE, Butcher BA, Denkers EY. Mouse neutrophils are professional antigen-presenting cells programmed to instruct Th1 and Th17 T-cell differentiation. Int Immunol (2011) 23:317–26. doi:10.1093/intimm/dxr007
61. Schuster S, Hurrell B, Tacchini-Cottier F. Crosstalk between neutrophils and dendritic cells: a context-dependent process. J Leukoc Biol (2013) 94:671–5. doi:10.1189/jlb.1012540
62. Tecchio C, Scapini P, Pizzolo G, Cassatella MA. On the cytokines produced by human neutrophils in tumors. Semin Cancer Biol (2013) 23:159–70. doi:10.1016/j.semcancer.2013.02.004
63. Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181:5791–802. doi:10.4049/jimmunol.181.8.5791
64. Youn J-I, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol (2012) 91:167–81. doi:10.1189/jlb.0311177
65. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69:1553–60. doi:10.1158/0008-5472.CAN-08-1921
66. Khaled YS, Ammori BJ, Elkord E. Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients. J Immunol Res (2014) 2014:879897. doi:10.1155/2014/879897
67. Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother (2012) 61:1155–67. doi:10.1007/s00262-012-1294-5
68. Fridlender ZG, Albelda SM, Granot Z. Promoting metastasis: neutrophils and T cells join forces. Cell Res (2015) 25:765–6. doi:10.1038/cr.2015.62
69. Brandau S, Dumitru CA, Lang S. Protumor and antitumor functions of neutrophil granulocytes. Semin Immunopathol (2013) 35:163–76. doi:10.1007/s00281-012-0344-6
70. Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol (2011) 54:948–55. doi:10.1016/j.jhep.2010.08.041
71. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest (2010) 120:1151–64. doi:10.1172/JCI37223
72. Bank U, Reinhold D, Schneemilch C, Kunz D, Synowitz HJ, Ansorge S. Selective proteolytic cleavage of IL-2 receptor and IL-6 receptor ligand binding chains by neutrophil-derived serine proteases at foci of inflammation. J Interferon Cytokine Res (1999) 19:1277–87. doi:10.1089/107999099312957
73. Bank U, Ansorge S. More than destructive: neutrophil-derived serine proteases in cytokine bioactivity control. J Leukoc Biol (2001) 69:197–206.
74. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–73. doi:10.1182/blood-2006-06-031856
75. Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol (2006) 176:2085–94. doi:10.4049/jimmunol.176.4.2085
76. Yachimovich-Cohen N, Even-Ram S, Shufaro Y, Rachmilewitz J, Reubinoff B. Human embryonic stem cells suppress T cell responses via arginase I-dependent mechanism. J Immunol (2010) 184:1300–8. doi:10.4049/jimmunol.0804261
77. Gelderman KA, Hultqvist M, Holmberg J, Olofsson P, Holmdahl R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci U S A (2006) 103:12831–6. doi:10.1073/pnas.0604571103
78. Malmberg KJ, Arulampalam V, Ichihara F, Petersson M, Seki K, Andersson T, et al. Inhibition of activated/memory (CD45RO(+)) T cells by oxidative stress associated with block of NF-kappaB activation. J Immunol (2001) 167:2595–601. doi:10.4049/jimmunol.167.5.2595
79. Jacobsen LC, Theilgaard-Mönch K, Christensen EI, Borregaard N. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood (2007) 109:3084–7. doi:10.1182/blood-2006-06-032599
80. Munder M, Mollinedo F, Calafat J, Canchado J, Gil-lamaignere C, Fuentes M, et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Immunobiology (2005) 105:2549–56. doi:10.1182/blood-2004-07-2521.Supported
81. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem (2002) 277:21123–9. doi:10.1074/jbc.M110675200
82. Feldmeyer N, Wabnitz G, Leicht S, Luckner-minden C, Schiller M, Franz T, et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int Immunol (2012) 24:303–13. doi:10.1093/intimm/dxs004
83. Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol (2008) 26:233–59. doi:10.1146/annurev.immunol.26.021607.090347
84. Klebanoff SJ, White LR. Iodination defect in the leukocytes of a patient with chronic granulomatous disease of childhood. N Engl J Med (1969) 280:460–6. doi:10.1056/NEJM196902272800902
85. Seger RA. Chronic granulomatous disease: recent advances in pathophysiology and treatment. Neth J Med (2010) 68:334–40.
86. Nathan C, Ding A. Snapshot: reactive oxygen intermediates (ROI). Cell (2010) 140:8–10. doi:10.1016/j.cell.2010.03.008
87. Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity (2008) 29:404–13. doi:10.1016/j.immuni.2008.06.016
88. Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood (2009) 113:3542–5. doi:10.1182/blood-2008-09-181040
89. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol (2010) 88:233–40. doi:10.1189/jlb.0110037
90. Matsushima H, Geng S, Lu R, Okamoto T, Yao Y, Mayuzumi N, et al. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood (2013) 121:1677–89. doi:10.1182/blood-2012-07-445189
91. Oehler L, Majdic O, Pickl WF, Stöckl J, Riedl E, Drach J, et al. Neutrophil granulocyte-committed cells can be driven to acquire dendritic cell characteristics. J Exp Med (1998) 187:1019–28. doi:10.1084/jem.187.7.1019
92. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519–31. doi:10.1038/nri3024
Keywords: inflammation, immune-regulation, neutrophil, myeloid-derived suppressor cells, immune-paralysis, neutrophil phenotypes, T-cells
Citation: Leliefeld PHC, Koenderman L and Pillay J (2015) How neutrophils shape adaptive immune responses. Front. Immunol. 6:471. doi: 10.3389/fimmu.2015.00471
Received: 14 July 2015; Accepted: 31 August 2015;
Published: 14 September 2015
Edited by:
Elisabetta Padovan, University of Basel, SwitzerlandReviewed by:
Matteo Iannacone, San Raffaele Scientific Institute, ItalyMarco A. Cassatella, University of Verona, Italy
Dieter Kabelitz, Christian-Albrechts University Kiel, Germany
Copyright: © 2015 Leliefeld, Koenderman and Pillay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janesh Pillay, Department of Anaesthesiology and Critical Care, University Medical Center Utrecht, Heidelberglaan 100, Utrecht GA 3508, Netherlands,ai5waWxsYXktMkB1bWN1dHJlY2h0Lm5sLA==anBpbGxheUBnbWFpbC5jb20=