 Chantelle L. Ahlenstiel
Chantelle L. Ahlenstiel Kazuo Suzuki1,2
Kazuo Suzuki1,2- 1The Kirby Institute, UNSW Australia, Sydney, NSW, Australia
- 2Immunovirology Laboratory, St. Vincent’s Centre for Applied Medical Research, Darlinghurst, NSW, Australia
- 3Calimmune Inc., Darlinghurst, NSW, Australia
The current treatment strategy for HIV-1 involves prolonged and intensive combined antiretroviral therapy (cART), which successfully suppresses plasma viremia. It has transformed HIV-1 infection into a chronic disease. However, despite the success of cART, a latent form of HIV-1 infection persists as integrated provirus in resting memory CD4+ T cells. Virus can reactivate from this reservoir upon cessation of treatment, and hence HIV requires lifelong therapy. The reservoir represents a major barrier to eradication. Understanding molecular mechanisms regulating HIV-1 transcription and latency are crucial to develop alternate treatment strategies, which impact upon the reservoir and provide a path toward a “functional cure” in which there is no detectable viremia in the absence of cART. Numerous reports have suggested ncRNAs are involved in regulating viral transcription and latency. This review will discuss the latest developments in ncRNAs, specifically short interfering (si)RNA and short hairpin (sh)RNA, targeting molecular mechanisms of HIV-1 transcription, which may represent potential future therapeutics. It will also briefly address animal models for testing potential therapeutics and current gene therapy clinical trials.
Introduction
Non-coding RNAs (ncRNAs) are widely accepted as important regulators of cellular processes acting through post-transcriptional control of protein expression. The coding regions translated into protein of the human genome account for ~2%, while over 90% of the non-coding genome is reported to be utilized for transcription (1–4). Consequently, ncRNAs make up the majority of the mammalian transcriptome (2) and are reported to function in the transcriptional regulation of gene expression during embryogenesis, cell differentiation (5) and in response to external stimuli, particularly virus infection (6). NcRNAs are classified as regulatory or infrastructural (ribosomal and transfer RNAs). The regulatory class can be divided into small ncRNAs (<200 nucleotides) and long ncRNAs (>200 nucleotides) (7), with small ncRNAs being further categorized into microRNA (miRNA), short interfering RNAs (siRNAs), and antisense RNAs (asRNAs). This review will primarily focus on siRNAs and short hairpin RNAs (shRNAs) as there have been several recent reviews of the roles of long ncRNAs and other small ncRNAs (8, 9).
The HIV-1 5′ long terminal repeat (LTR), which acts as the promoter for integrated virus, consists of ~450 base pairs. It includes multiple binding sites for host transcription factors, including NF-κB, NFAT, SP1, and AP-1, which act to enhance viral transcription. HIV-1 transcription from integrated provirus is a highly controlled process, regulated by a number of epigenetic modifications, with recent studies identifying the involvement of ncRNAs. The major HIV-1 transactivation protein, Tat, dramatically enhances viral transcription by binding to a dynamic stem loop structure in viral RNA, the transactivation response element (TAR), which is also coded within the 5′LTR. In contrast, in cells harboring latent HIV-1, transcription is severely restricted and the 5′LTR promoter region carries a specific epigenetic profile, which includes increased histone methylation, decreased histone acetylation, which follows the recruitment of histone deacetylases (HDACs). These biochemical changes involve the histones of two nucleosomes (nuc0 and nuc1) that associate with specific regions of the 5′LTR. In particular, these changes are associated with repositioning of nuc1 (10–15). In latent virus, nuc1 overlies the transcription start site, whereas in actively transcribing virus, the demethylated/acetylated nucleosome slides upstream of the transcription start site (13).
This review aims to highlight a highly innovative approach to current HIV-1 therapies that utilizes small RNAs, which may represent an alternative strategy to eradication through a functional cure. We will discuss the latest developments in ncRNAs, specifically siRNA and shRNA, for targeting the molecular mechanisms of HIV-1 transcription as well as briefly address humanized murine models, which provide a vehicle to assess potential therapeutics, current gene therapy clinical trials, and future directions of promising therapeutics.
Limitations of HIV-1 Therapy
Combined antiretroviral therapy (cART) has revolutionized the treatment of HIV-1 infection in the developed world, changing a fatally acute disease into a manageable chronic condition. However, there are significant caveats accompanying lifelong cART therapy, which is currently necessary to control HIV-1 replication. These include the ongoing burden of compliance, drug toxicities, and residual excess morbidity and mortality, mostly due to serious non-AIDS events (16). Whilst effective cART rapidly suppresses the plasma viral load (pVL) to near undetectable levels (17) and permits reconstitution of immune cells (18), the integrated HIV-1 provirus DNA remains essentially unaffected, and therefore provides a viral reservoir, from which recrudescence can occur upon interruption of cART (19, 20). The current barrier to HIV eradication is this reservoir that persists in resting memory CD4+ T cells and cells of myeloid lineage (17, 21–24). While multiple approaches have been explored to surmount this obstacle, including early intervention with cART (25), cART intensification (18, 26–30), and purging of the reservoir using reactivation strategies (31–35), the fundamental limitation remains that in the overwhelming majority of patients, the virus rebounds with cessation of cART (36).
Current Therapeutic Approaches to Achieve a Functional Cure
While the ultimate goal is often stated to be the eradication of HIV, that is achieving a sterilizing cure, recent studies have suggested a more realistic approach might be to achieve a functional cure, whereby therapeutic interventions mediate a clinical state of undetectable pVL in the absence of cART. So far, the “Kick and Kill Approach” to eradication is the most studied to date, with the objective of reactivating the latent reservoir while the patient continues cART. The expected outcome is that infected cells would be killed either directly by virus reactivation or through the cytotoxic T-lymphocyte immune response as viral proteins are expressed on the cell surface following reactivation. The presence of cART should prevent new cells becoming infected. The desired result is reduction in or elimination of the latent reservoir. However, despite various interventions including pan T cell activation through OKT3 (37), recombinant IL-2 (38), or IL-7 (34, 39), and activation of the protein kinase C (PKC) or protein kinase B pathways using prostatin, bryostatin, or disulfram, respectively (40), these approaches have had only limited or no success in achieving a reduction in the size of the latent reservoir.
Most “kick and kill” approaches have focused on driving virus reactivation by modifying the epigenetic profile of the virus in latent reservoir by using HDAC inhibitors (HDACi). These have included valporic acid (VPA), suberoylanilide hydroxamic acid (SAHA, vorinostat), romidepsin, and panobinostat (41–46). In vitro studies have shown substantial reactivation of integrated virus in cells from certain patients. However, in vivo the extent of viral reactivation has been limited and these drugs induce substantial off-target effects with significant non-specific host gene activation (47, 48). This has been most clearly demonstrated for SAHA, which was observed to inhibit CTL function (49), and would therefore impede CTL mediated “kill” of previously latently infected cells following the HDACi-induced “kick” (49). These findings all point to the need to dissect more precisely the molecular mechanisms involved in HIV-1 latency, particularly reactivation, to develop a more specific and targeted approach in manipulating the viral reservoir.
Post-Transcriptional Gene Silencing of HIV-1 by si/shRNAs
A major limitation of the post-transcriptional gene silencing (PTGS) approach is the opportunity for viral escape due to targeting at the mRNA level, which allows the transcription process to potentially incorporate resistance mutations in the targeted sequence. To address this limitation, a similar strategy to combat HIV-1 drug resistance has been adopted, using dual or triple combination therapy of anti-HIV-1 shRNAs and/or other anti-HIV-1 gene therapeutics (50–52).
The first Phase 2 cell-modified gene therapy clinical trial using a combination approach involved a tat-vpr-specific anti-HIV ribozyme, termed OZ1, delivered in autologous CD34+ HPSCs (53). Although there were no significant viral load differences reported between the OZ1 and placebo groups, this study demonstrated cell-modified gene therapy was safe and biologically active, with no adverse events and higher CD4+ cell counts in the OZ1 group (53). A substantially decrease in the blood therapeutic gene level throughout the 100-week trial (53) may provide an explanation for the lack of effect.
A combinatorial approach uses three anti-HIV-1 shRNAs, each specifically targeting highly conserved regions of the HIV Integrase, Protease and tat-rev genes, delivered in a single LV construct termed R3A (52). This study first demonstrated multiple shRNAs being efficiently expressed in a single LV construct, if expression is driven by different promoters for each shRNA, e.g., the human H1, 7SK, or U6 polymerase III promoters and the human U1 polymerase II promoter each driving one of four shRNAs (52). Potent inhibition of HIV-1 was reported in vitro, with combined LV shRNAs constructs showing greater inhibition compared to single LV shRNA constructs (52). More recently, a preclinical in vivo study demonstrated safety of the R3A, triple shRNA expressing construct, using a Balb/c Rag2(−/−) IL-2Rγc(−/−) (BRG) humanized mouse model (54). Future clinical trials are planned to develop this potential therapeutic.
Another combinatorial HIV-1 approach involves shRNA targeting the ccr5 gene and the C46 peptide fusion inhibitor in a LV construct termed LV sh5/C46 (or Cal-1), which has been developed by Calimmune Inc. It will be discussed further in the humanized murine model section.
Transcriptional Gene Silencing of HIV-1 by si/shRNAs
The transcriptional gene silencing (TGS) approach has several distinct advantages over a PTGS approach; first, it directly targets the integrated virus, locking transcription, thus the opportunity for virus escape is minimized; second, epigenetic changes induced during TGS are heritable resulting in daughter cells maintaining the suppressive phenotype, providing a prolonged therapeutic response; and third, the exquisite sequence specificity of promoter-targeted si/shRNAs reduces the chance of non-specific off-target effects and toxicities.
Following the first identification of small interfering RNA-induced TGS in tobacco plants just over 10 years ago (55), the field has rapidly developed with multiple studies now identifying the phenomena in other plant species (Arabidopsis) (56, 57), fission yeast (Schizosaccharomyces pombe) (58), flies (Drosophila) (59), and nematode worms (Caenorhabditis elegans). Most recently, we and others have demonstrated TGS also occurs in human cells (60–66). We were the first group to report TGS could be induced in active HIV-1 infection through siRNA targeting the tandem NF-κB binding motifs in the 5′LTR viral promoter region (67). Subsequently, several studies have also identified targets for siRNAs that induce TGS in the HIV-1 promoter, specifically LTR-247, LTR-362, LTR-366 (68), and S4-siRNA (69). Of note, promoter-targeted siRNAs LTR-366 and S4-siRNA also target sequences in the NF-κB binding motif.
Our extensive investigations have demonstrated TGS of HIV-1 using the promoter-targeted si/shRNA PromA sequence complementary to the 5′LTR tandem NF-κB motifs initially in vitro using cell lines (64, 67, 70–72) and more recently in an in vivo humanized murine model (73). Characterization of the effects of si/shPromA revealed prolonged and profound TGS with up to a 1000-fold decrease in viral replication after a single siRNA transfection or retroviral-delivered shRNA construct. We also reported si/shPromA suppressed viral mRNA expression and de novo virion formation, while proviral DNA was still detected, indicating that virus suppression occurs post-integration. In further studies, we confirmed si/shPromA-mediated suppression of HIV-1 occurred via the TGS pathway using nuclear run-on assays to confirm transcription initiation rates, which clearly distinguished transcriptional suppression in si/shPromA treated HIV-1 infected nuclei from potential post-transcriptional effects (67, 72). Similarly, our investigations using a 3′LTR HIV-1-driven luciferase reporter construct also confirmed a limited contribution of PTGS to the observed virus suppression (72). Finally, chromatin immunoprecipitation (ChIP) analyses provided insight into the mechanism underlying the TGS, demonstrating that silencing was associated with increased histone methylation (H3K9me2 and H3K27me3), decreased histone acetylation, and recruitment of HDAC1 in the 5′LTR promoter region (64, 67, 72, 74). These structural histone changes are consistent with epigenetic-induced latent HIV infection (10–15, 72).
A recent study by Singh et al. using S4-siRNA targeted to the unique triple repeat of NF-κB binding motifs found in subtype C virus demonstrated long-term suppression of HIV-1 (69). The mechanism of silencing was shown in vitro to act through TGS, as determined by ChIP analysis of histone methylation, which revealed enrichment of H3K9me2 and H3K27me3 in S4-siRNA-transfected reporter cell lines (containing the subtype C LTR, luciferase reporter, and subtype C Tat protein expression cassette), but no enrichment in mock- or control siRNA-transfected cells (69). The suppressive effect was confirmed ex vivo in human PBMCs using four different subtype C isolates, which were shown to produce significantly lower p24 levels when transfected with S4-siRNA compared to mock- or control siRNAs (69). Interestingly, while a single mismatch at position 15 of the S4-siRNA sequence and a double mismatch at positions 1 and 15 were sufficient to disrupt the suppressive effect, one particular single mismatch at position 1 of the target sequence showed no disruption of the suppressive effect (69). This is intriguing considering the highly sequence specific nature of siRNAs and our experience of single mismatches disrupting any prolonged suppressive effect with loss of responses by ~6 days (75). Nevertheless, the unique S4-siRNA presents an interesting potential therapeutic approach in the specific targeting of subtype C, which is present in ~50% of the world’s HIV-positive population and demonstrates that much is still to be learnt regarding the properties of a targeted sequence that allows effective induction of viral silencing by promoter-targeted siRNAs.
We have also recently discovered another novel highly conserved sequence in the 5′LTR promoter region (including subtype C), upstream of siPromA, which when targeted by an si/sh RNA dubbed 143, induces potent transcriptional suppression of HIV associated with epigenetic modifications similar to those induced by siPromA (75). ChIP analyses revealed that the epigenetic changes induced by siRNA143 consisted of increased levels of both H3K27me3 and H3K9me3, reduction in H3K9Ac, and recruitment of Argonaute-1 (75), which are all characteristic heterochromatin marks observed during shRNA-induced TGS. Further, these changes in histone methylation and acetylation are consistent with epigenetic modifications found in latent HIV-1 infection. Following both siRNA PromA and 143 transfections, we observed that silencing can be partially reversed by highly potent, but also highly toxic, HDACi, such as trichostatin A (TSA), but not by those undergoing clinical trial evaluation, such as vironostat (SAHA) (75). Further, TNF at supra-physiological concentrations partially reduced the silencing. Additionally, using the J-Lat 9.2 cell latency model transduced with shPromA and/or sh143, we also observed robust resistance to viral reactivation by various stimuli, including SAHA and/or TNF, used at pharmacological or physiological concentrations (75). These observations are important in the context of the proposed gene-therapy applications as successful therapy will require sustained viral silencing despite activation of CD4+ T cells by inflammatory, homeostatic, or immune response proteins.
Identification of a second effective TGS target provides the opportunity to combine the shRNA targets (shPromA and sh143) in a single therapeutic. This combinatorial approach addresses concerns of any sequence-based target by covering HIV sequence variability. The PromA and si143 sequence targets are both highly conserved, however where variability does occur, the two constructs are complementary; e.g., subtype C viruses have a single nucleotide deletion at position 14 of the PromA target, while the sh143 sequence target has minimal subtype C variability. Thus, a multiplexed approach, delivered by a single LV construct expressing multiple shRNAs simultaneously, would likely provide increased viral coverage and more entrenched enforcement of epigenetic changes, which more robustly resist viral reactivation from alterations in the host’s inflammatory or immunological status. We are currently testing these constructs in humanized mouse models, described below.
Humanized Mouse Models for Assessing Potential HIV-1 Therapeutics
Animal models for assessing HIV-1 therapeutics include various humanized murine models and non-human primates. Although the latter species contain host-restriction factors that impede HIV-1 replication and experiments performed using this model must instead use the closely related Simian immunodeficiency virus or chimeric Simian/HIV (SHIV) (76). We recently utilized a (NOD)/SCID/Janus kinase 3 (NOJ) knockout humanized mouse model to demonstrate in vivo TGS activity of shPromA, delivered via a LV (Figure 1) (73). NOJ knockout mice were reconstituted with human PBMCs transduced with the shPromA carrying lentiviral construct, which was processed into mature siPromA by cellular ribonucleases (77). HIV-1JRFL challenge of mice reconstituted with the PromA-M2 inactive control transduced PBMCs showed acute HIV-1 infection (Figure 1A) as determined by high pVL, CD4+ T cell depletion and extensive immunodeficiency (78, 79). In stark contrast, mice reconstituted with shPromA-transduced PBMCs demonstrated significantly lower pVL and normal human CD4+ to CD8+ T cell ratios in mononuclear cells recovered from the peritoneal cavity and spleen at sacrifice 14 days post HIV-1 challenge (Figure 1B) (73). This corresponds to a protective effect in the form of an induced HIV-1 “latent-like” state, which locks down active virus transcription even in this acute model of HIV-1 infection.
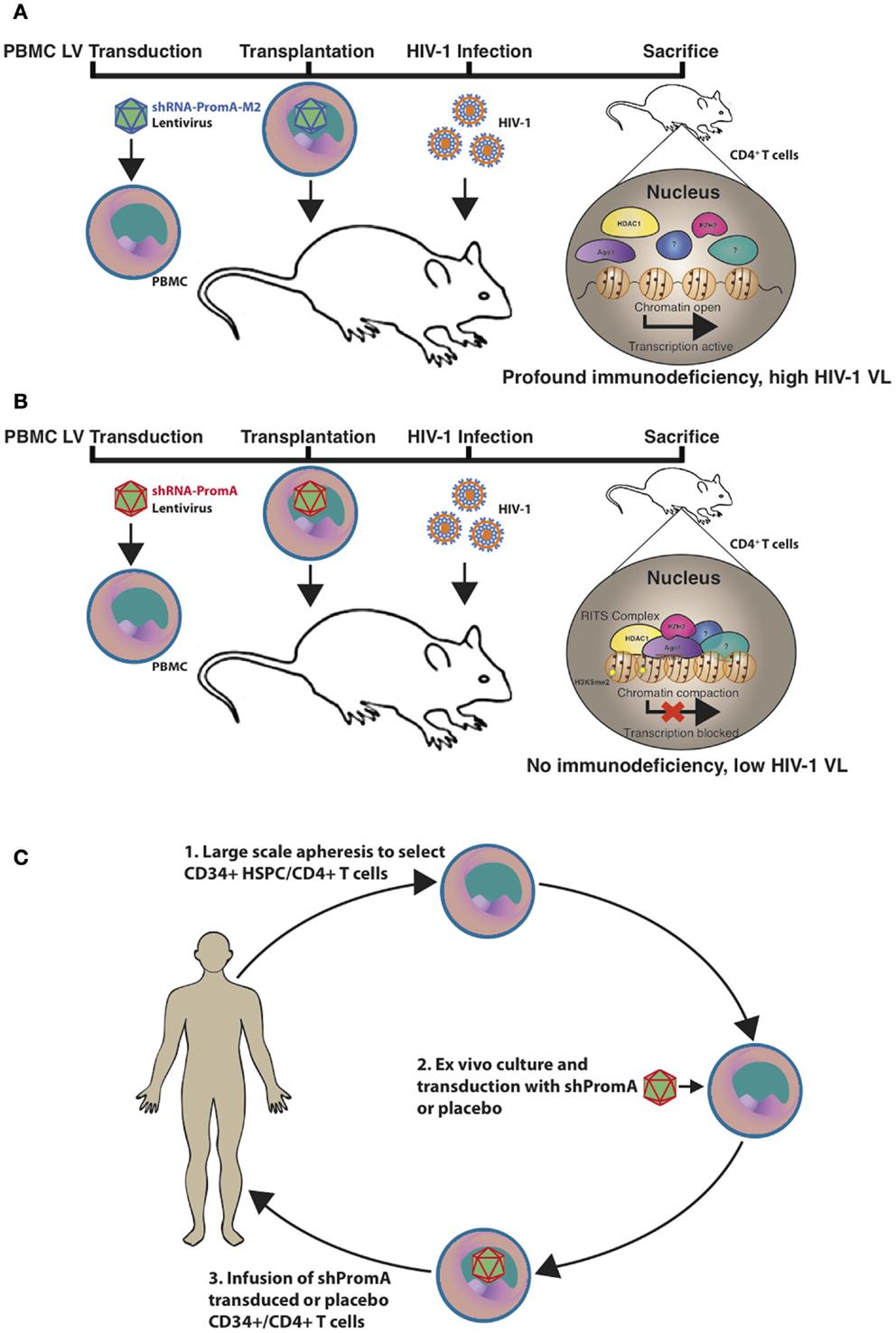
Figure 1. Schematic representation of the in vivo effects of a promoter-targeted siRNA approach in a humanized mouse model and envisaged gene therapy approach. Replication-incompetent lentivirus carrying (A) the inactive control shPromA-M2 or (B) active shPromA is transduced into healthy control human CD4+ T cells. Transduced CD4+ T cells are transplanted into (NOD)/SCID/Janus kinase 3 (NOJ) knockout mice and engraftment ensues. The humanized mice are then challenged with HIV-1 and sacrificed 14 days post challenge. The shPromA antisense strand (red), associates with Ago1 (purple) and other RITS-like complex components (HDAC – yellow and EZH2 – pink) and induces heterochromatin formation with methylation marks (H3K9me2, indicated by stars) in the targeted HIV-1 promoter region. This process suppresses HIV-1 transcription and results in protection of CD4+ T cells, which results in lower pVL in mice transplanted with shPromA compared to control shPromA-M2 lentivirus-transduced PBMCs. (C) Our envisaged gene therapy approach with the future shPromA and/or sh143 TGS-inducing targets involves initial apheresis to obtain and select CD34+ HSPC and/or CD4+ T cells, which are then cultured ex vivo and transduced with the multiplexed shRNAs. The transduced cells are then infused back into the patient, whereby HIV-1 will be locked down in a latent-like state.
Our aim is to generate sufficient data using the humanized mouse model to merit further therapeutic development of these TGS-inducing constructs. An alternative humanized murine model, which circumvents the highly acute infection reported in NOJ knockout mice, is the humanized BLT murine model. This model uses the non-obese diabetic (NOD)/SCID, common gamma chain −/− (NSG) mouse, humanized with implanted bone marrow, fetal liver, and thymus tissue (BLT) and results in systemic repopulation with human T cells, B cells, monocytes/macrophages, and dendritic cells (76, 80). This system has been used to perform in vivo studies of autologous CD34+ HSPCs transduced with the H1-CCR5 shRNA 1005 vector and showed effective down-regulation of the HIV-1 co-receptor, CCR5, which protected mouse-derived human splenocytes ex vivo (80) and CD4+ T cells in vivo (81) from CCR5-tropic HIV-1 infection. This CCR5-shRNA vector has also been analyzed in non-human primates by delivery through HSPC transplantation (82). Although this required a single nucleotide mutation in the human CCR5 shRNA 1005 sequence to match the rhesus macaque CCR5 target sequence, this analysis successfully showed specific inhibition of rhesus macaque CCR5 expression, with no modulation of human CCR5 expression observed (82).
The H1-CCR5 shRNA 1005 vector has been further developed to include an additional anti-HIV target in the form of a sequence that encodes for the C46 fusion inhibitor. This HIV-1 entry inhibitor is a mimetic derived from the peptide sequence of the C-terminal heptad repeat of HIV-1 gp41, which interacts with the N-terminal coiled-coil domain of the intermediate HIV-1 gp41 to block the six-helix bundle formation and subsequent fusion between HIV-1 envelope and the host cell membrane. C46 has been tested in Phase 1 clinical trials without any adverse effects in HIV-1 positive patients infused with autologous T cells transduced with C46 expressing retrovirus vector (83). Combination of H1-CCR5 shRNA 1005 and C46 into a single LV termed LVsh5/C46 (or Cal-1) has been tested in a preclinical trial discussed below and is currently being assessed in a Phase 1 trial in patients with chronic HIV-1 infection (NCT01734850). We are planning to assess the new sh143 target in combination with shPromA contained within the Cal-1 lentivirus construct backbone in the BLT model to determine whether a multiplexed approach can entrench enforced HIV-1 lockdown at the transcriptional level.
Gene Therapy Approaches to HIV-1 Treatment
In 2009, the extraordinary success of utilizing bone marrow transplant to deliver HPSC from a CCR5-Δ32 homozygous donor opened the door for gene therapy approaches to HIV-1 treatment. The “Berlin patient” effectively received a functional cure for HIV-1, and currently 6 years post-transplantation, does not require ART and has no detectable pVL or proviral DNA (84, 85). Although this truly represents a modern day medical success, the circumstances of aggressive malignant disease and the rarity of matching HLA/CCR5-Δ32 homozygous donors present a formidable challenge in repeating this feat [reviewed in Ref. (50)] Further, this approach has been tried in six other patients without success (86). There are, however, several gene therapy clinical trials in various stages of completion involving HIV-1 infected patients with non-malignancies using constructs aimed at knocking down CCR5 to protect cells from HIV-1 infection. These current gene therapy clinical trials are summarized in Table 1.
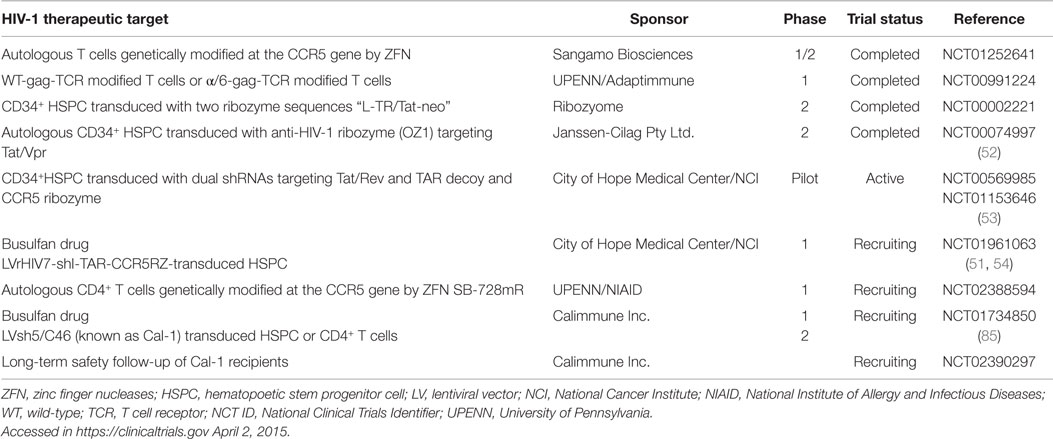
Table 1. Current HIV-1 gene therapy clinical trials.
One of the preclinical studies assessing safety and efficacy of an anti-HIV-1 lentiviral vector containing CCR5 shRNA and the C46 fusion inhibitor, termed Cal-1, has recently been reported (87). Effective delivery of LVsh5/C46 (Cal-1) was demonstrated in human T cell lines, PBMCs, CD4+ T cells, and CD34+ HSPCs, with both the CCR5-shRNA and C46 peptide being stably expressed in the target cells (87). Importantly, the study showed the Cal-1 construct was able to effectively protect gene-modified cells from both CXCR4- and CCR5-tropic HIV-1 strains (87). Further, the Cal-1 construct treatment was shown to be non-toxic, non-inflammatory, and had no adverse effect on HSPC differentiation (87). These encouraging data have led to the Cal-1 construct currently being tested in a Phase 1/2 clinical trial for the treatment of HIV-1 (Table 1).
Our envisaged future application of the TGS-inducing PromA and 143 sequences as a proposed therapeutic strategy is outlined in Figure 1C. The approach would be to use shPromA/sh143 TGS-inducing constructs to enforce latency in HIV-1 positive individuals who have cART-suppressed pVL and latent viral reservoirs. This strategy may provide a functional cure, by inducing and enforcing HIV-1 latency, thereby maintaining transcriptionally inactive virus and effectively render patients free from cART.
Conclusion
In terms of approaches to a functional cure for HIV-1, ncRNA-mediated transcriptional regulation of HIV-1, particularly in the context of the viral reservoir, is starkly juxtaposed to current activation approaches, which rely on pan T-cell activation or extensive histone acetylation modification and are associated with substantial off-target responses. Instead, the ncRNA approach relies on sequence specificity to provide a highly focused approach in manipulation of the latent reservoir. Developing ncRNA-therapeutic approaches to control HIV-1 may have the potential to enforce HIV-1 latency and block initial infection, allowing control of the viral reservoir, free from traditional antiretroviral therapies.
Author Contributions
CA, AK, KS, and GS wrote the manuscript; and KM prepared the figures.
Conflict of Interest Statement
We declare there is no conflict of interest. Chantelle L. Ahlenstiel, Anthony D. Kelleher, and Kazuo Suzuki hold a patent for si/shRNA sequences targeting HIV-1. Geoff P. Symonds works for Calimmune Inc.
Acknowledgments
This paper was funded by the following sources: grants from the National Health and Medical Research Council including a Practitioner Fellowship APP1020536 (AK) and a Project grant 1049473 (KS and CA).
Abbreviations
AP-1, activator protein 1; CTL, cytotoxic T lymphocyte; H3K27me3, histone 3 lysine 27 tri-methylation; H3K9me2, histone 3 lysine 9 di-methylation; HSPC, hematopoetic stem progenitor cell; LTR, long terminal repeat; LV, lentiviral vector; NF-κB, nuclear factor kappa B; NFAT, nuclear factor of activated T cells; SP1, specificity protein 1; TCR, T cell receptor; TGS, transcriptional gene silencing; WT, wild-type; ZFN, zinc finger nucleases.
References
1. Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature (2007) 447:799–816. doi: 10.1038/nature05874
2. Kaikkonen MU, Lam MT, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res (2011) 90:430–40. doi:10.1093/cvr/cvr097
3. Malecova B, Morris KV. Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. Curr Opin Mol Ther (2010) 12:214–22.
4. Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet (2014) 15:423–37. doi:10.1038/nrg3722
5. Amaral PP, Mattick JS. Noncoding RNA in development. Mamm Genome (2008) 19:454–92. doi:10.1007/s00335-008-9136-7
6. Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol (2008) 9:219–30. doi:10.1038/nrm2347
7. Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer (2011) 10:38. doi:10.1186/1476-4598-10-38
8. Méndez C, Ahlenstiel CL, Kelleher AD. Post-transcriptional gene silencing, transcriptional gene silencing and HIV. World J Virol (2015) 4(3):219–44. doi:10.5501/wjv.v4.i3.219
9. Suzuki K, Ahlenstiel C, Marks K, Kelleher AD. Promoter targeting RNAs: unexpected contributors to the control of HIV-1 transcription. Mol Ther Nucleic Acids (2015) 4:e222. doi:10.1038/mtna.2014.67
10. du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J (2007) 26:424–35. doi:10.1038/sj.emboj.7601517
11. Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J (2003) 22:1868–77. doi:10.1093/emboj/cdg188
12. Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J (1996) 15:1112–20.
13. Verdin E, Paras P Jr, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J (1993) 12:3249–59.
14. Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, Verdin E, et al. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem (2004) 279:42008–17. doi:10.1074/jbc.M402124200
15. Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J (2006) 25:139–49. doi:10.1038/sj.emboj.7600900
16. Hsu DC, Sereti I, Ananworanich J. Serious non-AIDS events: immunopathogenesis and interventional strategies. AIDS Res Ther (2013) 10:29. doi:10.1186/1742-6405-10-29
17. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature (1995) 373:123–6. doi:10.1038/373123a0
18. Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, Cranmer L, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A (2009) 106:9403–8. doi:10.1073/pnas.0903107106
19. Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science (1997) 278:1291–5. doi:10.1126/science.278.5341.1291
20. Chun TW, Justement JS, Moir S, Hallahan CW, Maenza J, Mullins JI, et al. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. J Infect Dis (2007) 195:1762–4. doi:10.1086/518250
21. Mellors JW, Rinaldo CR Jr, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science (1996) 272:1167–70. doi:10.1126/science.272.5265.1167
22. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science (1996) 271:1582–6. doi:10.1126/science.271.5255.1582
23. Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med (1997) 337:734–9. doi:10.1056/NEJM199709113371102
24. Palella FJ Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med (1998) 338:853–60.
25. Koelsch KK, Boesecke C, McBride K, Gelgor L, Fahey P, Natarajan V, et al. Impact of treatment with raltegravir during primary or chronic HIV infection on RNA decay characteristics and the HIV viral reservoir. AIDS (2011) 25:2069–78. doi:10.1097/QAD.0b013e32834b9658
26. Yukl SA, Shergill AK, McQuaid K, Gianella S, Lampiris H, Hare CB, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS (2010) 24:2451–60. doi:10.1097/QAD.0b013e32833ef7bb
27. McMahon D, Jones J, Wiegand A, Gange SJ, Kearney M, Palmer S, et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin Infect Dis (2010) 50:912–9. doi:10.1086/650749
28. Hammer SM, Ribaudo H, Bassett R, Mellors JW, Demeter LM, Coombs RW, et al. A randomized, placebo-controlled trial of abacavir intensification in HIV-1-infected adults with virologic suppression on a protease inhibitor-containing regimen. HIV Clin Trials (2010) 11:312–24. doi:10.1310/hct1106-312
29. Gandhi RT, Zheng L, Bosch RJ, Chan ES, Margolis DM, Read S, et al. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Med (2010) 7:e1000321. doi:10.1371/journal.pmed.1000321
30. Gandhi RT, Bosch RJ, Aga E, Albrecht M, Demeter LM, Dykes C, et al. No evidence for decay of the latent reservoir in HIV-1-infected patients receiving intensive enfuvirtide-containing antiretroviral therapy. J Infect Dis (2010) 201:293–6. doi:10.1086/649569
31. Archin NM, Cheema M, Parker D, Wiegand A, Bosch RJ, Coffin JM, et al. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PLoS One (2010) 5:e9390. doi:10.1371/journal.pone.0009390
32. Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature (2012) 487:482–5. doi:10.1038/nature11286
33. Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, et al. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem (2009) 284:6782–9. doi:10.1074/jbc.M807898200
34. Wang FX, Xu Y, Sullivan J, Souder E, Argyris EG, Acheampong EA, et al. IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J Clin Invest (2005) 115:128–37. doi:10.1172/JCI200522574
35. Wightman F, Ellenberg P, Churchill M, Lewin SR. HDAC inhibitors in HIV. Immunol Cell Biol (2012) 90:47–54. doi:10.1038/icb.2011.95
36. Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A (1999) 96:15109–14. doi:10.1073/pnas.96.26.15109
37. van Praag RM, Prins JM, Roos MT, Schellekens PT, Ten Berge IJ, Yong SL, et al. OKT3 and IL-2 treatment for purging of the latent HIV-1 reservoir in vivo results in selective long-lasting CD4+ T cell depletion. J Clin Immunol (2001) 21:218–26. doi:10.1023/A:1011091300321
38. Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med (2011) 1:a007096. doi:10.1101/cshperspect.a007096
39. Paiardini M. Editorial: hijacking the IL-7/IL-7R system in HIV infection. J Leukoc Biol (2011) 89:491–3. doi:10.1189/jlb.1110614
40. McKernan LN, Momjian D, Kulkosky J. Protein kinase C: one pathway towards the eradication of latent HIV-1 reservoirs. Adv Virol (2012) 2012:805347. doi:10.1155/2012/805347
41. Weissman D, Dybul M, Daucher MB, Davey RT Jr, Walker RE, Kovacs JA. Interleukin-2 up-regulates expression of the human immunodeficiency virus fusion coreceptor CCR5 by CD4+ lymphocytes in vivo. J Infect Dis (2000) 181:933–8. doi:10.1086/315303
42. Sanchez-Duffhues G, Vo MQ, Perez M, Calzado MA, Moreno S, Appendino G, et al. Activation of latent HIV-1 expression by protein kinase C agonists. A novel therapeutic approach to eradicate HIV-1 reservoirs. Curr Drug Targets (2011) 12:348–56. doi:10.2174/138945011794815266
43. Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood (2007) 109:31–9. doi:10.1182/blood-2006-06-025999
44. Edelstein LC, Micheva-Viteva S, Phelan BD, Dougherty JP. Short communication: activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res Hum Retroviruses (2009) 25:883–7. doi:10.1089/aid.2008.0294
45. Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I, et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol (2009) 82:440–9. doi:10.1111/j.1600-0609.2009.01239.x
46. Wei DG, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog (2014) 10:e1004071. doi:10.1371/journal.ppat.1004071
47. Battistini A, Sgarbanti M. HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses (2014) 6:1715–58. doi:10.3390/v6041715
48. Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell (2013) 155:540–51. doi:10.1016/j.cell.2013.09.020
49. Jones RB, O’Connor R, Mueller S, Foley M, Szeto GL, Karel D, et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog (2014) 10:e1004287. doi:10.1371/journal.ppat.1004287
50. Burke BP, Boyd MP, Impey H, Breton LR, Bartlett JS, Symonds GP, et al. CCR5 as a natural and modulated target for inhibition of HIV. Viruses (2014) 6:54–68. doi:10.3390/v6010054
51. DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med (2010) 2:36ra43. doi:10.1126/scitranslmed.3000931
52. ter Brake O, t Hooft K, Liu YP, Centlivre M, von Eije KJ, Berkhout B. Lentiviral vector design for multiple shRNA expression and durable HIV-1 inhibition. Mol Ther (2008) 16:557–64. doi:10.1038/sj.mt.6300382
53. Mitsuyasu RT, Merigan TC, Carr A, Zack JA, Winters MA, Workman C, et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med (2009) 15:285–92. doi:10.1038/nm.1932
54. Centlivre M, Legrand N, Klamer S, Liu YP, Jasmijn von Eije K, Bohne M, et al. Preclinical in vivo evaluation of the safety of a multi-shRNA-based gene therapy against HIV-1. Mol Ther Nucleic Acids (2013) 2:e120. doi:10.1038/mtna.2013.48
55. Matzke MA, Primig M, Trnovsky J, Matzke AJ. Reversible methylation and inactivation of marker genes in sequentially transformed tobacco plants. EMBO J (1989) 8:643–9.
56. Wassenegger M, Heimes S, Riedel L, Sanger HL. RNA-directed de novo methylation of genomic sequences in plants. Cell (1994) 76:567–76. doi:10.1016/0092-8674(94)90119-8
57. Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJ. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J (2000) 19:5194–201. doi:10.1093/emboj/19.19.5194
58. Lippman Z, May B, Yordan C, Singer T, Martienssen R. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol (2003) 1:E67. doi:10.1371/journal.pbio.0000067
59. Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science (2004) 305:1289–92. doi:10.1126/science.1101372
60. Castanotto D, Tommasi S, Li M, Li H, Yanow S, Pfeifer GP, et al. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol Ther (2005) 12:179–83. doi:10.1016/j.ymthe.2005.03.003
61. Hawkins PG, Santoso S, Adams C, Anest V, Morris KV. Promoter targeted small RNAs induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res (2009) 37:2984–95. doi:10.1093/nar/gkp127
62. Janowski BA, Huffman KE, Schwartz JC, Ram R, Nordsell R, Shames DS, et al. Involvement of AGO1 and AGO2 in mammalian transcriptional silencing. Nat Struct Mol Biol (2006) 13:787–92. doi:10.1038/nsmb1140
63. Kim DH, Saetrom P, Snove O Jr, Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci U S A (2008) 105:16230–5. doi:10.1073/pnas.0808830105
64. Suzuki K, Juelich T, Lim H, Ishida T, Watanebe T, Cooper DA, et al. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J Biol Chem (2008) 283:23353–63. doi:10.1074/jbc.M709651200
65. Ting AH, Schuebel KE, Herman JG, Baylin SB. Short double-stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nat Genet (2005) 37:906–10. doi:10.1038/ng1611
66. Weinberg MS, Villeneuve LM, Ehsani A, Amarzguioui M, Aagaard L, Chen ZX, et al. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA (2006) 12:256–62. doi:10.1261/rna.2235106
67. Suzuki K, Shijuuku T, Fukamachi T, Zaunders J, Guillemin G, Cooper D, et al. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J RNAi Gene Silencing (2005) 1:66–78.
68. Weinberg MS, Barichievy S, Schaffer L, Han J, Morris KV. An RNA targeted to the HIV-1 LTR promoter modulates indiscriminate off-target gene activation. Nucleic Acids Res (2007) 35:7303–12. doi:10.1093/nar/gkm847
69. Singh A, Palanichamy JK, Ramalingam P, Kassab MA, Bhagat M, Andrabi R, et al. Long-term suppression of HIV-1C virus production in human peripheral blood mononuclear cells by LTR heterochromatization with a short double-stranded RNA. J Antimicrob Chemother (2014) 69:404–15. doi:10.1093/jac/dkt348
70. Ahlenstiel CL, Lim HG, Cooper DA, Ishida T, Kelleher AD, Suzuki K. Direct evidence of nuclear Argonaute distribution during transcriptional silencing links the actin cytoskeleton to nuclear RNAi machinery in human cells. Nucleic Acids Res (2012) 40:1579–95. doi:10.1093/nar/gkr891
71. Suzuki K, Ishida T, Yamagishi M, Ahlenstiel C, Swaminathan S, Marks K, et al. Transcriptional gene silencing of HIV-1 through promoter targeted RNA is highly specific. RNA Biol (2011) 8:1035–46. doi:10.4161/rna.8.6.16264
72. Yamagishi M, Ishida T, Miyake A, Cooper DA, Kelleher AD, Suzuki K, et al. Retroviral delivery of promoter-targeted shRNA induces long-term silencing of HIV-1 transcription. Microbes Infect (2009) 11:500–8. doi:10.1016/j.micinf.2009.02.003
73. Suzuki K, Hattori S, Marks K, Ahlenstiel C, Maeda Y, Ishida T, et al. Promoter targeting shRNA suppresses HIV-1 infection in vivo through transcriptional gene silencing. Mol Ther Nucleic Acids (2013) 2:e137. doi:10.1038/mtna.2013.64
74. Lim HG, Suzuki K, Cooper DA, Kelleher AD. Promoter-targeted siRNAs induce gene silencing of simian immunodeficiency virus (SIV) infection in vitro. Mol Ther (2008) 16:565–70. doi:10.1038/sj.mt.6300380
75. Ahlenstiel C, Mendez C, Lim STH, Marks K, Turville SG, Cooper DA, et al. Novel RNA duplex locks HIV-1 in a latent state via chromatin-mediated transcriptional silencing. Mol Ther Nucleic Acids (2015).
76. Malim MH, Bieniasz PDHIV. Restriction factors and mechanisms of evasion. Cold Spring Harb Perspect Med (2012) 2:a006940. doi:10.1101/cshperspect.a006940
77. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science (2002) 296:550–3. doi:10.1126/science.1068999
78. Okada S, Harada H, Ito T, Saito T, Suzu S. Early development of human hematopoietic and acquired immune systems in new born NOD/Scid/Jak3null mice intrahepatic engrafted with cord blood-derived CD34 + cells. Int J Hematol (2008) 88:476–82. doi:10.1007/s12185-008-0215-z
79. Hattori S, Ide K, Nakata H, Harada H, Suzu S, Ashida N, et al. Potent activity of a nucleoside reverse transcriptase inhibitor, 4’-ethynyl-2-fluoro-2’-deoxyadenosine, against human immunodeficiency virus type 1 infection in a model using human peripheral blood mononuclear cell-transplanted NOD/SCID Janus kinase 3 knockout mice. Antimicrob Agents Chemother (2009) 53:3887–93. doi:10.1128/AAC.00270-09
80. Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N, et al. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood (2010) 115:1534–44. doi:10.1182/blood-2009-04-215855
81. Shimizu S, Ringpis GE, Marsden MD, Cortado RV, Wilhalme HM, Elashoff D, et al. RNAi-mediated CCR5 knockdown provides HIV-1 resistance to memory T cells in humanized BLT mice. Mol Ther Nucleic Acids (2015) 4:e227. doi:10.1038/mtna.2015.3
82. An DS, Donahue RE, Kamata M, Poon B, Metzger M, Mao SH, et al. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci U S A (2007) 104:13110–5. doi:10.1073/pnas.0705474104
83. van Lunzen J, Glaunsinger T, Stahmer I, von Baehr V, Baum C, Schilz A, et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol Ther (2007) 15:1024–33. doi:10.1038/mt.sj.6300124
84. Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med (2009) 360:692–8. doi:10.1056/NEJMoa0802905
85. Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood (2011) 117:2791–9. doi:10.1182/blood-2010-09-309591
86. Verheyen J, Esser S, Kordelas L. More on shift of HIV tropism in stem-cell transplantation with CCR5 delta32/delta32 mutation. N Engl J Med (2014) 371:2438. doi:10.1056/NEJMc1412279
Keywords: HIV-1 transcription, latency, non-coding RNA, siRNA, shRNA, humanized mouse model, gene therapy, clinical trials
Citation: Ahlenstiel CL, Suzuki K, Marks K, Symonds GP and Kelleher AD (2015) Controlling HIV-1: non-coding RNA gene therapy approaches to a functional cure. Front. Immunol. 6:474. doi: 10.3389/fimmu.2015.00474
Received: 08 April 2015; Accepted: 31 August 2015;
Published: 16 September 2015
Edited by:
Lucy Dorrell, University of Oxford, UKReviewed by:
Clive Maurice Gray, University of Cape Town, South AfricaAurelio Cafaro, Istituto Superiore di Sanità, Italy
Copyright: © 2015 Ahlenstiel, Suzuki, Marks, Symonds and Kelleher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chantelle L. Ahlenstiel, The Kirby Institute, University of New South Wales (UNSW) Australia, Sydney, NSW 2052, Australia,Y2FobGVuc3RpZWxAa2lyYnkudW5zdy5lZHUuYXU=