 Jia Nie
Jia Nie Yang Yang Li1
Yang Yang Li1 Bin Li
Bin Li- 1Key Laboratory of Molecular Virology and Immunology, Institut Pasteur of Shanghai, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
- 2Clinical Immunology Center, The Third Affiliated Hospital, Sun Yat-Sen University, Guangzhou, China
- 3Department of Medicine, Division of Rheumatology, Penn State Hershey College of Medicine, Hershey, PA, USA
- 4Innovent Biologics Inc., Suzhou, China
CD4+CD25+ regulatory T (Treg) cells play a pivotal role in the maintenance of immune homeostasis, where the X-linked master transcription factor forkhead box P3 (FOXP3) determines Treg cell development and function. Genetic deficiency of foxp3 induces dysfunction of Treg cells and immuno-dysregulation, polyendocrinopathy, enteropathy, and X-linked syndrome in humans. Functionally deficient Treg cells or the development of exTreg cells positively correlate with autoimmune diseases, such as systemic lupus erythematosus (SLE), multiple sclerosis (MS), and ankylosing spondylitis (AS). In general, females are more susceptible to SLE and MS but less susceptible to AS, where the expression of FOXP3 and its protein complex are perturbed by multiple factors, including hormonal fluctuations, inflammatory cytokines, and danger signals. Therefore, it is critical to explore the potential molecular mechanisms involved and these differences linked to gender. Here, we review recent findings on the regulation of FOXP3 activity in Treg cells and also discuss gender difference in the determination of Treg cell function in autoimmune diseases.
Introduction
Regulatory T (Treg) cells, via their immune suppressive capability, play an indispensable role in maintaining immune homeostasis and preventing autoimmunity induced by excessive, misdirected, or unnecessary immune activation. Surface-expressed cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) mediates suppression of target cells by cell–cell contact (1–4). Treg cells can also reduce T cell activation and proliferation through CD39–CD73-mediated production of metabolic adenosine (5). Meanwhile, Treg cells have been shown to harbor cytotoxic capacity and induce target cell apoptosis through release of granzymes A/B and perforin (4). Anti-inflammatory cytokines that are secreted by Treg cells can also induce immune tolerance (6, 7).
Under pathogenic conditions, such as systemic lupus erythematosus (SLE) and multiple sclerosis (MS), Treg cells exhibit plasticity to some extent and may mimic T helper-like phenotypes. Recent studies have provided insight into the understanding of the stability and activity of forkhead box P3 (FOXP3) in Treg cells regulated by T cell receptor (TCR) signaling, inflammatory cytokines, and danger signals. Here, we discuss the cellular and molecular mechanisms underlying FOXP3-mediated regulation of Treg cells and also the possible effect that gender difference has on Treg cells and autoimmune diseases.
FOXP3 Mutations and Autoimmunity
The transcription factor FOXP3 belongs to the fork-winged helix family and is encoded by the foxp3 gene on the X chromosome. Genetic deletion of the foxp3 gene and the loss of Treg cells promote the development of autoimmune and inflammatory syndromes (8–10). Ectopic expression of FOXP3 in CD4+CD25− T cells may endow CD4+CD25− T cells with Treg-like suppressive capability to prevent inflammatory bowel disease (IBD) and autoimmune gastritis (9). FOXP3-deficient Treg cells have decreased levels of Treg cell signature genes, including ctla4, ebi3, il10, and entpd1, and acquire the expression of T effector cytokine genes such as ifng, tnfα, il4, and il17 (11–14). A frame-shift mutation in the foxp3 gene locus in scurfy mice results in the expression of FOXP3 protein lacking its forkhead domain (15). Many other loss-of-function mutations at the foxp3 gene locus have also been identified in patients with immune-dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance syndrome (IPEX) (16, 17). Genetic mutations of the foxp3 gene are always accompanied by the lack of the functional Treg cells, therefore resulting in the development of diverse arrays of autoimmune diseases. A compilation of studies describing the role of genetic mutants of the foxp3 gene in autoimmune diseases is shown in Table 1.
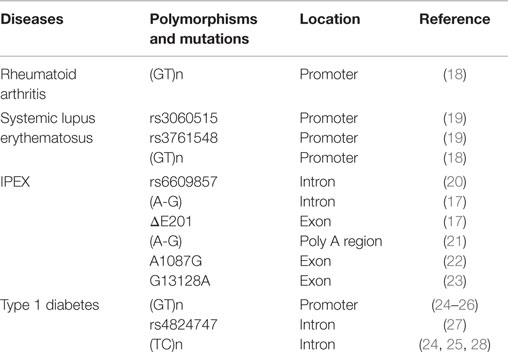
Table 1. The polymorphism of the foxp3 gene in autoimmune diseases.
FOXP3 and Treg Cell Development
Treg cells comprise approximately 5–15% of the CD4+ T cell compartment and can be subdivided into two subpopulations, including thymus-derived Treg (tTreg) cells and peripherally derived Treg (pTreg) cells. tTreg (also called natural Treg (nTreg)) cells are generated from Treg precursors at the immature HSAhi CD4SP stage when FOXP3 is induced and Treg lineage commitment established (29). pTreg cells are differentiated from naïve T cells at peripheral sites in the presence of IL-2 and TGF-β (Figure 1). Those generated in vitro through TGF-β signals are known as induced Treg (iTreg) cells (30).
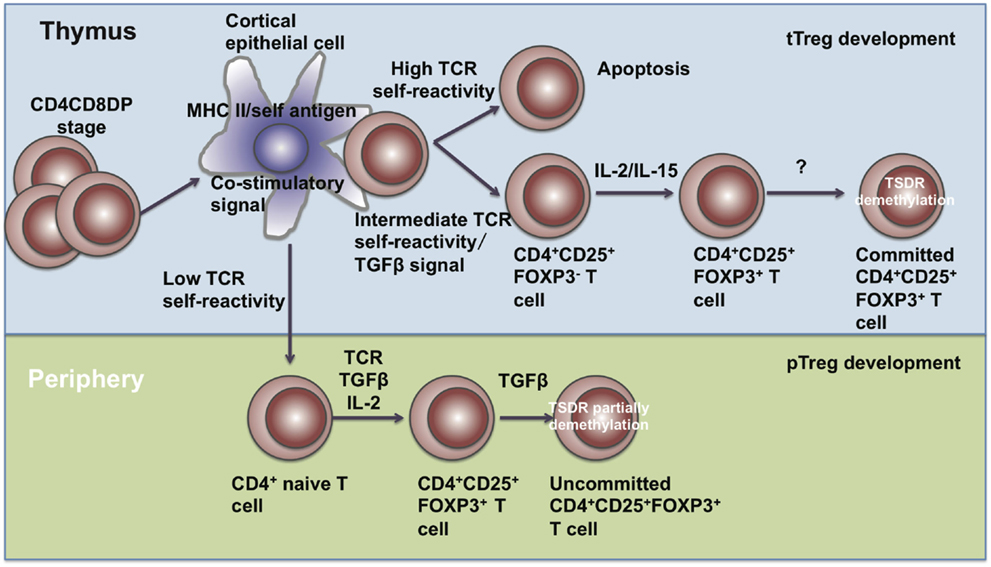
Figure 1. The development of Treg cells. Treg cells develop in the thymus and periphery. In the thymus, CD4+CD8+ T cells undergo negative selection and become mature tTreg cells through IL-2, IL-15, and TGF-β signals. In the periphery, naïve CD4+ T cells encounter antigen and differentiate into pTreg cells in the presence of TGF-β and IL-2.
In the thymus, the development of tTreg cells requires extracellular signals, including TCR-mediated self-antigen recognition, γ chain cytokines, and TGF-β etc. DO11.10 transgenic mice expressing transgenic TCRs specific for an OVA peptide had normal proportions of CD4+CD8−CD25+ thymocytes, while DO11.10 transgenic mice with a RAG-2 gene-deficient background had fewer CD4+CD8−CD25+ thymocytes (31), suggesting that TCR signaling is required for the development of tTreg cells. Also, transgenic mice harboring T cells specific for the major I-Ed determinant (S1) of influenza hemagglutinin (HA) exhibited higher percentages and numbers of FOXP3+ Treg cells recognizing HA (32, 33), showing that the TCRs of tTreg are biased toward self-antigens. Intermediate TCR strength has also been reported to be required for tTreg development. Sequencing of TCRs has showed that Treg cells share little similarity with naïve T cells. The diversity of TCRs on Treg cells surpasses the diversity of TCRs from naïve T cells (34). Although some studies have reported no substantial differences between the TCR repertories of Treg and non-Treg cells, their conclusions may only be based on the usage of the TCR variable region segments Vβ or Vα and size distribution of complementarity-determining region 3 (CDR3) (35, 36). These parameters are too limited to determine the identity of individual TCRs and reflect the differences only when a clonotypic, oligoclonal response occurs. CD4+CD25− T cells harboring the TCRα chains from Treg cells have been shown to expand faster when transferred into a lymphopenic host, suggesting that TCRs on Treg cells possess substantially higher affinity with MHC class II-bound self-peptides (37). In Nur77GFP mice, the mean fluorescence intensity (MFI) of GFP revealed that the TCR signal strength in tTreg and pTreg cells was almost two-fold compared with conventional CD4+ T cells (38). All these studies indicate that Treg cells are self-reactive.
Besides TCR signaling, γ chain cytokines are also required for FOXP3 expression, including IL-2, IL-7, and IL-15. Treg cells express high levels of the IL-2 receptor α chain (CD25) (39). il2rα- or il2rβ-deficient mice have decreased numbers of Treg cells in spleens and lymph nodes and develop autoimmunity around 4–8 weeks of age (40–46). Other non-IL-2 cytokines through γc partially compensate for IL-2 signaling. In il2−/− mice, CD4+FOXP3+ T cells were still detectable, but drastically reduced in il2−/−il7−/−, il2−/−il15−/−, il2rβ−/− and γc−/− mice (47). In the thymus, TGF-β signals prevent tTreg cell apoptosis. Conditional deletion of the TGF-β type I receptor (Tgfbr1) gene in T cells causes tTreg cells in the thymus to become more susceptible to apoptosis during negative selection, while bim ablation may restore TGF-β signal deficiency (48).
Recent studies showed that FOXP3 expression alone was not sufficient for Treg lineage commitment. The demethylation status of a Treg-specific demethylation region (TSDR) in the foxp3 promoter plays an essential role in Treg lineage maintenance where the demethylation of the TSDR correlates with stable Treg cell phenotype. Gene expression profile analysis in FOXP3-non-expressing T cells that lacked methylation of the TSDR, and FOXP3-expressing T cells that retained methylation of the TSDR, showed higher similarity to tTreg cells in the former in gene expression but lack of repression in the expression of il2, ifng, and zap70; however, the latter cells exhibited normal il2, ifng, and zap70 repression but upregulated a set of genes that were not expressed in tTreg cells. These results indicated that FOXP3 expression and the demethylation of the TSDR are both vital to establish Treg lineage commitment, but neither of them alone is sufficient (49).
In the periphery, combined TCR, TGF-β, and IL-2 signals polarize naïve CD4+ T cells into pTreg cells. These pTreg cells possess similar suppressive capacities as tTreg cells in vitro and in vivo (50, 51). Both tTreg and pTreg cells express FOXP3, CD25, CTLA-4, GITR, CD39, and CD73, along with low levels of IL-7Rα (CD127) (52). Current studies indicate that tTreg and pTreg cells play differential roles in different inflamed tissues. pTreg cells are more functional for maintaining mucosal tolerance, while tTreg cells are for maintaining immune tolerance. Due to the lack of specific lineage markers to distinguish between tTreg and pTreg cells in humans, it remains difficult to illustrate the different functions of tTreg and pTreg cells. Helios has been identified as a marker for tTreg cells (53). However, tTreg subsets have been found to contain both FOXP3+Helios+ and FOXP3+Helios− subpopulations, suggesting that Helios is not a specific marker for tTreg/pTreg cells (54). Other studies have identified Neuropilin 1 (NRP1) specifically and highly expressed on tTreg cells but not pTreg cells (55), and glycoprotein A repetitions predominant (GARP) expressed on activated human tTreg cells but not TGFβ-induced iTreg cells (56), but subsequent reports found that NRP1low pTreg cells could be converted into NRP1hi pTreg cells under inflammatory environments (57). Therefore, other surface markers need to be discovered for distinguishing between tTreg and pTreg cells.
The Stability of Treg Cells
As Treg cells have been identified as a specific cell population possessing suppressive capacity to maintain immune homeostasis, Treg cell therapy is seen as a promising method for treating autoimmune diseases. However, clinical trials for autoimmune disease indications thus far, via re-administration of expanded Treg cells into patients, have been far from satisfactory (58) as the phenotype and function of Treg cells may change in vivo. This raises the question of whether or not Treg cells are stable (59). Due to the ambiguity of specific Treg cell markers, FOXP3 is so far the most distinct marker to distinguish Treg cells from other T effector cells; therefore, most of the work aimed at elucidating the stability of Treg cells has been based on the expression of FOXP3.
Some investigations have shown that Treg cells are unstable and phenotypically flexible under certain inflammatory microenvironments, supported by evidence of how CD4+FOXP3+ Treg cells convert into T-helper-like cells with appropriate stimulation, including Th1-, Th2-, Th17-, and Tfh-like cells (60–63). Through adoptive transfer of CD4+EGFP+ and CD4+EGFP− T cells from the spleen and LN of Foxp3EGFP mice into rag2−/− mice, investigators found that over 90% of the transferred eGFP+ T cells maintained FOXP3 expression, and a minor fraction lost their FOXP3 expression. Analysis of the minor fraction of T cells identified a population limited to the FOXP3+CD25− subset that exhibits flexible responses to other cytokines, indicating that natural FOXP3+ T cells contained a committed Treg cell lineage and an uncommitted minor population (64).
Zhou et al. generated Foxp3–GFP–Cre × R26-YFP mice to track Foxp3+ T cells in vivo by crossing transgenic mice expressing a green fluorescent protein–Cre recombinase fusion protein (GFP–Cre) controlled by the foxp3 promoter on a bacterial artificial chromosome (BAC; Foxp3–GFP–Cre mice) with reporter mice that express yellow fluorescent protein (YFP) driven by the Rosa26 promoter only after excision of a loxP-flanked stop cassette (R26-YFP mice). YFP+GFP− T cells represented cells that had expressed FOXP3 at some point before loss of expression, while YFP+GFP+ T cells represented stable FOXP3-expressing cells. They found approximately 15% of the YFP+ cells lost FOXP3 expression, and coined these as “exFoxp3 cells.” Characteristic analysis found that these exFoxp3 cells exhibited an activated-memory T cell phenotype and expressed inflammatory cytokines. Adoptive transfer of these cells in vivo caused rapid onset of diabetes (65).
Meanwhile, other researchers have shown that Treg cells are very stable, and suggest that the unstable Treg cells that have been observed are not bona fide Treg cells but an uncommitted “pre”-Treg cell lineage. To avoid the occurrence of monitoring transiently expressed FOXP3 in effector T cells, Rubtsov et al. generated Foxp3GFP–Cre–ERT2 ROSA26YFP mice to distinguish cells that had only begun to express FOXP3 from those that expressed FOXP3 for a longer duration by detecting YFP intensity, and observed that only 3% of YFP+ cells had lost FOXP3 (66). Hori et al. carried out similar experiments with Foxp3GFP–CreROSA26RFP knock-in mice, and claimed that exFOXP3 T cells were generated from transiently induced FOXP3+ T cells in lymphopenic environments but not from committed Treg cells (67).
The Regulation of FOXP3 Expression
The significance of FOXP3 to Treg development and stability is well documented. Direct evidence that has shown FOXP3 protein to be important for Treg function has been provided by experiments that inserted a gene cassette co-expressing luciferase and enhanced green fluorescent protein (eGFP) into the 3′-untranslated region (UTR) of the endogenous foxp3 locus of C57BL/6 mice. This lead to FOXP3 mRNA instability, a 90% decrease of FOXP3 protein expression, and as a consequence these mice succumbed to aggressive lymphoproliferative autoimmune syndrome, indicating that Treg cell function directly correlates with the amount of FOXP3 protein expressed (12). Observations like this make it imperative to explore the molecular mechanisms regulating FOXP3 expression (Figure 2).
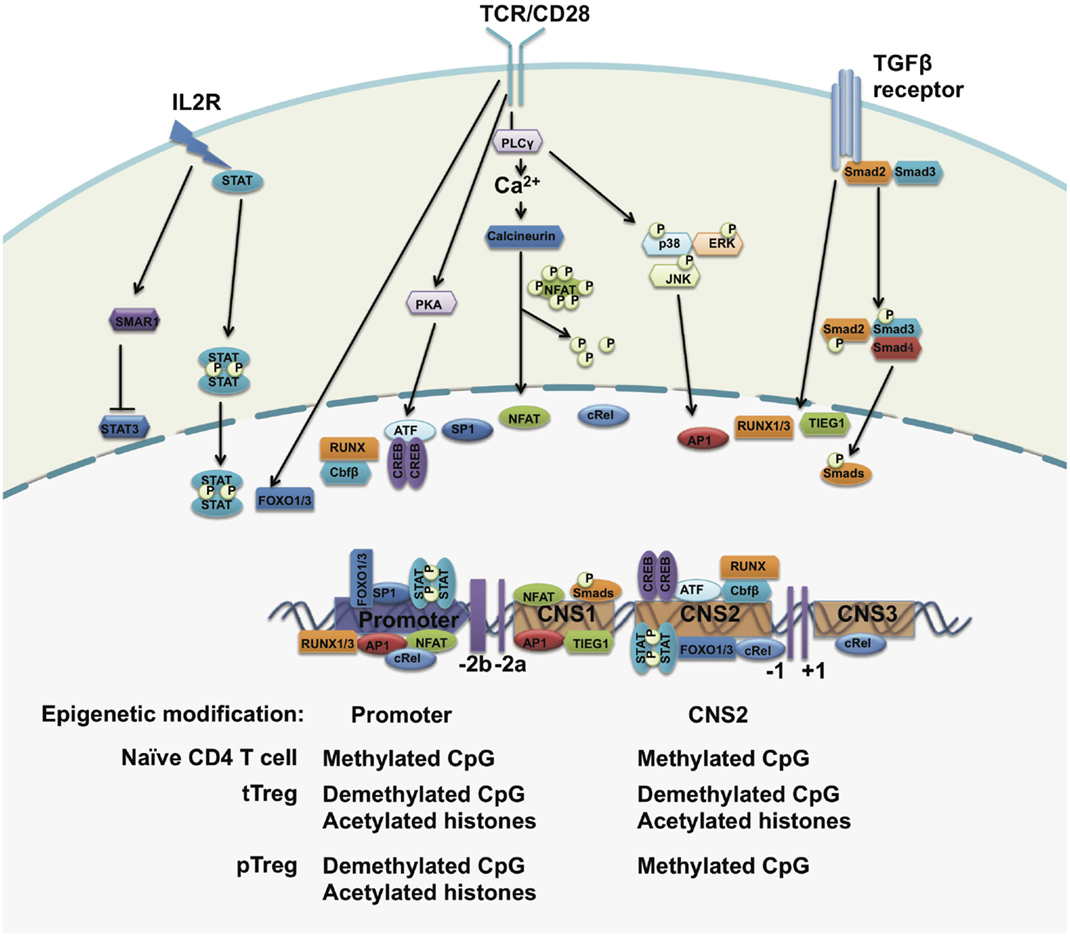
Figure 2. The regulation of FOXP3 expression. The foxp3 promoter, three conserved regulatory regions, and the epigenetic modification status of the foxp3 gene. Upon TCR stimulation, NFAT, AP1, Sp1, and CREB-ATF bind to the promoter of the foxp3 gene. STAT5 forms a dimer in response to IL-2 signals and translocates to the foxp3 promoter. In the periphery, TGF-β signals drive SMADs and NFAT occupancy at the CNS2 region and may induce FOXP3 expression. The CpG island within the foxp3 promoter region is demethylated in both tTreg cells and pTreg cells but not in naïve T cells. The histones bound to the foxp3 promoter region are hyperacetylated in both tTreg and pTreg cells. However, CNS2 is demethylated only in tTreg but not in pTreg cells.
Epigenetic Control of FOXP3 Expression
Epigenetic modifications of the foxp3 gene at its regulatory regions regulate chromatin accessibility for transcription factors and other transcriptional regulators to control FOXP3 expression and Treg cell stability. Chromatin immunoprecipitation (ChIP) assays have revealed higher levels of acetylated histone H4 within the foxp3 promoter in activated Treg cells (68). Treatment with histone deacetylase inhibitors leads to an increased expression of FOXP3 and percentages of FOXP3+ Treg cells in vivo (69), implying that the upregulation of FOXP3 expression is controlled by histone modifications. Both H3K4me2 and H3K4me3 are induced at the transcriptional start sites and regulatory regions at the foxp3 gene locus in both tTreg and iTreg cells upon TCR stimulation (70). Inhibition of H3K4me3 at the foxp3 gene locus impairs TGFβ-induced FOXP3 expression (71).
The methylation status of CpG islands within the foxp3 promoter and regulatory elements also regulates the expression of FOXP3 in Treg cells. Through bisulfite sequencing, investigators have identified a CpG-rich region upstream of exon-1 of the foxp3 gene locus and this region is highly conserved between human and mice. This evolutionarily conserved region is highly demethylated in tTreg cells, incompletely demethylated in iTreg cells, and methylated in naïve CD4+CD25− T cells. This demethylated region is correlated with stable FOXP3 expression and closely associated with modified histones, including acetylated and trimethylated histone H3 but not acetylated histone H4 (72). Genome-wide DNA methylation pattern analysis confirmed specific CpG methylation patterns at other Treg cell-associated gene regions, including il2ra, ctla4, tnfrsf18, ikzf4, and ikzf2 (49). Inhibition of DNA methylation by 5-aza-2′-deoxycytidine or deleting DNA methyltransferase-1 (DNMT-1) induces strong and stable expression of FOXP3 under TCR stimulation even in the absence of TGF-β, which further confirms that the TSDR methylation status of the foxp3 gene locus controls the expression of FOXP3 (73, 74).
Transcriptional Regulation of FOXP3
Upon TCR activation, AP1, CREB, NFAT, c-Rel and ATF bind to the promoter of the foxp3 gene and activate its gene transcription in Treg cells (68, 75–80). Foxo-binding sites were also found within the foxp3 basal promoter, where deficiency of Foxo1 and Foxo3 in Treg cells causes a loss of FOXP3 expression (81). IL-2 signaling is essential to maintain FOXP3 expression in a STAT5-dependent manner (47, 82, 83). Additionally, IL-2 may induce the expression of SMAR1 in Treg cells, while IL-6 does the opposite. SMAR1-bound STAT3 promoters can suppress its gene transcription. Deficiency of SMAR1 in Treg cells causes the upregulation of STAT3, which in turn converts Tregs into Th17-like cells and facilitates increased susceptibility to IBD (84).
In the periphery, naïve T cells can be converted into FOXP3+ Treg cells in the presence of TGF-β. TGF-β induces the occupancy of Runx1 and Runx3 on the promoter of foxp3, but also activates SMAD3 and NFAT binding to the conserved non-coding sequence 1(CNS1) of the foxp3 gene and induces FOXP3 expression (78, 85–90). Thus, CNS1 is considered to be involved in the development of pTreg cells in response to TGF-β signals. In CNS1-deficient mice, FOXP3+ Treg cells are markedly decreased in the gut-associated lymphoid tissue (GALT) and mesenteric lymph node (MLN), where TGF-β-dependent pTreg cells are generated, but not in the spleen and non-gut draining lymph nodes (91). In addition, RA was reported to be capable of augmenting the enrichment of SMADs to CNS1 and therefore enhances FOXP3 expression (88).
Conserved non-coding sequence 2(CNS2) was identified as a unique region containing CpG-rich islands to maintain stable FOXP3 expression in mature tTreg cells. In naïve T cells and pTreg cells, CNS2 is hypermethylated by DNMT-1 and occupied by HDACs and Mecp2 to repress the expression of FOXP3. Under the stimulation of TCR signals plus IL-2, DNMT-1 is released from CNS2 and induces demethylation (47, 77, 83, 92). The transcription factors CREB, STAT5, Est1, c-Rel, FOXP3, Runx–Cbfb heterodimer, and Foxo1/3 are recruited to this element to initiate FOXP3 transcription (77, 81, 86, 91, 93, 94). Deletion of CNS2 induces a loss of FOXP3 protein in mature Treg cells in the presence of IL-6, IFNγ, IL-12, and IL-4 (95, 96). However, a high amount of IL-2 rescues the loss of FOXP3 expression through enhancing STAT5 enrichment onto the foxp3 basal promoter (73, 74).
Conserved non-coding sequence 3(CNS3) is also responsible for the induction of FOXP3. Conditional knockouts of CNS3 in Treg cells can markedly decrease the frequency of tTreg cells and may impair TGF-β-mediated pTreg induction (91). c-Rel was found to bind to this region to drive FOXP3 expression (91).
The FOXP3 Protein Complex and Its Modifications
FOXP3 cooperates with various cofactors to induce the Treg cell gene expression signature and tailor their suppressive function. Biochemical and mass-spectrometric studies showed that FOXP3 could associate with several hundred partners to form a large multi-protein complex (97, 98). FOXP3 cooperates with NFAT and AML1/Runx1 to regulate the expression of IL-2, CD25, and CTLA4 through binding to their promoters and activating gene transcription. Disruption of their interaction would impair Treg suppressive function (99, 100). The association of FOXP3 with Eos–CtBP co-repressor complexes is required for FOXP3-mediated IL-2 repression in Treg cells. In a colitis mouse model, Eos-deficient Treg cells failed to repress the development of adoptive colitis (101). Additionally, a FOXP3–IRF4 complex contributes to establishing Treg-specific gene programs. A conditional knockout of IRF4 in Treg cells showed elevated Th2 responses (102). Deleted in breast cancer 1 (DBC1), a subunit of the FOXP3 complex, prevents FOXP3 degradation and maintains Treg cell stability under inflammatory conditions. Functional Dbc1−/− mice are more resistant to develop severe autoimmune disease symptoms during induction of experimental autoimmune encephalomyelitis (EAE) (103).
The transcription factor GATA3 is highly induced in Treg cells that reside in barrier sites, including the gastrointestinal tract and skin. GATA3 is required for maintaining high levels of FOXP3 expression by binding to and promoting the activity of cis-acting elements of FOXP3. GATA3-deficient Treg cells are more prone to acquire an effector T cell phenotype and express effector cytokines in inflamed tissues (104, 105). USP21 positively regulates and stabilizes GATA3, which can maintain FOXP3 expression. Furthermore, USP21-knockout mice show spontaneous T cell activation (106, 107). Tbet and RORgt have also been identified to be essential for Th1-like and Th17-like Treg cells in inflammatory microenvironments, respectively, and promote Treg cell homing to inflamed loci (108–110).
Post-Translational Modifications of FOXP3
The post-translational modifications of FOXP3 affect Treg differentiation, function, and phenotypic commitment through regulating FOXP3 protein stability and transcriptional activity (Figure 3). Several previous studies have reported that FOXP3 protein stability is controlled by ubiquitination-mediated degradation. Under inflammatory conditions, STUB1 was found recruited to FOXP3 by HSP70 to polyubiquitinate FOXP3 at its K227/250/263/268 sites in a K48-linked polyubiquitination manner. K48-linked polyubiquitinated FOXP3 is further led to proteasome-mediated degradation. Manipulating the level of STUB1 in Treg cells through ectopic expression or knockdown directly affected the protein levels of FOXP3, signature Treg gene expression and the ability to suppress inflammatory immune responses (111). On the other hand, the deubiquitinase USP7 is able to deubiquitinate FOXP3 in an HSP90-dependent manner and stabilizes FOXP3 to increase Treg number to enhance Treg suppressive activity (112). HIF1a and PKB/Akt1-mediated FOXP3 phosphorylation also affects FOXP3 stabilization through indirectly regulating FOXP3 ubiquitination levels (113–116).
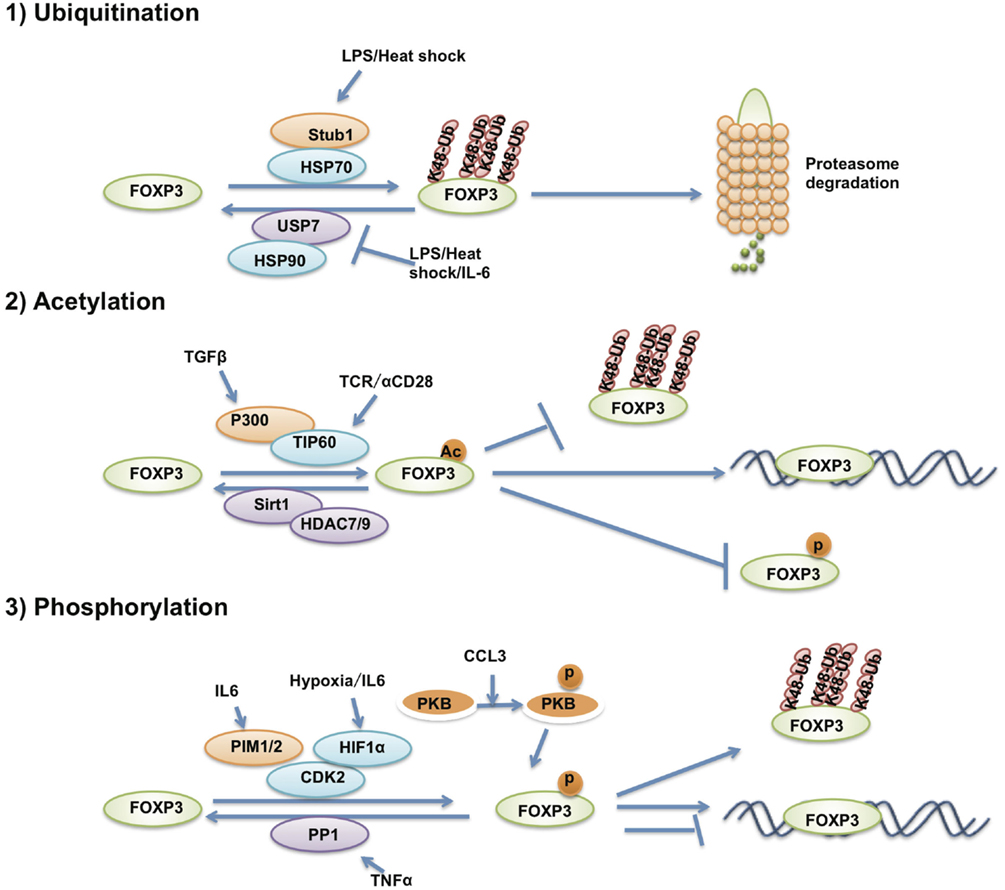
Figure 3. The post-translational modification of FOXP3. The post-translational modifications that affect FOXP3 stability and transcriptional activity. FOXP3 protein is ubiquitinated, acetylated and phosphorylated by various post-translational modification enzymes.
The transcriptional activity of FOXP3 is also regulated by post-translational modifications. Our previous results demonstrated that FOXP3 could associate with the histone acetyltransferase TIP60 and the class II histone deacetylases HDAC9 and HDAC7. TIP60 can acetylate FOXP3 and enhance FOXP3-mediated transcription repression of IL-2 expression through the FOXP3 N-terminal 106 to 109aa region (117). FOXP3 can also be acetylated by P300 and affects FOXP3 stability through impairing polyubiquitination of FOXP3, thus, blocking proteasome-mediated FOXP3 degradation (114, 115).
In addition to ubiquitination and acetylation, MS analysis has revealed that multiple residues of FOXP3 could be phosphorylated. Among these, only a small number have been further investigated. In the synovial fluid of rheumatoid arthritis patients, the pro-inflammatory cytokine TNFα induces the expression and enzymatic activation of protein phosphatase 1 (PP1) that dephosphorylates Ser418 of FOXP3. Subsequently, FOXP3 loses its transcription repression of IL-2 and Treg cells lose their suppressive function, causing increased numbers of IL-17+ and IFN-γ+CD4+ T cells within the inflamed synovium of rheumatoid arthritis patients (118). IL-6-induced PIM1 can phosphorylate Ser422 of FOXP3, which negatively regulates FOXP3 binding affinity on chromatin and also Treg function. Reversing PIM1-mediated FOXP3 phosphorylation through TCR stimulation, shRNA-mediated PIM1 depletion or by using a PIM1 inhibitor could enhance Treg suppressive function (119). Another member of the PIM kinase family named PIM2 was also reported to be able to phosphorylate multiple sites of FOXP3 at its N-terminal domain, leading to attenuated Treg suppressive function. Pim2−/− mice show more resistance to DSS-induced colitis (117). FOXP3 is also the target of CDK2, which phosphorylates FOXP3 at its Ser19 and Thr175 sites to negatively regulate the stability and transcriptional activity of FOXP3 (120). Although most investigations have reported that FOXP3 is strictly expressed in Treg cells, FOXP3 can also be expressed in cancer cells and acts as a cancer repressor (121, 122). Lck can also phosphorylate FOXP3 at Tyr342 in breast cancer cells and increase FOXP3 transcriptional repression of mmp9, skp, and vegfa, and thus suppresses cellular invasion (123).
Treg Cells and Gender Bias in Autoimmune Diseases
Females and males process basic immune responses rather differently. In response to infection, vaccination, or trauma, females exhibit stronger inflammation for protection against infection, while this characteristic also renders females more susceptible to autoimmune diseases. The factors that contribute to these disparate immune responses between males and females are mainly X-linked, which includes hormonal differences.
Current theories related to the pathogenesis of autoimmune diseases assume that the disrupted balance between effector T cells (that cause tissue damage) and Treg cells (that suppress self-reactive cells) correlates with the pathogenesis of autoimmune diseases. The number and function of Treg cells is affected by X-linked foxp3 and hormonal fluctuations. Thus, new insight into gender differences in autoimmune disease may reveal novel therapeutic avenues.
Treg and IPEX
The foxp3 gene is localized on the X chromosome, where mutations in this gene may cause IPEX. In females, there are two X chromosomes, where one undergoes random inactivation. If the foxp3 gene on one X chromosome is mutated, this would potentially produce functionally impaired Treg cells, whereas the other gene with the wild-type foxp3 gene would generate normal Treg cells to protect females from IPEX (124).
Treg Cells and MS
Multiple sclerosis is characterized by chronic inflammation, primary demyelination, and axonal damage. EAE is the animal model of MS. In adoptive transfer experiments, Treg cells may prevent the development of chronic EAE in recipient mice (125–127), implying that Treg cells contribute to protection against MS. Investigators have found no differences in the frequency of CD4+CD25hi Treg cells between patients with MS and healthy controls, while several groups revealed how CD4+CD25hi Treg cells in MS patients are functionally impaired (128–131). MS is more prevalent in females (132). In females, the symptoms of MS have been reported to correlate with hormonal levels. When estrogen (E2) and progesterone (P4) levels decrease during menstruation, disease relapses (133, 134); in turn, during the third trimester of pregnancy when estrogen and progesterone levels are at its highest, the symptoms of MS regress, followed by relapse until dropping at post-partum (135, 136). Treatment with ER ligand protected mice from the development of EAE (137, 138). The protective effect of ER ligand was blocked in estrogen receptor-α (Esr1−/−)- and estrogen receptor-β (Esr2−/−)-deficient mice (138). Both E2 and P4 have been reported to induce high numbers of Treg cells and enhance Treg function (139–142). E2 treatment increased Treg cell number and FOXP3 expression both in vitro and in vivo. In estrogen receptor-α-deficient mice, E2-induced expression of FOXP3 is abrogated (141, 143). E2 was reported to regulate Treg function partially through increasing intracellular levels of the checkpoint inhibitor PD-1. PD-1 expression and Treg suppressive function were attenuated in ER-KO mice. E2 pre-treatment could partially restore the suppressive function of Treg cells in PD-1 KO mice without affecting FOXP3 expression (144).
Other reports have revealed how 17β-estradiol enhances Treg suppressive function via promoting TGF-β and IL-10 secretion (145). P4 may drive cord blood fetal T cells but not adult peripheral blood T cells to differentiate into FOXP3+ Treg cells. These P4-induced Treg cells exhibit a memory phenotype and better suppressive activity. Mechanistically, P4 enhances IL-2-STAT5 signaling and represses IL-6-mediated STAT3 activation by downregulating the IL-6 receptor, facilitating Treg differentiation but suppression of Th17 differentiation (139). P4 could also suppress the mTOR pathway, and thus promote the generation of Treg cells (146) and these Treg express higher levels of ERβ compared with T-responder cells. In MS patients, Treg cells express lower levels of ERβ (147), thus implying that having Treg cells unresponsive to hormones might result in the dysregulation of immune homeostasis and contribute to the pathogenesis of MS.
Frequencies of Treg cells change during the course of pregnancy (148). During pregnancy, elevated E2 levels at early stages are important for CD4+CD25+ Treg cell expansion in mice and are required for embryo implantation (149). Estrogen-treated mice and pregnant mice share similarities in increases of FOXP3 expression and Treg function (150). E2 and P4 increase maintains the expansion of systemic and local uterine Treg cells (140). The correlation between pregnancy-induced fluctuations in Treg cells and MS amelioration remain unclear, which might be influenced by different flow-cytometric approaches and current lack of studies.
Treg Cells and SLE
The imbalance of Th17/Treg cells usually correlates with the pathogenesis of SLE (151, 152). For SLE, data have shown a gender bias toward prevalence in females, with the female:male ratio at almost 9:1 (132). IL-6 plays a very important role in regulating the balance between Th17 cells and Treg cells. In the presence of IL-6, naïve CD4+ T cells differentiate into Th17 cells (with TGF-β) rather than iTreg cells (153). IL-6 together with IL-1 induces the degradation of FOXP3 and deregulates Treg cells (61). Higher concentrations of IL-6 in sera and in urine have been detected in SLE patients; the concentration of IL-6 in SLE patient sera and urine is positively correlated with disease severity (154–157). The expression of IL-6 is upregulated by estrogens (158) and is dominant in females (159). In mice, blocking IL-6 could significantly increase FOXP3 expression and make animals resistant to ALD-DNA-induced SLE (160). IL-6 may affect Th17/Treg balance in males and females, and thus contributes to the prevalence of SLE in females. So far, related studies are limited and more evidence is required to further characterize this correlation.
Treg Cells and AS
Ankylosing spondylitis (AS) is a chronic inflammatory disease with strong genetic connections (161, 162). Patients with AS are two to three times higher in males than females, and suffer from inflammatory spinal pain that could lead to the pathogenesis of spondyloarthritis and spinal immobility (163). Treatment of AS by tumor necrosis factor α inhibitors seem effective, which leads to the reduction of disease progression (164). The imbalance of Treg cells and inflammatory Th17 cells in AS patients has been previously studied but the underlying mechanism remains unclear (165, 166). Small molecule inhibitors that promote Treg function could play a beneficial role in preventing the pathogenesis of AS (167, 168).
Conclusion
Accumulating experimental evidence has revealed the important role of Treg cells in maintaining immune homeostasis and preventing the occurrence of autoimmune diseases. Treg cells adopt multiple molecular mechanisms to maintain their lineage stability and obtain a certain degree of functional plasticity to adapt to various inflammatory conditions. However, inflammatory factors from the local microenvironment would interfere with the stability of Treg cells and promote the development of autoimmune diseases. Therefore, exploring the molecular mechanisms behind the function of the Treg cell-lineage transcription factor FOXP3 in autoimmunity would provide insight into the understanding of the stability and plasticity of Treg cells. Treg therapy could be an important tool for treating autoimmune disease in the future. Current reports describing the effect of gender differences on Treg cells and the contributions of Treg cells to the prevalence of autoimmune diseases in females are limited. The latest findings that Treg cells are regulated by hormonal fluctuations suggest that these risk factors that may disrupt the balance between T helper and Treg cells and induce autoimmune disease include birth control pills, stress, existence or development of ovarian cysts, and overuse of products containing xenoestrogens, etc., causing hormonal imbalance. Hence, it is significantly important to take sex-based differences into consideration when exploring the role of Treg cells in human illnesses and development of Treg cell therapies for treating autoimmune diseases.
Although Treg cells are well acknowledged as a potential and promising tool for the treatment of autoimmune diseases, there is still a large gap between theory and reality. To achieve the goal of successfully and effectively using Treg cells to restore tolerance and for treating autoimmune diseases, the following important questions in Treg cell biology still need to be further addressed:
1. Except for NRP1, Helios, and GARP, are there better surface makers for distinguishing between tTreg and pTreg cells, and what are the different physiological functions of tTreg and pTreg cells in the context of autoimmune disease?
2. How is the FOXP3 complex and post-translational modifications dynamically regulated in response to various physiological signals and how do they modify Treg cell function?
3. What is the role of Treg cells in the onset and progression of different autoimmune diseases?
4. What is the correlation between Treg cells and gender bias in different autoimmune diseases?
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our work was supported by National Basic Research Program of China Grants 2014CB541803 and 2014CB541903; National Natural Science Foundation of China Grants 31170825, 31200646, 31150110337, 30972702; and the National Science and Technology Major Project Grants 2012ZX10002007-003 and 2013ZX10003009-002. We thank our lab members for helpful discussions.
References
1. Sojka DK, Hughson A, Fowell DJ. CTLA-4 is required by CD4(+)CD25(+) Treg to control CD4(+) T-cell lymphopenia-induced proliferation. Eur J Immunol (2009) 39:1544–51. doi:10.1002/eji.200838603
2. Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA (2008) 105:19396–401. doi:10.1073/pnas.0806855105
3. Ji HB, Liao G, Faubion WA, Abadia-Molina AC, Cozzo C, Laroux FS, et al. Cutting edge: the natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J Immunol (2004) 172:5823–7. doi:10.4049/jimmunol.172.10.5823
4. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27:635–46. doi:10.1016/j.immuni.2007.08.014
5. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi:10.1182/blood-2006-12-064527
6. Collison LW, Pillai MR, Chaturvedi V, Vignali DA. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J Immunol (2009) 182:6121–8. doi:10.4049/jimmunol.0803646
7. Beissert S, Schwarz A, Schwarz T. Regulatory T cells. J Invest Dermatol (2006) 126:15–24. doi:10.1038/sj.jid.5700004
8. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4:337–42. doi:10.1038/ni909
9. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
10. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
11. Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol (2007) 8:277–84. doi:10.1038/ni1437
12. Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445:766–70. doi:10.1038/nature05479
13. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453:236–40. doi:10.1038/nature06878
14. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445:771–5. doi:10.1038/nature05543
15. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27:68–73. doi:10.1038/83784
16. Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol (2003) 15:430–5. doi:10.1097/00002281-200307000-00010
17. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest (2000) 106:R75–81. doi:10.1172/JCI11679
18. Sanchez E, Rueda B, Orozco G, Oliver J, Vilchez JR, Paco L, et al. Analysis of a GT microsatellite in the promoter of the foxp3/scurfin gene in autoimmune diseases. Hum Immunol (2005) 66:869–73. doi:10.1016/j.humimm.2005.06.001
19. Lin YC, Lee JH, Wu AS, Tsai CY, Yu HH, Wang LC, et al. Association of single-nucleotide polymorphisms in FOXP3 gene with systemic lupus erythematosus susceptibility: a case-control study. Lupus (2011) 20:137–43. doi:10.1177/0961203310382428
20. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi:10.1038/83713
21. Bennett CL, Brunkow ME, Ramsdell F, O’Briant KC, Zhu Q, Fuleihan RL, et al. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA – >AAUGAA) leads to the IPEX syndrome. Immunogenetics (2001) 53:435–9. doi:10.1007/s002510100358
22. Kobayashi I, Shiari R, Yamada M, Kawamura N, Okano M, Yara A, et al. Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). J Med Genet (2001) 38:874–6. doi:10.1136/jmg.38.12.874
23. An YF, Zhao XD, Xu F, Yang XQ. [A novel missense mutation of FOXP3 causes immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome in a Chinese child]. Zhonghua Er Ke Za Zhi (2009) 47:824–8. doi:10.3760/cma.j.issn.0578-1310.2009.11.007
24. Bassuny WM, Ihara K, Sasaki Y, Kuromaru R, Kohno H, Matsuura N, et al. A functional polymorphism in the promoter/enhancer region of the FOXP3/Scurfin gene associated with type 1 diabetes. Immunogenetics (2003) 55:149–56. doi:10.1007/s00251-003-0559-8
25. Zavattari P, Deidda E, Pitzalis M, Zoa B, Moi L, Lampis R, et al. No association between variation of the FOXP3 gene and common type 1 diabetes in the Sardinian population. Diabetes (2004) 53:1911–4. doi:10.2337/diabetes.53.7.1911
26. Owen CJ, Eden JA, Jennings CE, Wilson V, Cheetham TD, Pearce SH. Genetic association studies of the FOXP3 gene in Graves’ disease and autoimmune Addison’s disease in the United Kingdom population. J Mol Endocrinol (2006) 37:97–104. doi:10.1677/jme.1.02072
27. Howson JMM, Walker NM, Smyth DJ, Todd J, Consortium TIDG. Analysis of 19 genes for association with type I diabetes in the Type I Diabetes Genetics Consortium families. Genes Immun (2009) 10:S74–84. doi:10.1038/gene.2009.96
28. Bjornvold M, Amundsen SS, Stene LC, Joner G, Dahl-Jorgensen K, Njolstad PR, et al. FOXP3 polymorphisms in type 1 diabetes and coeliac disease. J Autoimmun (2006) 27:140–4. doi:10.1016/j.jaut.2006.06.007
29. Lee HM, Hsieh CS. Rare development of Foxp3+ thymocytes in the CD4+CD8+ subset. J Immunol (2009) 183:2261–6. doi:10.4049/jimmunol.0901304
30. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14:307–8. doi:10.1038/ni.2554
31. Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, et al. Thymus and autoimmunity: production of CD25(+)CD4(+) naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol (1999) 162:5317–26.
32. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol (2001) 2:301–6. doi:10.1038/86302
33. Lerman MA, Larkin J III, Cozzo C, Jordan MS, Caton AJ. CD4+ CD25+ regulatory T cell repertoire formation in response to varying expression of a neo-self-antigen. J Immunol (2004) 173:236–44. doi:10.4049/jimmunol.173.1.236
34. Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity (2006) 25:249–59. doi:10.1016/j.immuni.2006.05.016
35. Fujishima M, Hirokawa M, Fujishima N, Sawada K. TCRalphabeta repertoire diversity of human naturally occurring CD4+CD25+ regulatory T cells. Immunol Lett (2005) 99:193–7. doi:10.1016/j.imlet.2005.02.011
36. Kasow KA, Chen X, Knowles J, Wichlan D, Handgretinger R, Riberdy JM. Human CD4+CD25+ regulatory T cells share equally complex and comparable repertoires with CD4+CD25- counterparts. J Immunol (2004) 172:6123–8. doi:10.4049/jimmunol.172.10.6123
37. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity (2004) 21:267–77. doi:10.1016/j.immuni.2004.07.009
38. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med (2011) 208:1279–89. doi:10.1084/jem.20110308
39. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
40. Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity (1995) 3:521–30. doi:10.1016/1074-7613(95)90180-9
41. Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science (1995) 268:1472–6. doi:10.1126/science.7770771
42. Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell (1993) 75:253–61. doi:10.1016/0092-8674(93)80067-O
43. Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature (1991) 352:621–4. doi:10.1038/352621a0
44. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity (2002) 17:167–78. doi:10.1016/S1074-7613(02)00367-9
45. Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol (2002) 169:4850–60. doi:10.4049/jimmunol.169.9.4850
46. Papiernik M, de Moraes ML, Pontoux C, Vasseur F, Penit C. Regulatory CD4 T cells: expression of IL-2R alpha chain, resistance to clonal deletion and IL-2 dependency. Int Immunol (1998) 10:371–8. doi:10.1093/intimm/10.4.371
47. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol (2007) 178:280–90. doi:10.4049/jimmunol.178.1.280
48. Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity (2010) 32:642–53. doi:10.1016/j.immuni.2010.04.012
49. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37:785–99. doi:10.1016/j.immuni.2012.09.010
50. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
51. Park HB, Paik DJ, Jang E, Hong S, Youn J. Acquisition of anergic and suppressive activities in transforming growth factor-beta-costimulated CD4+CD25- T cells. Int Immunol (2004) 16:1203–13. doi:10.1093/intimm/dxh123
52. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203:1701–11. doi:10.1084/jem.20060772
53. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184:3433–41. doi:10.4049/jimmunol.0904028
54. Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK. Helios+ and Helios- cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol (2013) 190:2001–8. doi:10.4049/jimmunol.1201379
55. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209:S1–19. doi:10.1084/jem.20120822
56. Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci USA (2009) 106:13439–44. doi:10.1073/pnas.0901965106
57. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med (2012) 209:1723–42,S1. doi:10.1084/jem.20120914
58. Tang Q, Bluestone JA. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med (2013) 3:ii:a015552. doi:10.1101/cshperspect.a015552
59. Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol (2013) 13:461–7. doi:10.1038/nri3464
60. Yurchenko E, Shio MT, Huang TC, Da Silva Martins M, Szyf M, Levings MK, et al. Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS One (2012) 7:e35572. doi:10.1371/journal.pone.0035572
61. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29:44–56. doi:10.1016/j.immuni.2008.05.007
62. Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science (2009) 323:1488–92. doi:10.1126/science.1169152
63. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol (2007) 178:6725–9. doi:10.4049/jimmunol.178.11.6725
64. Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA (2009) 106:1903–8. doi:10.1073/pnas.0811556106
65. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10:1000–7. doi:10.1038/ni.1774
66. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (2010) 329:1667–71. doi:10.1126/science.1191996
67. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity (2012) 36:262–75. doi:10.1016/j.immuni.2011.12.012
68. Mantel PY, Ouaked N, Ruckert B, Karagiannidis C, Welz R, Blaser K, et al. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol (2006) 176:3593–602. doi:10.4049/jimmunol.176.6.3593
69. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13:1299–307. doi:10.1038/nm1652
70. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA (2008) 105:7797–802. doi:10.1073/pnas.0800928105
71. de Almeida Nagata DE, Ting HA, Cavassani KA, Schaller MA, Mukherjee S, Ptaschinski C, et al. Epigenetic control of Foxp3 by SMYD3 H3K4 histone methyltransferase controls iTreg development and regulates pathogenic T-cell responses during pulmonary viral infection. Mucosal Immunol (2015) 8(5):1131–43. doi:10.1038/mi.2015.4
72. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5:e38. doi:10.1371/journal.pbio.0050038
73. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38:1654–63. doi:10.1002/eji.200838105
74. Josefowicz SZ, Wilson CB, Rudensky AY. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J Immunol (2009) 182:6648–52. doi:10.4049/jimmunol.0803320
75. Kitoh A, Ono M, Naoe Y, Ohkura N, Yamaguchi T, Yaguchi H, et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity (2009) 31:609–20. doi:10.1016/j.immuni.2009.09.003
76. Rudra D, Egawa T, Chong MM, Treuting P, Littman DR, Rudensky AY. Runx-CBFbeta complexes control expression of the transcription factor Foxp3 in regulatory T cells. Nat Immunol (2009) 10:1170–7. doi:10.1038/ni.1795
77. Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med (2007) 204:1543–51. doi:10.1084/jem.20070109
78. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol (2008) 9:194–202. doi:10.1038/ni1549
79. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity (2009) 31:932–40. doi:10.1016/j.immuni.2009.10.006
80. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med (2009) 206:3001–14. doi:10.1084/jem.20091411
81. Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol (2010) 11:618–27. doi:10.1038/ni.1884
82. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity (2007) 26:371–81. doi:10.1016/j.immuni.2007.02.009
83. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4(+)CD25(+) regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108:1571–9. doi:10.1182/blood-2006-02-004747
84. Mirlekar B, Ghorai S, Khetmalas M, Bopanna R, Chattopadhyay S. Nuclear matrix protein SMAR1 control regulatory T-cell fate during inflammatory bowel disease (IBD). Mucosal Immunol (2015). doi:10.1038/mi.2015.42
85. Klunker S, Chong MM, Mantel PY, Palomares O, Bassin C, Ziegler M, et al. Transcription factors RUNX1 and RUNX3 in the induction and suppressive function of Foxp3+ inducible regulatory T cells. J Exp Med (2009) 206:2701–15. doi:10.1084/jem.20090596
86. Bruno L, Mazzarella L, Hoogenkamp M, Hertweck A, Cobb BS, Sauer S, et al. Runx proteins regulate Foxp3 expression. J Exp Med (2009) 206:2329–37. doi:10.1084/jem.20090226
87. Schlenner SM, Weigmann B, Ruan Q, Chen Y, von Boehmer H. Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J Exp Med (2012) 209:1529–35. doi:10.1084/jem.20112646
88. Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity (2010) 33:313–25. doi:10.1016/j.immuni.2010.09.001
89. Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol (2010) 185:842–55. doi:10.4049/jimmunol.0904100
90. Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci (2001) 114:4359–69.
91. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463:808–12. doi:10.1038/nature08750
92. Burchill MA, Yang J, Vang KB, Farrar MA. Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol Lett (2007) 114:1–8. doi:10.1016/j.imlet.2007.08.005
93. Polansky JK, Schreiber L, Thelemann C, Ludwig L, Kruger M, Baumgrass R, et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (2010) 88:1029–40. doi:10.1007/s00109-010-0642-1
94. Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31:921–31. doi:10.1016/j.immuni.2009.09.022
95. Li X, Liang Y, LeBlanc M, Benner C, Zheng Y. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell (2014) 158:734–48. doi:10.1016/j.cell.2014.07.030
96. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell (2014) 158:749–63. doi:10.1016/j.cell.2014.07.031
97. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol (2012) 13:1010–9. doi:10.1038/ni.2402
98. Hori S. The Foxp3 interactome: a network perspective of T(reg) cells. Nat Immunol (2012) 13:943–5. doi:10.1038/ni.2424
99. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell (2006) 126:375–87. doi:10.1016/j.cell.2006.05.042
100. Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature (2007) 446:685–9. doi:10.1038/nature05673
101. Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science (2009) 325:1142–6. doi:10.1126/science.1176077
102. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458:351–6. doi:10.1038/nature07674
103. Gao Y, Tang J, Chen W, Li Q, Nie J, Lin F, et al. Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc Natl Acad Sci USA (2015) 112(25):E3246–54. doi:10.1073/pnas.1421463112
104. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest (2011) 121:4503–15. doi:10.1172/JCI57456
105. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity (2011) 35:337–48. doi:10.1016/j.immuni.2011.08.012
106. Pannu J, Belle JI, Forster M, Duerr CU, Shen S, Kane L, et al. Ubiquitin specific protease 21 is dispensable for normal development, hematopoiesis and lymphocyte differentiation. PLoS One (2015) 10:e0117304. doi:10.1371/journal.pone.0117304
107. Zhang J, Chen C, Hou X, Gao Y, Lin F, Yang J, et al. Identification of the E3 deubiquitinase ubiquitin-specific peptidase 21 (USP21) as a positive regulator of the transcription factor GATA3. J Biol Chem (2013) 288:9373–82. doi:10.1074/jbc.M112.374744
108. Kornete M, Mason ES, Girouard J, Lafferty EI, Qureshi S, Piccirillo CA. Th1-like ICOS+ Foxp3+ Treg cells preferentially express CXCR3 and home to beta-islets during pre-diabetes in BDC2.5 NOD mice. PLoS One (2015) 10:e0126311. doi:10.1371/journal.pone.0126311
109. Zheng J, Liu Y, Qin G, Lam KT, Guan J, Xiang Z, et al. Generation of human Th1-like regulatory CD4+ T cells by an intrinsic IFN-gamma- and T-bet-dependent pathway. Eur J Immunol (2011) 41:128–39. doi:10.1002/eji.201040724
110. Tartar DM, VanMorlan AM, Wan X, Guloglu FB, Jain R, Haymaker CL, et al. FoxP3+RORgammat+ T helper intermediates display suppressive function against autoimmune diabetes. J Immunol (2010) 184:3377–85. doi:10.4049/jimmunol.0903324
111. Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity (2013) 39:272–85. doi:10.1016/j.immuni.2013.08.006
112. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity (2013) 39:259–71. doi:10.1016/j.immuni.2013.05.018
113. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
114. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115:965–74. doi:10.1182/blood-2009-02-207118
115. van Loosdregt J, Brunen D, Fleskens V, Pals CE, Lam EW, Coffer PJ. Rapid temporal control of Foxp3 protein degradation by sirtuin-1. PLoS One (2011) 6:e19047. doi:10.1371/journal.pone.0019047
116. Chen L, Wu J, Pier E, Zhao Y, Shen Z. mTORC2-PKBalpha/Akt1 Serine 473 phosphorylation axis is essential for regulation of FOXP3 Stability by chemokine CCL3 in psoriasis. J Invest Dermatol (2013) 133:418–28. doi:10.1038/jid.2012.333
117. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci USA (2007) 104:4571–6. doi:10.1073/pnas.0700298104
118. Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med (2013) 19:322–8. doi:10.1038/nm.3085
119. Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S, et al. PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem (2014) 289:26872–81. doi:10.1074/jbc.M114.586651
120. Morawski PA, Mehra P, Chen C, Bhatti T, Wells AD. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem (2013) 288:24494–502. doi:10.1074/jbc.M113.467704
121. Wang LZ, Liu RH, Li WQ, Chen C, Katoh H, Chen GY, et al. Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer Cell (2009) 16:336–46. doi:10.1016/j.ccr.2009.08.016
122. Zuo T, Wang LZ, Morrison C, Chang X, Zhang HM, Li WQ, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell (2007) 129:1275–86. doi:10.1016/j.cell.2007.04.034
123. Nakahira K, Morita A, Kim NS, Yanagihara I. Phosphorylation of FOXP3 by LCK downregulates MMP9 expression and represses cell invasion. PLoS One (2013) 8:e77099. doi:10.1371/journal.pone.0077099
124. Tommasini A, Ferrari S, Moratto D, Badolato R, Boniotto M, Pirulli D, et al. X-chromosome inactivation analysis in a female carrier of FOXP3 mutation. Clin Exp Immunol (2002) 130:127–30. doi:10.1046/j.1365-2249.2002.01940.x
125. Kohm AP, Carpentier PA, Miller SD. Regulation of experimental autoimmune encephalomyelitis (EAE) by CD4+CD25+ regulatory T cells. Novartis Found Symp (2003) 252:45–52;discussion52–4, 106–14. doi:10.1002/0470871628.ch4
126. McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol (2005) 175:3025–32. doi:10.4049/jimmunol.175.5.3025
127. Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol (2005) 174:6772–80. doi:10.4049/jimmunol.174.11.6772
128. Haas J, Hug A, Viehover A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol (2005) 35:3343–52. doi:10.1002/eji.200526065
129. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med (2004) 199:971–9. doi:10.1084/jem.20031579
130. Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H. Increased frequency of CD4+ CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol (2007) 147:412–8. doi:10.1111/j.1365-2249.2006.03271.x
131. Putheti P, Pettersson A, Soderstrom M, Link H, Huang YM. Circulating CD4+CD25+ T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J Clin Immunol (2004) 24:155–61. doi:10.1023/B:JOCI.0000019780.93817.82
132. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol (2014) 35:347–69. doi:10.1016/j.yfrne.2014.04.004
133. Holmqvist P, Wallberg M, Hammar M, Landtblom AM, Brynhildsen J. Symptoms of multiple sclerosis in women in relation to sex steroid exposure. Maturitas (2006) 54:149–53. doi:10.1016/j.maturitas.2005.10.003
134. Zorgdrager A, De Keyser J. The premenstrual period and exacerbations in multiple sclerosis. Eur Neurol (2002) 48:204–6. doi:10.1159/000066166
135. Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med (1998) 339:285–91. doi:10.1056/NEJM199807303390501
136. Korn-Lubetzki I, Kahana E, Cooper G, Abramsky O. Activity of multiple sclerosis during pregnancy and puerperium. Ann Neurol (1984) 16:229–31. doi:10.1002/ana.410160211
137. Du S, Sandoval F, Trinh P, Umeda E, Voskuhl R. Estrogen receptor-beta ligand treatment modulates dendritic cells in the target organ during autoimmune demyelinating disease. Eur J Immunol (2011) 41:140–50. doi:10.1002/eji.201040796
138. Polanczyk M, Zamora A, Subramanian S, Matejuk A, Hess DL, Blankenhorn EP, et al. The protective effect of 17beta-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-alpha. Am J Pathol (2003) 163:1599–605. doi:10.1016/S0002-9440(10)63516-X
139. Lee JH, Ulrich B, Cho J, Park J, Kim CH. Progesterone promotes differentiation of human cord blood fetal T cells into T regulatory cells but suppresses their differentiation into Th17 cells. J Immunol (2011) 187:1778–87. doi:10.4049/jimmunol.1003919
140. Mao G, Wang J, Kang Y, Tai P, Wen J, Zou Q, et al. Progesterone increases systemic and local uterine proportions of CD4+CD25+ Treg cells during midterm pregnancy in mice. Endocrinology (2010) 151:5477–88. doi:10.1210/en.2010-0426
141. Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, et al. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J Immunol (2004) 173:2227–30. doi:10.4049/jimmunol.173.4.2227
142. Prieto GA, Rosenstein Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology (2006) 118:58–65. doi:10.1111/j.1365-2567.2006.02339.x
143. Bebo BF Jr, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol (2001) 166:2080–9. doi:10.4049/jimmunol.166.3.2080
144. Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int Immunol (2007) 19:337–43. doi:10.1093/intimm/dxl151
145. Luo CY, Wang L, Sun C, Li DJ. Estrogen enhances the functions of CD4(+)CD25(+)Foxp3(+) regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol (2011) 8:50–8. doi:10.1038/cmi.2010.54
146. Lee JH, Lydon JP, Kim CH. Progesterone suppresses the mTOR pathway and promotes generation of induced regulatory T cells with increased stability. Eur J Immunol (2012) 42:2683–96. doi:10.1002/eji.201142317
147. Aristimuno C, Teijeiro R, Valor L, Alonso B, Tejera-Alhambra M, de Andres C, et al. Sex-hormone receptors pattern on regulatory T-cells: clinical implications for multiple sclerosis. Clin Exp Med (2012) 12:247–55. doi:10.1007/s10238-011-0172-3
148. Arruvito L, Sanz M, Banham AH, Fainboim L. Expansion of CD4+CD25+and FOXP3+ regulatory T cells during the follicular phase of the menstrual cycle: implications for human reproduction. J Immunol (2007) 178:2572–8. doi:10.4049/jimmunol.178.4.2572
149. Tai P, Wang J, Jin H, Song X, Yan J, Kang Y, et al. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol (2008) 214:456–64. doi:10.1002/jcp.21221
150. Haghmorad D, Amini AA, Mahmoudi MB, Rastin M, Hosseini M, Mahmoudi M. Pregnancy level of estrogen attenuates experimental autoimmune encephalomyelitis in both ovariectomized and pregnant C57BL/6 mice through expansion of Treg and Th2 cells. J Neuroimmunol (2014) 277:85–95. doi:10.1016/j.jneuroim.2014.10.004
151. Dal Ben ER, do Prado CH, Baptista TS, Bauer ME, Staub HL. Patients with systemic lupus erythematosus and secondary antiphospholipid syndrome have decreased numbers of circulating CD4(+)CD25(+)Foxp3(+) Treg and CD3(-)CD19(+) B cells. Revista Bras Reumatol (2014) 54:241–6. doi:10.1016/j.rbre.2013.09.001
152. Szmyrka-Kaczmarek M, Kosmaczewska A, Ciszak L, Szteblich A, Wiland P. Peripheral blood Th17/Treg imbalance in patients with low-active systemic lupus erythematosus. Postepy Hig Med Dosw (2014) 68:893–8. doi:10.5604/17322693.1111127
153. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol (2010) 40:1830–5. doi:10.1002/eji.201040391
154. Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol (1991) 147:117–23.
155. Sabry A, Elbasyouni SR, Sheashaa HA, Alhusseini AA, Mahmoud K, George SK, et al. Correlation between levels of TNF-alpha and IL-6 and hematological involvement in SLE Egyptian patients with lupus nephritis. Int Urol Nephrol (2006) 38:731–7. doi:10.1007/s11255-006-0047-9
156. Ripley BJ, Goncalves B, Isenberg DA, Latchman DS, Rahman A. Raised levels of interleukin 6 in systemic lupus erythematosus correlate with anaemia. Ann Rheum Dis (2005) 64:849–53. doi:10.1136/ard.2004.022681
157. Li Y, Tucci M, Narain S, Barnes EV, Sobel ES, Segal MS, et al. Urinary biomarkers in lupus nephritis. Autoimmun Rev (2006) 5:383–8. doi:10.1016/j.autrev.2005.10.006
158. Isse K, Specht SM, Lunz JG III, Kang LI, Mizuguchi Y, Demetris AJ. Estrogen stimulates female biliary epithelial cell interleukin-6 expression in mice and humans. Hepatology (2010) 51:869–80. doi:10.1002/hep.23386
159. Olivieri F, Bonafe M, Cavallone L, Giovagnetti S, Marchegiani F, Cardelli M, et al. The -174 C/G locus affects in vitro/in vivo IL-6 production during aging. Exp Gerontol (2002) 37:309–14. doi:10.1016/S0531-5565(01)00197-8
160. Mao X, Wu Y, Diao H, Hao J, Tian G, Jia Z, et al. Interleukin-6 promotes systemic lupus erythematosus progression with Treg suppression approach in a murine systemic lupus erythematosus model. Clin Rheumatol (2014) 33:1585–93. doi:10.1007/s10067-014-2717-9
161. International Genetics of Ankylosing Spondylitis C, Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet (2013) 45:730–8. doi:10.1038/ng.2667
162. Bowness P. Hla-B27. Annu Rev Immunol (2015) 33:29–48. doi:10.1146/annurev-immunol-032414-112110
163. Tam LS, Gu J, Yu D. Pathogenesis of ankylosing spondylitis. Nat Rev Rheumatol (2010) 6:399–405. doi:10.1038/nrrheum.2010.79
164. Haroon N, Inman RD, Learch TJ, Weisman MH, Lee M, Rahbar MH, et al. The impact of tumor necrosis factor alpha inhibitors on radiographic progression in ankylosing spondylitis. Arthritis Rheum (2013) 65:2645–54. doi:10.1002/art.38070
165. Ciccia F, Accardo-Palumbo A, Giardina A, Di Maggio P, Principato A, Bombardieri M, et al. Expansion of intestinal CD4+CD25(high) Treg cells in patients with ankylosing spondylitis: a putative role for interleukin-10 in preventing intestinal Th17 response. Arthritis Rheum (2010) 62:3625–34. doi:10.1002/art.27699
166. Liao HT, Lin YF, Tsai CY, Chou CT. Regulatory T cells in ankylosing spondylitis and the response after adalimumab treatment. Joint Bone Spine (2015). doi:10.1016/j.jbspin.2015.03.003
167. Lin F, Luo X, Tsun A, Li Z, Li D, Li B. Kaempferol enhances the suppressive function of Treg cells by inhibiting FOXP3 phosphorylation. Int Immunopharmacol (2015). doi:10.1016/j.intimp.2015.03.044
Keywords: Treg cells, FOXP3, gender, autoimmunity, inflammation
Citation: Nie J, Li YY, Zheng SG, Tsun A and Li B (2015) FOXP3+ Treg cells and gender bias in autoimmune diseases. Front. Immunol. 6:493. doi: 10.3389/fimmu.2015.00493
Received: 04 July 2015; Accepted: 09 September 2015;
Published: 28 September 2015
Edited by:
Xin M. Luo, Virginia Polytechnic Institute and State University, USAReviewed by:
Nathan Karin, Technion – Israel Institute of Technology, IsraelYisong Wan, University of North Carolina at Chapel Hill, USA
Copyright: © 2015 Nie, Li, Zheng, Tsun and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andy Tsun, Innovent Biologics Inc., 168 Dongping Street, Suzhou, Jiangsu 215123, China,YW5keS50c3VuQGlubm92ZW50YmlvLmNvbQ==;
Bin Li, Unit of Molecular Immunology, Insitut Pasteur of Shanghai, No. 411, Hefei Road (South), Shanghai 200031, China,YmlubGlAc2licy5hYy5jbg==