 Martin James Stebbing
Martin James Stebbing Jennifer Marie Cottee
Jennifer Marie Cottee Indrajeetsinh Rana
Indrajeetsinh Rana- 1Health Innovations Research Institute and School of Medical Sciences, RMIT University, Bundoora, VIC, Australia
- 2School of Health Sciences, Federation University Australia, Ballarat, VIC, Australia
Microglia are often referred to as the immune cells of the brain. They are most definitely involved in immune responses to invading pathogens and inflammatory responses to tissue damage. However, recent results suggest microglia are vital for normal functioning of the brain. Neuroinflammation, as well as more subtle changes, in microglial function has been implicated in the pathogenesis of many brain diseases and disorders. Upon sensing alterations in their local environment, microglia change their shape and release factors that can modify the excitability of surrounding neurons. During neuroinflammation, microglia proliferate and release NO, reactive oxygen species, cytokines and chemokines. If inflammation resolves then their numbers normalize again via apoptosis. Microglia express a wide array of ion channels and different types are implicated in all of the cellular processes listed above. Modulation of microglial ion channels has shown great promise as a therapeutic strategy in several brain disorders. In this review, we discuss recent advances in our knowledge of microglial ion channels and their roles in responses of microglia to changes in the extracellular milieu.
Microglia Express a Variety of Ion Channels
For cells that were, until relatively recently, thought to be quiescent in the normal brain and simply surveilling the surrounding tissue for damage or infection, microglia are exquisitely sensitive to their environment and express a remarkable array of different ion channels. In addition to many neurotransmitter sensitive and other ligand-gated ion channels, there is evidence for the presence of H+ channels, Na+ channels, voltage-gated Ca2+ channels, Ca2+-release-activated Ca2+ channels, voltage-dependent and voltage-independent Cl− channels, and at least six different types of K+ channels in microglia (1). The later include inward rectifier, delayed rectifier, HERG-like, G-protein-activated, as well as voltage-dependent and voltage-independent Ca2+-activated K+ channels. Some of these channels are species specific, whereas others are commonly expressed between species, but the majority of these appear to be expressed in human microglia (1). For cells that not “excitable” in the neuronal sense, this large array of different channel types channels might seem superfluous, yet a substantial body of evidence suggests these channels regulate all of the important functions of microglia, including activation, chemotaxis, secretion, proliferation, the respiratory burst (2, 3), and phagocytic activity (4). In addition, the expression level of most of these ion channels depends on the functional state of the microglial cell and several have been proposed as therapeutic targets in neurological diseases.
Ligand-Gated Ion Channels Help Microglia to Monitor Their Local Environment
Microglia express a variety of receptors that detect changes in their local environment, many of which are ligand-gated ion channels, including purinergic receptors, glutamate receptors, and other neurotransmitter gated channels (5–14). For example, microglial glutamate receptors include ionotropic receptors N-methyl-d-aspartic acid (NMDA), (S)-a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainic acid (KA) receptors in addition to metabotropic glutamate receptors. Many of these receptors are sensitive to microglial secretions acting in an autocrine manner. For instance, neurotransmitters can induce microglial glutamate release, which can then act back on microglia to induce more neurotransmitter release. Microglia express receptors for ATP and cultured microglia release ATP following application of glutamate. This release is prevented by AMPA receptor blockers but not NMDA receptor inhibitors and is dependent on PKC activation and release of Ca2+ from stores, but not on extracellular Ca2+ (15). This type of autocrine action of microglial secretions has been reported by various in vitro studies (8, 11, 15, 16) and it may provide positive feedback to allow rapid responses to danger signals and recruitment of surrounding microglia in vivo. However, there is also evidence that microglia express only some of these receptors in their resting condition. Upon activation, they increase their expression of some receptors and begin to express others de novo. For instance, microglial P2X4 purinoreceptor expression is increased both by lipopolysaccharide (LPS) treatment in vitro (17) and in spinal microglia following a peripheral nerve injury in vivo (5).
Among microglial receptors, P2X purinergic receptors have been extensively investigated due to their reported roles in microglial activation in various pathological conditions linked to injury and inflammation (5, 18–20). As distinct from P2Y receptors that are G-protein coupled, P2X receptors are cation-permeable ligand-gated ion channels that open in response to the binding of extracellular purines, such as ATP, which are thought to be released from damaged/degenerating neuronal tissue. In microglial cells, activation of these receptors results in entry of Ca2+, which subsequently causes various cellular responses associated with microglial function both in vitro and in vivo. For instance, several studies suggest a vital role for microglial P2X receptors (5, 21) in generating neuropathic pain following nerve injuries, although microglial P2Y receptors may also be involved (22, 23). Some reports have suggested involvement of P2X receptors in microglial cytokine release and P2Y receptors in microglial chemotaxis (24). As with other immune cells, P2X7 receptors have been implicated not only in microglial inflammatory responses and inflammasome activation (25) but also in microglial proliferation (26, 27). Genetic variants of P2X receptors are associated with neurodegenerative disease and appear to also play a role in microglial phagocytosis (28).
One of the most interesting consequences of microglial P2X receptor activation is the formation of membrane pores large enough to admit molecules, such as NMDG+ and DNA binding dyes. This process has been studied extensively for P2X7 receptors and may result in pro-inflammatory actions and cytolysis, but is also apparently required for the proliferative actions of P2X7 in microglia (26). A similar process was recently reported to occur following P2X4 receptor activation in microglial cells without consequent cell death (29). The formation of these pores may enable the release of factors that have paracrine and autocrine functions, such as glutamate (30), but will also pass Ca2+ and lead to a sustained increase in intracellular Ca2+, which in turn may underlie many of the downstream effects of P2X receptor activation on microglial function.
Role of Increased Intracellular Ca2+ in Microglial Function
Microglial responses, including morphological changes, migration, proliferation, and secretion of cytokines and reactive oxygen species, are usually associated with an increase in intracellular Ca2+ concentration ([Ca2+]i) (31, 32). The majority of receptor agonists known to cause microglial activation also cause an increase in [Ca2+]i, although often via different mechanisms and with markedly different consequences. For example, both ATP and LPS are capable of activating microglia in vitro and in vivo (33–37) and both cause an increase in [Ca2+]i. LPS does not produce an immediate Ca2+ response, but is reported to cause a sustained increase in basal [Ca2+]i in microglia after a 24-h treatment (38), although this increase is evident as early as 1 h after LPS application (Figure 1). An intracellular Ca2+ chelator prevented the LPS-stimulated increase in microglial NO, cytokines and chemokine release, but the Ca2+ ionophore ionomycin caused none of these effects. This suggests that elevated [Ca2+]i is necessary but not sufficient for the pro-inflammatory actions of LPS on microglia (38). By contrast, ionomycin can mimic the stimulation of c-fos expression in microglia via glutamate receptor-mediated calcium influx (13).
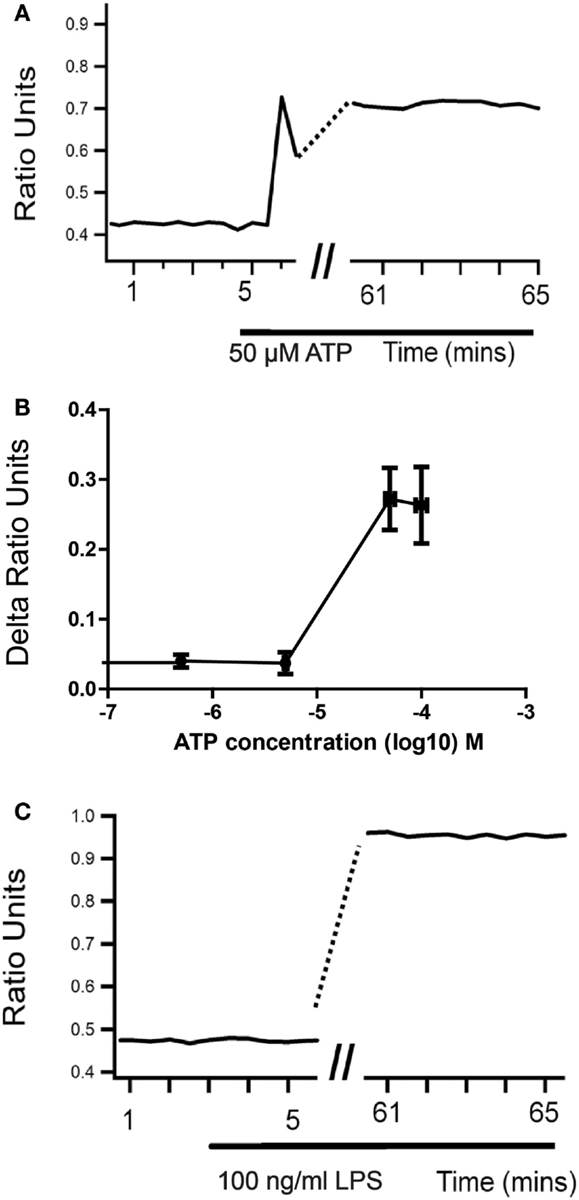
Figure 1. Sustained changes in intracellular Ca2+ concentration in microglia upon activation. When cultured neonatal rat microglia are stimulated with either ATP (A,B) or LPS (C), there is a sustained changed in intracellular Ca2+ levels as measured using Fura-2 ratiometric Ca2+ imaging that develops over 1 hour.
Unlike LPS, Ca2+ imaging experiments show that ATP induces an immediate, transient increase in [Ca2+]i in microglia that is biphasic in many cells. The amplitude of this response is dose dependent and maximal at 300 μM. However, when ATP is applied in the absence of extracellular Ca2+, microglia commonly show no response or a smaller monophasic increase in [Ca2+]i (39). In the presence of extracellular Ca2+ and thapsigargin to block the endoplasmic reticulum calcium pump, microglia either responded to ATP or displayed an increased basal calcium (39), which has been shown to prevent ATP responses (38, 39). These results support the involvement of ionotropic receptors in the response to ATP, although metabotropic receptors and Ca2+ induced Ca2+ release may also be involved. These responses to ATP are not blocked by 100 μM suramin and similar [Ca2+]i increases are seen in response to the selective P2X receptor agonist 2-methylthio ATP, but not αβ-methylene ATP (40) suggesting P2X4 or P2X7 but not P2X1 and P2X3 are involved (41, 42). Although the concentration dependence of the ATP-induced Ca2+ influx would suggest mediation by P2X4 receptors, the fact that similar responses are seen to the selective P2X7 receptor agonist 2′- and 3′-O-(4-benzoylbenzoyl) adenosine 5′-triphosphate (BzATP) indicates that functional P2X7 receptors are also present. Interestingly, pretreatment with oxidized ATP (oATP; 100 μM), which blocks P2X7 but not P2X4-mediated currents in microglia (43) prevented the increase in [Ca2+]i induced by ATP concentrations (10 and 100 μM) that activate P2X4 but not P2X7 receptors (40). One possible explanation for this might be autocrine release of ATP (44), although other actions of oATP cannot be discounted.
A later more sustained increase in [Ca2+]i similar to that reported following exposure to LPS can be seen in microglia following prolonged exposure to ATP at 30 μM or more (Figure 1). The time course and concentration dependence of this response are consistent with those of macropore formation in response to P2X4 activation measured via dye uptake (29). In contrast to LPS, however, application of 30–100 μM ATP does not cause release of cytokines, NO or activate a respiratory burst in microglia, again emphasizing that an increase in [Ca2+]i is not sufficient to stimulate these pro-inflammatory actions. Indeed, there is evidence that a prior sustained increase in [Ca2+]i can inhibit responses of microglia to further stimulation (38, 39). Changes in microglial morphology are seen when microglia are treated with 50–100 μM ATP (45); however, these changes are more subtle that those seen in response to LPS. The activation state induced in microglia by LPS is widely believed to be analogous to the M1 or pro-inflammatory activation state seen in macrophages. Whether or not the state induced by low micromolar concentrations of ATP in microglia is analogous to the M2, “alternate” or anti-inflammatory activation state of macrophages is less certain. It is clear, however, that P2X4 receptors on microglia and their activation by ATP can contribute to neuronal hyperexcitability and pathological changes within the nervous system (5, 40). Higher concentrations of ATP that would activate P2X7 receptors are reported to cause pro-inflammatory actions, such as generation of a respiratory burst (46), but this is not the case in all studies, and may depend on the conditions of the experiment.
Other agents known to increase [Ca2+]i in microglia include glutamate (15), complement (38), interferon gamma and BDNF (47). BDNF release from microglia has been implicated in causing the increased excitability seen in dorsal horn neurons following peripheral nerve injury (48, 49). BDNF can also act back on microglial TrkB receptors to induce a sustained elevation of [Ca2+]i via a complex mechanism, but BDNF also suppresses IFN-γ-induced calcium influx and the associated increase in iNOS expression and nitric oxide release (47, 50). These results again illustrate that an increase in [Ca2+]i by itself is not enough to activate the overt inflammatory actions of microglia and may even inhibit them. They also emphasize that overt inflammatory responses are not required for microglia to participate in pathological processes and nervous system disorders (Figure 2).
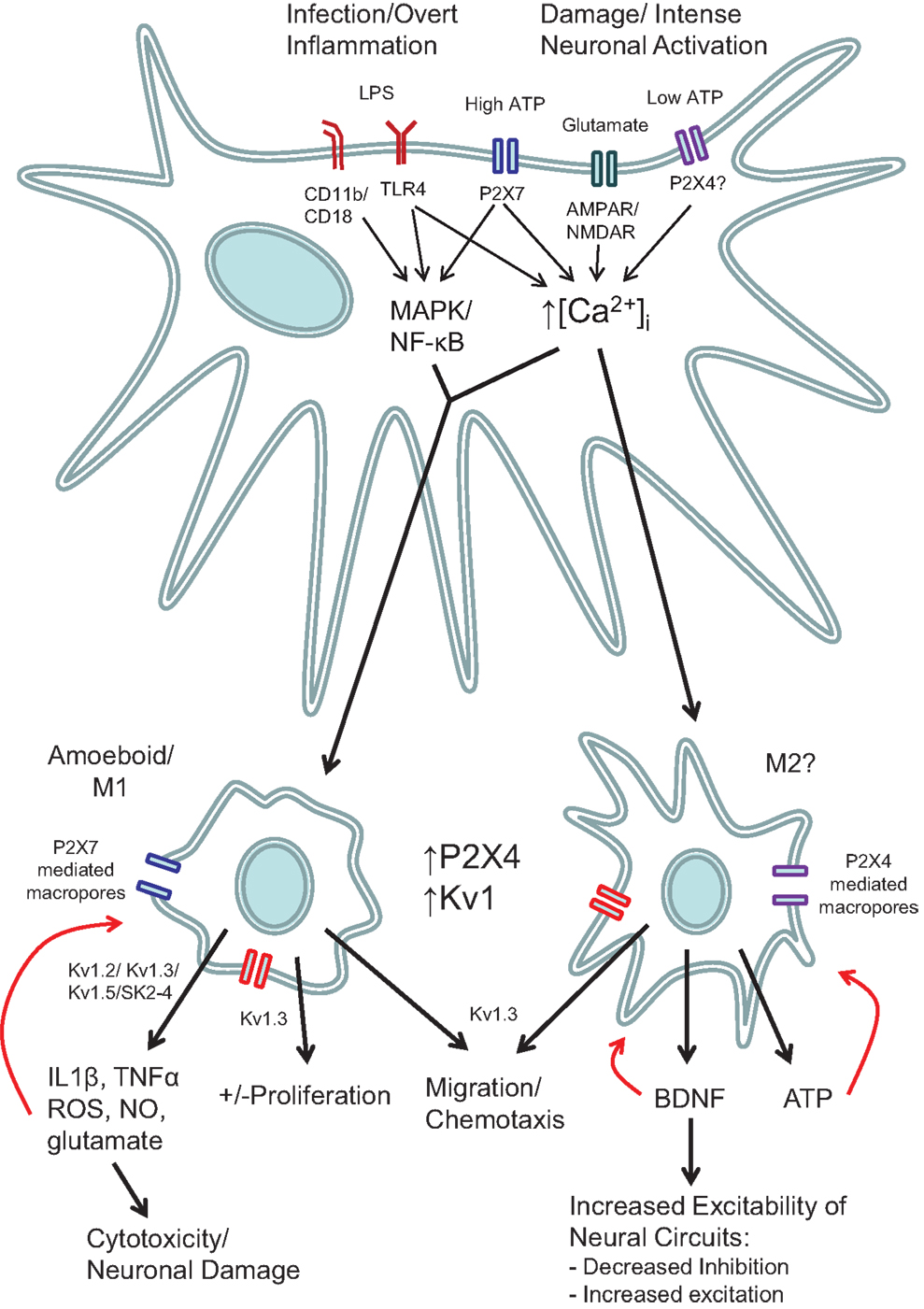
Figure 2. Schematic showing the various roles of ion channels in microglial function. Stimulation of microglia with either LPS or high concentrations of ATP (e.g., 3 mM) act via increased intracellular Ca2+ and intracellular signaling pathways such as MAP kinases and NF-κB. Low concentrations of ATP that stimulate P2X4 receptors (e.g., 100 μM), do not cause cytokine release or ROS production, but nevertheless do increase intracellular Ca2+ and produce morphological and functional changes that can lead to pathological changes in the function of surrounding neurons. Red arrows indicate potential autocrine and positive feedback mechanisms.
Role of Voltage-Gated and Store-Operated Ca2+ Channels in Microglial Function
As discussed earlier, rat, mouse, and human microglia express a variety of Ca2+ channels, including voltage-gated Ca2+ channels and Ca2+-release-activated Ca2+ channels (1, 51). A study by Tokuhara et al. demonstrated involvement of N-type calcium channels in monocyte chemoattractant protein-1 (MCP-1) production from microglia. MCP-1 released from glial cells acts as a chemokine and attracts a variety of cells, including monocytes, T lymphocytes, and dendritic cells, to the brain (52). Store-operated Ca2+ entry (SOCE) has been demonstrated as another mechanism that supplies Ca2+ for intracellular processes in microglia via Orai1/ICRAC channels (53). More recent evidence suggests that Ca2+ entry via SOCE plays an important role in responses to purines and BDNF and contributes to the activation of intracellular pathways causing cytokine secretion, phagocytosis, and chemotaxis (54, 55). In general, however, the role of these sources of Ca2+ in microglial signaling has not been investigated as thoroughly as have glutamate and purinergic receptors.
Microglial Expression of K+ Channels – Kv1.3 and 1.5
In addition to the role of ligand-gated ion channels and calcium influx, many studies have also demonstrated a critical role of K+ channels in various pro-inflammatory microglial functions. This is presumably because K+ channels prevent depolarization of the membrane and maintain the driving force for Ca2+ entry as is proposed to occur in peripheral immune cells (56). The variety of these channels, however, and their roles in different aspects of microglial function have proven complex to unravel.
Initial reports suggested that, in culture, unstimulated microglia express only inward rectifying K+ channels (57), but that purine-, LPS-, gamma interferon-, or granulocyte macrophage colony stimulating factor-stimulated microglia display an outward K+ conductance (57–60). For instance, Norenberg et al. demonstrated a protein synthesis-dependent expression of outward K+ conductance peaking at 3 h after LPS exposure, which then gradually decreased despite the continued presence of LPS (57, 58). By contrast, Khanna et al. reported the presence both Kv1.5 and Kv1.3 channel protein in unstimulated microglial cell lysates, but saw only Kv1.3 like-current as demonstrated by the complete blockade of the voltage-dependant current by agitoxin-2. Immunohistochemical analysis demonstrated that Kv1.3 was present on the microglial cell membrane, whereas Kv1.5 was found mainly intracellularly (2). Another in vitro study demonstrated a functional role for Kv1.5 in microglia (61). Unlike control/wildtype microglia, LPS-stimulated production of nitric oxide was not seen in microglia isolated from Kv1.5−/− knockout mice or microglia pre-treated with antisense oligonucleotide (AO) for Kv1.5, whereas LPS-stimulated chemokine release remained intact. By contrast, AO for Kv1.3 or pharmacological blockade of Kv1.3 did not inhibit LPS-stimulated NO production, suggesting that KV1.5, but not Kv1.3 is required for the microglial NO release function (61, 62). Pannasch et al. also reported that decreasing expression of either Kv1.5 or Kv1.3 channels can prevent the LPS-mediated decrease in microglial proliferation. Moreover, increased microglial proliferation was observed in Kv1.5−/− mice after facial nerve injury in vivo (61). Collectively, these studies suggest that inflammatory stimuli increase expression of microglial Kv 1.5 channels, which is required for increased NO production and inhibition of the cell cycle and but not chemokine release.
As mentioned above, Kv1.3 channels do appear to have a role in microglial proliferation (3), as well as cytokine release (63), morphological changes, and the NADPH oxidase-dependent respiratory burst response to PMA (2) as demonstrated by pharmacological blockade. Adhesion has also been suggested to regulate both microglia responses and expression of microglial K+ channels. Integrins are transmembrane glycoproteins responsible for regulation of adhesion and the ability of immune cells to migrate. Microglia express several different integrin, including α4, α5, α6, β1, lymphocyte function-associated antigen-1, and Mac 1 β2-integrin. Interestingly, the pharmacological inhibition of Kv1.3 channels or of β-integrin also inhibits microglial migration toward various chemo attractants (64), although ATP was not tested. These results clearly indicate that Kv1.3 channels are also involved in microglial migration.
Fordyce et al. also demonstrated that blockers of Kv1.3 channels, but not Kv1.2, Kv1.5, and Kv1.6, inhibited the respiratory bust and consequent killing of neurons by microglia in co-culture (62), suggesting that Kv1.3 blockers may be useful as neuroprotective agents. With the development of potential therapeutic agents targeting this channel, their protective properties have been confirmed in a rat model of radiation-induced brain injury (65). The potential importance of these findings is emphasized by the recent demonstration of high levels of Kv1.3 in microglia in human brains from Alzheimer’s disease patients (66).
Other K+ Channels with Roles in Microglial Cytokines, Nitric Oxide, and ROS Production
Some studies have also suggested a functional role for Kv1.1 and Kv1.2 shaker-like voltage-gated K+ channels in microglia. These channels are present in early postnatal microglia but disappear in ramified microglia present in adult rat brain. In vitro studies have reported induced expression (mRNA as well as protein) of Kv1.1 and Kv1.2 channels in microglia upon activation with LPS, hypoxia (67, 68), or ATP (68). Application of neutralizing antibodies to Kv1.1 is reported to reduce microglial cytokine (TNF-α, IL-1β) and nitric oxide production upon exposure to LPS or hypoxia (67). In contrast to Fordyce et al., Li et al. reported that an inhibitor of Kv1.2 channels reduced microglial cytokine mRNA production (TNF-α and IL1-β) and completely prevented ROS production in microglia and BV-2 cells exposed to LPS or hypoxia (68). Notably, none of the studies on Kv1.1 and Kv1.2 channels have reported complete inhibition of cytokine production suggesting the involvement of other pathways or ion channels.
Several studies have reported a functional role of Ca2+-activated K+ channels in microglial function. For instance, the respiratory burst, but not the morphological changes in response to PMA were shown to be dependent on the Ca2+/calmodulin-gated channels SK2, SK3, and SK4 (2). These investigators have now elaborated on this mechanism in a study published as part of this research topic (69).
Conclusion
In conclusion, microglial activation occurs via a cascade of extracellular and intracellular signaling events beginning with cell surface receptor and ligand-gated ion channel activation followed by short-term and long-term changes in intracellular Ca2+. These Ca2+ signals can cause release of other factors in autocrine paracrine feedback loops and are necessary, although not sufficient for various subsequent cellular responses in microglia. The roles of various K+ channels in enabling the different subsequent pro-inflammatory responses of microglia have been clearly demonstrated. Their role in more subtle microglial responses, in particular “alternate activation” and P2X-mediated events require more careful investigation. These more subtle events are potentially just as clinically relevant as the more overt inflammatory functions of microglia and so a deeper understanding of the role of ion channels in these responses will no doubt have major consequences for human health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Khanna R, Roy L, Zhu X, Schlichter LC. K+ channels and the microglial respiratory burst. Am J Physiol Cell Physiol (2001) 280:C796–806.
3. Kotecha SA, Schlichter LC. A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci (1999) 19:10680–93.
4. Black JA, Liu S, Waxman SG. Sodium channel activity modulates multiple functions in microglia. Glia (2009) 57:1072–81. doi: 10.1002/glia.20830
5. Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature (2003) 424:778–83. doi:10.1038/nature01786
6. Wu X, Zhu D, Jiang X, Okagaki P, Mearow K, Zhu G, et al. AMPA protects cultured neurons against glutamate excitotoxicity through a phosphatidylinositol 3-kinase-dependent activation in extracellular signal-regulated kinase to upregulate BDNF gene expression. J Neurochem (2004) 90:807–18. doi:10.1111/j.1471-4159.2004.02526.x
7. Wu Y, Willcockson HH, Maixner W, Light AR. Suramin inhibits spinal cord microglia activation and long-term hyperalgesia induced by formalin injection. J Pain (2004) 5:48–55. doi:10.1016/j.jpain.2003.09.006
8. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem (2006) 281:21362–8. doi:10.1074/jbc.M600504200
9. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature (2007) 446:1091–5. doi:10.1038/nature05704
10. Illes P, Nieber K, Frohlich R, Norenberg W. P2 purinoceptors and pyrimidinoceptors of catecholamine-producing cells and immunocytes. Ciba Found Symp (1996) 198:110–25; discussion 125–9.
11. Seo DR, Kim SY, Kim KY, Lee HG, Moon JH, Lee JS, et al. Cross talk between P2 purinergic receptors modulates extracellular ATP-mediated interleukin-10 production in rat microglial cells. Exp Mol Med (2008) 40:19–26. doi:10.3858/emm.2008.40.1.19
12. Morigiwa K, Fukuda Y, Yamashita M. [Neurotransmitter ATP and cytokine release]. Nippon Yakurigaku Zasshi (2000) 115:185–92. doi:10.1254/fpj.115.185
13. Eun SY, Hong YH, Kim EH, Jeon H, Suh YH, Lee JE, et al. Glutamate receptor-mediated regulation of c-fos expression in cultured microglia. Biochem Biophys Res Commun (2004) 325:320–7. doi:10.1016/j.bbrc.2004.10.035
14. Noda M, Nakanishi H, Nabekura J, Akaike N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci (2000) 20:251–8.
15. Liu GJ, Kalous A, Werry EL, Bennett MR. Purine release from spinal cord microglia after elevation of calcium by glutamate. Mol Pharmacol (2006) 70:851–9. doi:10.1124/mol.105.021436
16. Jantaratnotai N, Choi HB, McLarnon JG. ATP stimulates chemokine production via a store-operated calcium entry pathway in C6 glioma cells. BMC Cancer (2009) 9:442. doi:10.1186/1471-2407-9-442
17. Raouf R, Chabot-Dore AJ, Ase AR, Blais D, Seguela P. Differential regulation of microglial P2X4 and P2X7 ATP receptors following LPS-induced activation. Neuropharmacology (2007) 53:496–504. doi:10.1016/j.neuropharm.2007.06.010
18. Abbracchio MP, Verderio C. Pathophysiological roles of P2 receptors in glial cells. Novartis Found Symp (2006) 276:91–103; discussion 103–12, 275–81. doi:10.1002/9780470032244.ch8
19. Inoue K. Purinergic systems in microglia. Cell Mol Life Sci (2008) 65:3074–80. doi:10.1007/s00018-008-8210-3
20. Koles L, Furst S, Illes P. P2X and P2Y receptors as possible targets of therapeutic manipulations in CNS illnesses. Drug News Perspect (2005) 18:85–101. doi:10.1358/dnp.2005.18.2.886479
21. Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Collo G, Buell G, et al. ATP-mediated cytotoxicity in microglial cells. Neuropharmacology (1997) 36:1295–301. doi:10.1016/S0028-3908(97)00137-8
22. Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, et al. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci (2001) 21:1975–82.
23. Inoue K, Koizumi S, Kataoka A, Tozaki-Saitoh H, Tsuda M. P2Y(6)-evoked microglial phagocytosis. Int Rev Neurobiol (2009) 85:159–63. doi:10.1016/S0074-7742(09)85012-5
24. James G, Butt AM. P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur J Pharmacol (2002) 447:247–60. doi:10.1016/S0014-2999(02)01756-9
25. Shieh CH, Heinrich A, Serchov T, van Calker D, Biber K. P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-alpha in cultured mouse microglia. Glia (2014) 62:592–607. doi:10.1002/glia.22628
26. Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci (2009) 29:3781–91. doi:10.1523/JNEUROSCI.5512-08.2009
27. Zou J, Vetreno RP, Crews FT. ATP-P2X7 receptor signaling controls basal and TNFalpha-stimulated glial cell proliferation. Glia (2012) 60:661–73. doi:10.1002/glia.22302
28. Gu BJ, Baird PN, Vessey KA, Skarratt KK, Fletcher EL, Fuller SJ, et al. A rare functional haplotype of the P2RX4 and P2RX7 genes leads to loss of innate phagocytosis and confers increased risk of age-related macular degeneration. FASEB J (2013) 27:1479–87. doi:10.1096/fj.12-215368
29. Bernier LP, Ase AR, Boue-Grabot E, Seguela P. P2X4 receptor channels form large noncytolytic pores in resting and activated microglia. Glia (2012) 60:728–37. doi:10.1002/glia.22301
30. Marcoli M, Cervetto C, Paluzzi P, Guarnieri S, Alloisio S, Thellung S, et al. P2X7 pre-synaptic receptors in adult rat cerebrocortical nerve terminals: a role in ATP-induced glutamate release. J Neurochem (2008) 105:2330–42. doi:10.1111/j.1471-4159.2008.05322.x
31. Eder C. Regulation of microglial behavior by ion channel activity. J Neurosci Res (2005) 81:314–21. doi:10.1002/jnr.20476
32. Farber K, Kettenmann H. Functional role of calcium signals for microglial function. Glia (2006) 54:656–65. doi:10.1002/glia.20412
33. Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci USA (2005) 102:9936–41. doi:10.1073/pnas.0502552102
34. Zheng LT, Ock J, Kwon BM, Suk K. Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol (2008) 8:484–94. doi:10.1016/j.intimp.2007.12.012
35. Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem (2000) 75:965–72. doi:10.1046/j.1471-4159.2000.0750965.x
36. Buttini M, Limonta S, Boddeke HW. Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem Int (1996) 29:25–35. doi:10.1016/0197-0186(95)00141-7
37. Montero-Menei CN, Sindji L, Garcion E, Mege M, Couez D, Gamelin E, et al. Early events of the inflammatory reaction induced in rat brain by lipopolysaccharide intracerebral injection: relative contribution of peripheral monocytes and activated microglia. Brain Res (1996) 724:55–66. doi:10.1016/0006-8993(96)00268-5
38. Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK, Kettenmann H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J Neurosci (2003) 23:4410–9.
39. Moller T, Kann O, Verkhratsky A, Kettenmann H. Activation of mouse microglial cells affects P2 receptor signaling. Brain Res (2000) 853:49–59. doi:10.1016/S0006-8993(99)02244-1
40. Inoue K. The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol Ther (2006) 109:210–26. doi:10.1016/j.pharmthera.2005.07.001
41. Volonte C, Amadio S, D’Ambrosi N, Colpi M, Burnstock G. P2 receptor web: complexity and fine-tuning. Pharmacol Ther (2006) 112:264–80. doi:10.1016/j.pharmthera.2005.04.012
42. North RA, Surprenant A. Pharmacology of cloned P2X receptors. Annu Rev Pharmacol Toxicol (2000) 40:563–80. doi:10.1146/annurev.pharmtox.40.1.563
43. Visentin S, Renzi M, Frank C, Greco A, Levi G. Two different ionotropic receptors are activated by ATP in rat microglia. J Physiol (1999) 519 Pt 3:723–36. doi:10.1111/j.1469-7793.1999.0723n.x
44. Dou Y, Wu HJ, Li HQ, Qin S, Wang YE, Li J, et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res (2012) 22:1022–33. doi:10.1038/cr.2012.10
45. McIlvain HB, Ma L, Ludwig B, Manners MT, Martone RL, Dunlop J, et al. Purinergic receptor-mediated morphological changes in microglia are transient and independent from inflammatory cytokine release. Eur J Pharmacol (2010) 643:202–10. doi:10.1016/j.ejphar.2010.06.046
46. Mead EL, Mosley A, Eaton S, Dobson L, Heales SJ, Pocock JM. Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. J Neurochem (2012) 121:287–301. doi:10.1111/j.1471-4159.2012.07659.x
47. Mizoguchi Y, Monji A, Kato T, Seki Y, Gotoh L, Horikawa H, et al. Brain-derived neurotrophic factor induces sustained elevation of intracellular Ca2+ in rodent microglia. J Immunol (2009) 183:7778–86. doi:10.4049/jimmunol.0901326
48. Biggs JE, Lu VB, Stebbing MJ, Balasubramanyan S, Smith PA. Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Mol Pain (2010) 6:44. doi:10.1186/1744-8069-6-44
49. Lu VB, Biggs JE, Stebbing MJ, Balasubramanyan S, Todd KG, Lai AY, et al. Brain-derived neurotrophic factor drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. J Physiol (2009) 587:1013–32. doi:10.1113/jphysiol.2008.166306
50. Nakajima K, Kikuchi Y, Ikoma E, Honda S, Ishikawa M, Liu Y, et al. Neurotrophins regulate the function of cultured microglia. Glia (1998) 24:272–89. doi:10.1002/(SICI)1098-1136(199811)24:3<272::AID-GLIA2>3.0.CO;2-4
51. Tokuhara N, Namiki K, Uesugi M, Miyamoto C, Ohgoh M, Ido K, et al. N-type calcium channel in the pathogenesis of experimental autoimmune encephalomyelitis. J Biol Chem (2010) 285:33294–306. doi:10.1074/jbc.M109.089805
52. Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol (1998) 84:238–49. doi:10.1016/S0165-5728(97)00208-7
53. Ohana L, Newell EW, Stanley EF, Schlichter LC. The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels (Austin) (2009) 3:129–39. doi:10.4161/chan.3.2.8609
54. Heo DK, Lim HM, Nam JH, Lee MG, Kim JY. Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell Signal (2015) 27:177–86. doi:10.1016/j.cellsig.2014.11.003
55. Michaelis M, Nieswandt B, Stegner D, Eilers J, Kraft R. STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia (2015) 63:652–63. doi:10.1002/glia.22775
56. Panyi G. Biophysical and pharmacological aspects of K+ channels in T lymphocytes. Eur Biophys J (2005) 34:515–29.
57. Norenberg W, Gebicke-Haerter PJ, Illes P. Inflammatory stimuli induce a new K+ outward current in cultured rat microglia. Neurosci Lett (1992) 147:171–4. doi:10.1016/0304-3940(92)90587-W
58. Norenberg W, Appel K, Bauer J, Gebicke-Haerter PJ, Illes P. Expression of an outwardly rectifying K+ channel in rat microglia cultivated on teflon. Neurosci Lett (1993) 160:69–72. doi:10.1016/0304-3940(93)9001-0
59. Langosch JM, Gebicke-Haerter PJ, Norenberg W, Illes P. Characterization and transduction mechanisms of purinoceptors in activated rat microglia. Br J Pharmacol (1994) 113:29–34. doi:10.1111/j.1476-5381.1994.tb16169.x
60. Fischer HG, Eder C, Hadding U, Heinemann U. Cytokine-dependent K+ channel profile of microglia at immunologically defined functional states. Neuroscience (1995) 64:183–91. doi:10.1016/0306-4522(94)00398-O
61. Pannasch U, Farber K, Nolte C, Blonski M, Yan Chiu S, Messing A, et al. The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol Cell Neurosci (2006) 33:401–11. doi:10.1016/j.mcn.2006.08.009
62. Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci (2005) 25:7139–49. doi:10.1523/JNEUROSCI.1251-05.2005
63. Charolidi N, Schilling T, Eder C. Microglial Kv1.3 channels and P2Y12 receptors differentially regulate cytokine and chemokine release from brain slices of young adult and aged mice. PLoS One (2015) 10:e0128463. doi:10.1371/journal.pone.0128463
64. Nutile-McMenemy N, Elfenbein A, Deleo JA. Minocycline decreases in vitro microglial motility, beta1-integrin, and Kv1.3 channel expression. J Neurochem (2007) 103:2035–46. doi:10.1111/j.1471-4159.2007.04889.x
65. Peng Y, Lu K, Li Z, Zhao Y, Wang Y, Hu B, et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro Oncol (2014) 16:528–39. doi:10.1093/neuonc/not221
66. Rangaraju S, Gearing M, Jin LW, Levey A. Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer’s disease. J Alzheimers Dis (2015) 44:797–808. doi:10.3233/JAD-141704
67. Wu CY, Kaur C, Sivakumar V, Lu J, Ling EA. Kv1.1 expression in microglia regulates production and release of proinflammatory cytokines, endothelins and nitric oxide. Neuroscience (2009) 158:1500–8. doi:10.1016/j.neuroscience.2008.11.043
68. Li F, Lu J, Wu CY, Kaur C, Sivakumar V, Sun J, et al. Expression of Kv1.2 in microglia and its putative roles in modulating production of proinflammatory cytokines and reactive oxygen species. J Neurochem (2008) 106:2093–105. doi:10.1111/j.1471-4159.2008.05559.x
Keywords: microglia, P2X receptors, ion channels, K+ channels, neuroinflammation
Citation: Stebbing MJ, Cottee JM and Rana I (2015) The role of ion channels in microglial activation and proliferation – a complex interplay between ligand-gated ion channels, K+ channels, and intracellular Ca2+. Front. Immunol. 6:497. doi: 10.3389/fimmu.2015.00497
Received: 14 June 2015; Accepted: 14 September 2015;
Published: 22 October 2015
Edited by:
Amanda MacKenzie, University of Bath, UKReviewed by:
Fulvio D’Acquisto, Queen Mary University of London, UKPablo Pelegrin, Fundación para la Formación e Investigación Sanitarias de la Región de Murcia, Spain
Copyright: © 2015 Stebbing, Cottee and Rana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin James Stebbing, Health Innovations Research Institute and School of Medical Sciences, RMIT University, Bundoora West Campus, P.O. Box 71, Bundoora, VIC 3083, Australia,bWFydGluLnN0ZWJiaW5nQHJtaXQuZWR1LmF1