 Kelly Kersten
Kelly Kersten Camilla Salvagno
Camilla Salvagno Karin E. de Visser
Karin E. de Visser- Division of Immunology, Netherlands Cancer Institute, Amsterdam, Netherlands
Cancer immunotherapy is gaining momentum in the clinic. The current challenge is to understand why a proportion of cancer patients do not respond to cancer immunotherapy, and how this can be translated into the rational design of combinatorial cancer immunotherapy strategies aimed at maximizing success of immunotherapy. Here, we discuss how tumors orchestrate an immunosuppressive microenvironment, which contributes to their escape from immune attack. Relieving the immunosuppressive networks in cancer patients is an attractive strategy to extend the clinical success of cancer immunotherapy. Since the clinical availability of drugs specifically targeting immunosuppressive cells or mediators is still limited, an alternative strategy is to use conventional chemotherapy drugs with immunomodulatory properties to improve cancer immunotherapy. We summarize the preclinical and clinical studies that illustrate how the anti-tumor T cell response can be enhanced by chemotherapy-induced relief of immunosuppressive networks. Treatment strategies aimed at combining chemotherapy-induced relief of immunosuppression and T cell-boosting checkpoint inhibitors provide an attractive and clinically feasible approach to overcome intrinsic and acquired resistance to cancer immunotherapy, and to extend the clinical success of cancer immunotherapy.
Introduction
Cancer immunotherapy – harnessing the patient’s immune system against cancer – is currently gaining momentum in the clinic. Clinical trials with immune checkpoint inhibitors show remarkable success in patients with advanced metastatic melanoma, non-small cell lung cancer, renal cancer, bladder cancer, and Hodgkin’s lymphoma (1–6). As a result, the journal Science proclaimed cancer immunotherapy as the breakthrough of 2013 (7). Furthermore, these encouraging results led to FDA approval of the immune checkpoint inhibitors ipilimumab (anti-CTLA-4), nivolumab, and pembrolizumab (anti-PD-1) in the past few years. Although cancer immunotherapy was proclaimed a breakthrough, a significant proportion of cancer patients do not show clinical benefit. There are various cancer cell-intrinsic and cancer cell-extrinsic processes that regulate intrinsic or acquired resistance to cancer immunotherapy. Cancer cell-intrinsic characteristics like the mutational load have been reported to affect responsiveness to immunotherapy (8, 9). In terms of cancer cell-extrinsic processes, tumors exploit different strategies to induce immune escape by hampering the recruitment and activation of effector T cells, and by creating a local immunosuppressive environment through recruitment of suppressive myeloid and regulatory T cells that dampen T cell effector functions. Which of these immune escape mechanisms are active in a certain tumor depends on the tumor type, tumor stage, and therapy history. A deeper understanding of the molecular mechanisms underlying these processes will contribute to the identification of biomarkers that can predict therapeutic efficacy of immunotherapy and to the design of combinatorial strategies aimed at maximizing the success of immunotherapy.
In this review, we discuss how tumor-induced immunosuppressive networks counteract efficacious anti-tumor immune responses, and how disruption of these networks can increase the anti-cancer efficacy of cancer immunotherapy with immune checkpoint inhibitors. Development and clinical testing of novel drugs specifically targeting immunosuppressive networks are ongoing and preliminary results are promising (10). An alternative strategy to relieve tumor-induced immunosuppressive states is to use conventional, and more easily accessible, anti-cancer treatment strategies with known immunomodulatory properties, such as chemotherapy, radiotherapy, and targeted therapy (11–15). Here, we focus on the immunomodulatory properties of conventional chemotherapy, and how these properties can be exploited to improve the anti-cancer efficacy of immune checkpoint inhibitors.
Cancer Immunotherapy: Opportunities and Challenges
Tumor-Induced Mechanisms of Immune Escape
Cancers do not merely consist of tumor cells, but comprise a variety of cell types that together form the tumor microenvironment (TME) (Figures 1 and 2). Infiltrating immune cells are of special interest because of their paradoxical role in cancer progression. While some immune cell populations have pro-tumorigenic properties, others counteract tumorigenesis (16–18). Many tumors are characterized by an immunosuppressive TME, which makes it unfavorable for anti-tumor immunity. To mount effective anti-tumor immunity, tumor-associated antigens need to be sampled and processed by antigen-presenting cells (APCs). After receiving specific maturation signals, these APCs migrate to tumor-draining lymphoid organs where antigens are presented to T cells. Upon activation and proliferation, tumor antigen-specific T cells migrate to the tumor bed where they exert their cytotoxic function. At every step of this T cell priming and effector process, tumors employ strategies to hamper anti-cancer immunity.
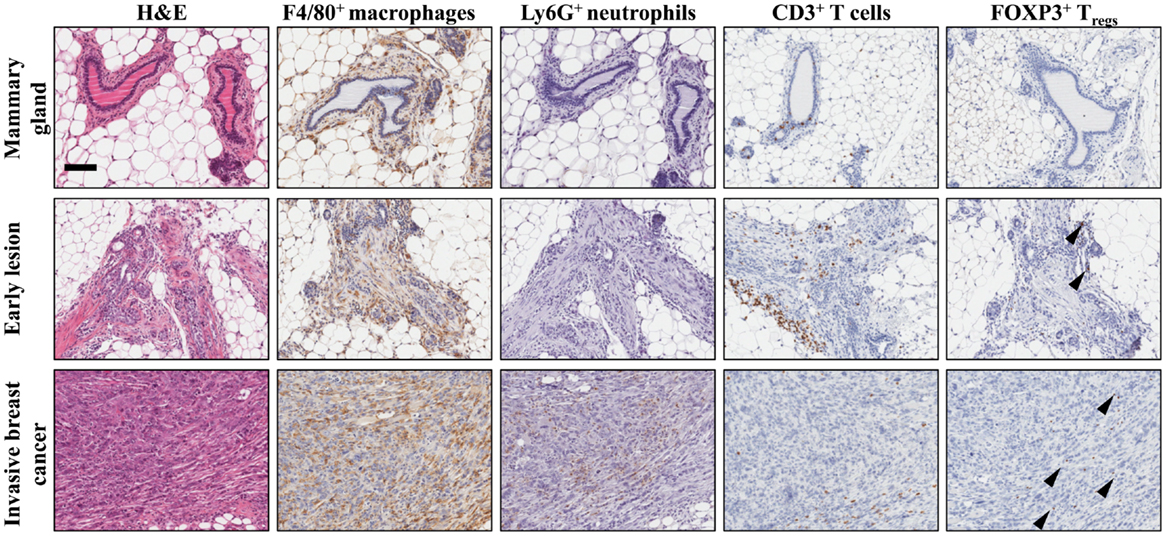
Figure 1. Establishment of the immune microenvironment during breast cancer progression in a conditional mouse model for mammary tumorigenesis. Female K14Cre;Cdh1F/F;Trp53F/F mice develop de novo invasive mammary tumors that closely resemble human invasive lobular carcinoma (19). Immunohistochemical staining on mammary tissue from K14Cre;Cdh1F/F;Trp53F/F mice obtained during different stages of mammary tumor progression. From top to bottom are represented wild-type mammary gland (top), early lesion (middle), established mammary tumor (bottom). From left to right, identification of different immune cell populations by H&E, F4/80 (macrophages), Ly6G (neutrophils), CD3 (total T cells), and FOXP3 (regulatory T cells) staining showing the dynamics of the tumor microenvironment. Arrowheads indicate FOXP3+ nuclei. Scale bar 100 μm.
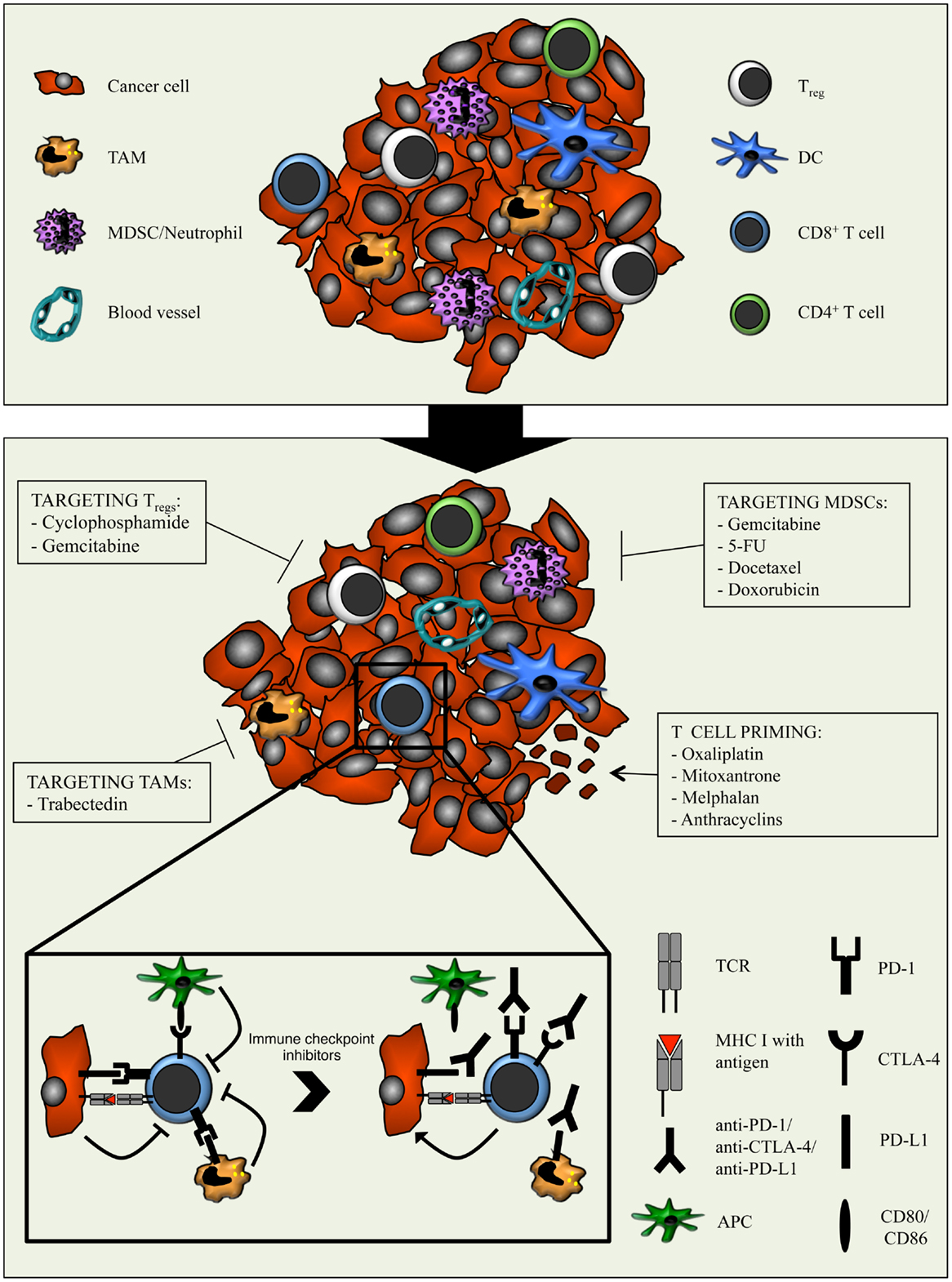
Figure 2. Combination strategies aimed at relieving the immunosuppressive tumor microenvironment with chemotherapy and potentiating cytotoxic T cells with immune checkpoint inhibitors. The tumor microenvironment is characterized by the presence of various immune cell types, including different subsets of adaptive immune cells and TAMs, MDSCs, and Tregs. The latter dampens the anti-cancer activity of T cells through several mechanisms. Moreover, cancer cells and myeloid cells express PD-L1/PD-L2 and APCs express CD80/CD86. Binding of these molecules to PD-1 and CTLA-4 respectively, expressed on T cells, results in inhibitory signals that counteract T cell activation and function. The immunomodulatory properties of different types of chemotherapeutic drugs can be exploited to enhance anti-tumor immunity. By optimally matching the immunomodulatory features of specific chemotherapeutic drugs with the T cell-boosting effect of immune checkpoint inhibitors, the efficacy of immunotherapy might be improved.
Tumors often show dysfunctional recruitment and activation of dendritic cells (DCs), which are the most potent APCs for initiating immune responses. Several studies show that tumor-infiltrating DCs display an immature phenotype (20, 21). Tumor-derived factors like IL10, IL6, CSF1, and VEGF interfere with DC maturation, causing failure to migrate to the tumor-draining lymphoid organs, and to provide the appropriate co-stimulatory signals required to stimulate T cells (21). Although a thorough analysis of the antigen-presenting myeloid immune cell compartment in the MMTV-PyMT mammary tumor model showed that intratumoral DCs are able to ingest and present tumor antigens to T cells, they fail to activate them (22). Nevertheless, even in these immunoevasive tumors, a rare population of IL12-expressing CD103+ DCs exists that is able to prime tumor antigen-specific T cells (23). Besides hampered T cell priming, the recruitment of activated T cells and their access into the tumor bed is often disrupted by the disorganized tumor vasculature and impaired expression of adhesion molecules on endothelial cells (24, 25). Some studies suggest that tumor-derived chemokines may cause selective trapping of T cells in the tumor stroma preventing access into the tumor bed (26). When tumor-specific T cells do succeed to reach the tumor, downregulation of MHC class I expression on tumor cells renders them invisible to T cell attack (27). Additionally, T cells face systemic and local tumor-induced immunosuppression, which limits their activation and function (28). Tumor-associated immunosuppression can be caused by tumor-infiltrating or systemically expanded myeloid cells or regulatory T cells (Tregs) that – directly or indirectly via secretion of soluble mediators – hamper T cell priming and effector function or even induce T cell death (28). These mechanisms will be discussed in more detail later.
Enhancing Anti-Tumor Immunity by Immune Checkpoint Inhibitors
To improve anti-tumor T cell immunity, different types of cancer immunotherapy approaches exist. While passive immunotherapy is based on adoptive transfer of (genetically engineered) autologous T cells, active immunotherapy boosts the endogenous immune response via cancer vaccines or inhibitors of immune checkpoints. The therapeutic effect of the latter is aimed at inhibition of negative immune regulatory pathways including cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) and the programed cell death protein-1 (PD-1) receptor and one of its ligands, PD-L1 (B7-H1; CD274) (29). CTLA-4 is a member of the CD28 immunoglobulin superfamily and is expressed mainly on the surface of activated CD4+ T cells and Tregs, while absent on naïve T cells (30). CTLA-4 plays a central role in maintaining immune tolerance by competing with CD28 to bind the ligands CD80 and CD86 present on activated APCs to inhibit T cell co-stimulation. The PD-1/PD-L1 axis shows similarities to that of CTLA-4. PD-1 is mainly expressed on activated T cells upon T cell receptor (TCR) engagement and on Tregs, while naïve and memory T cells do not usually express this surface marker. Recent studies suggest that PD-1, rather than being a marker of activated T cells, identifies exhausted T cells (31). PD-L1 is expressed on multiple cell types, whereas expression of PD-L2 (B7-DC; CD273) seems to be restricted to APCs (32, 33). Like CTLA-4, binding of PD-L1/PD-L2 to its receptor results in an inhibitory signal that prevents T cell activation. While CTLA-4 blockade is hypothesized to act mainly in secondary lymphoid organs during the T cell priming phase, it is believed that blockade of PD-1 or PD-L1 targets the TME during the T cell effector phase (34). However, PD-1 can also play a role in the early T cell response as a regulator of CD8+ T cell expansion upon antigen recognition (35). In addition to its role in T cell priming, CTLA-4 also regulates the suppressive function of tumor-infiltrating Tregs (36, 37). In line with this, blockade of CTLA-4 in the B16 melanoma model acts locally in the TME by inactivating Tregs in an Fc-dependent manner resulting in a favorable shift in the effector T cell/Treg ratio (38). The exact mechanisms of action of anti-CTLA-4 and anti-PD-1/PD-L1 are not completely clear. Just recently, the combination of anti-CTLA-4 and anti-PD-1 was reported to significantly increase the fraction of melanoma patients responding to immunotherapy compared to anti-CTLA-4 monotherapy-treated patients (39), emphasizing the different modes of action of CTLA-4 and PD-1.
The rational of using CTLA-4 blockade in cancer therapy is to release the brake on pre-existing tumor-reactive T cells and to generate new T cell responses. Ipilimumab (anti-CTLA-4) was the first immune checkpoint inhibitor that yielded a significant increase in survival of patients with metastatic melanoma, for which all conventional therapeutic options had failed (1). Interestingly, a broadening of the tumor-reactive T cell repertoire was reported upon ipilimumab treatment (40). In a second clinical study, ipilimumab was combined with dacarbazine in metastatic melanoma patients resulting in prolonged survival compared to dacarbazine alone (41). In both studies, a fraction of patients showed long-term durable responses (42). Similarly, clinical trials with anti-PD-1 have shown tumor regression in a substantial fraction of cancer patients (3). These initial results lead to an immense increase in clinical trials with drugs targeting the PD-1/PD-L1 axis in different cancer types, and many report anti-tumor efficacy (3–6, 43). Recent clinical observations show that the combination of anti-CTLA-4 and anti-PD-1 is more effective than either monotherapy (39). Although very successful and promising, a significant proportion of cancer patients do not show long-term benefit of immune checkpoint inhibitors. Therefore, it is of utmost importance to mechanistically understand intrinsic and acquired resistance to cancer immune checkpoint inhibitors, in order to identify biomarkers that can be used to pre-select those patients that will or will not benefit from cancer immunotherapy and to develop therapeutic strategies to overcome or bypass resistance mechanisms.
What are the Requirements for Therapeutic Response to Checkpoint Inhibitors?
To predict the response to immunotherapy per patient and tumor type, several variables should be taken into account. For successful activation of a T cell-mediated anti-tumor immune response, T cells need to “see” the cancer cells with their TCR. In general, there are three classes of tumor antigens that can potentially be recognized by T cells: viral antigens, self-antigens, and neo-antigens. Our T cell repertoire is basically built to recognize and respond to viral antigens because these antigens are perceived as foreign or non-self. However, only a subset of established human cancers expresses viral antigens. During the T cell maturation process, thymic selection eliminates maturing lymphocytes that display a high avidity for self-antigens. As a consequence, only low-avidity self-specific T cells can be found in the peripheral T cell repertoire, which may not be ideal for cancer immunotherapy. Non-synonymous somatic mutations can give rise to neo-antigens toward which no central T cell tolerance is present. Recently, neo-antigen-specific T cell responses have been reported in melanoma patients (44–46), indicating that these mutations can be recognized by T cells and induce tumor-specific T cell responses. In line with this, the number of predicted neo-antigens is linked with a metric for immune cytolytic activity based on gene expression in a large panel of cancer types (47). Thus, the extent of the mutational load of a certain tumor would serve – albeit at a low resolution – as a predictor of response to cancer immunotherapy. Indeed, a growing body of data supports this hypothesis (48). Whole-exome sequencing analyses revealed that melanoma and lung cancer – the two cancer types that show promising responses to immunotherapy – bear relatively high mutational loads compared to other types of cancer due to their exposure to DNA damaging insults like UV radiation and tobacco smoke, respectively (49). Recent studies uncovered that a high mutational load is associated with long-term clinical benefit to checkpoint inhibitors (8, 9). However, not all cancer patients with tumors bearing a high mutational load respond to checkpoint inhibitors, and some patients bearing tumors with low mutational load do (8, 9). Together, these results suggest that the mutational load of tumors is correlated with response to immune checkpoint inhibitors, but it cannot solely be used to predict response.
A growing body of clinical observations suggests that the intratumoral presence of pre-existing T cells is required for clinical benefit of immunotherapy (50). PD-1 expression on tumor-infiltrating CD8+ T cells has been suggested to identify the repertoire of clonally expanded tumor-reactive T cells (51). In addition, T cell infiltration correlates with PD-L1 expression in tumors and is associated with increased responsiveness to drugs targeting the PD-1/PD-L1 axis in melanoma patients (50, 52, 53). Expression of PD-L1 in tumors is one of the main characteristics pursued as a potential biomarker for response to PD-1/PD-L1 blockade. However, there are examples of tumors with high expression of PD-L1 that do not respond to PD-1 blockade, and PD-L1 negative tumors that do respond (53). Why certain tumors express PD-L1 and others do not remains to be elucidated.
Interestingly, expression of PD-L1 and responsiveness to immune checkpoint blockade is associated with genomic instability in different tumor types (54). Patients bearing mismatch-repair-deficient colorectal cancer (CRC) respond better to anti-PD-1 therapy than mismatch-repair-proficient CRC patients (54). In line with this, a microsatellite instable (MSI) subset of CRC patients shows high T cell influx (55). However, this is counterbalanced by simultaneous upregulation of checkpoint molecules including PD-1, PD-L1, and CTLA-4 leaving T cells dysfunctional (55). Moreover, in breast cancer, the expression of PD-L1 is correlated with TIL infiltration, and is mostly prevalent in basal-like, hormone-receptor-negative, and triple-negative tumors (56, 57). Furthermore, in glioma patients increased expression of PD-L1 in tumors was correlated with PTEN loss (58), suggesting that patients bearing genetically unstable cancer types might benefit from treatment with checkpoint inhibitors. Intriguingly, not only cancer cells, but also tumor-infiltrating myeloid cells express PD-L1, and counteract anti-tumor immunity in ovarian carcinoma and MSI-CRC (55, 59). Actually, PD-L1 expression on tumor-infiltrating immune cells has been suggested to be a better predictor of clinical response to anti-PD-L1 therapy than PD-L1 expression on cancer cells (52). It will be interesting to explore, which other cancer types are characterized by the influx of PD-L1-expressing myeloid cells.
In conclusion, to increase the efficacy of immunotherapy in different types of cancer, we could consider manipulating the many variables that determine intrinsic and acquired resistance. While altering cancer cell-intrinsic characteristics, such as mutational load or genomic instability, might be challenging, cancer cell-extrinsic characteristics, like an immunosuppressive TME, are easier to manipulate.
Evasion from Cancer Immunotherapy: Relieving Immunosuppression as an Attractive Strategy to Improve the Efficacy of Immune Checkpoint Blockade
Established tumors are characterized by an abundant influx of a variety of immune cells with immunosuppressive activity, including Tregs, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) (Figures 1 and 2). There is accumulating evidence that interference with these immunosuppressive networks can improve anti-tumor immunity. Here, we discuss the different types of immunosuppressive immune cells present in the TME, and how blockade or reprograming of these cells or their downstream effects can enhance anti-tumor immunity and the efficacy of immune checkpoint blockade.
Regulatory T Cells
Regulatory T cells play an important role in maintaining homeostasis during infections and in preventing the development of autoimmune diseases by blocking proliferation and cytotoxic activity of effector T cells. The history of Tregs goes back to the 1970s, when it was discovered that a subpopulation of thymocytes induced tolerance to certain antigens in mice (60). A turning point in the research of these “suppressor cells” came in 1995. Tregs, phenotyped as CD4+CD25+ cells, were shown to be important for self-tolerance in mice, as inoculation of CD4+ cells depleted of CD4+CD25+ cells resulted in autoimmunity in nude mice (61). Another big step forward in the characterization of Tregs was the identification of FOXP3, a member of the fork-head/winged-helix family of transcription factors and a key regulator of Treg development and function (62). In the following years, the knowledge of Tregs expanded enormously. Two subpopulations of Tregs were identified: natural Tregs and induced Tregs (or adaptive Tregs), which are formed in the thymus and in the periphery, respectively. Regardless of their origin, both natural and induced Tregs inhibit effector T cells (63).
In 1980, it was hypothesized that a T cell population in tumors suppresses anti-tumor immune responses (64). Indeed, many experimental studies support the notion that tumor-associated Tregs contribute to immune escape via suppression of anti-tumor CD8+ T cells. For example, elimination of Tregs in MO4 melanoma cell line-bearing mice results in T cell-dependent tumor rejection (65). Moreover, in a xenotransplant model for HER2+ ovarian cancer, adoptive transfer of autologous CD3+CD25− T cells and DCs loaded with HER2+ antigen results in T cell-mediated tumor regression, whereas concomitant transfer of Tregs blocks this antigen-specific immune response (66). Tregs not only suppress CD8+ T cells, but also CD4+ T cells, NK, NKT, and B cells (67). Tregs exert their immunosuppressive function either by direct suppression of effector cells, or indirectly by affecting the activation state of APCs. Importantly, in order to exert their functions, Tregs need to be activated via their TCR, but once activated their suppressive function is non-specific (68, 69). The direct T cell-suppressive functions are mediated by release of cytokines, serine proteases and the expression of enzymes that catabolize ATP. For example, Tregs inhibit T cells via secretion of cytokines like TGFβ, IL10, and IL35 (70–72) or even induce T cell apoptosis by the release of granzyme B (GRZMB) or perforin (73–75). In addition, Tregs express CD39 and CD73, two ectoenzymes that generate the immunosuppressive molecule adenosine from extracellular ATP (76). It has been shown that Tregs from CD39 knock-out mice fail to inhibit CD4+CD25− cell proliferation (76). Finally, CTLA-4+ Tregs can indirectly impair T cells by reducing the CD80/CD86 levels on APCs (36).
Supporting these data, increased numbers of intratumoral Tregs correlate with worse overall survival in patients with ovarian cancer, breast cancer, gastric cancer, and hepatocellular carcinoma (66, 77–81). Interestingly, this is not true for CRC in which a high number of CD8+ cells and FOXP3+ cells correlates with a good prognosis (82). This may be explained by the fact that Tregs in CRC attenuate inflammation against gut microbiota that would otherwise enhance tumor growth (82). These findings illustrate that the tumor context dictates the function of associated immune cells. Although strategies targeting CD25 (like the neutralizing monoclonal antibody daclizumab and the recombinant interleukin 2/diphtheria toxin conjugate Ontak) showed transient depletion of peripheral Tregs and increased activity of CD8+ T cells, these approaches only result in a modest clinical benefit in cancer patients (83, 84). This might be explained by the fact that CD25 is also expressed on active effector T cells, so the lack of specificity for Tregs might complicate their clinical applicability. Therefore, a mechanistic understanding of the role of Tregs in different tumor contexts will be important for the design of therapeutic strategies aimed at suppressing the downstream effects of Tregs.
Myeloid-Derived Suppressor Cells
The first report describing the existence of MDSCs showed that bone marrow-derived cells were able to suppress the killing activity of splenocytes in vitro (85). These cells were called “natural suppressor cells” or “null cells” because they did not express markers of B, T, or NK cells or macrophages (86). Subsequently, these cells were found to expand in inflammatory conditions and in tumor-bearing hosts (85, 87). In tumor-bearing mice, tumor-derived growth factors trigger the accumulation of T cell suppressive myeloid cells in the bone marrow and spleen (87, 88). The identification of these cells was hampered by the lack of clear markers, which caused variation in terminology and ambiguity among researchers. In order to bring some clarity into the field, Gabrilovich and colleagues published a consensus paper in 2007 in which they coined the term “MDSC” to refer to a heterogeneous population of myeloid cells with the ability to suppress T cell activity (89). MDSCs consist of a group of immature and mature myeloid cells that are defined by their immunosuppressive function. Within the MDSC population, two subpopulations can be distinguished based on the expression of Ly6G and Ly6C: Ly6ChighLy6G− monocytic-MDSC and Ly6ClowLy6G+ granulocytic-MDSC. In humans, MDSCs are defined as CD11b+CD33+HLA-DR−Lin− cells with the addition of CD14 or CD15 to discriminate between monocytic- or granulocytic-MDSCs, respectively (90).
In patients with various cancer types, including melanoma, gastric, breast, and CRC, increased numbers of MDSCs in the circulation correlate with poor survival (91–93). Numerous cytokines have been implicated in the expansion of MDSCs during cancer progression, including G-CSF, GM-CSF, and stem-cell factor (SCF or KIT ligand) (94–96).
Myeloid-derived suppressor cells exert their immunosuppressive function by different mechanisms, one of which is the consumption of essential amino acids from the environment. MDSCs frequently express high levels of arginase I, which catabolizes arginine, thereby depriving T cells from arginine, which is essential for their metabolism and function (97, 98). l-Arginine is also the substrate of another enzyme highly expressed in MDSCs, called iNOS. The release of reactive oxygen species (ROS) and nitric oxide (NO) by iNOS can lead to the inhibition of MHC class II expression on APCs causing impaired antigen presentation to CD4+ T cells (99). Moreover, NO can cause apoptosis of CD8+ T cells (100). Another amino acid is tryptophan, whose breakdown by the enzyme IDO suppresses T cell proliferation. MDSCs isolated from human breast cancer tissues inhibit T cell proliferation and induce T cell apoptosis in an IDO-dependent manner (101). Moreover, IDO inhibitors enhance the therapeutic efficacy of anti-CTLA-4 treatment leading to intratumoral accumulation of T cells and improved survival in the B16 melanoma model (102). Additionally, the amino acid cysteine is also important for T cell activation and function. T cells depend on other cells (macrophages and DCs) for cysteine metabolism. MDSCs internalize cystine (formed of two cysteines linked via a disulfide bond), catabolize it to cysteine and, unlike macrophages and DCs, do not release it into the environment. Therefore, MDSCs limit the amount of cystine that macrophages and DCs can metabolize to activate T cells (103). Finally, MDSCs contribute to an immunosuppressive TME by inducing the development of Tregs in tumor-bearing mice, as adoptive transfer of MDSCs and CD4+ T cells in MCA26 colon carcinoma cell line-bearing irradiated mice, induces expression of FOXP3 in transferred T cells (104). Thus, these data suggest that MDSCs play an important role in creating an immunosuppressive network in tumors, supporting the idea that reprograming or depletion of MDSCs could benefit immunotherapy strategies. Strategies to inhibit MDSCs include blocking their development or recruitment, targeting their immunosuppressive molecules or depleting them.
Tumor-Induced Neutrophils
In various cancer patients, a high neutrophil to T lymphocyte ratio in blood is associated with poor disease outcome (105, 106). Recent studies have reported that neutrophils also expand in experimental mouse tumor models, and that they exert immunosuppressive activity. A distinguishing feature of murine neutrophils is the expression of Ly6G, a surface marker shared with granulocytic-MDSC. When the T cell suppressive ability of neutrophils is confirmed, they can be categorized into the granulocytic-MDSC population (107). We recently showed in a mouse model for de novo breast cancer metastasis that neutrophils have a pro-metastatic phenotype and exert their function through suppression of CD8+ T cells. While depletion of Ly6G+ neutrophils results in decreased multi-organ metastasis, double depletion of neutrophils and CD8+ T cells reverses this phenotype (108). In line with this, chemotherapy-induced neutropenia correlates with improved overall survival in breast cancer patients (109). The metastasis-promoting role of neutrophils has also been demonstrated in UV-induced melanoma and in tumor inoculation models (110, 111). It would be interesting to study whether – as in the experimental tumor models – T cells in neutropenic cancer patients are more active. Interestingly, in 4T1-tumor-bearing mice, neutrophils inhibit the seeding of metastatic cells in the lung by the release of hydrogen peroxide (112). These data indicate a controversial role of neutrophils in metastasis that might be explained by the differences in tumor subtype or tumor model.
We and others have shown that T cell-suppressive neutrophils accumulate systemically during cancer progression in a G-CSF-dependent fashion (108, 113). In the transgenic MMTV-PyMT mammary tumor mouse model, tumor-derived G-CSF skews hematopoietic cell differentiation toward the granulocytic lineage in the bone marrow, resulting in increased numbers of immunosuppressive neutrophils in the circulation (113). In 4T1 mammary tumor-bearing mice, TGFβ polarizes mature neutrophils from cytotoxic anti-tumor activity toward pro-tumor immature immunosuppressive neutrophils (114). This is in line with previous findings identifying TGFβ as one of the drivers of pro-tumor polarized neutrophils (115). As such, it is tempting to speculate that for those tumors characterized by pro-metastatic neutrophils, inhibition of these cells – either by targeting upstream or downstream molecules – may be an interesting strategy for therapeutic intervention, in particular when combined with cancer immunotherapy.
Tumor-Associated Macrophages
Macrophages are frequently the most predominant immune cell type in tumors. In the past, macrophages were subdivided into classically activated macrophages (M1) exerting microbicidal and anti-tumor activity, or alternatively activated macrophages (M2) exerting pro-tumoral, immunosuppressive, and tissue repair functions (116, 117). TAMs are frequently classified as M2 macrophages. However, there is a growing realization that this black and white distinction of macrophage subsets is too simplistic and does not accurately reflect the heterogeneity, plasticity, and versatility of macrophages (118). Transcriptome and bioinformatic analyses of cultured macrophages exposed to different stimuli revealed a spectrum of activation programs for each stimulus that goes beyond the M1 and M2 model (119). Based on these data, it is to be expected that TAMs will also change their phenotype and function according to the cytokine milieu present in a specific tumor type. In the vast majority of cancers, high intratumoral macrophage density correlates with poor prognosis (120, 121). However, macrophages in CRC are associated with good prognosis, and in other types of cancers, like prostate and lung cancer, their role is still controversial (122). Depletion of macrophages by genetic ablation of CSF-1 in the MMTV-PyMT mammary tumor model reduces metastasis formation without affecting primary tumor growth (123). Likewise, several other experimental studies have reported a pro-metastatic role of macrophages (124, 125). TAMs produce a variety of factors that foster tumor growth and invasiveness, angiogenesis, and immunosuppression (120, 124, 126).
Tumor-associated macrophages exert their immunosuppressive activity in a similar fashion as that of MDSCs. TAMs can express various enzymes like arginase 1, IDO (127–129), and cytokines like IL10 (130). Another mechanism by which TAMs suppress T cells is the upregulation of PD-L1. In hepatocellular carcinoma, high density of peritumoral macrophages that express PD-L1 correlates with worse overall survival (131). Co-culture experiments showed that PD-L1+ macrophages suppress T cell activity unless anti-PD-L1 antibody is added in the culture (131). Based on these immunosuppressive properties, it is tempting to speculate that interference with TAMs will unleash anti-tumor immunity. Indeed, this idea has recently been supported by experimental studies in mouse models for glioblastoma and pancreatic cancer showing that CSF-1/CSF-1R pathway blockade can shift TAM polarization toward an anti-tumor phenotype, resulting in enhanced CD8+ T cell-mediated anti-tumor immunity (132, 133). Similarly, targeting the CCL2/CCR2 chemokine pathway – involved in recruitment of monocytes and macrophages – relieves the immunosuppressive phenotype of TAMs and enhances anti-tumor CD8+ T cell responses (134, 135). Based on these encouraging results, clinical trials are ongoing in which compounds targeting TAMs are being tested in cancer patients. Preliminary results of a clinical trial with anti-CSF-1R in patients with various types of solid malignancies showed a decrease in TAMs and an increase in intratumoral CD8/CD4 ratio (10).
Blocking the Suppressors to Release Anti-Tumor T Cells
As discussed above, many immunosuppressive cells and mediators can be identified in the TME that dampen anti-tumor T cell responses and may contribute to immune escape upon cancer immunotherapy. The combination of compounds that relieve immunosuppression with T cell-boosting therapy seems attractive to overcome immune tolerance toward the tumor.
Regulatory T cells seem to be interesting targets, since, as discussed earlier in this review, these cells suppress the functionality of CD4+ and CD8+ effector cells. In line with this, in the transgenic TRAMP prostate cancer model – engineered to express prostate-specific antigen (PSA) – Treg-depletion enhances IFNγ production by PSA-specific CD8+ T cells (136). This augmented effect of anti-tumor immunity is further enhanced by CTLA-4 blockade, and results in delayed tumor growth. Interestingly, the same experiments performed in the parental TRAMP model show only a modest activation of PSA-specific T cells upon anti-CD25 and anti-CTLA-4, and no survival benefit, suggesting the requirement of a tumor-specific antigen for this anti-tumor response (136). In the ID8 ovarian cancer model, tumor-infiltrating Tregs – which express both CTLA-4 and PD-1 – are reduced upon CTLA-4 and PD-1 dual blockade coinciding with increased tumor-infiltrating CD8+ T cells (137). However, additional depletion of Tregs does not further enhance this effect. In the same model, blockade of PD-L1, expressed on tumor cells and tumor-infiltrating immune cells, reduces the number of MDSCs and Tregs and enhances the frequency of effector T cells, resulting in prolonged survival (138). Furthermore, in a mouse model for rhabdomyosarcoma, PD-1 blockade increases the number of tumor-infiltrating CD8+ T cells, but does not change their activation status. Upon interference with the chemokine receptor CXCR2, which prevents MDSC trafficking into the tumor, enhanced activation of CD8+ T cells is observed (139). Blockade of CXCR2 improves the therapeutic efficacy of anti-PD-1 treatment resulting in a significant survival benefit (139). Moreover, in a mouse model of pancreatic ductal adenocarcinoma, blockade of CSF-1/CSF-1R signaling results in macrophage reprograming to support anti-tumor immune function and modestly delays tumor growth (133). TAMs obtained from anti-CSF1 treated mice are impaired in suppressing CD8+ T cell proliferation compared to control TAMs. The induction of CTLA-4 expression on CD8+ T cells and PD-L1 expression on tumor cells suggests the onset of acquired resistance to effective anti-tumor immune responses. Combining anti-CTLA-4 and anti-PD-1 with a CSF-1R inhibitor shows profound synergy with a significant reduction in tumor burden (133). Thus, together these results indicate that alleviation of immunosuppression reactivates anti-tumor immunity, which can be further enhanced by checkpoint inhibition.
Immunomodulatory Properties of Chemotherapeutic Drugs
Although various novel compounds targeting tumor-associated myeloid cells and their immunosuppressive mediators are being developed and tested, their clinical availability is still limited. An alternative and clinically available strategy is to relieve immunosuppression by exploiting the immunomodulatory effects of conventional anti-cancer strategies like chemotherapy (Figure 2). The impact of chemotherapeutic drugs on the proportion and phenotypic and functional characteristics of immune cells is to a great extent dictated by the type of drug and the dosing scheme: while high-dose chemotherapy usually results in lympho- or myelodepletion, low-dose (metronomic) treatment has more subtle anti-angiogenic and immunomodulatory effects (140, 141). In this section, we discuss the effects of chemotherapy on the immunosuppressive TME.
The Impact of Chemotherapy on T Cell Priming
Optimal T cell priming is dependent on antigen processing, presentation, and co-stimulation by properly matured and activated DCs. As discussed, impaired DC function and T cell priming are important mechanisms of immune escape by tumors. Certain chemotherapeutics induce anti-cancer immune responses by improving the recruitment and functionality of intratumoral DCs (142, 143). For example, low-dose cyclophosphamide promotes DC maturation (144). Besides the enhanced release of tumor antigens through induction of cancer cell death, chemotherapeutics, including oxaliplatin, doxorubicin, mitoxantrone, and melphalan, induce HMGB1 release and calreticulin translocation in cancer cells, facilitating antigen uptake by DCs and subsequent T cell stimulation (145–147). In addition, in the MCA205 fibrosarcoma model, anthracyclins induce the differentiation of myeloid cells in the tumor bed toward a DC-like phenotype in an ATP-dependent manner (142). In these relatively high immunogenic tumor models, the activated T cells subsequently enhance the anti-cancer efficacy of chemotherapy (142, 143, 145).
In less immunogenic models, such as de novo tumorigenesis models, an important role for T cells in chemotherapy efficacy is lacking (120, 148, 149). One possible explanation is that spontaneously arising tumors are characterized by local and systemic immunosuppression, which may overrule any chemotherapy-induced T cell responses. Indeed, in the MMTV-PyMT mammary tumor model, TAM-derived IL10 indirectly blocks anti-tumor CD8+ T cell activity by suppressing IL12 expression by intratumoral DCs upon paclitaxel treatment (149). These results apply to human breast cancer patients since low CD68+ macrophage over CD8+ T cell ratio prior to neo-adjuvant chemotherapy correlates with a better pathologic response (120). Moreover, high levels of IL12A mRNA in human breast cancer samples correlates with expression of DC-related transcription factors and GRZMB, CD8A, and IFNγ expression, suggesting an active anti-tumor T cell response (149). However, the role of TAMs and their potential suppressive function in cancer patients was not evaluated. Together, these results suggest that therapeutic targeting of TAMs could enhance the functionality of intratumoral DCs and anti-tumor T cell responses in chemotherapy treatment.
Impact of Chemotherapy on Tregs
With the knowledge that Tregs play an important role in suppressing effector T cell responses, a lot of effort has been put into the identification of chemotherapeutic drugs that target these cells. The best studied is cyclophosphamide, an alkylating agent, which crosslinks DNA, thus interfering with replication. Cyclophosphamide is known for its dose-dependent effect on the immune system. High doses of cyclophosphamide result in immunosuppression by reducing T cell proliferation and inducing apoptosis, thus making it useful for the prevention of graft-versus-host disease or rejection of transplanted organs (150, 151). In contrast, low doses selectively ablate Tregs and dampen their T cell suppressive ability (152). While the anti-tumor effect of high-dose cyclophosphamide is mainly due to its cytotoxic activity against cancer cells, the anti-tumor effect of low-dose cyclophosphamide depends on its immune-modulatory effects (153). Indeed, studies in T cell-deficient mice bearing inoculated tumors show loss of the anti-cancer activity of low-dose cyclophosphamide (153, 154). Moreover, reinfusion of CD4+CD25+ T cells in tumor-bearing mice, pre-treated with low-dose cyclophosphamide, abrogated the anti-tumor effect of the drug, emphasizing that Tregs counteract the therapeutic efficacy of the drug (153). In line with this, patients with different types of metastasized solid tumors receiving low-dose metronomic cyclophosphamide show a specific decrease of Tregs in the periphery with concomitant enhancement of NK lytic activity and T cell proliferation (155). In cancer patients receiving higher doses of metronomic cyclophosphamide, all lymphocyte populations were depleted, emphasizing the importance of accurate drug dosing to achieve selective Treg depletion (155). It has been proposed that the increased sensitivity of Tregs for cyclophosphamide is linked to their low ATP levels. Low levels of ATP result in decreased synthesis of glutathione, which is important for cyclophosphamide detoxification (156).
Another chemotherapeutic drug affecting Tregs is gemcitabine, a nucleoside analog interfering with DNA replication. In an orthotopic pancreatic cancer model, gemcitabine reduces the percentage of Tregs in the tumor resulting in a small but significant survival benefit (157). Whether this also results in improved CD8+ and CD4+ T cell activity remains unknown. A study performed in cancer patients showed that the percentage of Tregs in blood was decreased after gemcitabine treatment (158). Among the CD4+ cells, Tregs were identified as the most proliferative cells, which may explain the selectivity of gemcitabine for these cells. However, the effect of gemcitabine on other T cell populations was not assessed in this study (158). Also, other (combinations of) chemotherapy drugs have been reported to influence the presence or function of Tregs (159, 160).
Chemotherapeutics with Inhibitory Activity toward Tumor-Associated Myeloid Cells
Several chemotherapy drugs have been implicated in the selective reduction of MDSCs in the tumor and spleen of tumor-bearing mice (161, 162). In an EL4 inoculation tumor model, a set of chemotherapy drugs was tested for their influence on the number of splenic and intratumoral MDSCs (161). This study showed that high-dose gemcitabine and 5-fluorouracil (5-FU), two anti-metabolite drugs that interfere with DNA replication, reduce MDSC accumulation (161). Consequently, 5-FU-mediated MDSC depletion results in increased IFNγ-producing intratumoral CD8+ T cells. This effect is reverted by adoptive transfer of MDSCs, suggesting that the effect of 5-FU is exerted through MDSCs (161). Similar results were obtained in the MCA203 cell line inoculation sarcoma model combined with cytotoxic T cell transfer (163), highlighting the critical role of MDSCs in dampening T cell activity upon 5-FU treatment. While the exact mechanisms underlying the selectivity of 5-FU for MDSCs are unknown, it has been proposed that 5-FU inhibits the enzyme thymidylate synthase and that the resistance to 5-FU is due to insufficient inhibition of this enzyme (164). Indeed, low levels of thymidylate synthase are found in MDSCs compared to splenocytes and EL4 tumor cells, suggesting that 5-FU selectivity for MDSCs could be due to this low enzymatic expression (161).
High-dose gemcitabine induces similar effects on MDSCs as 5-FU (162). In vitro analyses of splenocytes from TC-1 lung cancer-bearing mice showed the cytotoxic specificity of gemcitabine for MDSCs, while CD4+, CD8+ T cells, and B cells are unaffected (162). Although the exact mechanism underlying this specificity has not been identified, it has been hypothesized that gemcitabine induces apoptosis in MDSCs (162). Yet, a thorough mechanistic analysis of gemcitabine-induced apoptotic cell death in various immune cell populations has not been performed. In the 4T1 breast cancer mouse model, gemcitabine treatment also reduces splenic MDSC accumulation, which results in increased proliferation and IFNγ production by splenic lymphocytes upon antigen stimulation compared to untreated mice (165). However, no difference in anti-cancer efficacy of gemcitabine was observed between immunocompetent and nude mice, indicating a T cell-independent mechanism of 4T1 tumor control by gemcitabine (165). Perhaps, this observation might be explained by the presence of other immunosuppressive cells in the TME, like Tregs or macrophages.
The beneficial effect of chemotherapeutic drugs on the immunosuppressive TME is not only a direct result of reduced MDSC numbers, but also a result of a more favorable phenotype of the remaining MDSCs. For example, in the 4T1-Neu mammary tumor model, docetaxel reduces splenic granulocytic-MDSCs and enhances CD8+ and CD4+ cytotoxic activity (166). The remaining MDSCs exhibit a different phenotypic profile compared to MDSCs from untreated mice. In line with these in vivo findings, MDSCs pre-treated with docetaxel induce the proliferation of OVA-exposed OT-II CD4+ T cells compared to untreated MDSCs in vitro, suggesting that docetaxel treatment induces a phenotypical switch to a more favorable state (166). Likewise, doxorubicin selectively decreases the proportion of MDSCs in the 4T1 breast tumor model via apoptosis and subdues the immunosuppressive phenotype of the remaining MDSCs. The remaining MDSCs have a lower expression of immunosuppressive molecules like ROS, ARG-1, and IDO (167). This less suppressive environment caused by doxorubicin enhanced the activity of adoptively transferred T helper cells (167). Interestingly, some subpopulations of MDSCs may be more susceptible to chemotherapy than others. Whether chemotherapy selectively depletes pro-tumorigenic MDSCs or skews them toward an anti-tumor phenotype is unknown. Future studies using lineage tracing methodologies would provide more insight into this topic.
Besides the favorable immunomodulatory “off-target” effects of various chemotherapeutic drugs, these drugs can at the same time exert less desirable functions. For instance, in addition to its inhibitory effect on Tregs, cyclophosphamide increases the number of CD11b+Gr1+ MDSCs. In a transgenic mouse model for melanoma, a single injection of low-dose cyclophosphamide increases the accumulation of MDSCs in the tumor and spleen, stimulates their immunosuppressive ability by inducing NO and ROS production, and reduces splenocyte proliferation (168). In line with these findings, MDSCs accumulate in the blood of breast cancer patients after treatment with doxorubicin or cyclophosphamide (169). This may be due to IFNγ release by CD4+ and CD8+ T cells that promotes survival of MDSCs (170). Based on these data, a combination of cyclophosphamide and cancer immunotherapy might not work; however, additional studies in other tumor models should be performed to test this.
Another study underscoring the complex impact of chemotherapy on myeloid cells shows that in EL4-tumor-bearing mice 5-FU induces IL1β secretion in MDSCs in an Nlrp3 inflammasome-dependent manner (171). Using depletion experiments and knock-out mice, it was shown that the MDSC-derived IL1β triggers IL17 production by CD4+ T cells, which limits the anti-cancer efficacy of 5-FU (171). These data highlight that the effect of certain chemotherapy drugs is not simply limited to depletion of immunosuppressive cells but these drugs also change the functionality of cells that may impair their efficacy. These results suggest that the combination of chemotherapeutic and immunomodulatory compounds must be chosen carefully to increase their anti-cancer efficacy (172).
While several chemotherapy drugs have been reported to target MDSCs, thus far only one drug seems to strongly affect TAMs. Trabectedin, a drug that binds DNA and affects transcription and DNA repair pathways, depletes macrophages, and suppresses the differentiation of monocytes in the tumor bed in the transplantable MN/MCA1 fibrosarcoma tumor model through a TRAIL-dependent mechanism (173). Importantly, this macrophage selectivity is also observed in sarcoma patients after trabectedin neo-adjuvant treatment (173). It would be interesting to assess whether the anti-cancer activity of trabectedin is CD8+ T cell mediated. The macrophage-depleting effect of trabectedin makes it an interesting candidate for combination strategies with immunotherapy.
As discussed before, many studies illustrate the complexity of immunomodulation by conventional chemotherapeutics, which is highly context-dependent. The differential effect on specific immune cells of different types of chemotherapeutics is to a large extent dependent on the timing and dosing schedule. While high-dose chemotherapy often depletes immune cell subsets, low-dose metronomic chemotherapy exerts a more subtle anti-angiogenic and immunomodulatory mode of action (140, 141). It will be interesting to perform a side-by-side comparison of various types of chemotherapies administered at high versus low (metronomic) dose and evaluate their immunomodulatory effects, followed by more mechanistic studies. Ideally, these types of experiments would be performed in clinically relevant mouse models that faithfully recapitulate human cancer (Box 1) to facilitate clinical translation.
Box 1. Experimental mouse models to study the anti-tumor immune response.
Understanding the complex crosstalk between innate and adaptive immune cells and (disseminated) cancer cells requires the use of preclinical mouse models that faithfully recapitulate human cancer. The most widely used experimental mouse models are carcinogen-induced cancer models and cell line inoculation models. The latter is based on inoculation of large numbers of (genetically modified) homogenous cancer cells grown in 2D conditions. Implantation of these cells often results in massive cell death, thereby priming an effective anti-tumor immune response. Shaping of the tumor immune microenvironment during cancer progression in these models can hardly take place in the short amount of time that it takes for transplanted tumors to grow to their maximum tolerated size. Of notice, when implanting human cancer cells, either patient-derived tumor material or established human cancer cell lines, immunocompromised mice are used, thereby excluding the important role of the adaptive immune system.
While cell line inoculation models proved useful to decipher some aspects of the anti-tumor immune response, we should keep in mind that these models do not reflect physiological processes as they occur in human patients. Genetically engineered mouse (GEM) models, which develop de novo cancers, generally mimic human cancer genetically – because of the introduction of specific driver mutations – and histopathologically (180). In addition, tumor progression occurs in a multi-step nature in their natural microenvironment shaping the local immune responses (Figure 1), therefore mimicking the human setting. In contrast to inoculation models expressing known tumor antigens, the anti-tumor immune response in GEM models can be considered a black box. Due to their cellular and genetic heterogeneity, GEM models induce a variety of T cell responses directed against multiple unknown tumor neo-antigens, which faithfully reflects human cancer. Interestingly, comparative studies have shown that inoculation models greatly differ from GEM models in terms of response to anti-cancer therapies and endogenous T cell responses (181, 182). The advantages and disadvantages of different experimental mouse models in studying responsiveness to anti-cancer therapy have been recently discussed (14, 183).
Future Perspectives: Exploiting the Immunomodulatory Properties of Chemotherapeutic Drugs to Improve Cancer Immunotherapy
Given their immunomodulatory properties, conventional chemotherapy drugs are interesting candidates to combine with T cell-boosting immunotherapy – a concept termed chemo-immunotherapy (174). Clinical trials report enhanced anti-tumor T cell responses in cancer patients treated with chemotherapy in combination with cancer vaccines (13). Moreover, clinical testing of chemotherapy combined with other immunotherapy approaches like adoptive transfer of (genetically engineered) autologous T cells or toll-like receptor (TLR) agonists are likely to be explored in the near future. Indeed, various experimental studies support the concept that chemotherapy-induced relief of immunosuppression could improve cancer immunotherapy. In a passive immunotherapy setting, in the MC203 fibrosarcoma and TC-1 lung cancer cell line inoculation models, low-dose gemcitabine and 5-FU reduced the splenic population of CD11b+Gr1+ MDSCs, resulting in enhanced anti-tumor activity of adoptively transferred tumor-specific CTL (163).
The results obtained in preclinical models combining chemotherapeutics with immune checkpoint inhibitors are promising. The immunomodulatory effects of melphalan – administered in a subtherapeutic dose – synergizes with CTLA-4 blockade in a plasmacytoma model (175). In vitro assays revealed that splenocytes obtained from melphalan-treated mice co-cultured with anti-CTLA-4 induced tumor cell cytotoxicity, while splenocytes from non-treated mice – irrespective of CTLA-4 blockade – did not (175). Furthermore, in the poorly immunogenic AB-1 malignant mesothelioma and Lewis lung cancer (LLC) inoculation tumor models, a combination therapy of gemcitabine and CTLA-4 blockade synergizes, inducing potent anti-tumor immune responses and subsequent regression of tumors in a CD4- and CD8-dependent manner (176). In addition, in a subcutaneous murine mesothelioma model, synergy is observed between cisplatin and CTLA-4 blockade, resulting in a profound anti-tumor effect that is characterized by increased influx and activation of CD4+ and CD8+ T cells in the tumor (177). Moreover, preclinical studies in mice show that doxorubicin, cisplatin, and paclitaxel in addition to their immunomodulatory role, can sensitize tumor cells for CTL attack in a direct manner (178). Here, chemotherapy causes increased permeability of tumor cell membranes to GRZMB, which sensitizes cancer cells to the cytotoxic effects of T cells and improved different cancer immunotherapy strategies (178). Together, these preclinical studies – albeit limited numbers – show the potential to exploit immunomodulatory chemotherapeutic drugs to improve the efficacy of checkpoint blockade.
Clinical trials that evaluate the combination of chemotherapeutic drugs and checkpoint inhibitors in cancer patients are still limited. Some studies in melanoma and lung cancer have used chemotherapeutics in combination with checkpoint blockade resulting in improved survival compared to chemotherapy alone (41, 179). However, the rational of these studies was not to evaluate the effect of treatment on the immunosuppressive microenvironment. Moreover, the design of clinical trials makes it impossible to perform a structural comparison in patients to study the effect of the immunosuppressive microenvironment on immunotherapy efficacy and whether this efficacy can be enhanced by adding chemotherapeutics to the treatment regimen. Therefore, we need to rely on preclinical research in mouse tumor models that faithfully recapitulate human cancer in terms of the genetic composition, anti-tumor immunity, and the immunosuppressive TME (Box 1). Results obtained in mouse models that mimic human cancer might shape the design of clinical trials and guide toward interesting treatment strategies. There are still various important questions that need to be addressed to maximally exploit the therapeutic efficacy of chemotherapy and immunotherapy combinations, like the determination of the most optimal combinations. Based on preclinical findings, different cancer types will likely require different combinations of therapy. In addition, despite the devastating effects of metastatic disease, mechanistic insights into the site-specific therapeutic response profiles and resistance mechanisms of cancer immunotherapy are completely lacking. Moreover, it is critical to gain insights into the mechanisms underlying intrinsic and acquired resistance to cancer immunotherapy. To answer these questions within the next decade, it is critical that basic researchers and clinicians intensify their efforts to join forces, so that results from preclinical research can guide the design of clinical trials, and the results from clinical trials, in turn, can guide mechanistic studies in mouse models. Together, these efforts will improve treatment strategies using chemotherapeutics to alleviate immunosuppression and enhance cancer immunotherapy.
Author Contributions
KK, CS, and KV reviewed relevant literature and drafted the manuscript. KV revised the manuscript and supervised KK and CS. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Seth Coffelt for insightful comments on the manuscript. Work in the lab is supported by grants from the European Union (FP7 MCA-ITN 317445 TIMCC), the European Research Council (ERC consolidator award INFLAMET 615300), the Beug Foundation for Metastasis Research, the Dutch Cancer Society (2011-5004), and the Netherlands Organization for Scientific Research (NWO VIDI 917.96.307).
References
1. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
2. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) 515(7528):558–62. doi:10.1038/nature13904
3. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
4. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
5. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372(4):311–9. doi:10.1056/NEJMoa1411087
6. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol (2015) 33(13):1430–7. doi:10.1200/JCO.2014.59.0703
7. Couzin-Frankel J. Cancer immunotherapy. Science (2013) 342(6165):1432–3. doi:10.1126/science.342.6165.1432
8. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi:10.1056/NEJMoa1406498
9. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi:10.1126/science.aaa1348
10. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25(6):846–59. doi:10.1016/j.ccr.2014.05.016
11. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12(4):237–51. doi:10.1038/nrc3237
12. Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res (2012) 72(13):3163–74. doi:10.1158/0008-5472.CAN-12-0210
13. Chen G, Emens LA. Chemoimmunotherapy: reengineering tumor immunity. Cancer Immunol Immunother (2013) 62(2):203–16. doi:10.1007/s00262-012-1388-0
14. Coffelt SB, de Visser KE. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol (2015) 36(4):198–216. doi:10.1016/j.it.2015.02.006
15. Wargo JA, Reuben A, Cooper ZA, Oh KS, Sullivan RJ. Immune effects of chemotherapy, radiation, and targeted therapy and opportunities for combination with immunotherapy. Semin Oncol (2015) 42(4):601–16. doi:10.1053/j.seminoncol.2015.05.007
16. DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res (2007) 9(4):212. doi:10.1186/bcr1746
17. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21(3):309–22. doi:10.1016/j.ccr.2012.02.022
18. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi:10.1038/nrc1782
19. Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, et al. Somatic inactivation of E-Cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell (2006) 10(5):437–49. doi:10.1016/j.ccr.2006.09.013
20. Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med (1999) 190(10):1417–26. doi:10.1084/jem.190.10.1417
21. Pinzon-Charry A, Maxwell T, López JA. Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol Cell Biol (2005) 83(5):451–61. doi:10.1111/j.1440-1711.2005.01371.x
22. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell (2012) 21(3):402–17. doi:10.1016/j.ccr.2012.01.008
23. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T Cell immunity. Cancer Cell (2014) 26(5):638–52. doi:10.1016/j.ccell.2014.09.007
24. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res (2010) 70(15):6171–80. doi:10.1158/0008-5472.CAN-10-0153
25. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi:10.1038/nm1699
26. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi:10.1084/jem.20101956
27. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
28. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi:10.1126/science.aaa6204
29. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348(6230):56–61. doi:10.1126/science.aaa8172
30. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. doi:10.1016/1074-7613(94)90071-X
31. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted cd8 t cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi:10.1038/nature04444
32. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol (2007) 19(7):813–24. doi:10.1093/intimm/dxm057
33. Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol (2015) 33(April):23–35. doi:10.1016/j.coi.2015.01.006
34. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27(4):450–61. doi:10.1016/j.ccell.2015.03.001
35. Goldberg MV, Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM, et al. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood (2007) 110(1):186–92. doi:10.1182/blood-2006-12-062422
36. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi:10.1126/science.1160062
37. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206(8):1717–25. doi:10.1084/jem.20082492
38. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells Co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi:10.1084/jem.20130579
39. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med (2015) 372(21):2006–17. doi:10.1056/NEJMoa1414428
40. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med (2014) 6(254):254ra128. doi:10.1126/scitranslmed.3008918
41. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi:10.1056/NEJMoa1104621
42. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol (2015) 33(17):1889–94. doi:10.1200/JCO.2014.56.2736
43. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
44. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi:10.1038/nature13988
45. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med (2015) 21(1):81–5. doi:10.1038/nm.3773
46. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31(32):e439–42. doi:10.1200/JCO.2012.47.7521
47. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160(1–2):48–61. doi:10.1016/j.cell.2014.12.033
48. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi:10.1126/science.aaa4971
49. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi:10.1038/nature12477
50. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi:10.1038/nature13954
51. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124(5):2246–59. doi:10.1172/JCI73639
52. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi:10.1038/nature14011
53. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res (2014) 20(19):5064–74. doi:10.1158/1078-0432.CCR-13-3271
54. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi:10.1056/NEJMoa1500596
55. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov (2015) 5(1):43–51. doi:10.1158/2159-8290.CD-14-0863
56. Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, et al. PD-L1 expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol Res (2015) 3(4):326–32. doi:10.1158/2326-6066.CIR-14-0133
57. Ali HR, Glont S-E, Blows FM, Provenzano E, Dawson S-J, Liu B, et al. PD-L1 protein Expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann Oncol (2015) 26(7):1488–93. doi:10.1093/annonc/mdv192
58. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13(1):84–8. doi:10.1038/nm1517
59. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi:10.1038/nm863
60. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology (1970) 18(5):723–37.
61. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
62. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
63. Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol (2013) 4:190. doi:10.3389/fimmu.2013.00190
64. Berendt MJ, North RJ. T-cell-mediated suppression of anti-tumor immunity. An explanation for progressive growth of an immunogenic tumor. J Exp Med (1980) 151(1):69–80. doi:10.1084/jem.151.1.69
65. Li X, Kostareli E, Suffner J, Garbi N, Hämmerling GJ. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur J Immunol (2010) 40(12):3325–35. doi:10.1002/eji.201041093
66. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
67. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30(5):636–45. doi:10.1016/j.immuni.2009.04.010
68. Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol (2000) 164(1):183–90. doi:10.4049/jimmunol.164.1.183
69. Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med (1998) 188(2):287–96. doi:10.1084/jem.188.2.287
70. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi:10.1084/jem.20030152
71. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389(6652):737–42. doi:10.1038/39614
72. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450(7169):566–9. doi:10.1038/nature06306
73. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27(4):635–46. doi:10.1016/j.immuni.2007.08.014
74. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21(4):589–601. doi:10.1016/j.immuni.2004.09.002
75. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol (2005) 174(4):1783–6. doi:10.4049/jimmunol.174.4.1783
76. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi:10.1084/jem.20062512
77. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol (2006) 24(34):5373–80. doi:10.1200/JCO.2006.05.9584
78. Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer (2003) 98(5):1089–99. doi:10.1002/cncr.11618
79. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol (2007) 25(18):2586–93. doi:10.1200/JCO.2006.09.4565
80. Li YQ, Liu FF, Zhang XM, Guo XJ, Ren MJ, Fu L. Tumor secretion of CCL22 activates intratumoral Treg infiltration and is independent prognostic predictor of breast cancer. PLoS One (2013) 8(10):e76379. doi:10.1371/journal.pone.0076379
81. Huang Y, Liao H, Zhang Y, Yuan R, Wang F, Gao Y, et al. Prognostic value of tumor-infiltrating FoxP3+ T cells in gastrointestinal cancers: a meta analysis. PLoS One (2014) 9(5):e94376. doi:10.1371/journal.pone.0094376
82. Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother (2011) 60(7):909–18. doi:10.1007/s00262-011-1046-y
83. Rasku MA, Clem AL, Telang S, Taft B, Gettings K, Gragg H, et al. Transient T cell depletion causes regression of melanoma metastases. J Transl Med (2008) 6:12. doi:10.1186/1479-5876-6-12
84. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med (2012) 4(134):134ra62. doi:10.1126/scitranslmed.3003330
85. Bennett JA, Rao VS, Mitchell MS. Systemic Bacillus Calmette-Guérin (BCG) activates natural suppressor cells. Proc Natl Acad Sci U S A (1978) 75(10):5142–4. doi:10.1073/pnas.75.10.5142
86. Oseroff A, Okada S, Strober S. Natural suppressor (NS) cells found in the spleen of neonatal mice and adult mice given total lymphoid irradiation (TLI) express the null surface phenotype. J Immunol (1984) 132(1):101–10.
87. Tsuchiya Y, Igarashi M, Suzuki R, Kumagai K. Production of colony-stimulating factor by tumor cells and the factor-mediated induction of suppressor cells. J Immunol (1988) 141(2):699–708.
88. Lee MY, Rosse C. Depletion of lymphocyte subpopulations in primary and secondary lymphoid organs of mice by a transplanted granulocytosis-inducing mammary carcinoma. Cancer Res (1982) 42(4):1255–60.
89. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425. doi:10.1158/0008-5472.CAN-06-3037
90. Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother (2012) 61(8):1155–67. doi:10.1007/s00262-012-1294-5
91. Chevolet I, Speeckaert R, Schreuer M, Neyns B, Krysko O, Bachert C. Clinical significance of plasmacytoid dendritic cells and myeloid-derived suppressor cells in melanoma. J Transl Med (2015) 13(1):9. doi:10.1186/s12967-014-0376-x
92. Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol (2013) 190(2):794–804. doi:10.4049/jimmunol.1202088
93. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood (2011) 118(8):2254–65. doi:10.1182/blood-2010-12-325753
94. Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One (2011) 6(11):e27690. doi:10.1371/journal.pone.0027690
95. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res (2004) 64(17):6337–43. doi:10.1158/0008-5472.CAN-04-0757
96. Pan PY, Wang GX, Yin B, Ozao J, Ku T, Divino CM, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood (2008) 111(1):219–28. doi:10.1182/blood-2007-04-086835
97. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202(7):931–9. doi:10.1084/jem.20050715
98. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69(4):1553–60. doi:10.1158/0008-5472.CAN-08-1921
99. Harari O, Liao JK. Inhibition of MHC II gene transcription by nitric oxide and antioxidants. Curr Pharm Des (2004) 10(8):893–8. doi:10.2174/1381612043452893
100. Saio M, Radoja S, Marino M, Frey AB. Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide. J Immunol (2001) 167(10):5583–93. doi:10.4049/jimmunol.167.10.5583
101. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190(7):3783–97. doi:10.4049/jimmunol.1201449
102. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med (2013) 210(7):1389–402. doi:10.1084/jem.20130066
103. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi:10.1158/0008-5472.CAN-09-2587
104. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi:10.1158/0008-5472.CAN-05-1299
105. Noh H, Eomm M, Han A. Usefulness of pretreatment neutrophil to lymphocyte ratio in predicting disease-specific survival in breast cancer patients. J Breast Cancer (2013) 16(1):55–9. doi:10.4048/jbc.2013.16.1.55
106. Tomita M, Shimizu T, Ayabe T, Yonei A, Onitsuka T. Preoperative neutrophil to lymphocyte ratio as a prognostic predictor after curative resection for non-small cell lung cancer. Anticancer Res (2011) 31(9):2995–8.
107. Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell Mol Life Sci (2013) 70(20):3813–27. doi:10.1007/s00018-013-1286-4
108. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522(7556):345–8. doi:10.1038/nature14282
109. Han Y, Yu Z, Wen S, Zhang B, Cao X, Wang X. Prognostic value of chemotherapy-induced neutropenia in early-stage breast cancer. Breast Cancer Res Treat (2012) 131(2):483–90. doi:10.1007/s10549-011-1799-1
110. Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature (2014) 507(7490):109–13. doi:10.1038/nature13111
111. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A (2010) 107(50):21248–55. doi:10.1073/pnas.1015855107
112. Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell (2011) 20(3):300–14. doi:10.1016/j.ccr.2011.08.012
113. Casbon AJ, Reynaud D, Park C, Khuc E, Gan DD, Schepers K. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A (2015) 112(6):E566–75. doi:10.1073/pnas.1424927112
114. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep (2015) 10(4):562–73. doi:10.1016/j.celrep.2014.12.039
115. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L. Polarization of tumor-associated neutrophil phenotype by TGF-beta: ‘N1’ versus ‘N2’ TAN. Cancer Cell (2009) 16(3):183–94. doi:10.1016/j.ccr.2009.06.017
116. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi:10.1016/j.it.2004.09.015
117. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi:10.1016/j.immuni.2014.06.008
118. Schultze JL. Transcriptional programming of human macrophages: on the way to systems immunology. J Mol Med (Berl) (2015) 93(6):589–97. doi:10.1007/s00109-015-1286-y
119. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40(2):274–88. doi:10.1016/j.immuni.2014.01.006
120. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov (2011) 1:54–67. doi:10.1158/2159-8274.CD-10-0028
121. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res (1996) 56(20):4625–9.
122. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27(4):462–72. doi:10.1016/j.ccell.2015.02.015
123. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med (2001) 193(6):727–40. doi:10.1084/jem.193.6.727
124. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell (2009) 16(2):91–102. doi:10.1016/j.ccr.2009.06.018
125. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475(7355):222–5. doi:10.1038/nature10138
126. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66(23):11238–46. doi:10.1158/0008-5472.CAN-06-1278
127. Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, et al. Regulation of macrophage arginase expression and tumor growth by the ron receptor tyrosine kinase. J Immunol (2011) 187(5):2181–92. doi:10.4049/jimmunol.1003460
128. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol (2003) 171(3):1232–9. doi:10.4049/jimmunol.171.3.1232
129. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189(9):1363–72. doi:10.1084/jem.189.9.1363
130. Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme J, et al. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol (2000) 164(2):762–7. doi:10.4049/jimmunol.164.2.762
131. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206(6):1327–37. doi:10.1084/jem.20082173
132. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19(10):1264–72. doi:10.1038/nm.3337
133. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74(18):5057–69. doi:10.1158/0008-5472.CAN-13-3723
134. Fridlender ZG, Buchlis G, Kapoor V, Cheng G, Sun J, Singhal S, et al. CCL2 blockade augments cancer immunotherapy. Cancer Res (2010) 70(1):109–18. doi:10.1158/0008-5472.CAN-09-2326
135. Fridlender ZG, Kapoor V, Buchlis G, Cheng G, Sun J, Wang LC. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am J Respir Cell Mol Biol (2011) 44(2):230–7. doi:10.1165/rcmb.2010-0080OC
136. Klyushnenkova EN, Riabov VB, Kouiavskaia DV, Wietsma A, Zhan M, Alexander RB. Breaking immune tolerance by targeting CD25+ regulatory T cells is essential for the anti-tumor effect of the CTLA-4 blockade in an HLA-DR transgenic mouse model of prostate cancer. Prostate (2014) 74(14):1423–32. doi:10.1002/pros.22858
137. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res (2013) 73(12):3591–603. doi:10.1158/0008-5472.CAN-12-4100
138. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res (2013) 73(23):6900–12. doi:10.1158/0008-5472.CAN-13-1550
139. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6(237):237ra67. doi:10.1126/scitranslmed.3007974
140. Nars MS, Kaneno R. Immunomodulatory effects of low dose chemotherapy and perspectives of its combination with immunotherapy. Int J Cancer (2013) 132(11):2471–8. doi:10.1002/ijc.27801
141. Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer (2004) 4(6):423–36. doi:10.1038/nrc1369
142. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38(4):729–41. doi:10.1016/j.immuni.2013.03.003
143. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15(10):1170–8. doi:10.1038/nm.2028
144. Wada S, Yoshimura K, Hipkiss EL, Harris TJ, Yen HR, Goldberg MV, et al. Cyclophosphamide augments antitumor immunity: studies in an autochthonous prostate cancer model. Cancer Res (2009) 69(10):4309–18. doi:10.1158/0008-5472.CAN-08-4102
145. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13(9):1050–9. doi:10.1038/nm1622
146. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi:10.1038/nm1523
147. Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol (2015) 6:187. doi:10.3389/fimmu.2015.00187
148. Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE. Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med (2012) 18(3):344–6. doi:10.1038/nm.2652
149. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell (2014) 26(5):623–37. doi:10.1016/j.ccell.2014.09.006
150. Luznik L, Jones RJ, Fuchs EJ. High-dose cyclophosphamide for graft-versus-host disease prevention. Curr Opin Hematol (2010) 17(6):493–9. doi:10.1097/MOH.0b013e32833eaf1b
151. Luznik L, O’Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant (2008) 14(6):641–50. doi:10.1016/j.bbmt.2008.03.005
152. Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood (2005) 105(7):2862–8. doi:10.1182/blood-2004-06-2410
153. Motoyoshi Y, Kaminoda K, Saitoh O, Hamasaki K, Nakao K, Ishii N, et al. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol Rep (2006) 16(1):141–6. doi:10.3892/or.16.1.141
154. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol (2004) 34(2):336–44. doi:10.1002/eji.200324181
155. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother (2007) 56(5):641–8. doi:10.1007/s00262-006-0225-8
156. Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B. Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res (2010) 70(12):4850–8. doi:10.1158/0008-5472.CAN-10-0283
157. Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, et al. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic panc02 model of pancreatic cancer. Int J Cancer (2013) 133(1):98–107. doi:10.1002/ijc.27990
158. Rettig L, Seidenberg S, Parvanova I, Samaras P, Curioni A, Knuth A, et al. Gemcitabine depletes regulatory T-cells in human and mice and enhances triggering of vaccine-specific cytotoxic T-cells. Int J Cancer (2011) 129(4):832–8. doi:10.1002/ijc.25756
159. Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood (2005) 106(6):2018–25. doi:10.1182/blood-2005-02-0642
160. Chen C, Chen Z, Chen D, Zhang B, Wang Z, Le H. Suppressive effects of gemcitabine plus cisplatin chemotherapy on regulatory T cells in nonsmall-cell lung cancer. J Int Med Res (2015) 43(2):180–7. doi:10.1177/0300060514561504
161. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70(8):3052–61. doi:10.1158/0008-5472.CAN-09-3690
162. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res (2005) 11(18):6713–21. doi:10.1158/1078-0432.CCR-05-0883
163. Ugel S, Peranzoni E, Desantis G, Chioda M, Walter S, Weinschenk T, et al. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep (2012) 2(3):628–39. doi:10.1016/j.celrep.2012.08.006
164. Peters GJ, van der Wilt CL, van Triest B, Codacci-Pisanelli G, Johnston PG, van Groeningen CJ, et al. Thymidylate synthase and drug resistance. Eur J Cancer (1995) 31A(7–8):1299–305. doi:10.1016/0959-8049(95)00172-F
165. Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol (2009) 9(7–8):900–9. doi:10.1016/j.intimp.2009.03.015
166. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res (2010) 16(18):4583–94. doi:10.1158/1078-0432.CCR-10-0733
167. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res (2014) 74(1):104–18. doi:10.1158/0008-5472.CAN-13-1545
168. Sevko A, Sade-Feldman M, Kanterman J, Michels T, Falk CS, Umansky L, et al. Cyclophosphamide promotes chronic inflammation-dependent immunosuppression and prevents antitumor response in melanoma. J Invest Dermatol (2013) 133(6):1610–9. doi:10.1038/jid.2012.444
169. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol Immunother (2009) 58(1):49–59. doi:10.1007/s00262-008-0523-4
170. Guo Q, Lv Z, Fu Q, Jiang C, Liu Y, Lai L, et al. IFN-γ producing T cells contribute to the increase of myeloid derived suppressor cells in tumor-bearing mice after cyclophosphamide treatment. Int Immunopharmacol (2012) 12(2):425–32. doi:10.1016/j.intimp.2011.12.016
171. Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med (2013) 19(1):57–64. doi:10.1038/nm.2999
172. Shurin MR. Dual role of immunomodulation by anticancer chemotherapy. Nat Med (2013) 19(1):20–2. doi:10.1038/nm.3045
173. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23(2):249–62. doi:10.1016/j.ccr.2013.01.008
174. Lake RA, Robinson BW. Immunotherapy and chemotherapy – a practical partnership. Nat Rev Cancer (2005) 5(5):397–405. doi:10.1038/nrc1613
175. Mokyr MB, Kalinichenko T, Gorelik L, Bluestone JA. Realization of the therapeutic potential of CTLA-4 blockade in low-dose chemotherapy-treated tumor-bearing mice. Cancer Res (1998) 58(23):5301–4.
176. Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One (2013) 8(4):e61895. doi:10.1371/journal.pone.0061895
177. Wu L, Yun Z, Tagawa T, Rey-McIntyre K, de Perrot M. CTLA-4 blockade expands infiltrating T cells and inhibits cancer cell repopulation during the intervals of chemotherapy in murine mesothelioma. Mol Cancer Ther (2012) 11(8):1809–19. doi:10.1158/1535-7163.MCT-11-1014
178. Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, et al. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest (2010) 120(4):1111–24. doi:10.1172/JCI40269
179. Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol (2012) 30(17):2046–54. doi:10.1200/JCO.2011.38.4032
180. Singh M, Murriel CL, Johnson L. Genetically engineered mouse models: closing the gap between preclinical data and trial outcomes. Cancer Res (2012) 72(11):2695–700. doi:10.1158/0008-5472.CAN-11-2786
181. DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell (2011) 19(1):72–85. doi:10.1016/j.ccr.2010.11.011
182. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (2009) 324(5933):1457–61. doi:10.1126/science.1171362
Keywords: cancer immunotherapy, immune checkpoint blockade, chemotherapy, tumor microenvironment, immunosuppression, anti-tumor immunity
Citation: Kersten K, Salvagno C and de Visser KE (2015) Exploiting the immunomodulatory properties of chemotherapeutic drugs to improve the success of cancer immunotherapy. Front. Immunol. 6:516. doi: 10.3389/fimmu.2015.00516
Received: 17 July 2015; Accepted: 22 September 2015;
Published: 07 October 2015
Edited by:
Abhishek D. Garg, KU Leuven – University of Leuven, BelgiumReviewed by:
Marie-Lise Gougeon, Institut Pasteur, FranceShubhada Vivek Chiplunkar, Advanced Centre for Treatment, Research & Education in Cancer, India
Copyright: © 2015 Kersten, Salvagno and de Visser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin E. de Visser, Division of Immunology, Netherlands Cancer Institute, Plesmanlaan 121, Amsterdam 1066 CX, Netherlands,ay5kLnZpc3NlckBua2kubmw=
†Kelly Kersten and Camilla Salvagno have contributed equally to this work.