 Kevin Blauth
Kevin Blauth Gregory P. Owens1
Gregory P. Owens1 Jeffrey L. Bennett
Jeffrey L. Bennett- 1Department of Neurology, University of Colorado Denver, Aurora, CO, USA
- 2Department of Ophthalmology, University of Colorado Denver, Aurora, CO, USA
- 3Program in Neuroscience, University of Colorado Denver, Aurora, CO, USA
B cells play a central role in multiple sclerosis (MS) pathology. B and plasma cells may contribute to disease activity through multiple mechanisms: antigen presentation, cytokine secretion, or antibody production. Molecular analyses of B cell populations in MS patients have revealed significant overlaps between peripheral lymphoid and clonally expanded central nervous system (CNS) B cell populations, indicating that B cell trafficking may play a critical role in driving MS exacerbations. In this review, we will assess our current knowledge of the mechanisms and pathways governing B cell migration into the CNS and examine evidence for and against a compartmentalized B cell response driving progressive MS pathology.
Introduction
In recent decades, accumulating evidence has brought B cells into focus as critical players in multiple sclerosis (MS) pathogenesis. B cells are present at elevated levels in inflamed MS central nervous system (CNS) tissue and are significantly increased in MS cerebrospinal fluid (CSF) (1, 2). Furthermore, IgG is synthesized intrathecally in MS patients (3), and IgG and complement are characteristic features of both type 2 and active MS lesions (4–6). In the CSF, the presence of oligoclonal IgG bands (OCBs) are a long-standing hallmark of MS diagnosis, and in the meninges, B cell-predominant lymphoid aggregations [germinal center (GC)-like structures] are observed in some relapsing and secondary progressive patients (7, 8). Finally, clinical trials of the anti-CD20 monoclonal antibodies rituximab (9), and ocrelizumab (10), have demonstrated beneficial effects on MRI lesion load and relapse activity in MS patients.
Many questions about the role of B and plasma cells in MS remain unanswered. What factors drive B cells into the CNS, through which pathways do they travel, and are these cells persistent or transient? When during the course of disease do B cells populate the CNS and are there particular CNS niches in which B cells thrive? How may (GC)-like structures contribute to MS pathology? In this review, we will examine the chemotactic cues, migratory pathways, and CNS factors that facilitate B cells trafficking and survival in the inflamed CNS, and evaluate evidence supporting a compartmentalized B cell response in MS pathogenesis.
B Cell Migration into the CNS in Health and Disease
B Cells are Directed into the CNS by Chemokine Signaling
B cells may be observed in the healthy brain but are sparse in number, and increase drastically during neuroinflammation (11, 12). B cells express a robust array of chemokine receptors that largely dictate their movement, and the B cell chemokine receptor profile is dependent upon their state of differentiation and external microenvironment. The local milieu of cytokines in the inflamed CNS may also promote B cell migration by enhancing B cell chemoattraction and lymphoid organization. For instance, lymphotoxin-α expressed along the outer layer of inflamed vessel walls may facilitate lymphoid organogenesis and the formation of meningeal B cell GC-like structures (13).
Several chemokines and their receptors (in parentheses) have been shown to influence CNS B cell trafficking: CCL2 (CCR2, CCR3), CCL3 (CCR1, CCR5), CCL20 (CCR6), CXCL10 (CXCR3), CXCL12 (CXCR4, CXCR7), and CXCL13 (CXCR5) (Table 1). Among these factors, CXCL13 may play a central role. The CSF concentration of CXCL13 is elevated in MS patients (14), correlates with conversion from clinically isolated syndrome (CIS) to definite MS (15), and shows a strong correlation with B cell numbers in the CSF of MS and other neuroinflammatory diseases (14, 16). Indeed, nearly all CD19+ CSF B cells express the CXCL13 receptor, CXCR5 (14). Elevated CSF CXCL13 correlates strongly with the CNS accumulation of class-switched CD27+ memory B cells, CD27−IgD− B cells, and unswitched CD27+ memory B cells, but bears no relationship to the numbers of CD138 + CD38+ antibody-secreting plasmablasts and plasma cells (17). The ability of CXCL13 blockade to disrupt the formation of GC-like structures in the pancreatic islets of NOD mice suggests that meningeal B cell aggregates in MS patients may also develop from migrating memory B cells that differentiate intrathecally to plasmablasts and plasma cells (18).

Table 1. B cell chemokines in multiple sclerosis.
As short-lived plasmablasts comprise a significant proportion of the CSF B cell population in MS (28), the chemokines CXCL10 and CXCL12 may also act as chemoattractants for CXCR3+ and CXCR4+ plasmablasts and additionally regulate the dynamics of CNS B cell trafficking in disease. Since CXCL10 is constitutively expressed by a subset of cells in the CNS subventricular zone, the gradient of CXCL10 may be a potent chemo-attractant signal for both activated T cells and antibody-secreting cells (29).
Adhesion Molecules, B Cells, and the Blood–Brain Barrier
B cells follow chemokine gradients into the CNS via one of several anatomical pathways: (1) through the choroid plexus into the CSF; (2) through parenchymal vessels into the perivascular space; or (3) or through the post-capillary venules into the subarachnoid and Virchow–Robin spaces (30). B cells entering into the CNS through the choroid plexus must traverse apical tight junctions between epithelial cells composing the blood–CSF barrier, whereas B cells trafficking through parenchymal vessels or stromal venules ultimately need to traverse the tight junctions of the microvascular endothelial cells composing the blood–brain barrier (BBB). While the stages of lymphocyte transmigration across the blood–CSF barrier have yet to be described in detail, the sequence of leukocyte rolling, activation, arrest, crawling, and migration has been defined in great detail for blood–brain barrier trafficking (30). Basic adhesion molecule interactions important for T cell transmigration across the BBB include selectins during rolling (31), leukocyte very late antigen-4 (VLA-4) and endothelial vascular cell adhesion molecule-1 (VCAM-1) during the rolling and arrest, leukocyte lymphocyte function associated antigen-1 (LFA-1), and endothelial intercellular adhesion molecule-1 (ICAM-1) during arrest and migration (32, 33), as well as activated leukocyte cell adhesion molecule (ALCAM), and CD6 in migration (34).
The specific molecules required for B cell transmigration, however, are less clearly understood. Similar to requirements for T cell BBB transit, ex vivo studies using human adult brain-derived endothelial cells (HBECs) show that blockade of VLA-4, but not VCAM-1, inhibits B cell transmigration (35). Consistent with these findings, mice lacking the VLA-4 α-4 subunit specifically on B cells but not on other lymphocyte populations reduced disease severity significantly, and inhibited the recruitment of B cells into the CNS in an experimental autoimmune encephalitis model (36). In natalizumab-treated MS patients, CSF B and plasma cells are decreased in concert with the reduction in intrathecal CD4+ and CD8+ T cells (37). Complete (55%) or partial (27%) loss of CSF OCBs was observed in a natatlizumab-treated patient cohort following 2 years of therapy, suggesting that continuous trafficking of B cells to the CNS may be required to maintain the plasma cell niches producing intrathecal oligoclonal IgG (38). Antibody blockade of ICAM-1 and ALCAM also result in reduced migration of CD19+ B cells in ex vivo transmigration assays using HBECs as an artificial BBB (34, 35). The exact roles of ICAM-1 and ALCAM in CNS B cell trafficking in vivo, however, remain to be determined.
Recently, CNS meningeal lymphatic vessels containing T lymphocytes were discovered running parallel to the dural sinuses (39). These vessels drain to the deep cervical lymph nodes and may provide a novel route for trafficking B and T cells into or out of the CNS. This pathway may involve similar or distinct chemokine and adhesion molecules in the transit of various B cell populations that may infiltrate into the brain parenchyma, circulate in the CSF, populate GC-like structures, and transit back to the peripheral lymphoid compartment (39).
Bidirectional B Cell Trafficking in MS
In general, lymphocytic surveillance of the healthy CNS is significantly lower than that of other peripheral organs (40). The majority of data, particularly in humans and mice, indicate that activated antigen-experienced T and B cells constitute almost the entirety (41) or the vast majority (17, 42) of the infiltrating lymphocytes. Whether activated lymphocytes return from the CNS compartment to the peripheral circulation has remained uncertain.
Recently, the ability of B cells to exit the CNS compartment and re-enter the peripheral circulation and, potentially germinal center responses, has been investigated by deep sequencing (43). Deep, or next-generation sequencing, allows for high-throughput recovery of B cell IgG heavy-chain variable region (VH) repertoires from patient fluids and tissues. When compared to single-cell methods, the large number of VH sequences analyzed by deep sequencing provides a more complete representation of the B cell Ig repertoire contained in a biological sample and substantially increase the likelihood of observing identical or related VH sequences between samples. This enhanced sensitivity likely accounts for the frequent identification of common peripheral and CNS B cell clones with deep sequencing (43–45) and the rare identification of those with single-cell analyses (46, 47).
Using diverse strategies, patient populations, and methods, the VH repertoire from the peripheral blood, cervical lymph nodes, meninges, parenchyma, and CSF have been compared within the same MS patient (43–45). A common finding of each investigation was overlapping clonal B cell populations common to both the peripheral and CNS compartments. Overlapping peripheral blood and CSF B cell clones were observed among multiple subsets of Ig class-switched and post-germinal center B cells: CD27(+)IgD(−) memory B cells, CD27(hi)CD38(hi) plasma cells/plasmablasts, and CD27(−)IgD(−) negative memory B cells (44, 48, 49). While the number of overlapping sequences observed in each study varied due to technique and disease activity, lineage analysis of bi-compartmental B cell clones demonstrated patterns of somatic hypermutation consistent with bidirectional exchange (43–45). Some lineages showed a balanced distribution of peripheral and CNS compartment clones; while other lineages exhibited isolated CNS clones that were closely related to germline sequences. The pattern of overlapping B cell clones in these lineage trees suggest that B cells may travel back and forth across the BBB and re-enter germinal centers to undergo further somatic hypermutation (43–45) (Figure 1). In-depth analysis of the relationship between overlapping B cell clones in the cervical lymph nodes and CNS compartment of the same patient revealed that most of the shared VH clones were less mature sequences that originated, more often, in the periphery (45). More mature B cell clones tended to be restricted to either the peripheral lymph node or CNS compartment. Permutation testing supported a model in which B cell maturation into antibody-secreting cells occurs in both the periphery and CNS with antigen-specific maturation occurring in the periphery.
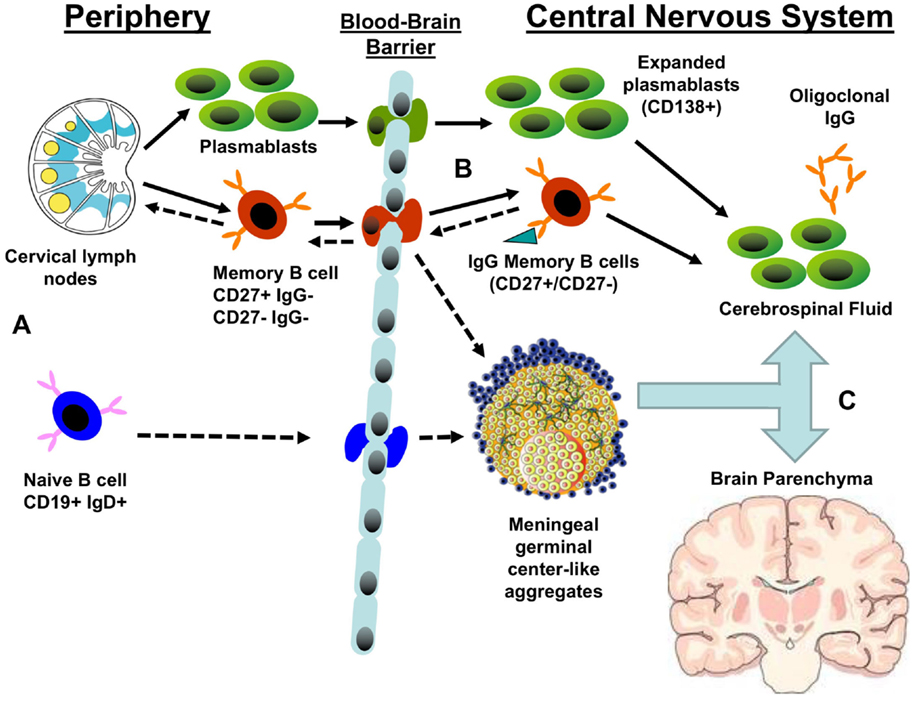
Figure 1. Potential patterns of B cell trafficking in multiple sclerosis. (A) The predominant stream of migratory B cells from the periphery to the CNS are likely to consist of either memory B cells or plasmablasts produced in the germinal centers of cervical lymph nodes. The presence of CSF B cell clones closely related to germline sequences suggests that naïve B cells may transit the blood–brain barrier to populate meningeal germinal center-like structures and produce CNS-restricted memory B cells. (B) Both migratory plasmablasts and memory B cells may contribute to the pool of central nervous system (CNS) antibody-secreting cells that produce the oligoclonal bands. Memory B cells may also enter germinal centers in meningeal lymphoid aggregates or draining cervical lymph nodes, resulting in further clonal expansion and affinity maturation. (C) A significant fraction of expanded B cell clones circulates between CNS compartments: cerebrospinal fluid, meningeal lymphoid aggregates, parenchymal lesions, and normal white matter. Solid arrows represent established pathways; dashed arrows represent putative pathways.
Compartmentalization of the CNS B Cell Response in MS
A key question related to MS pathogenesis is whether B cell-mediated antigen-driven responses are generated, supported, and sustained within the CNS (43)? CNS B cells show evidence of clonal expansion (50, 51), and express somatically mutated, class-switched Ig transcripts (46, 52–55). As noted previously, B cells with clonally related VH sequences are recovered on both sides of the BBB; however, CNS B cells may eventually form a compartmentalized population that is independent of the peripheral B cell pool as disease progresses. Interestingly, compartmentalized CNS inflammation has been hypothesized to drive treatment-resistant progressive disease (56).
Oligoclonal CSF IgG (OCBs) are observed in over 95% of MS patients. The CSF Ig proteome and B cell Ig transcriptome show strong overlap, indicating that CSF B cell clones are a major contributor to MS intrathecal IgG (57). In a subsequent study, peptide sequences from the CSF Ig proteome were also found to match heavy- (VH) and light-chain (VL) transcriptome sequences recovered from the CNS parenchyma and CSF of the same individual (58). The CSF Ig proteome covered high percentages of VH (CNS-77%; CSF-84%) and VL (CNS-39%; CSF-60%) transcriptome sequences in one patient and were somewhat limited in a second due to low CSF Ig quotient (58). The results indicate that B cells and IgG in MS CSF accurately mirror the humoral immunity present at the site of brain tissue damage (Figure 1). Indeed, 39–62% of the B cell transcriptome sequences recovered from the meninges, demyelinating plaques, normal appearing white matter, and CSF of the same MS patient were shared between intrathecal compartments, indicating that a significant fraction of intrathecal B cells trafficked through the CNS (59). Some expanded B cells clones, however, appear restricted to regions of MS plaque and meninges, suggesting some potential for localized tissue injury (59).
Interestingly, recent studies have questioned whether the CSF-restricted OCBs identified by isoelectric focusing are truly exclusive to the CNS (60). While the majority of CSF OCBs matched IgG-VH transcripts only recovered from the CSF B cell transcriptome, several OCB peptides matched bi-compartmental peripheral blood and CSF VH sequences. Although the type of MS and disease therapies were not reported, lineage tree analysis of bi-compartmental B cell populations suggested that these B cell groups underwent immune stimulation on both sides of the BBB (Figure 1). As a result, there remains the possibility that CNS immune populations may maintain molecular links with the periphery despite contrary data from isoelectric focusing.
Ig VH gene usage from the periphery and CNS provides additional data supporting compartmentalization of the humoral immune response in MS patients. The analysis of Ig VH sequences from demyelinating plaques and CSF of affected individuals reveal substantial VH4 family bias compared to normal VH4 prevalence (61, 62). Similar to patients with viral meningitis, CNS VH4 germline sequences displayed evidence of clonal expansion and extensive somatic mutations consistent with antigen selection (53, 54). MS patient with the longest disease course had the largest number of distinct IgG clonal populations, while the patients with recent diagnoses had limited clonal populations. CSF B cells from patients with a single demyelinating event (clinically isolated syndrome) also showed clonally expanded, somatically hypermutated VH genes (63, 64). Interestingly, both the overrepresentation of VH4 family sequences (65) and a unique pattern of somatic hypermutation “antibody gene signature” (66) within the CSF Ig VH transcriptome predicted transition to clinically definite MS. Recent deep sequencing of MS CSF VH repertoires from six MS patients has also revealed an overrepresentation of VH4-39, VH4-59, and VH4-61 heavy-chain sequences. The bias of MS CSF B cell heavy chains to VH4 germline sequences suggests that their basic structure may define an antigen-binding pocket that favors interaction with target antigen(s). As a result, a compartmental CNS humoral immune response may be able to drive CNS injury independent of peripheral immune activity.
Lastly, the GC-like structures or lymphoid infiltration have been noted in a large proportion of meningeal tissue from secondary progressive early stage cortical biopsies (7, 8, 67, 68). These CNS-specific immune infiltrates correlate with the severity of disease progression (8) and are associated with cortical neuronal loss in adjacent gray matter (69). The composition of these infiltrates included B cells, T cells, and dendritic cells, whose organization may resemble lymphoid follicles (7, 67). In addition, the presence of IgG and CXCL13 (7, 67) provide additional information, suggesting the active attraction and maintenance of B cells in MS meninges. The identification of CD19 + CD38hiCD77 + Ki67 + Bcl2− centroblasts in the CSF but not the peripheral blood of MS patients suggests that a compartmental humoral immune response in the MS CNS recapitulates all stages of B cell differentiation and may create a self-sufficient CNS response that is independent of the immune activity in the periphery (13). Additional data, however, are required to establish the relationship between the generation and maintenance of meningeal GC-like structures, intrathecal B cell clonal populations, and progressive disease (Figure 1). Peripheral B cell depletion, effective in early phase clinical trials in relapsing MS (9, 10), has not delivered similar efficacy for the treatment of primary progressive disease (70). This could be directly related to the inefficient depletion of the intrathecal B cell population in progressive (71) versus relapsing MS (72) due to compartmentalization of the B cell response in progressive disease and inefficient transit of anti-CD20 monoclonal antibody across the BBB. Interestingly, intrathecal administration of anti-CD20 monoclonal antibody rapidly depleted both peripheral and CD19+ B cells within days of delivery (73). Therefore, intrathecal anti-CD20 therapy may offer a novel avenue to evaluate the role of intrathecal B cell inflammation in progressive disease. The recent development of novel MRI techniques to identify meningeal follicles may offer a non-invasive tool to correlate therapeutic response with changes in meningeal inflammation (74).
Conclusion
Molecular analysis of the B cell response in MS has demonstrated that antigen-experienced B cells are shared between multiple CNS compartments and the peripheral immune response. Several features of CNS clonal B cell populations suggest that B cell subsets may not be shared between the CNS and periphery as disease progresses and that meningeal GC-like structures may support an independent, compartmentalized immune response that is correlative with measures of CNS injury. The data supporting the trafficking of B cells back and forth across the BBB are undermined by the technical constraints of single-cell PCR, deep sequencing, and sampling errors. For instance, the VH sequences defining the bi-compartmental B cell clones may be skewed by errors in PCR sequencing, multiple cDNA copies from the same cell, errors in flow cytometry, or limited blood and CSF sampling. Future studies are needed to confirm present data using defined MS cohorts at multiple stages of disease. The influence of current MS therapeutics on B cell trafficking and survival may be critical for understanding MS pathogenesis and establishing biomarkers of disease activity and therapeutic efficacy.
Author Contributions
Drs. KB, GO, and JB participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Support was provided by the NIH EY022936 (JLB), NS072141 (GPO), NS007321 (KB); the Guthy-Jackson Charitable Foundation (JLB, GPO); and the National Multiple Sclerosis Society (JLB, GPO).
References
1. Cepok S, Jacobsen M, Schock S, Omer B, Jaekel S, Boddeker I, et al. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain (2001) 124(Pt 11):2169–76. doi: 10.1093/brain/124.11.2169
2. Esiri MM. Multiple sclerosis: a quantitative and qualitative study of immunoglobulin-containing cells in the central nervous system. Neuropathol Appl Neurobiol (1980) 6(1):9–21. doi:10.1111/j.1365-2990.1980.tb00199.x
3. Kabat EA, Moore DH, Landow H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J Clin Invest (1942) 21(5):571–7. doi:10.1172/JCI101335
4. Lumsden CE. The immunogenesis of the multiple sclerosis plaque. Brain Res (1971) 28(3):365–90. doi:10.1016/0006-8993(71)90052-7
5. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol (2000) 47(6):707–17. doi:10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
6. Breij EC, Brink BP, Veerhuis R, van den Berg C, Vloet R, Yan R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol (2008) 63(1):16–25. doi:10.1002/ana.21311
7. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain (2007) 130(Pt 4):1089–104. doi:10.1093/brain/awm038
8. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain (2011) 134(Pt 9):2755–71. doi:10.1093/brain/awr182
9. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med (2008) 358(7):676–88. doi:10.1056/NEJMoa0706383
10. Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet (2011) 378(9805):1779–87. doi:10.1016/S0140-6736(11)61649-8
11. Anthony IC, Crawford DH, Bell JE. B lymphocytes in the normal brain: contrasts with HIV-associated lymphoid infiltrates and lymphomas. Brain (2003) 126(Pt 5):1058–67. doi:10.1093/brain/awg118
12. Monson NL, Ortega SB, Ireland SJ, Meeuwissen AJ, Chen D, Plautz EJ, et al. Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke. J Neuroinflammation (2014) 11:22. doi:10.1186/1742-2094-11-22
13. Corcione A, Casazza S, Ferretti E, Giunti D, Zappia E, Pistorio A, et al. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc Natl Acad Sci U S A (2004) 101(30):11064–9. doi:10.1073/pnas.0402455101
14. Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisakk P, Ransohoff RM, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain (2006) 129(Pt 1):200–11. doi:10.1093/brain/awh680
15. Brettschneider J, Czerwoniak A, Senel M, Fang L, Kassubek J, Pinkhardt E, et al. The chemokine CXCL13 is a prognostic marker in clinically isolated syndrome (CIS). PLoS One (2010) 5(8):e11986. doi:10.1371/journal.pone.0011986
16. Kowarik MC, Cepok S, Sellner J, Grummel V, Weber MS, Korn T, et al. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflammation (2012) 9:93. doi:10.1186/1742-2094-9-93
17. Haas J, Bekeredjian-Ding I, Milkova M, Balint B, Schwarz A, Korporal M, et al. B cells undergo unique compartmentalized redistribution in multiple sclerosis. J Autoimmun (2011) 37(4):289–99. doi:10.1016/j.jaut.2011.08.003
18. Henry RA, Kendall PL. CXCL13 blockade disrupts B lymphocyte organization in tertiary lymphoid structures without altering B cell receptor bias or preventing diabetes in nonobese diabetic mice. J Immunol (2010) 185(3):1460–5. doi:10.4049/jimmunol.0903710
19. Mahad DJ, Howell SJ, Woodroofe MN. Expression of chemokines in the CSF and correlation with clinical disease activity in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry (2002) 72(4):498–502. doi:10.1136/jnnp.72.4.498
20. Malmestrom C, Andersson BA, Haghighi S, Lycke J. IL-6 and CCL2 levels in CSF are associated with the clinical course of MS: implications for their possible immunopathogenic roles. J Neuroimmunol (2006) 175(1–2):176–82. doi:10.1016/j.jneuroim.2006.03.004
21. McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol (1998) 86(1):20–9. doi:10.1016/S0165-5728(98)00002-2
22. Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol (1998) 84(2):238–49. doi:10.1016/S0165-5728(97)00208-7
23. Kalinowska-Lyszczarz A, Szczucinski A, Pawlak MA, Losy J. Clinical study on CXCL13, CCL17, CCL20 and IL-17 as immune cell migration navigators in relapsing-remitting multiple sclerosis patients. J Neurol Sci (2011) 300(1–2):81–5. doi:10.1016/j.jns.2010.09.026
24. Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest (1999) 103(6):807–15. doi:10.1172/JCI5150
25. Sellebjerg F, Bornsen L, Khademi M, Krakauer M, Olsson T, Frederiksen JL, et al. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology (2009) 73(23):2003–10. doi:10.1212/WNL.0b013e3181c5b457
26. Blauth K, Zhang X, Chopra M, Rogan S, Markovic-Plese S. The role of fractalkine (CX3CL1) in regulation of CD4(+) cell migration to the central nervous system in patients with relapsing-remitting multiple sclerosis. Clin Immunol (2015) 157(2):121–32. doi:10.1016/j.clim.2015.01.001
27. Broux B, Pannemans K, Zhang X, Markovic-Plese S, Broekmans T, Eijnde BO, et al. CX(3)CR1 drives cytotoxic CD4(+)CD28(-) T cells into the brain of multiple sclerosis patients. J Autoimmun (2012) 38(1):10–9. doi:10.1016/j.jaut.2011.11.006
28. Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, et al. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain (2005) 128(Pt 7):1667–76. doi:10.1093/brain/awh486
29. Muzio L, Cavasinni F, Marinaro C, Bergamaschi A, Bergami A, Porcheri C, et al. Cxcl10 enhances blood cells migration in the sub-ventricular zone of mice affected by experimental autoimmune encephalomyelitis. Mol Cell Neurosci (2010) 43(3):268–80. doi:10.1016/j.mcn.2009.11.008
30. Takeshita Y, Ransohoff RM. Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunol Rev (2012) 248(1):228–39. doi:10.1111/j.1600-065X.2012.01127.x
31. Kivisakk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A (2003) 100(14):8389–94. doi:10.1073/pnas.1433000100
32. Wong D, Dorovini-Zis K. Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J Neuroimmunol (1992) 39(1–2):11–21. doi:10.1016/0165-5728(92)90170-P
33. Wong D, Prameya R, Dorovini-Zis K. In vitro adhesion and migration of T lymphocytes across monolayers of human brain microvessel endothelial cells: regulation by ICAM-1, VCAM-1, E-selectin and PECAM-1. J Neuropathol Exp Neurol (1999) 58(2):138–52. doi:10.1097/00005072-199902000-00004
34. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol (2008) 9(2):137–45. doi:10.1038/ni1551
35. Alter A, Duddy M, Hebert S, Biernacki K, Prat A, Antel JP, et al. Determinants of human B cell migration across brain endothelial cells. J Immunol (2003) 170(9):4497–505. doi:10.4049/jimmunol.170.9.4497
36. Lehmann-Horn K, Sagan SA, Bernard CC, Sobel RA, Zamvil SS. B-cell very late antigen-4 deficiency reduces leukocyte recruitment and susceptibility to central nervous system autoimmunity. Ann Neurol (2015) 77(5):902–8. doi:10.1002/ana.24387
37. Stuve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol (2006) 59(5):743–7. doi:10.1002/ana.20858
38. Mancuso R, Franciotta D, Rovaris M, Caputo D, Sala A, Hernis A, et al. Effects of natalizumab on oligoclonal bands in the cerebrospinal fluid of multiple sclerosis patients: a longitudinal study. Mult Scler (2014) 20(14):1900–3. doi:10.1177/1352458514538111
39. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi:10.1038/nature14432
40. Flugel A, Willem M, Berkowicz T, Wekerle H. Gene transfer into CD4+ T lymphocytes: green fluorescent protein-engineered, encephalitogenic T cells illuminate brain autoimmune responses. Nat Med (1999) 5(7):843–7. doi:10.1038/10567
41. Flugel A, Schwaiger FW, Neumann H, Medana I, Willem M, Wekerle H, et al. Neuronal FasL induces cell death of encephalitogenic T lymphocytes. Brain Pathol (2000) 10(3):353–64. doi:10.1111/j.1750-3639.2000.tb00267.x
42. Cepok S, von Geldern G, Grummel V, Hochgesand S, Celik H, Hartung H, et al. Accumulation of class switched IgD-IgM- memory B cells in the cerebrospinal fluid during neuroinflammation. J Neuroimmunol (2006) 180(1–2):33–9. doi:10.1016/j.jneuroim.2006.06.031
43. von Budingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest (2012) 122(12):4533–43. doi:10.1172/JCI63842
44. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med (2014) 6(248):248ra106. doi:10.1126/scitranslmed.3008930
45. Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med (2014) 6(248):248ra107. doi:10.1126/scitranslmed.3008879
46. Colombo M, Dono M, Gazzola P, Roncella S, Valetto A, Chiorazzi N, et al. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. J Immunol (2000) 164(5):2782–9. doi:10.4049/jimmunol.164.5.2782
47. Monson NL, Brezinschek HP, Brezinschek RI, Mobley A, Vaughan GK, Frohman EM, et al. Receptor revision and atypical mutational characteristics in clonally expanded B cells from the cerebrospinal fluid of recently diagnosed multiple sclerosis patients. J Neuroimmunol (2005) 158(1–2):170–81. doi:10.1016/j.jneuroim.2004.04.022
48. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol (2007) 178(10):6624–33. doi:10.4049/jimmunol.178.10.6624
49. Mahmood Z, Muhammad K, Schmalzing M, Roll P, Dorner T, Tony HP. CD27-IgD- memory B cells are modulated by in vivo interleukin-6 receptor (IL-6R) blockade in rheumatoid arthritis. Arthritis Res Ther (2015) 17:61. doi:10.1186/s13075-015-0580-y
50. Baranzini SE, Jeong MC, Butunoi C, Murray RS, Bernard CC, Oksenberg JR. B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J Immunol (1999) 163(9):5133–44.
51. Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol (2003) 171(5):2725–33. doi:10.4049/jimmunol.171.5.2725
52. Ritchie AM, Gilden DH, Williamson RA, Burgoon MP, Yu X, Helm K, et al. Comparative analysis of the CD19+ and CD138+ cell antibody repertoires in the cerebrospinal fluid of patients with multiple sclerosis. J Immunol (2004) 173(1):649–56. doi:10.4049/jimmunol.173.1.649
53. Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest (1998) 102(5):1045–50. doi:10.1172/JCI3568
54. Smith-Jensen T, Burgoon MP, Anthony J, Kraus H, Gilden DH, Owens GP. Comparison of immunoglobulin G heavy-chain sequences in MS and SSPE brains reveals an antigen-driven response. Neurology (2000) 54(6):1227–32. doi:10.1212/WNL.54.6.1227
55. Sandberg-Wollheim M, Turesson I. Lymphocyte subpopulations in the cerebrospinal fluid and peripheral blood in patients with multiple sclerosis. Scand J Immunol (1975) 4(8):831–6. doi:10.1111/j.1365-3083.1975.tb02693.x
56. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain (2009) 132:1175–89. doi:10.1093/brain/awp070
57. Obermeier B, Mentele R, Malotka J, Kellermann J, Kumpfel T, Wekerle H, et al. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat Med (2008) 14(6):688–93. doi:10.1038/nm1714
58. Obermeier B, Lovato L, Mentele R, Bruck W, Forne I, Imhof A, et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J Neuroimmunol (2011) 233(1–2):245–8. doi:10.1016/j.jneuroim.2011.01.010
59. Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, et al. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain (2011) 134(Pt 2):534–41. doi:10.1093/brain/awq350
60. Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol (2014) 75(2):266–76. doi:10.1002/ana.24088
61. Owens GP, Winges KM, Ritchie AM, Edwards S, Burgoon MP, Lehnhoff L, et al. VH4 gene segments dominate the intrathecal humoral immune response in multiple sclerosis. J Immunol (2007) 179(9):6343–51. doi:10.4049/jimmunol.179.9.6343
62. Owens GP, Kraus H, Burgoon MP, Smith-Jensen T, Devlin ME, Gilden DH. Restricted use of VH4 germline segments in an acute multiple sclerosis brain. Ann Neurol (1998) 43(2):236–43. doi:10.1002/ana.410430214
63. Qin Y, Duquette P, Zhang Y, Olek M, Da RR, Richardson J, et al. Intrathecal B-cell clonal expansion, an early sign of humoral immunity, in the cerebrospinal fluid of patients with clinically isolated syndrome suggestive of multiple sclerosis. Lab Invest (2003) 83(7):1081–8. doi:10.1097/01.LAB.0000077008.24259.0D
64. Haubold K, Owens GP, Kaur P, Ritchie AM, Gilden DH, Bennett JL. B-lymphocyte and plasma cell clonal expansion in monosymptomatic optic neuritis cerebrospinal fluid. Ann Neurol (2004) 56(1):97–107. doi:10.1002/ana.20152
65. Bennett JL, Haubold K, Ritchie AM, Edwards SJ, Burgoon M, Shearer AJ, et al. CSF IgG heavy-chain bias in patients at the time of a clinically isolated syndrome. J Neuroimmunol (2008) 199(1–2):126–32. doi:10.1016/j.jneuroim.2008.04.031
66. Cameron EM, Spencer S, Lazarini J, Harp CT, Ward ES, Burgoon M, et al. Potential of a unique antibody gene signature to predict conversion to clinically definite multiple sclerosis. J Neuroimmunol (2009) 213(1–2):123–30. doi:10.1016/j.jneuroim.2009.05.014
67. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol (2004) 14(2):164–74. doi:10.1111/j.1750-3639.2004.tb00049.x
68. Prineas JW. Multiple sclerosis: presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science (1979) 203(4385):1123–5. doi:10.1126/science.424741
69. Kooi EJ, Geurts JJ, van Horssen J, Bo L, van der Valk P. Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol (2009) 68(9):1021–8. doi:10.1097/NEN.0b013e3181b4bf8f
70. Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol (2009) 66(4):460–71. doi:10.1002/ana.21867
71. Monson NL, Cravens PD, Frohman EM, Hawker K, Racke MK. Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch Neurol (2005) 62(2):258–64. doi:10.1001/archneur.62.2.258
72. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol (2006) 180(1–2):63–70. doi:10.1016/j.jneuroim.2006.06.029
73. Svenningsson A, Bergman J, Dring A, Vagberg M, Birgander R, Lindqvist T, et al. Rapid depletion of B lymphocytes by ultra-low-dose rituximab delivered intrathecally. Neurol Neuroimmunol Neuroinflamm (2015) 2(2):e79. doi:10.1212/NXI.0000000000000079
Keywords: multiple sclerosis, B cells, lymphocyte trafficking, chemokines, blood–brain barrier
Citation: Blauth K, Owens GP and Bennett JL (2015) The ins and outs of B cells in multiple sclerosis. Front. Immunol. 6:565. doi: 10.3389/fimmu.2015.00565
Received: 03 September 2015; Accepted: 23 October 2015;
Published: 05 November 2015
Edited by:
Jorge Ivan Alvarez, University of Pennsylvania, USAReviewed by:
Nancy Monson, University of Texas Southwestern Medical Center, USAEdgar Meinl, Ludwig Maximilian University of Munich, Germany
Roberta Magliozzi, Verona University, Italy
Copyright: © 2015 Blauth, Owens and Bennett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey L. Bennett, amVmZnJleS5iZW5uZXR0QHVjZGVudmVyLmVkdQ==