 Tali Lang
Tali Lang Andrew Foote
Andrew Foote Jacinta P. W. Lee
Jacinta P. W. Lee Eric F. Morand
Eric F. Morand James Harris
James Harris- Lupus Research Group, Monash Centre for Inflammatory Diseases, School of Clinical Sciences at Monash Health, Faculty of Medicine, Nursing and Health Sciences, Monash Medical Centre, Clayton, VIC, Australia
Macrophage migration Inhibitory factor (MIF) was one of the earliest pro-inflammatory cytokines to be identified. Increasing interest in this cytokine in recent decades has followed the cloning of human MIF and the generation of Mif−/− mice. Deepening understanding of signaling pathways utilized by MIF and putative receptor mechanisms have followed. MIF is distinct from all other cytokines by virtue of its unique induction by and counter regulation of glucocorticoids (GCs). MIF is further differentiated from other cytokines by its structural homology to specific tautomerase and isomerase enzymes and correlative in vitro enzymatic functions. The role of MIF in immune and inflammatory states, including a range of human autoimmune diseases, is now well established, as are the relationships between MIF polymorphisms and a number of inflammatory diseases. Here, we review the known pleiotropic activities of MIF, in addition to novel functions of MIF in processes including autophagy and autophagic cell death. In addition, recent developments in the understanding of the role of MIF in systemic lupus erythematosus (SLE) are reviewed. Finally, we discuss the potential application of anti-MIF strategies to treat human diseases such as SLE, which will require a comprehensive understanding of the unique and complex activities of this ubiquitously expressed cytokine.
Introduction
Macrophage migration inhibitory factor (MIF) also known as glycosylation-inhibiting factor (GIF) is a multifunctional protein with a broad range of immunomodulatory properties. The existence of a MIF has been hypothesized since the experiments of Rich and Lewis (1), where it was shown that tuberculin-induced delayed-type hypersensitivity reactions (DTH) were associated with the inhibition of macrophage migration. However, it was not until 1966 that MIF was first described as a soluble factor responsible for the inhibition of emigration of macrophages during DTH (2, 3). MIF acts as a mediator of innate immunity by promoting host inflammatory responses through induction of pro-inflammatory cytokines, including TNF-α and IL-6. MIF can also modulate host inflammatory responses by regulating cellular processes such as T-cell proliferation, suppression of p53-dependent apoptosis, and counter regulation of the immunosuppressive actions of glucocorticoids (GCs). Human MIF cDNA was first isolated in 1989 (4), although both human and murine forms of MIF were not cloned and functionally tested until the early 1990s by Bernhagen and colleagues (5–7). The first MIF knockout mice (Mif−/−), reported in 1999, were generated through disruption and deletion of exon 3 in the MIF gene (8). Since then there has been significant scientific interest in MIF, which has been shown to function not only as a pro-inflammatory protein but also as a stress factor and a growth factor (9) released by cells of the anterior pituitary gland, similar to a hormone (10).
MIF Genetics and Protein Structure
Migration inhibitory factor is a non-glycosylated 12.5-kDa protein composed of 114 amino acids, highly conserved across species with murine MIF showing 90% homology to human MIF (11). Within the human genome, the MIF gene is located on chromosome 22 (22q11.23). MIF is composed of three short exons of 107, 172, and 66 bp and two introns of 188 and 94 bp (11). Crystal structures demonstrate that MIF is a homotrimer with structural homology to three bacterial enzymes; oxalocrotonate tautomerase, 5-carboxymethyl-2-hydroxymuconate isomerase, and chorismate mutase (12–16). Within recent years, a gene homologous to MIF, which encodes the protein d-dopachrome tautamerase (D-DT), has been included in the MIF superfamily (17). MIF and D-DT are located within close proximity on chromosome 22, and are nearly identical in exon lengths with variable non-coding intron regions.
MIF and D-DT gene expression are both regulated by transcription factors. MIF is regulated by ten known polymorphic sites, as previously described within the MIF gene (18). Two of these polymorphisms have been demonstrated to have functional impact and to influence susceptibility to and/or severity of a number of diseases (Table 1). The first is a short-tandem repeat (STR), which is a microsatellite repetition consisting of cytosine–adenine–thymine–thymine (CATT) at position −794 bp, −794 CATT5−8 (rs5844572) within the 5′ promoter region (19). High expression alleles such as −794 CATT7 have been associated with an increase in MIF gene expression, increased levels of circulating MIF (20), and severity in clinical phenotypes (20). Conversely, the sub-Saharan, low expression −794 CATT5 allele is associated with reduced levels of circulating MIF (21). The second polymorphism is a single nucleotide polymorphism (SNP) in which guanine (G) is replaced with cytosine (C) in the MIF gene at position −173 bp, −173 G > C (rs755662) (22). The −173*C allele has also been shown to correlate with increased levels of circulating MIF, as identified in several populations (20, 23). Based on findings published to date, it can be postulated that MIF promoter polymorphisms and consequent changes in MIF expression contribute to the susceptibility and clinical severity of many inflammatory and autoimmune disorders where MIF has been implicated. However, one should be cautious about associations made between expression of MIF alleles and clinical severity and/or susceptibility, and study limitations, such as ethnic populations recruited as well as overall cohort size, which may influence outcomes in gene association studies due to population stratification of the MIF gene locus, need to be considered.
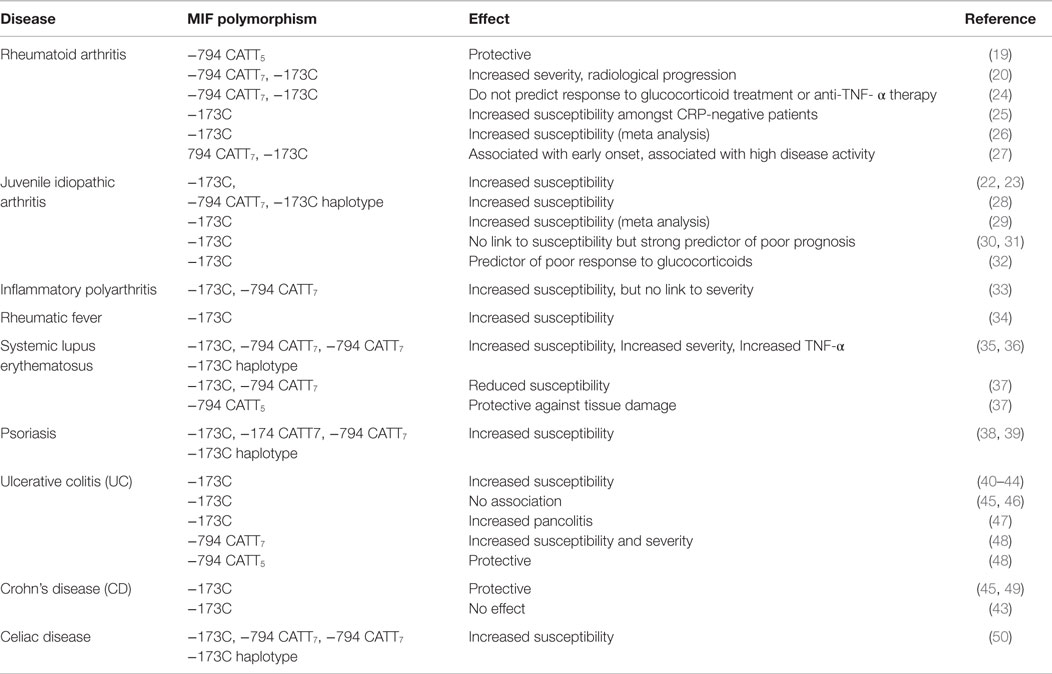
Table 1. Associations between MIF −173*C and −794 CATT5−8 polymorphisms and autoimmune disease.
Signal Transduction and Regulation of Cell Activation
Migration inhibitory factor is a highly pleiotropic cytokine, as reflected by the complexity of its involvement in regulating multiple signal transduction pathways (Figure 1). Cellular activation by MIF is reportedly initiated through interactions with its proposed receptor, CD74 – the cell surface form of the MHC class II invariant chain – which subsequently forms a signaling complex with the accessory protein CD44 (51). MIF has also been reported to interact with the chemokine receptors CXCR2 and CXCR4 in complexes involving CD74 (52–54).
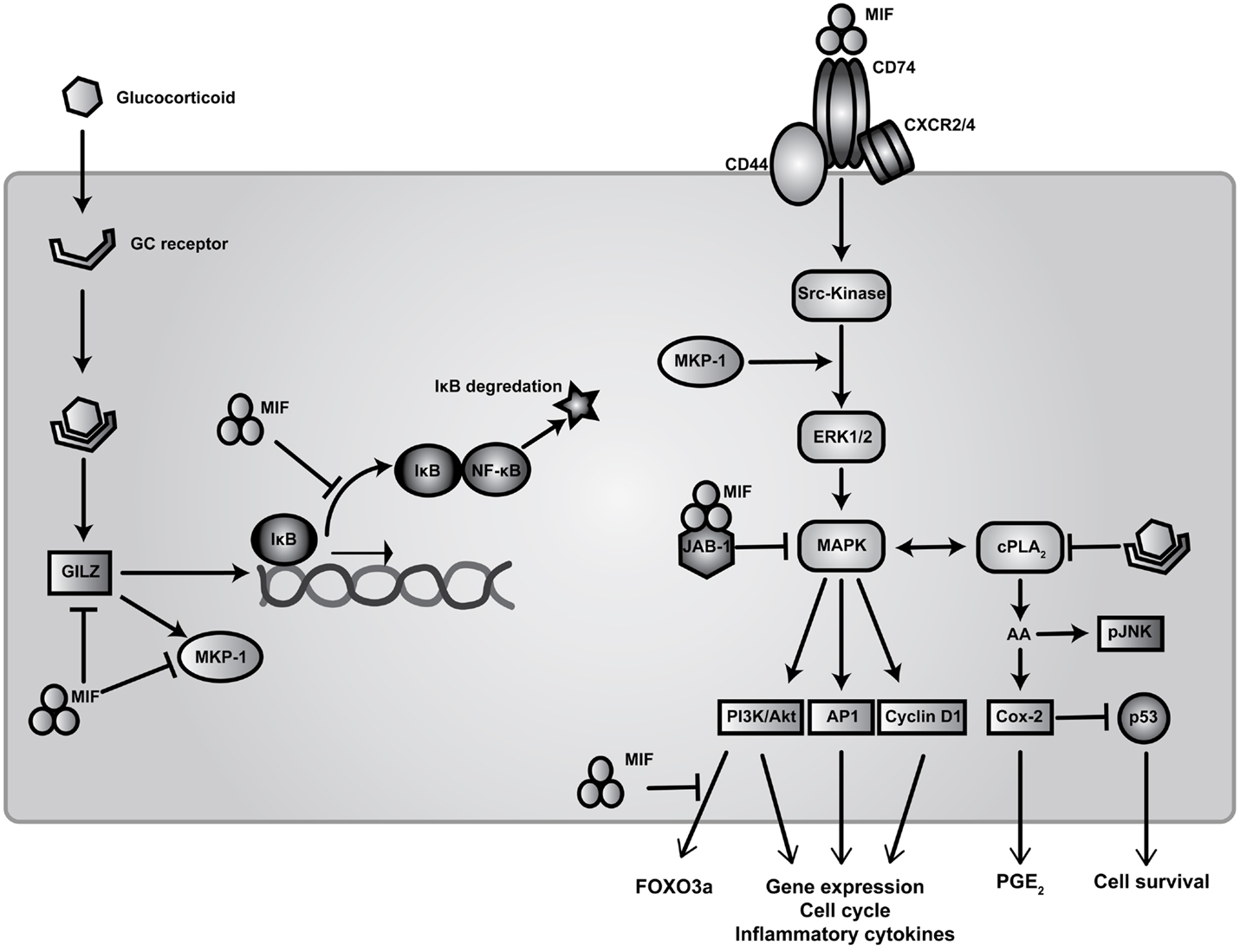
Figure 1. Signal transduction pathways of MIF and regulation of glucocorticoid immunosuppression. Extracellular MIF binds to the transmembrane receptor complex CD74 and CD44 to activate downstream Src-family kinase resulting in the subsequent phosphorylation of ERK/MAPK. This facilitates activation of transcription elements AP1, PI3K/Akt, and Cyclin D1 leading to expression of pro-inflammatory cytokines, cell cycle regulators, and co-stimulatory genes. MIF also binds to G-protein-coupled chemokine receptors CXCR2 and CXCR4 to promote calcium influx and integrin activation. Following endocytosis, MIF interaction with JAB-1 down-regulates MAPK thereby modulating cellular redox homeostasis. Elevated levels of MIF inhibit p53-mediated apoptosis through enhanced cPLA2 activity resulting in increased AA production and PGE2 release. Intracellular AA is required for the activation of pJNK for transcriptional stability of mRNAs for TNF-α and other cytokines. These signaling events can be inhibited by GCs via modulation of cPLA2 activity. GCs prevent expression of NF-ĸB-dependent genes primarily by increasing expression of IĸB. GC induced expression of MKP-1, which inactivates MAPK activity in response to pro-inflammatory stimuli, is inhibited by MIF through the dephosphorylation of multiple MAPK members. Finally, MIF can counter-regulate the expression of both MKP-1 and GILZ through blocking Akt-dependent activation and translocation of FoxO3a (55).
Migration inhibitory factor activates Src-family tyrosine kinases downstream of extracellular signal regulated kinase (ERK1/2) and p38, both members of the mitogen-activated protein kinase (MAPK) family (56, 57). Sustained activation of ERK is attained via c-Jun activation domain binding protein (JAB-1) (58). ERK activation leads to the phosphorylation of cytosolic proteins as well as up-regulation of phospholipase A2 (cPLA2) activity to produce prostaglandin precursors from arachidonic acid (AA) (56). AA activates JUN-terminal kinase (JNK), which is in turn required for activation of downstream regulatory elements ETS, PI3K/Akt, and AP1 (56, 59). These pathways lead to the transcription of pro-inflammatory cytokines, such as TNF-α, as well as chemokines such as CCL2 (MCP-1), implicated in the development of many autoimmune diseases, including SLE (60).
Elevated levels of MIF also result in reduced p53 accumulation in the cytoplasm, thereby blocking p53-mediated cell death and leading to continuous production of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and prostaglandins (11, 56, 61). This process occurs in an autocrine manner, whereby MIF phosphorylates ERK1/2 and activates cPLA2 and cyclooxygenase-2 (Cox-2), blocking p53-induced apoptosis. MIF has also been shown to counter-regulate GC-induced expression of MAPK phosphatase-1 (MKP-1), a critical MAPK signaling inhibitor, through which GCs signal to suppress pro-inflammatory cytokine secretion (62, 63). It was shown by Roger and others that MIF targets MKP-1 in an autocrine manner to prevent GC-induced MKP-1 expression, thereby tempering the post-transcriptional inhibition of cytokine production by GCs (62, 64). Moreover, MIF-deficient macrophages show increased sensitivity to GCs following LPS stimulation, with higher levels of MKP-1 expression and reduced activation and phosphorylation of p38-MAPK (62, 64). However, in this study no effect of GCs on IĸBα levels was observed. Correspondingly, there was no interference in the ability of NF-ĸB to translocate to the nucleus and bind the TNF-α promoter, which correlates with previous findings from earlier studies (63, 65, 66). It has been proposed that decreased levels of MIF leads to increased MKP-1, resulting in destabilization of AU-rich elements found in mRNA of multiple cytokines, via phosphorylation and activation of the downstream target MAPK-activated protein kinase-2 (MAPKAPK2) (67). Therefore, MIF counter regulation of anti-inflammatory actions by GCs results in the reduced expression of MKP-1, increased activity of p38 and MAPKAPK2 and greater stability of mRNAs conferring AU-rich element-dependent translation.
Studies from own lab have identified a novel molecular mechanism through which MIF can regulate the expression of MKP-1 and activation of MAPK through the GC-responsive protein, GC-induced leucine zipper (GILZ, also known as TSC22 domain family protein 3) (68). GILZ interacts with numerous signaling pathways relevant to inflammatory diseases (69). We recently demonstrated that exogenous MIF inhibited both GILZ and MKP-1 expression in fibroblasts and macrophages. MIF regulation of GILZ was shown to occur through inhibition of the Akt-dependent nuclear translocation of the transcription factor, FoxO3a. Moreover, MIF inhibition of MKP-1 expression was dependent on this inhibition of GILZ, suggesting a novel mechanism through which MIF impairs GC sensitivity (17).
MIF and Immunity
Migration inhibitory factor is produced by most cells of the immune system, including monocytes, macrophages, blood dendritic cells, B-cells, T-cells, neutrophils, eosinophils, mast cells, and basophils (1). MIF is constitutively expressed and stored in intracellular pools, so does not require de novo synthesis for secretion. MIF is secreted by macrophages following stimulation with LPS, or other pro-inflammatory cytokines such as TNF and IFN-γ (6). Moreover, macrophage-derived MIF can stimulate the synthesis of other pro-inflammatory mediators via autocrine and paracrine effects, enhancing macrophage functions, including phagocytosis, reactive oxygen species (ROS) production, and nitric oxide (NO) production (6, 70–72).
Migration inhibitory factor is secreted by pituitary cells following LPS stimulation in vivo and this contributes significantly to circulating MIF in the post-acute phase of LPS-induced endotoxemia. Furthermore, co-injection of MIF with LPS increases lethality, while anti-MIF antibody protects mice against LPS-induced endotoxemia (10). The small molecule MIF antagonist, ISO-1 ((S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester), also protects mice against LPS-induced endotoxemia and reduces TNF-α production by peritoneal macrophages (73). Similarly, MIF-deficient mice are protected against lethal sepsis induced by LPS or Staphylococcus aureus enterotoxin B (SEB) with d-galactosamine (8).
While MIF clearly has pathogenic roles to play in responses to bacterial products, it also facilitates the detection of endotoxin-containing bacteria through the up-regulation of TLR4 in macrophages, allowing rapid and protective pro-inflammatory responses to these pathogens (11). Consistent with this, MIF-deficient mice were more susceptible to infection with Salmonella typhimurium, producing lower levels of IL-12, IFN-γ, and TNF-α (74). More recently, MIF has been shown to play a role in protection against Mycobacterium tuberculosis, which does not express LPS and has a more complex relationship with TLR4 (75). Mif−/− mice are more susceptible to infection with either M. tuberculosis or Mycobacterium bovis BCG and demonstrate inhibited secretion of TNF-α, IL-12, and IL-10 (76). Moreover, the low expresser MIF genotype −794 CATT5/5 is enriched in a cohort of Ugandan patients with HIV and disseminated tuberculosis (TB) (76).
Migration inhibitory factor is constitutively expressed by T cells and secreted in response to mitogenic or antigenic stimulation (77, 78); treatment with anti-MIF antibody reduces T cell IL-2 production and proliferation (78–81). In vivo, MIF has a well-defined role in DTH, which is inhibited by MIF neutralization or deficiency, leading to decreased antigen-specific T cell proliferation, IgG production and IFN-γ secretion (78, 80, 82). MIF can stimulate secretion of both Th1 and Th2 cytokines by T cells (78), as well as IL-17 by lymph node cells (83), suggesting no single clear role in T cell polarization. MIF can also facilitate leukocyte recruitment and trafficking through the up-regulation of classical chemokines, such as CXCL8 (IL-8), CCL-5 (RANTES), and CCL-2 (MCP-1) (57, 84, 85). Moreover, MIF is now suggested to be a non-cognate ligand of the chemokine receptors CXCR2 and CXCR4, through which it can influence chemotaxis of monocytes, T cells, and B cells directly (52, 86). Interestingly, in the context of tumor immunology it has been demonstrated that MIF promotes the infiltration of immune-suppressive cells, including myeloid-derived suppressor cells (MDSCs) and Tregs (87, 88). These cell sub-populations have been implicated in tumor progression and metastasis by limiting anti-tumor immunity, as well as inducing immune tolerance. It is unknown whether MIF similarly modulates immune-suppressive cells in the context of other inflammatory diseases, including SLE.
MIF and Autophagy
Autophagy is a catabolic pathway for the delivery of cytosolic constituents, including long-lived proteins, protein aggregates and organelles, to lysosomes for degradation. Activated during nutrient deprivation, autophagy acts as a cytoprotective mechanism for amino acid recycling (89). In addition, autophagy has been shown by many groups to regulate the transcription, processing and secretion of pro-inflammatory cytokines (90). Inhibition of autophagy increases the secretion of IL-1α, IL-1β, IL-18, and IL-23 by macrophages and dendritic cells in response to TLR agonists (91–94). This process is dependent on the accumulation of ROS and mitochondrial DNA in the cytosol (95). Conversely, induction of autophagy inhibits the secretion of IL-1β and IL-23 (92, 96). Recent studies have demonstrated that MIF can regulate autophagy. In one study, MIF was shown to suppress a phenomenon termed autophagic cell death in the human MCF-7 breast cancer cell line (97). This is likely due to activation of the PI3K/Akt pathway, which inhibits autophagy (98, 99). In contrast, other studies have suggested that MIF induces or facilitates autophagy in mouse myoblasts, cardiomyocytes and human HuH-7 hepatoma cells (100–103). MIF-induced autophagy in HuH-7 cells was dependent on the generation of ROS and, interestingly, starvation induced MIF secretion, again dependent on ROS (100). However, it is not clear whether this is an autophagy-dependent process, or a side effect of amino acid starvation, independent of autophagy induction.
Given that autophagy has been linked to a number of inflammatory diseases (104) and there is evidence to suggest that autophagy is dysregulated in SLE patients (105–108), a better understanding of how MIF intersects with this important cellular process could prove highly significant.
MIF and Systemic Lupus Erythematosus
Given its pleiotropic role in the regulation of inflammatory cytokines and leukocyte trafficking, it is perhaps unsurprising that MIF has been linked with a number of autoimmune and inflammatory diseases. Genetic studies have identified associations between MIF polymorphisms and autoimmune diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), type I diabetes, and autoimmune liver disease (Table 1). Here, we will discuss the association of MIF polymorphisms and their relevance to disease progression, severity and clinical outcomes in SLE.
Systemic lupus erythematosus is complex chronic multi-organ autoimmune disease of unknown etiology with significant heterogeneity in clinical manifestations. It is generally considered a multifactorial disease, as a combination of genetics, environmental triggers, sex hormones, and other factors are thought to be involved (109). SLE is most prevalent within African-American and Asian populations, typically in females of childbearing age (110, 111). SLE is characterized by loss of tolerance to nucleic acids and their interacting proteins, resulting in the development of autoantibodies, inflammation, and tissue damage (109). Our own lab has demonstrated that levels of circulating MIF are raised in patients with SLE and are positively associated with disease damage (measured by SLICC/ACR index) and, interestingly, GC use (112). Similarly, renal MIF is increased in patients with lupus (and non-lupus) proliferative glomerulonephritis, correlating with leukocyte infiltration, tissue damage and impairment of renal function (113). To date, it is unknown whether kidney injury associated with SLE contributes to elevated serum MIF, and thus future studies examining levels of serum and urine MIF in relation to lupus nephritis are needed.
In the lupus-prone MRL/lpr mouse strain, MIF expression has been demonstrated to be increased in both skin and kidney lesions. Correspondingly, Mif−/− mice showed prolonged survival, reduced renal and skin lesions, as well as reduced proteinuria and glomerular injury; MIF deficiency was associated with almost complete protection from crescentic nephritis in this model (114). Complementary to this study, treatment of either MRL/lpr mice and another lupus-prone strain, NZB/NZW F1 mice, with the MIF antagonist ISO-1, reduced functional and histological indices of glomerulonephritis, inhibited CD74+ and CXCR4+ leukocyte recruitment, and lowered levels of circulating TNF-α in MRL/lpr mice and CCL2 in NZB/NZW F1 mice (115). Expression of mRNA for TNF-α, IL-1β, and CCL2 in kidney tissue was reduced in both strains of lupus-prone mice following treatment with ISO-1. Neither autoantibody production nor T and B cell activation was significantly affected (115), suggesting that the protective effect of MIF inhibition in SLE is dependent on the regulation of innate inflammation rather that autoimmunity. This conclusion is aligned with findings in MIF-deficient MRL/lpr mice, in which protection from renal damage was not accompanied by any change in systemic autoantibody levels or local autoantibody deposition (114). In humans, serum MIF levels are increased in patients with SLE, although this can be partly explained by increased GC use (112). However, independent of GC-induced MIF, high serum levels have been positively associated with SLE disease damage (SLICC/ACR index) (112).
To date, few studies have comprehensively investigated the role of MIF polymorphisms in the susceptibility and severity of SLE, thus leaving many questions to be answered as to how MIF alleles contribute to pathogenesis. Findings from one study with a multinational cohort of 1369 SLE patients showed that both Caucasian and African-American patients with the high expression haplotype −794 CATT7/−173*C had a lower incidence of SLE with higher levels of circulating MIF (37). Moreover, when they looked at the relationship between MIF alleles and anti-nuclear antibody (ANA) status, both healthy controls and SLE patients with the high expression CATT7 or −173*C alleles or the CATT7/−173*C haplotype were less likely to be ANA positive. These findings possibly suggest that high expression MIF alleles confer some protection from autoimmunity in SLE. One possible explanation for this is that high expression MIF alleles confer protection against infections, such as community-acquired pneumonia (18), which may be mechanistic triggers for SLE through antigenic mimicry. Conversely, patients within the cohort, who had established SLE with end-organ complications, such as serositis, nephritis, and cerebritis, had lower frequencies of the low expression MIF −794 CATT5 allele (37). This would suggest that, in patients with established disease, higher levels of MIF are associated with greater pathology, a finding consistent with the murine studies described above.
In contrast to this study, a report on a Mexican SLE cohort showed both the −173*C and −794 CATT7 polymorphisms increased susceptibility to SLE (35). In this study, both serum MIF and TNF-α were significantly increased in SLE patients and in patients with the high expression polymorphisms. Similarly, Sánchez et al. (36), reported that the −173*C allele was associated with increased susceptibility to SLE in a Spanish cohort and that homozygosity (−173C/C) increased susceptibility further. Moreover, the −173*C haplotype with the −794 CATT7 allele conferred a twofold increase in susceptibility to SLE. In this study, none of the MIF polymorphisms were significantly associated with specific clinical manifestations.
MIF as a Potential Therapeutic Target
In healthy individuals, MIF is typically found circulating in plasma at a range between 2 and 8 ng/ml. However, in autoimmune disease MIF concentrations can fluctuate to markedly higher levels. As such, MIF is commonly seen as a hallmark of disease progression and chronicity, even if increased levels of MIF are the consequence of exacerbated inflammatory cascades, rather than a primary cause of disease (116). By virtue of its breadth of activities, MIF is an essential regulator of innate and inflammatory responses (11). Conversely, MIF can also regulate physiological cell activities enzymatically, as a d-dopachrome tautomerase, phenylpyruvate tautomerase, or a thiol-protein oxidoreductase (14, 117, 118). Given MIF is a pluripotent protein with a range of biological functions, it has become an attractive small molecule and antibody target for therapeutic intervention in autoimmune inflammatory disorders. Currently, there are several classes of small molecule inhibitors of MIF that are designed to interact with MIF at its tautomerase active site and attenuate its pro-inflammatory activities. Most of these inhibitors, including ISO-1 and related molecules, work through direct binding to the active site, allosteric inhibition, modification of residues within the active site or disturbance to the tautomerase trimer (119–121). However, the majority of the reported compounds are not suitable candidates for pharmaceutical development due to the high concentrations (micromolar) required for activity (122). Currently, there are 11 classes of MIF small molecule inhibitors described within the literature [previously comprehensively reviewed in Ref. (123)]; one recently reported novel compound class was shown to have protective effects in a model of myocardial infarction (124), which is the most common cause of death in SLE patients (125).
To date, only a handful of MIF inhibitors have been found to disrupt MIF-CD74 interactions with IC50 values of less than 5 μM (120). More promisingly, AV411 (ibudilast; 3-isobutyryl-2-isopropylpyrazolo-[1,5-a]pyridine), is a non-selective inhibitor of phosphodiesterases that is used clinically as an anti-inflammatory drug to treat bronchial asthma and post-stroke complications. Cho and colleagues demonstrated that AV411 was able to allosterically inhibit MIF’s catalytic capabilities in vitro via conversion/substitution of a methyl-group to an amine group, which induces conformational changes in pockets adjacent to the active site (126). Furthermore, AV411 was shown to significantly inhibit chemotactic capabilities of PBMCs at clinically relevant concentrations (10 nM) (127).
Migration inhibitory factor is highly stable in its trimer conformation, but relatively unstable as a monomer. Ebselen, a compound known for its anti-inflammatory and anti-oxidant properties, was reported as the first small molecule inhibitor to interfere with MIF oligomerization through interactions with cysteine residues (119). This results in changes to the structural conformation of MIF, consequently inhibiting its ability to induce AKT phosphorylation and induction of pro-inflammatory cytokines. Furthermore, Ebselen was shown to reduce chemotactic activities of epithelial progenitor cells in the presence of recombinant MIF. More recently, Bai and colleagues have reported on a novel allosteric MIF inhibitor, p425, which occupies the interface of two MIF trimers (128). p425 was shown not only to potently inhibit MIF’s ability to tautomerize 4-hyrdoxy-phenyl pyruvate but also block the interaction between MIF and CD74, thereby hampering its pro-inflammatory actions (128). Molecular docking and modeling techniques have also been extensively used to characterize potential compounds that will specifically interact with MIF to block its tautomerase activities. Most compounds generated following computational analysis are tested in vitro using a variety of cell-based assays. However, very few of these in silico modeled compounds have been successfully translated for clinical use, due to very high IC50 values required for desired inhibitory effects (120, 129, 130). It is important to note that the connection, if any, between MIF’s tautomerase activity and pro-inflammatory actions, is unclear [see Ref. (131)]. Drugs which inhibit tautomerase activity might also induce conformational changes that alter MIF signaling and vice versa.
The use of anti-MIF neutralizing antibodies has be shown to be therapeutically efficacious in several models of inflammatory and autoimmune diseases (82, 132–135). In a recent study, Tarasuk and colleagues employed the human single-chain variable fragment (HuScFv) monoclonal antibody no. 22 to illustrate binding capabilities specific to MIF using a variety of in vitro based assays (136). Moreover, the tautomerase activity of MIF was dose-dependently reduced in the presence of HuScFv antibody through binding of the antibody to catalyitic residues within the tautomerase active site (136). Anti-MIF monoclonal antibody therapy is currently in phase I trials both for solid tumors (NCT01765790) and for lupus nephritis (NCT01541670).
Future Perspectives
Given the abundance of studies implicating MIF as a fundamental participant in the pathogenesis and progression in autoimmune diseases, MIF may represent a therapeutic target with untapped potential for benefits in the clinic. The effects of MIF to amplify pathogenic pathways, including cytokine expression, T cell activation, and macrophage function, as well as its effects to hinder the efficacy of GCs, mean that antagonizing MIF could have broad application in immune disease. The development of therapeutics using small molecule inhibitors that abrogate tautomerase activity has been limited, as many reported compound classes are not practical for pharmaceutical development. However, the development of anti-MIF monoclonal antibodies has opened new avenues. Understanding the precise mechanisms by which MIF regulates signaling cascades involved in inflammatory conditions will provide new and important insights into the potential to exploit inhibition of MIF to regulate inflammation and immunopathogenesis in autoimmune disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Ms. Vanessa Orlowski for her artistic contribution to this manuscript. This work is funded in part by a National Health & Medical Research Council (Australia) Project grant (1068040).
References
1. Rich AR, Lewis MR. The nature of allergy in tuberculosis as revealed by tissue culture studies. Bull Johns Hopkins Hosp (1932) 50:115–31.
2. Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science (1966) 153:80–2. doi:10.1126/science.153.3731.80
3. David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci U S A (1966) 56:72–7. doi:10.1073/pnas.56.1.72
4. Weiser WY, Temple PA, Witek-Giannotti JS, Remold HG, Clark SC, David JR. Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A (1989) 86:7522–6. doi:10.1073/pnas.86.19.7522
5. Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry (1994) 33:14144–55. doi:10.1021/bi00251a025
6. Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med (1994) 179:1895–902. doi:10.1084/jem.179.6.1895
7. Bernhagen J, Calandra T, Cerami A, Bucala R. Macrophage migration inhibitory factor is a neuroendocrine mediator of endotoxaemia. Trends Microbiol (1994) 2:198–201. doi:10.1016/0966-842X(94)90111-H
8. Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med (1999) 189:341–6. doi:10.1084/jem.189.2.341
9. Lan HY. Role of macrophage migration inhibition factor in kidney disease. Nephron Exp Nephrol (2008) 109:e79–83. doi:10.1159/000145463
10. Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature (1993) 365:756–9. doi:10.1038/365756a0
11. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol (2003) 3:791–800. doi:10.1038/nri1200
12. Subramanya HS, Roper DI, Dauter Z, Dodson EJ, Davies GJ, Wilson KS, et al. Enzymatic ketonization of 2-hydroxymuconate: specificity and mechanism investigated by the crystal structures of two isomerases. Biochemistry (1996) 35:792–802. doi:10.1021/bi951732k
13. Sugimoto H, Suzuki M, Nakagawa A, Tanaka I, Nishihira J. Crystal structure of macrophage migration inhibitory factor from human lymphocyte at 2.1 A resolution. FEBS Lett (1996) 389:145–8. doi:10.1016/0014-5793(96)00553-4
14. Sugimoto H, Taniguchi M, Nakagawa A, Tanaka I, Suzuki M, Nishihira J. Crystal structure of human D-dopachrome tautomerase, a homologue of macrophage migration inhibitory factor, at 1.54 A resolution. Biochemistry (1999) 38:3268–79. doi:10.1021/bi982184o
15. Sun HW, Bernhagen J, Bucala R, Lolis E. Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A (1996) 93:5191–6. doi:10.1073/pnas.93.11.5191
16. Suzuki M, Sugimoto H, Nakagawa A, Tanaka I, Nishihira J, Sakai M. Crystal structure of the macrophage migration inhibitory factor from rat liver. Nat Struct Biol (1996) 3:259–66. doi:10.1038/nsb0396-259
17. Merk M, Mitchell RA, Endres S, Bucala R. D-dopachrome tautomerase (D-DT or MIF-2): doubling the MIF cytokine family. Cytokine (2012) 59:10–7. doi:10.1016/j.cyto.2012.03.014
18. Yende S, Angus DC, Kong L, Kellum JA, Weissfeld L, Ferrell R, et al. The influence of macrophage migration inhibitory factor gene polymorphisms on outcome from community-acquired pneumonia. FASEB J (2009) 23:2403–11. doi:10.1096/fj.09-129445
19. Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Plant BJ, Wolfe F, et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun (2002) 3:170–6. doi:10.1038/sj.gene.6363867
20. Radstake TR, Sweep FC, Welsing P, Franke B, Vermeulen SH, Geurts-Moespot A, et al. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum (2005) 52:3020–9. doi:10.1002/art.21285
21. Awandare GA, Martinson JJ, Were T, Ouma C, Davenport GC, Ong’echa JM, et al. MIF (macrophage migration inhibitory factor) promoter polymorphisms and susceptibility to severe malarial anemia. J Infect Dis (2009) 200:629–37. doi:10.1086/600894
22. Donn RP, Shelley E, Ollier WE, Thomson W; British Paediatric Rheumatology Study Group. A novel 5’-flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum (2001) 44:1782–5. doi:10.1002/1529-0131(200108)44:8<1782::AID-ART314>3.0.CO;2-#
23. Donn R, Alourfi Z, De Benedetti F, Meazza C, Zeggini E, Lunt M, et al. Mutation screening of the macrophage migration inhibitory factor gene: positive association of a functional polymorphism of macrophage migration inhibitory factor with juvenile idiopathic arthritis. Arthritis Rheum (2002) 46:2402–9. doi:10.1002/art.10492
24. Radsake TR, Fransen J, Toonen EJ, Coenen MJ, Donn R, Van den Hoogen FH, et al. Macrophage migration inhibitory factor polymorphisms do not predict therapeutic response to glucocorticoids or to tumour necrosis factor alpha-neutralising treatments in rheumatoid arthritis. Ann Rheum Dis (2007) 66:1525–30. doi:10.1136/ard.2006.064394
25. Liu R, Xu N, Wang X, Shen L, Zhao G, Zhang H, et al. Influence of MIF, CD40, and CD226 polymorphisms on risk of rheumatoid arthritis. Mol Biol Rep (2012) 39:6915–22. doi:10.1007/s11033-012-1518-y
26. Xie Q, Wang SC, Bian G, Zhan FL, Xie JK, Li J. Association of MIF-173G/C and MBL2 codon 54 gene polymorphisms with rheumatoid arthritis: a meta-analysis. Hum Immunol (2012) 73:966–71. doi:10.1016/j.humimm.2012.07.043
27. Llamas-Covarrubias MA, Valle Y, Bucala R, Navarro-Hernández RE, Palafox-Sánchez CA, Padilla-Gutiérrez JR, et al. Macrophage migration inhibitory factor (MIF): genetic evidence for participation in early onset and early stage rheumatoid arthritis. Cytokine (2013) 61:759–65. doi:10.1016/j.cyto.2012.12.032
28. Donn R, Alourfi Z, Zeggini E, Lamb R, Jury F, Lunt M, et al. A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum (2004) 50:1604–10. doi:10.1002/art.20178
29. Lee YH, Bae SC, Song GG. The association between the functional PTPN22 1858 C/T and MIF -173 C/G polymorphisms and juvenile idiopathic arthritis: a meta-analysis. Inflamm Res (2012) 61:411–5. doi:10.1007/s00011-012-0447-5
30. Berdeli A, Ozyürek AR, Ulger Z, Gürses D, Levent E, Salar K, et al. Association of macrophage migration inhibitory factor gene -173 G/C polymorphism with prognosis in Turkish children with juvenile rheumatoid arthritis. Rheumatol Int (2006) 26:726–31. doi:10.1007/s00296-005-0062-7
31. De Benedetti F, Meazza C, Vivarelli M, Rossi F, Pistorio A, Lamb R, et al. Functional and prognostic relevance of the -173 polymorphism of the macrophage migration inhibitory factor gene in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum (2003) 48:1398–407. doi:10.1002/art.10882
32. Vivarelli M, D’Urbano LE, Insalaco A, Lunt M, Jury F, Tozzi AE, et al. Macrophage migration inhibitory factor (MIF) and oligoarticular juvenile idiopathic arthritis (o-JIA): association of MIF promoter polymorphisms with response to intra-articular glucocorticoids. Clin Exp Rheumatol (2007) 25:775–81.
33. Barton A, Lamb R, Symmons D, Silman A, Thomson W, Worthington J, et al. Macrophage migration inhibitory factor (MIF) gene polymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes Immun (2003) 4:487–91. doi:10.1038/sj.gene.6364014
34. Col-Araz N, Pehlivan S, Baspinar O, Sever T, Oguzkan-Balci S, Balat A. Association of macrophage migration inhibitory factor and mannose-binding lectin-2 gene polymorphisms in acute rheumatic fever. Cardiol Young (2013) 23:486–90. doi:10.1017/S1047951112000972
35. De la Cruz-Mosso U, Bucala R, Palafox-Sanchez CA, Parra-Rojas I, Padilla-Gutierrez JR, Pereira-Suarez AL, et al. Macrophage migration inhibitory factor: association of -794 CATT5-8 and -173 G>C polymorphisms with TNF-alpha in systemic lupus erythematosus. Hum Immunol (2014) 75:433–9. doi:10.1016/j.humimm.2014.02.014
36. Sánchez E, Gómez LM, Lopez-Nevot MA, González-Gay MA, Sabio JM, Ortego-Centeno N, et al. Evidence of association of macrophage migration inhibitory factor gene polymorphisms with systemic lupus erythematosus. Genes Immun (2006) 7:433–6. doi:10.1038/sj.gene.6364310
37. Sreih A, Ezzeddine R, Leng L, LaChance A, Yu G, Mizue Y, et al. Dual effect of the macrophage migration inhibitory factor gene on the development and severity of human systemic lupus erythematosus. Arthritis Rheum (2011) 63:3942–51. doi:10.1002/art.30624
38. Donn RP, Plant D, Jury F, Richards HL, Worthington J, Ray DW, et al. Macrophage migration inhibitory factor gene polymorphism is associated with psoriasis. J Invest Dermatol (2004) 123:484–7. doi:10.1111/j.0022-202X.2004.23314.x
39. Wu J, Chen F, Zhang X, Li Y, Ma H, Zhou Y, et al. Association of MIF promoter polymorphisms with psoriasis in a Han population in Northeastern China. J Dermatol Sci (2009) 53:212–5. doi:10.1016/j.jdermsci.2008.11.002
40. Falvey JD, Bentley RW, Merriman TR, Hampton MB, Barclay ML, Gearry RB, et al. Macrophage migration inhibitory factor gene polymorphisms in inflammatory bowel disease: an association study in New Zealand Caucasians and meta-analysis. World J Gastroenterol (2013) 19:6656–64. doi:10.3748/wjg.v19.i39.6656
41. Fei BY, Lv HX, Yang JM, Ye ZY. Association of MIF-173 gene polymorphism with inflammatory bowel disease in Chinese Han population. Cytokine (2008) 41:44–7. doi:10.1016/j.cyto.2007.10.010
42. Hao NB, He YF, Luo G, Yong X, Zhang Y, Yang SM. Macrophage migration inhibitory factor polymorphism and the risk of ulcerative colitis and Crohn’s disease in Asian and European populations: a meta-analysis. BMJ open (2013) 3:e003729. doi:10.1136/bmjopen-2013-003729
43. Przybyłowska K, Mrowicki J, Sygut A, Narbutt P, Dziki Ł, Dziki A, et al. Contribution of the -173 G/C polymorphism of macrophage migration inhibitory factor gene to the risk of inflammatory bowel diseases. Pol Przegl Chir (2011) 83:76–80. doi:10.2478/v10035-011-0012-x
44. Shen Y, Guo S, Yang T, Jia L, Chen L, An J, et al. The -173 G/C polymorphism of the MIF Gene and inflammatory bowel disease risk: a meta-analysis. Int J Mol Sci (2013) 14:11392–401. doi:10.3390/ijms140611392
45. Griga T, Wilkens C, Wirkus N, Epplen J, Schmiegel W, Klein W. A polymorphism in the macrophage migration inhibitory factor gene is involved in the genetic predisposition of Crohn’s disease and associated with cumulative steroid doses. Hepatogastroenterology (2007) 54:784–6.
46. Sivaram G, Tiwari SK, Bardia A, Anjum F, Vishnupriya S, Habeeb A, et al. Macrophage migration inhibitory factor, Toll-like receptor 4, and CD14 polymorphisms with altered expression levels in patients with ulcerative colitis. Hum Immunol (2012) 73:201–5. doi:10.1016/j.humimm.2011.12.006
47. Nohara H, Okayama N, Inoue N, Koike Y, Fujimura K, Suehiro Y, et al. Association of the -173 G/C polymorphism of the macrophage migration inhibitory factor gene with ulcerative colitis. J Gastroenterol (2004) 39:242–6. doi:10.1007/s00535-003-1284-7
48. Shiroeda H, Tahara T, Nakamura M, Shibata T, Nomura T, Yamada H, et al. Association between functional promoter polymorphisms of macrophage migration inhibitory factor (MIF) gene and ulcerative colitis in Japan. Cytokine (2010) 51:173–7. doi:10.1016/j.cyto.2010.05.003
49. Dambacher J, Staudinger T, Seiderer J, Sisic Z, Schnitzler F, Pfennig S, et al. Macrophage migration inhibitory factor (MIF) -173G/C promoter polymorphism influences upper gastrointestinal tract involvement and disease activity in patients with Crohn’s disease. Inflamm Bowel Dis (2007) 13:71–82. doi:10.1002/ibd.20008
50. Núñez C, Rueda B, Martínez A, López-Nevot MA, Fernández-Arquero M, de la Concha EG, et al. Involvement of macrophage migration inhibitory factor gene in celiac disease susceptibility. Genes Immun (2007) 8:168–70. doi:10.1038/sj.gene.6364365
51. Shi X, Leng L, Wang T, Wang W, Du X, Li J, et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity (2006) 25:595–606. doi:10.1016/j.immuni.2006.08.020
52. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007) 13:587–96. doi:10.1038/nm1567
53. Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med (2003) 197:1467–76. doi:10.1084/jem.20030286
54. Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, et al. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett (2009) 583:2749–57. doi:10.1016/j.febslet.2009.07.058
55. Flaster H, Bernhagen J, Calandra T, Bucala R. The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol (2007) 21:1267–80. doi:10.1210/me.2007-0065
56. Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem (1999) 274:18100–6.
57. Santos LL, Lacey D, Yang Y, Leech M, Morand EF. Activation of synovial cell p38 MAP kinase by macrophage migration inhibitory factor. J Rheumatol (2004) 31:1038–43.
58. Lue H, Thiele M, Franz J, Dahl E, Speckgens S, Leng L, et al. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene (2007) 26:5046–59. doi:10.1038/sj.onc.1210318
59. Cui XL, Douglas JG. Arachidonic acid activates c-jun N-terminal kinase through NADPH oxidase in rabbit proximal tubular epithelial cells. Proc Natl Acad Sci U S A (1997) 94:3771–6. doi:10.1073/pnas.94.8.3771
60. Swantek JL, Cobb MH, Geppert TD. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol (1997) 17:6274–82. doi:10.1128/MCB.17.11.6274
61. Leech M, Lacey D, Xue JR, Santos L, Hutchinson P, Wolvetang E, et al. Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum (2003) 48:1881–9. doi:10.1002/art.11165
62. Roger T, Chanson AL, Knaup-Reymond M, Calandra T. Macrophage migration inhibitory factor promotes innate immune responses by suppressing glucocorticoid-induced expression of mitogen-activated protein kinase phosphatase-1. Eur J Immunol (2005) 35:3405–13. doi:10.1002/eji.200535413
63. Aeberli D, Yang Y, Mansell A, Santos L, Leech M, Morand EF. Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett (2006) 580:974–81. doi:10.1016/j.febslet.2006.01.027
64. Aeberli D, Yamana J, Yang Y, Morand E. Modulation of glucocorticoid sensitivity by MIF via MAP kinase phosphatase-1 is dependent on p53. Arthritis Rheum (2006) 54:S94.
65. Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature (2000) 408:211–6. doi:10.1038/35041591
66. Lacey D, Sampey A, Mitchell R, Bucala R, Santos L, Leech M, et al. Control of fibroblast-like synoviocyte proliferation by macrophage migration inhibitory factor. Arthritis Rheum (2003) 48:103–9. doi:10.1002/art.10733
67. Van Molle W, Libert C. How glucocorticoids control their own strength and the balance between pro- and anti-inflammatory mediators. Eur J Immunol (2005) 35:3396–9. doi:10.1002/eji.200535556
68. Fan H, Kao W, Yang YH, Gu R, Harris J, Fingerle-Rowson G, et al. Macrophage migration inhibitory factor inhibits the anti-inflammatory effects of glucocorticoids via glucocorticoid-induced leucine zipper. Arthritis Rheumatol (2014) 66(8):2059–70. doi:10.1002/art.38689
69. Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat Rev Rheumatol (2011) 7:340–8. doi:10.1038/nrrheum.2011.59
70. Cunha FQ, Moss DW, Leal LM, Moncada S, Liew FY. Induction of macrophage parasiticidal activity by Staphylococcus aureus and exotoxins through the nitric oxide synthesis pathway. Immunology (1993) 78:563–7.
71. Froidevaux C, Roger T, Martin C, Glauser MP, Calandra T. Macrophage migration inhibitory factor and innate immune responses to bacterial infections. Crit Care Med (2001) 29:S13–5. doi:10.1097/00003246-200107001-00006
72. Onodera S, Suzuki K, Matsuno T, Kaneda K, Takagi M, Nishihira J. Macrophage migration inhibitory factor induces phagocytosis of foreign particles by macrophages in autocrine and paracrine fashion. Immunology (1997) 92:131–7. doi:10.1046/j.1365-2567.1997.00311.x
73. Al-Abed Y, Dabideen D, Aljabari B, Valster A, Messmer D, Ochani M, et al. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J Biol Chem (2005) 280:36541–4. doi:10.1074/jbc.C500243200
74. Koebernick H, Grode L, David JR, Rohde W, Rolph MS, Mittrücker HW, et al. Macrophage migration inhibitory factor (MIF) plays a pivotal role in immunity against Salmonella typhimurium. Proc Natl Acad Sci U S A (2002) 99:13681–6. doi:10.1073/pnas.212488699
75. Killick KE, Ní Cheallaigh C, O’Farrelly C, Hokamp K, MacHugh DE, Harris J. Receptor-mediated recognition of mycobacterial pathogens. Cell Microbiol (2013) 15:1484–95. doi:10.1111/cmi.12161
76. Das R, Koo MS, Kim BH, Jacob ST, Subbian S, Yao J, et al. Macrophage migration inhibitory factor (MIF) is a critical mediator of the innate immune response to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2013) 110:E2997–3006. doi:10.1073/pnas.1301128110
77. Altboum I, Pick E. Antigen and mitogen induced production of macrophage migration inhibitory factor in the mouse. Int Arch Allergy Appl Immunol (1979) 60:29–43. doi:10.1159/000232320
78. Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, et al. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci U S A (1996) 93:7849–54. doi:10.1073/pnas.93.15.7849
79. Mizue Y, Ghani S, Leng L, McDonald C, Kong P, Baugh J, et al. Role for macrophage migration inhibitory factor in asthma. Proc Natl Acad Sci U S A (2005) 102:14410–5. doi:10.1073/pnas.0507189102
80. Santos L, Hall P, Metz C, Bucala R, Morand EF. Role of macrophage migration inhibitory factor (MIF) in murine antigen-induced arthritis: interaction with glucocorticoids. Clin Exp Immunol (2001) 123:309–14. doi:10.1046/j.1365-2249.2001.01423.x
81. Santos LL, Dacumos A, Yamana J, Sharma L, Morand EF. Reduced arthritis in MIF deficient mice is associated with reduced T cell activation: down-regulation of ERK MAP kinase phosphorylation. Clin Exp Immunol (2008) 152:372–80. doi:10.1111/j.1365-2249.2008.03639.x
82. Bernhagen J, Bacher M, Calandra T, Metz CN, Doty SB, Donnelly T, et al. An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J Exp Med (1996) 183:277–82. doi:10.1084/jem.183.1.277
83. Stojanovic I, Cvjeticanin T, Lazaroski S, Stosic-Grujicic S, Miljkovic D. Macrophage migration inhibitory factor stimulates interleukin-17 expression and production in lymph node cells. Immunology (2009) 126:74–83. doi:10.1111/j.1365-2567.2008.02879.x
84. Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, et al. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol (2006) 177:8072–9. doi:10.4049/jimmunol.177.11.8072
85. Onodera S, Nishihira J, Koyama Y, Majima T, Aoki Y, Ichiyama H, et al. Macrophage migration inhibitory factor up-regulates the expression of interleukin-8 messenger RNA in synovial fibroblasts of rheumatoid arthritis patients: common transcriptional regulatory mechanism between interleukin-8 and interleukin-1beta. Arthritis Rheum (2004) 50:1437–47. doi:10.1002/art.20190
86. Klasen C, Ohl K, Sternkopf M, Shachar I, Schmitz C, Heussen N, et al. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. J Immunol (2014) 192:5273–84. doi:10.4049/jimmunol.1302209
87. Simpson KD, Cross JV. MIF: metastasis/MDSC-inducing factor? Oncoimmunology (2013) 2:e23337. doi:10.4161/onci.23337
88. Choi S, Kim HR, Leng L, Kang I, Jorgensen WL, Cho CS, et al. Role of macrophage migration inhibitory factor in the regulatory T cell response of tumor-bearing mice. J Immunol (2012) 189:3905–13. doi:10.4049/jimmunol.1102152
89. Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol (2013) 15:713–20. doi:10.1038/ncb2788
91. Crişan TO, Plantinga TS, van de Veerdonk FL, Farcaş MF, Stoffels M, Kullberg BJ, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One (2011) 6:e18666. doi:10.1371/journal.pone.0018666
92. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286:9587–97. doi:10.1074/jbc.M110.202911
93. Peral de Castro C, Jones SA, Ní Cheallaigh C, Hearnden CA, Williams L, Winter J, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol (2012) 189:4144–53. doi:10.4049/jimmunol.1201946
94. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456:264–8. doi:10.1038/nature07383
95. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi:10.1038/ni.1980
96. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13:255–63. doi:10.1038/ni.2215
97. Wu MY, Fu J, Xu J, O’Malley BW, Wu RC. Steroid receptor coactivator 3 regulates autophagy in breast cancer cells through macrophage migration inhibitory factor. Cell Res (2012) 22:1003–21. doi:10.1038/cr.2012.44
98. Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem (2001) 276:35243–6. doi:10.1074/jbc.C100319200
99. Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity (2007) 27:505–17. doi:10.1016/j.immuni.2007.07.022
100. Chuang YC, Su WH, Lei HY, Lin YS, Liu HS, Chang CP, et al. Macrophage migration inhibitory factor induces autophagy via reactive oxygen species generation. PLoS One (2012) 7:e37613. doi:10.1371/journal.pone.0037613
101. Xu X, Bucala R, Ren J. Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J Am Heart Assoc (2013) 2:e000439. doi:10.1161/JAHA.113.000439
102. Xu X, Hua Y, Nair S, Bucala R, Ren J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension (2014) 63:490–9. doi:10.1161/HYPERTENSIONAHA.113.02219
103. Xu X, Pacheco BD, Leng L, Bucala R, Ren J. Macrophage migration inhibitory factor plays a permissive role in the maintenance of cardiac contractile function under starvation through regulation of autophagy. Cardiovasc Res (2013) 99:412–21. doi:10.1093/cvr/cvt116
104. Jones SA, Mills KH, Harris J. Autophagy and inflammatory diseases. Immunol Cell Biol (2013) 91:250–8. doi:10.1038/icb.2012.82
105. Lopez P, Alonso-Perez E, Rodriguez-Carrio J, Suarez A. Influence of Atg5 mutation in SLE depends on functional IL-10 genotype. PLoS One (2013) 8:e78756. doi:10.1371/journal.pone.0078756
106. Alessandri C, Barbati C, Vacirca D, Piscopo P, Confaloni A, Sanchez M, et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J (2012) 26:4722–32. doi:10.1096/fj.12-206060
107. Gros F, Arnold J, Page N, Décossas M, Korganow AS, Martin T, et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy (2012) 8:1113–23. doi:10.4161/auto.20275
108. Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis (2015) 74:912–20. doi:10.1136/annrheumdis-2013-204343
109. Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med (2012) 18:871–82. doi:10.1038/nm.2752
110. Vina ER, Utset TO, Hannon MJ, Masi CM, Roberts N, Kwoh CK. Racial differences in treatment preferences among lupus patients: a two-site study. Clin Exp Rheumatol (2014) 32:680–8.
111. Elfving P, Puolakka K, Kautiainen H, Virta LJ, Pohjolainen T, Kaipiainen-Seppänen O. Incidence of systemic lupus erythematosus in Finland, 2000-2007, a nationwide study. Clin Exp Rheumatol (2014) 32:953–5.
112. Foote A, Briganti EM, Kipen Y, Santos L, Leech M, Morand EF. Macrophage migration inhibitory factor in systemic lupus erythematosus. J Rheumatol (2004) 31:268–73.
113. Lan HY, Yang N, Nikolic-Paterson DJ, Yu XQ, Mu W, Isbel NM, et al. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int (2000) 57:499–509. doi:10.1046/j.1523-1755.2000.00869.x
114. Hoi AY, Hickey MJ, Hall P, Yamana J, O’Sullivan KM, Santos LL, et al. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol (2006) 177:5687–96. doi:10.4049/jimmunol.177.8.5687
115. Leng L, Chen L, Fan J, Greven D, Arjona A, Du X, et al. A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. J Immunol (2011) 186:527–38. doi:10.4049/jimmunol.1001767
116. Grieb G, Merk M, Bernhagen J, Bucala R. Macrophage migration inhibitory factor (MIF): a promising biomarker. Drug News Perspect (2010) 23:257–64. doi:10.1358/dnp.2010.23.4.1453629
117. Kleemann R, Kapurniotu A, Frank RW, Gessner A, Mischke R, Flieger O, et al. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol (1998) 280:85–102. doi:10.1006/jmbi.1998.1864
118. Rosengren E, Aman P, Thelin S, Hansson C, Ahlfors S, Björk P, et al. The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett (1997) 417:85–8. doi:10.1016/S0014-5793(97)01261-1
119. Ouertatani-Sakouhi H, El-Turk F, Fauvet B, Cho MK, Pinar Karpinar D, Le Roy D, et al. Identification and characterization of novel classes of macrophage migration inhibitory factor (MIF) inhibitors with distinct mechanisms of action. J Biol Chem (2010) 285:26581–98. doi:10.1074/jbc.M110.113951
120. Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen WL. Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J Med Chem (2009) 52:416–24. doi:10.1021/jm801100v
121. Senter PD, Al-Abed Y, Metz CN, Benigni F, Mitchell RA, Chesney J, et al. Inhibition of macrophage migration inhibitory factor (MIF) tautomerase and biological activities by acetaminophen metabolites. Proc Natl Acad Sci U S A (2002) 99:144–9. doi:10.1073/pnas.011569399
122. Lubetsky JB, Dios A, Han J, Aljabari B, Ruzsicska B, Mitchell R, et al. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J Biol Chem (2002) 277:24976–82. doi:10.1074/jbc.M203220200
123. Garai J, Lorand T. Macrophage migration inhibitory factor (MIF) tautomerase inhibitors as potential novel anti-inflammatory agents: current developments. Curr Med Chem (2009) 16:1091–114. doi:10.2174/092986709787581842
124. White DA, Fang L, Chan W, Morand EF, Kiriazis H, Duffy SJ, et al. Pro-inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PLoS One (2013) 8:e76206. doi:10.1371/journal.pone.0076206
125. Suissa S, Bernatsky S, Hudson M. Antirheumatic drug use and the risk of acute myocardial infarction. Arthritis Rheum (2006) 55:531–6. doi:10.1002/art.22094
126. Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME, et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci U S A (2010) 107:11313–8. doi:10.1073/pnas.1002716107
127. Rolan P, Gibbons JA, He L, Chang E, Jones D, Gross MI, et al. Ibudilast in healthy volunteers: safety, tolerability and pharmacokinetics with single and multiple doses. Br J Clin Pharmacol (2008) 66:792–801. doi:10.1111/j.1365-2125.2008.03270.x
128. Bai F, Asojo OA, Cirillo P, Ciustea M, Ledizet M, Aristoff PA, et al. A novel allosteric inhibitor of macrophage migration inhibitory factor (MIF). J Biol Chem (2012) 287:30653–63. doi:10.1074/jbc.M112.385583
129. El Turk F, Fauvet B, Ouertatani-Sakouhi H, Lugari A, Betzi S, Roche P, et al. An integrative in silico methodology for the identification of modulators of macrophage migration inhibitory factor (MIF) tautomerase activity. Bioorg Med Chem (2010) 18:5425–40. doi:10.1016/j.bmc.2010.05.010
130. Winner M, Meier J, Zierow S, Rendon BE, Crichlow GV, Riggs R, et al. A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res (2008) 68:7253–7. doi:10.1158/0008-5472.CAN-07-6227
131. Cooke G, Armstrong ME, Donnelly SC. Macrophage migration inhibitory factor (MIF), enzymatic activity and the inflammatory response. Biofactors (2009) 35:165–8. doi:10.1002/biof.27
132. Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hültner L, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med (2000) 6:164–70. doi:10.1038/72262
133. Greven D, Leng L, Bucala R. Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets (2010) 14:253–64. doi:10.1517/14728220903551304
134. Kerschbaumer RJ, Rieger M, Völkel D, Le Roy D, Roger T, Garbaraviciene J, et al. Neutralization of macrophage migration inhibitory factor (MIF) by fully human antibodies correlates with their specificity for the beta-sheet structure of MIF. J Biol Chem (2012) 287:7446–55. doi:10.1074/jbc.M111.329664
135. Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol (1997) 158:5514–7.
Keywords: MIF, SLE, therapeutics, autophagy, innate immunity and responses
Citation: Lang T, Foote A, Lee JPW, Morand EF and Harris J (2015) MIF: Implications in the Pathoetiology of Systemic Lupus Erythematosus. Front. Immunol. 6:577. doi: 10.3389/fimmu.2015.00577
Received: 06 August 2015; Accepted: 28 October 2015;
Published: 11 November 2015
Edited by:
Luis Graca, University of Lisbon, PortugalReviewed by:
Bin Li, Chinese Academy of Sciences, ChinaDennis O. Adeegbe, Harvard Medical School, USA
Copyright: © 2015 Lang, Foote, Lee, Morand and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tali Lang, dGFsaS5sYW5nQG1vbmFzaC5lZHU=