 Ravi Starzl1,2,3*
Ravi Starzl1,2,3* Dolores Wolfram4
Dolores Wolfram4 Ruben Zamora5,6Bahiyyah Jefferson5Derek Barclay5Chien Ho7
Ruben Zamora5,6Bahiyyah Jefferson5Derek Barclay5Chien Ho7 Vijay Gorantla2Gerald Brandacher3Stefan Schneeberger3W. P. Andrew Lee3Jaime Carbonell1
Vijay Gorantla2Gerald Brandacher3Stefan Schneeberger3W. P. Andrew Lee3Jaime Carbonell1 Yoram Vodovotz5,6
Yoram Vodovotz5,6
- 1Language Technologies Institute, Carnegie Mellon University, Pittsburgh, PA, USA
- 2Department of Plastic and Reconstructive Surgery, University of Pittsburgh, Pittsburgh, PA, USA
- 3Department of Plastic and Reconstructive Surgery, Johns Hopkins Medicine, Baltimore, MD, USA
- 4Department of Plastic and Reconstructive Surgery, Innsbruck Medical University, Innsbruck, Austria
- 5Department of Surgery, University of Pittsburgh, Pittsburgh, PA, USA
- 6Center for Inflammation and Regenerative Modeling, McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA, USA
- 7Department of Biological Sciences, Carnegie Mellon University, Pittsburgh, PA, USA
Background: Trauma often cooccurs with cardiac arrest and hemorrhagic shock. Skin and muscle injuries often lead to significant inflammation in the affected tissue. The primary mechanism by which inflammation is initiated, sustained, and terminated is cytokine-mediated immune signaling, but this signaling can be altered by cardiac arrest. The complexity and context sensitivity of immune signaling in general has stymied a clear understanding of these signaling dynamics.
Methodology/principal findings: We hypothesized that advanced numerical and biological function analysis methods would help elucidate the inflammatory response to skin and muscle wounds in rats, both with and without concomitant shock. Based on the multiplexed analysis of inflammatory mediators, we discerned a differential interleukin (IL)-1α and IL-18 signature in skin vs. muscle, which was suggestive of inflammasome activation in the skin. Immunoblotting revealed caspase-1 activation in skin but not muscle. Notably, IL-1α and IL-18, along with caspase-1, were greatly elevated in the skin following cardiac arrest, consistent with differential inflammasome activation.
Conclusion/significance: Tissue-specific activation of caspase-1 and the NLRP3 inflammasome appear to be key factors in determining the type and severity of the inflammatory response to tissue injury, especially in the presence of severe shock, as suggested via data-driven modeling.
Introduction
Healthy skin and muscle tissue exist in a steady-state equilibrium that is characterized in large part by the absence of acute inflammation (1). When injured, the tissue’s steady state is upset, inducing a cascade of responses that include inflammation, repair, and remodeling of the affected region. The inflammatory response is an essential part of successful wound healing and sets the stage for effective repair and remodeling (2). The composition of the underlying inflammatory/immune signaling that drives the inflammatory response to injury is therefore critical in determining whether the inflammation ultimately leads to successful healing or instead leads to additional damage and dysfunctional tissue healing. However, when inflammatory signaling proportional to an injury is altered, the inflammatory response does not successfully transition to subsequent stages of tissue healing (3). Significant systemic insults, such as cardiac arrest, have the potential to drastically alter local immune signaling activity and possibly disrupt wound healing. To the best of our knowledge, the effect of cardiac arrest on these local wound-healing-associated immune signaling processes has not been elucidated.
In the setting of both civilian and military trauma, severe local tissue injury can often be accompanied by cardiac arrest (4–7). Cardiac arrest affects numerous physiological processes, as well as setting in motion systemic inflammation (8–11). Although short-term hypoxia may be an important part of stimulating wound healing (12), severe or extended disruption of oxygen supply interferes with successful wound healing (13). We hypothesize that the specific local inflammatory mediator network patterns expressed during the initial inflammatory response contain information about how the immune system is responding to a localized injury, and whether the response is leading to successful or dysfunctional healing outcome.
Cardiac arrest, even with eventual sudden restoration of blood flow, is known to cause significant cellular stress, the buildup of toxins, and the release of endogenous danger signals that can promote inflammation. In the neurological context, cardiac arrest is particularly well understood to cause a great deal of damage to nerves. Therefore, we further hypothesize that the eventual sudden restoration of blood flow will in essence create an ischemia/reperfusion milieu characterized by the further release of inflammatory mediators, which will have a further deleterious effect on properly localized and moderated wound healing.
The domain of wound healing has been extensively studied with many excellent articles and reviews that provide an overview of the phases, cellular processes, and molecular signals that have been observed in various wound-healing settings (2, 14–18). The effect of the insult of cardiac arrest on patient outcomes has been studied with respect to the impact on patient survival and neurological damage, with significant focus on the potential protective effects of induced mild hypothermia (19–22). However, to the best of our knowledge, no prior studies examine the effect that cardiac arrest may have on the local immune signaling processes essential to inflammation and wound healing.
Analysis and interpretation of the immune signaling process that drives inflammation is challenging because of the complex interplay among inflammatory mediators and their sensitive dependence on local and systemic conditions. Consequently, analysis of the progression of inflammation must leverage analytic methods and the insights that are capable of robustly assessing multiple dimensions of variance simultaneously, and of obtaining relevant information from high-dimensional data matrices (23).
A variety of data analysis methods suitable to exposing the internal structure of such noisy, high-dimensional data have been developed and utilized extensively for the analysis of complex systems in areas such as computational linguistics and machine learning (24). We and others have suggested the need to employ data-driven computational modeling in order to derive insights from the types of high-dimensional datasets obtained when studying complex biological system, such as the inflammatory response (25–27). We have explored multiple techniques for visualizing and enumerating salient features of acute inflammation in both preclinical and clinical settings, thus providing some insight into the types of analytic methods likely to be effective in elucidating key aspects of the observed inflammatory response (25, 28–37).
In the present study, we investigated patterns of immune signaling induced in vivo in rat skin and muscle following tissue injury in the form of excisional wounding. We then carried out in silico analyses to define principal drivers of local inflammatory responses, in the presence or absence of cardiac arrest. We demonstrate that tissue-specific immune signaling patterns are modified by cardiac arrest (also a paradigm of severe hemorrhagic shock) and suggest that inflammasome activity may govern the type of inflammation initiated.
Materials and Methods
Rat Model of Tissue Injury
To simulate tissue injury, we carried out deep tissue excisional biopsies of skin and muscle (38, 39). All animal procedures, care, and housing were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee and followed the National Institutes of Health guidelines for the care and use of laboratory animals. We divided the study into two experimental groups: injury only (injury group) and injury with cardiac arrest (cardiac arrest group). In the “injury group,” four Lewis rats were anesthetized, and an excision biopsy was taken from the lateral aspect of the thigh on one of the hind limbs in each of the rats. Tissue was drawn away from the body and held in forceps while surgical scissors cut 15 mm × 10 mm of tissue from the lateral aspect of the thigh. In the “cardiac arrest group,” four Lewis rats were sacrificed with a fatal sodium pentobarbital (Lundbeck Inc., Deerfield, IL, USA) overdose, and excision biopsy taken 15–30 s after cessation of heartbeat.
Protein Isolation and Sample Preparation
We have previously shown the preservation of animal and human tissues in RNALater™ (Ambion, Austin, TX, USA) is a method compatible with subsequent Luminex™ analysis (40–42). Accordingly, all tissue samples were sectioned into ≤0.5 cm3 pieces and placed into individual sample tubes filled with RNALater™ and stored as per manufacturer instructions and as determined empirically in our prior study (40). For tissue processing, approximately 50 mg of the tissue was transferred to a 2-ml microcentrifuge tube containing 0.6 ml of 1× BioSource™ (Invitrogen, San Diego, CA, USA) tissue extraction reagent supplemented with 10 μl of 100mM phenylmethanesulfonyl fluoride in ethanol as a protease inhibitor. The tissues were then homogenized using a tissue homogenizer, then centrifuged at 4°C for 10 min at 10,000 × g. After centrifugation, the supernatant were collected and assayed for protein content using the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL, USA) as per manufacturer’s protocol.
Assays for Inflammation Biomarkers
All samples were assayed for inflammatory cytokines and chemokines using the Luminex™ multiplexing platform (100 IS; MiraiBio, Alameda, CA, USA) and a Millipore™ 14-plex rat cytokine bead set (Millipore, Billerica, MA, USA) that included interferon (IFN) γ, interleukin (IL)-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p70, IL-18, monocyte chemotactic protein (MCP-1), GRO/KC, TNFα, and granulocyte-macrophage colony-stimulating factor (GM-CSF). Results were read in picogram per milliliter, then subsequently normalized to total mass of sample protein (picogram of cytokine/milligram of protein) for each of the 14 cytokines by the formula x = (a/b) × 1000, where x = (picogram of cytokine/milligram of protein), a = (picogram per milliliter of cytokine), and b = (microgram per milliliter of protein).
For immunodetection of caspase-1, protein samples (25 μg) were separated on 12% SDS-polyacrylamide gels, and the gels were electroblotted onto PVDF membranes. After overnight blocking, the membranes were incubated overnight with a rabbit polyclonal antibody from Abcam (Cambridge, MA, USA) at 4°C followed by 1 h incubation with a goat antirabbit secondary antibody from Pierce (Rockford, IL, USA) at room temperature. Bands were detected using the Supersignal™ West Dura Extended Duration Substrate Chemiluminescent kit as per the manufacturer instructions. All readings are for active caspase-1 (20 kDa).
For Coomassie blue staining, the same procedure was followed as for acrylamide gels (4–15%), then stained with Bio-safe™ (BioRad, Hercules, CA, USA) stain. After washing, 50 ml of Bio-safe™ Coomassie stain was added. After shaking for 1 h, the protein bands became visible within 20 min and reached maximum intensity within 1 h. Skin to muscle caspase-1 expression was calculated by dividing the measured band relative density (BRD) ratio of skin with the measured BRD ratio of muscle: r = s/m, where s = skin BRD, m = muscle BRD, and r = ratio of skin BRD to muscle BRD. SEM was calculated for each group.
Statistical and Computational Analyses
Statistical analyses were performed using Math Works MatLab™, Microsoft Excel™, and SAS Stat View™. Western blot quantifications were compared using a balanced one-way ANOVA with 5% significance. Cytokine quantifications were compared utilizing an unpaired one-tailed heteroscedastic t-test, again with a 5% significance level.
Principal component analysis (PCA) (26) was used to help discern which of the 14 proinflammatory cytokines and chemokines measured by Luminex™ are the most informative with regards to the observed immune response, similar to methodology we have used previously in the context of murine trauma/hemorrhage (25). Data were grouped by tissue (all skin samples and all muscle samples), and linear combinations of the original 14 dimensions (cytokines/chemokines) in each group were created in order to produce synthesized latent variables that explain >95% of the variance observed. The strength of each inflammatory mediator’s contribution to each of the principal components was also examined, in order to provide additional evidence for determining which cytokines/chemokines could be considered to be driving the observed immune reactions (25).
Immune signaling is driven to a large extent by the local inflammatory mediator milieu. While methods, such as PCA, are able to identify mediators that contribute the most variance to the observed immune response, it is essential to place these numerical results in biological context. Therefore, the PCA was combined with a confirmatory factor analysis (FAN) to elucidate the relevant pathways of the observed immune response. As a part of the FAN, the published literature was examined for descriptions of the similarities and differences in the known biological properties of the cytokines identified through PCA, in order to find common or complimentary patterns of function. Groupings of inflammatory mediator contributions to components one and two in the PCA were interpreted as latent factors and used as the basis for FAN. FAN seeks to model the observed variables as linear combinations of the provided potential factors. This form of analysis is based on regression modeling, and therefore provides additional weight to similar evidence found through PCA when there is similarity in the results.
While factor analysis is related to PCA, the two analyses are not identical. PCA takes a linear combination of the observed variables to derive synthesized latent variables; however, factor analysis takes the conceptually opposite approach. Using regression techniques, this method models the observed variables through linear combinations of potential latent variables that are provided. When the two methods produce models that are in agreement or similar, we interpret this as suggestive evidence that the latent variables are indeed playing an influential role in the observed pathology.
The literature-based analysis is a process by which we have utilized previously published studies that elucidate the mechanisms or behaviors of the signaling proteins under analysis. We extract the biological functions described as associated with the proteins and use those functions as labels in our feature transformation and factor analysis work. We believe that this is an efficient evidence-based method to describe what signaling proteins the transformation and factor analysis methods are emphasizing as important and putting them into a narrative that describes what role they are likely fulfilling in context.
Results
As a paradigm of localized tissue injury, excisional biopsies were made on the lateral aspect of the thigh on Lewis rat hind limbs, as described in Section “Materials and Methods.” To elucidate changes in the immune signaling profile that occur at the cessation of heartbeat, cardiac arrest was induced as described in Section “Materials and Methods.” The testing of the second hypothesis stated in the introduction is through samples collected within 30 s of cardiac arrest, within a timeframe that is common in trauma-associated cardiac arrest events.
Samples taken from the wounded areas of skin and muscle were found to have significantly different levels and patterns of inflammatory mediator expression. These differences were evident across groups as well as across tissue.
In skin, the levels of IL-18 and IL-1α present in “cardiac arrest group” animals (Figure 1A) were much higher than those seen in any other groups. MCP-1, GM-CSF, IL-10, IL-6, and IL-1β were all also elevated in the “injury group” animals (Figure 1B). In muscle, similar differences between “cardiac arrest group” animals (Figure 1C) and “injury group” (Figure 1D) were also seen, along with marked increases in IL-18, IL-1α, IL-6, IFNγ, and MCP-1.
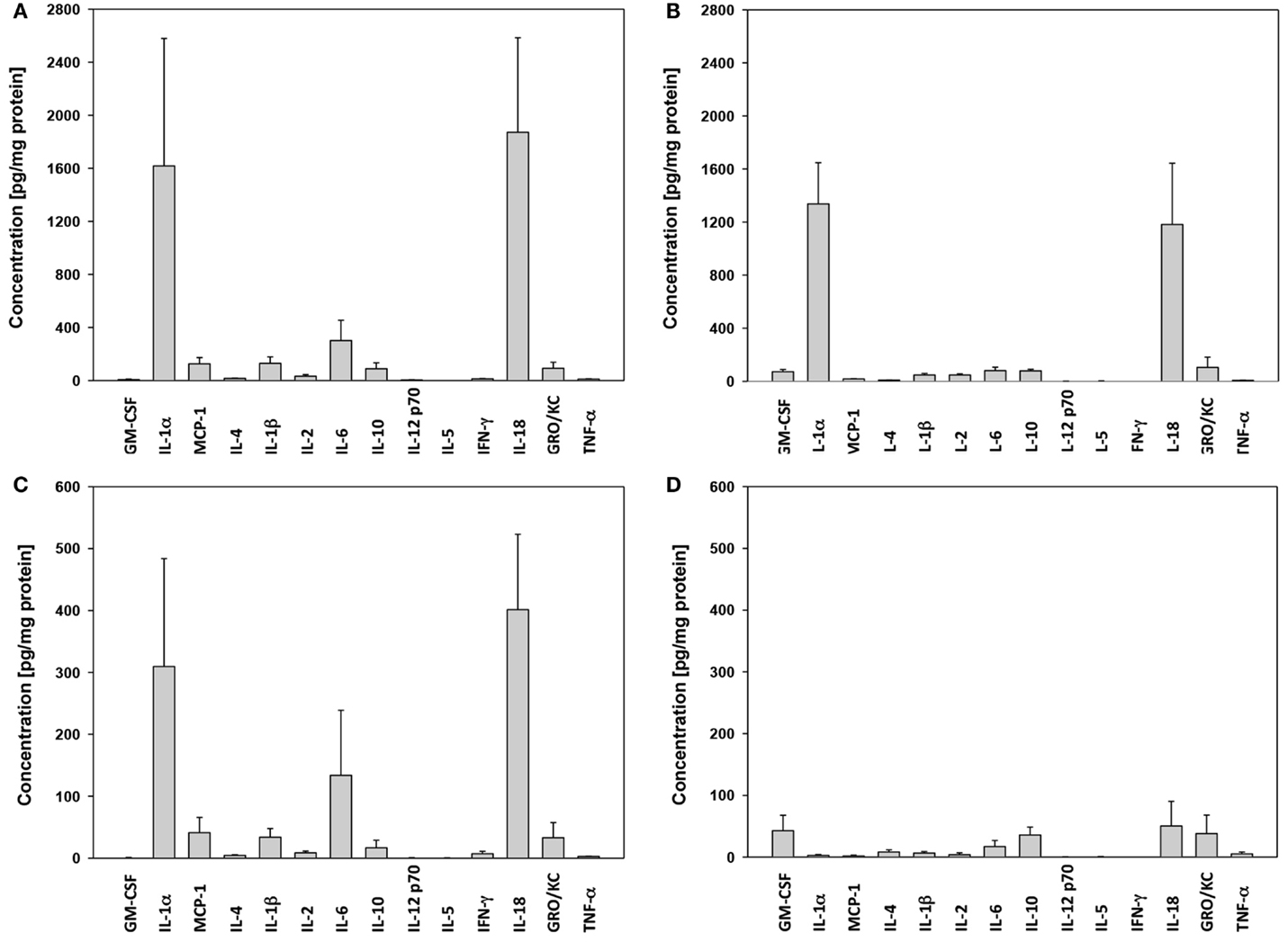
Figure 1. Cytokine expression in skin and muscle tissue: “cardiac arrest group” vs. “wound group.” The concentration of each cytokine in skin tissue in the “cardiac arrest group” (A) and the “wound group” (B) and in muscle tissue [“shock” (C) and the “wound group” (D)] across all time points, measured by Luminex™ as described in Section “Materials and Methods.” Cytokine concentrations are expressed in picogram per milligram total protein.
The presence of IL-18 and IL-1β led us to hypothesize the presence and activation of the NLRP3 inflammasome in response to localized tissue injury and cardiac arrest. To test this hypothesis, we examined the expression of caspase-1, which is required for inflammasome activity (43). As determined by Western blotting analysis, caspase-1 was present in both skin and muscle in all study groups (Figures 2A,B). This finding, in combination with the presence of IL-18 and IL-1β, strongly indicates NLRP3 inflammasome activity. Similar levels of caspase-1 expression were found in both the skin (Figure 2C) and muscle (Figure 2D) of “injury group” animals, implying relatively equal levels of inflammasome activation. In contrast, “cardiac arrest group” animals expressed significantly more caspase-1 in skin than muscle, by approximately a factor of 12.
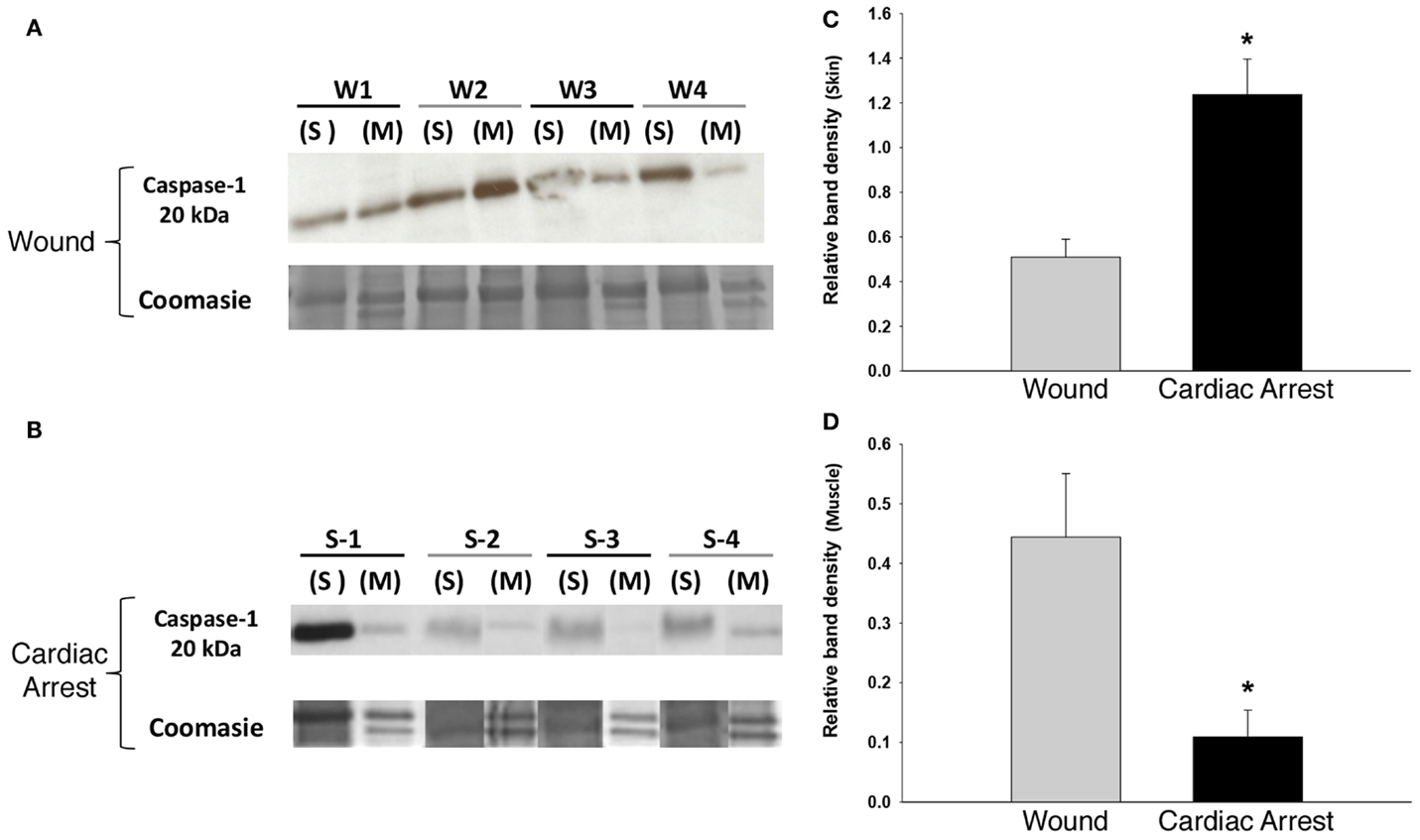
Figure 2. Caspase-1 expression in skin and muscle tissue: “cardiac arrest group” vs. “wound group.” Skin and muscle samples from both experimental groups (“cardiac arrest group” and “wound group”) were processed for protein isolation followed by Western blotting and analysis for active caspase-1 protein and Coomassie blue staining for loading control (see Materials and Methods). (A,B) Show a Western blot for caspase-1 in the “wound group” (W) and the “cardiac arrest group” (S), respectively. The numbers represent the sample from the individual animals in each group (n-4). Densitometric analysis of Western blots for active caspase-1 in skin (C) and muscle (D) (P < 0.05, as determined by one-way ANOVA).
“Cardiac arrest group” skin was associated with elevated levels of caspase-1 and IL-18. In “cardiac arrest group” muscle, IL-18 levels were also markedly increased; however, caspase-1 levels were lower than in “injury group” muscle.
Whereas significant inflammatory mediator concentration differences in the skin were observed between the “injury group” and “cardiac arrest group,” the levels were consistently higher in the “cardiac arrest group” animals (Figures 3B–G). The single exception was GM-CSF, which was present at a much higher concentration in the skin of “injury group” animals (Figure 3A). In “cardiac arrest group” animals, the cytokine with the highest differential elevation relative to “injury group” animals was IFNγ. The only cytokine elevated differentially between groups within muscle tissue was IL-18, whose expression was considerably higher in “cardiac arrest group” animals (Figure 3N).
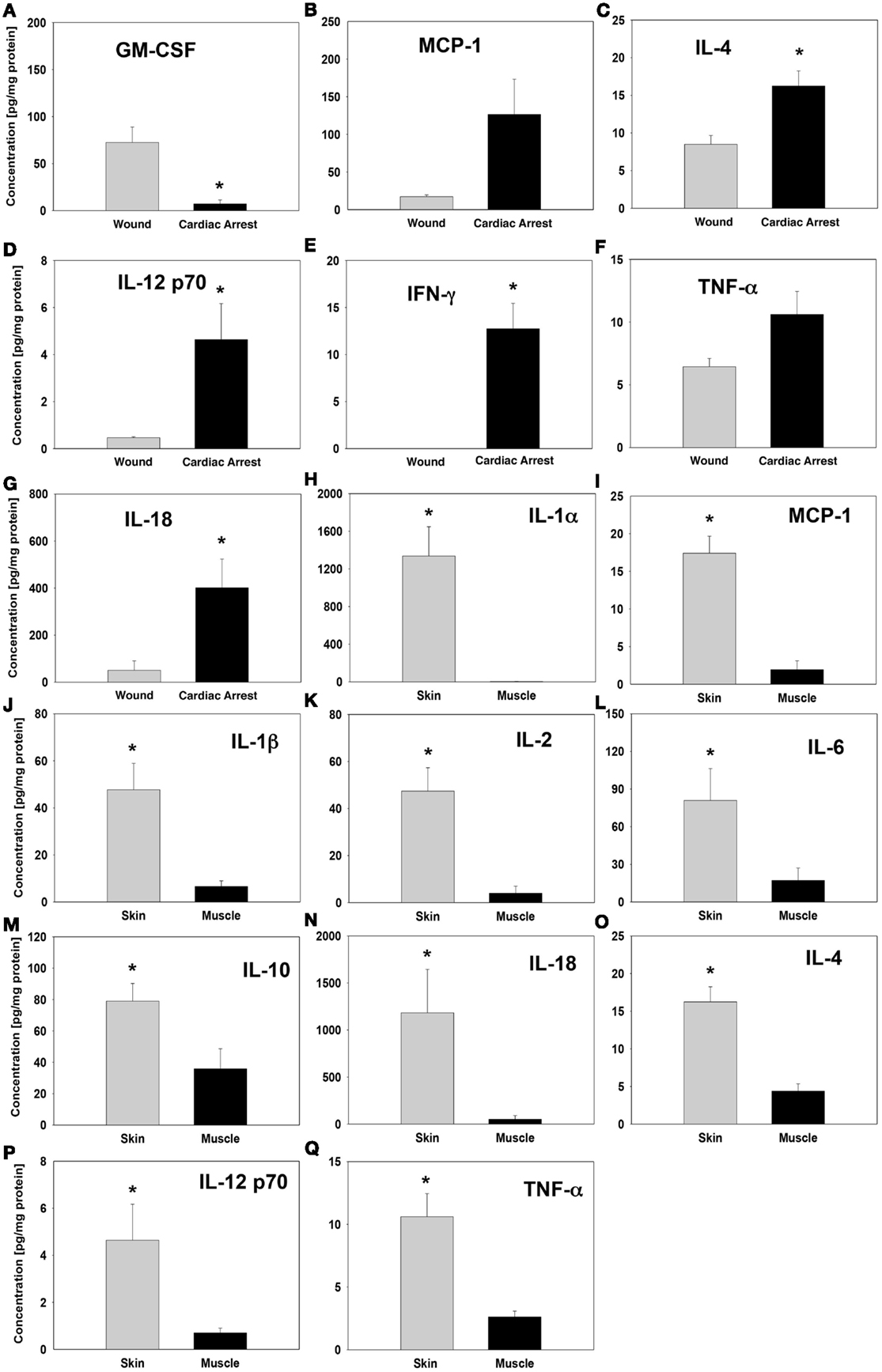
Figure 3. Cytokine levels in “wound group” vs. “cardiac arrest group” and in skin vs. muscle tissue. Tissue protein-normalized cytokine concentrations in “wound group” vs. “cardiac arrest group” (A–G) and in skin vs. muscle tissue (H–Q). The concentrations of each cytokine were measured by Luminex™ as described in Section “Materials and Methods” and are expressed in picogram per milligram total protein. *P < 0.05 by one-tail heteroscedastic t-test.
Also notable was the contrast between skin and muscle tissue inflammatory mediator profiles, particularly within the “injury group” (Figures 3H–Q). Interestingly, the “injury group” animals displayed a much wider range of statistically significant differences in the expression of cytokines and chemokines between skin and muscle tissue as compared to “cardiac arrest group” animals. Although the concentration of many of these inflammatory mediators was actually higher in “cardiac arrest group” animals, the difference between skin and muscle tissue expression of each cytokine was much smaller. IL-4, IL-12p70, and TNFα were expressed at significantly different levels between skin and muscle tissues within the “cardiac arrest group” animals (Figures 3O–Q). These expression levels are also distinct from any inflammatory mediators within the “injury group,” revealing distinct immune signaling patterns in the two groups.
Principal component analysis synthesized two latent variables in skin (Figure 4A) and in muscle (Figure 4B). In skin, the first principal component was comprised mostly of IL-18 and IL-1α, while the second component was comprised of IL-18, IL-6, MCP-1, IL-1β, and GRO/KC (Figure 4C). The first principal component of muscle was also primarily made up of IL-18 and IL-1α, whereas the second principal component was mostly comprised of IL-6, IL-18, MCP-1, and GRO-KC (Figure 4D). TNFα and IL-4 were identified as important cytokines in both “cardiac arrest group” skin and muscle and are known to play a role in many immune regulation or intercellular signaling contexts (44–46). Although the contribution of each of these cytokines is similar in the first principal component, their contributions were substantially different in the second principal component. This finding suggests that the cytokines are complementary in some respects but have distinct roles in the inflammatory mediator network.
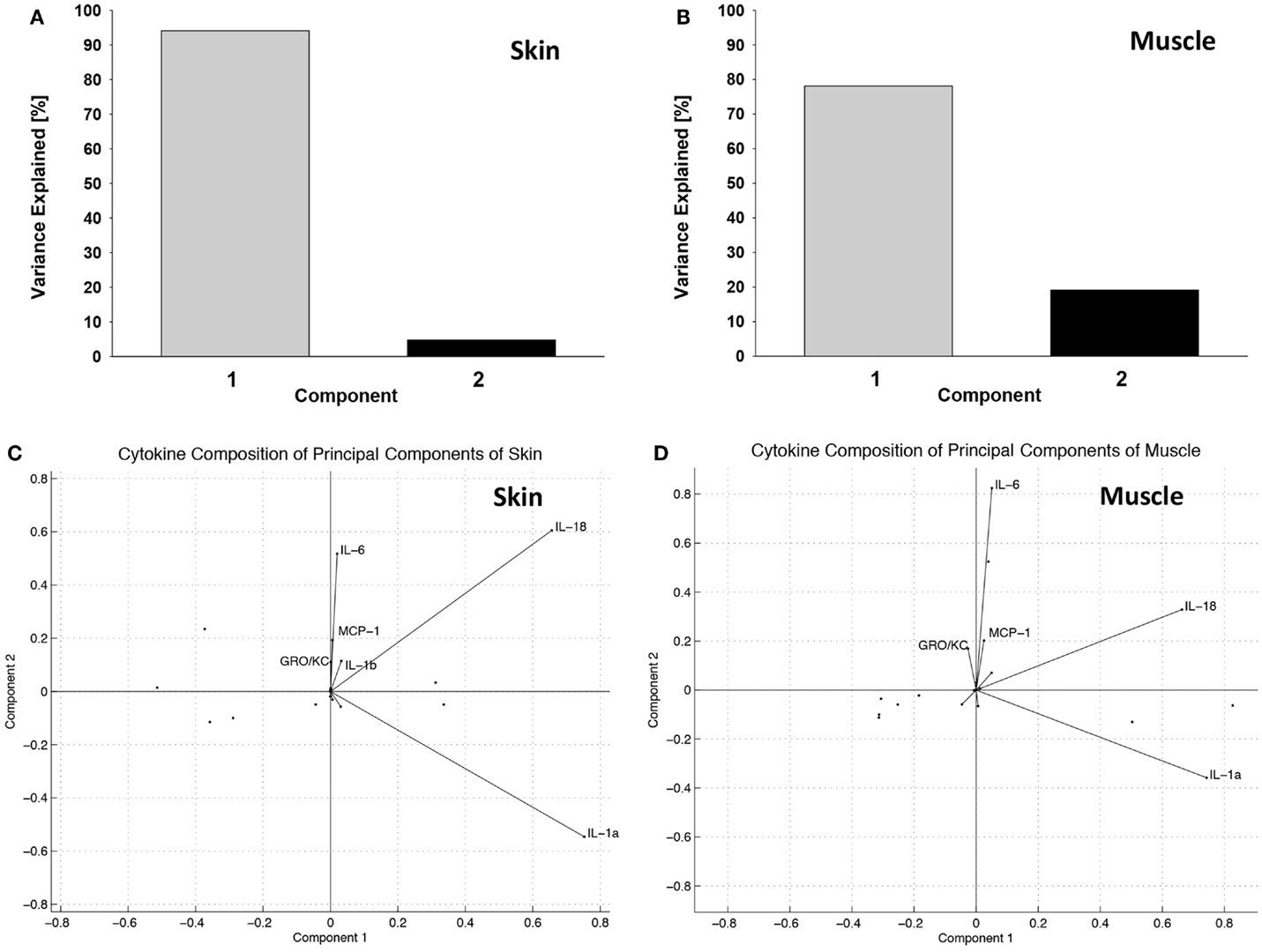
Figure 4. Principal component analysis suggests distinct groups of inflammatory mediators induced in the “wound group” vs. the “cardiac arrest group” in skin and muscle. (A) Percentage of variance explained by principal components in skin. Over 95% of the variance observed in skin can be explained by two latent variables, formed through linear combination of the original 14 variables (cytokines). Latent variables eliminate redundant information and identify the cytokines that are most informative about the observed immune response. (B) Percentage of variance explained by principal components in muscle. Over 95% of the variance observed in muscle can be explained by two latent variables, formed through linear combination of the original 14 variables (cytokines). (C) Principal component variable composition in skin. Each vector represents a cytokine’s contribution to each of the first and the second principal components. The lengths of the vectors indicate the strength of that cytokines contribution, and the direction indicates the proportion of principal component it is contributing to. This figure represents these relationships in the skin. (D) Principal component variable composition in muscle. This figure represents the strength of each measured cytokine’s contribution to each of the first and the second principal components, in muscle.
Principal component analysis suggested a potential role for GM-CSF in tissue injury (but not shock) skin. The converse was observed in the case of the chemokine MCP-1 (CCL2), which was highly expressed in the skin of “cardiac arrest group” animals, but expressed at far lower levels in the skin of the “injury group” animals. Furthermore, high levels of IFNγ were observed in the “cardiac arrest group” animals, while IFNγ was entirely absent from the “injury group” animals.
Two-factor analysis of the skin generated a model of cytokine/chemokine contributions to the latent variables that was very similar to the representation derived by PCA (Figure 5A). Two-factor analysis of the muscle yielded a model that has similarities with the model derived by PCA; however, the overall agreement of the two models is not as strong as found in skin (Figure 5B). The functions of inflammatory mediators most heavily influencing each factor were correlated with descriptions of their biological function from the literature, and through this analysis labels for the latent variables, which were established as “macrophage activation” and “cell-mediated cytotoxic response.”
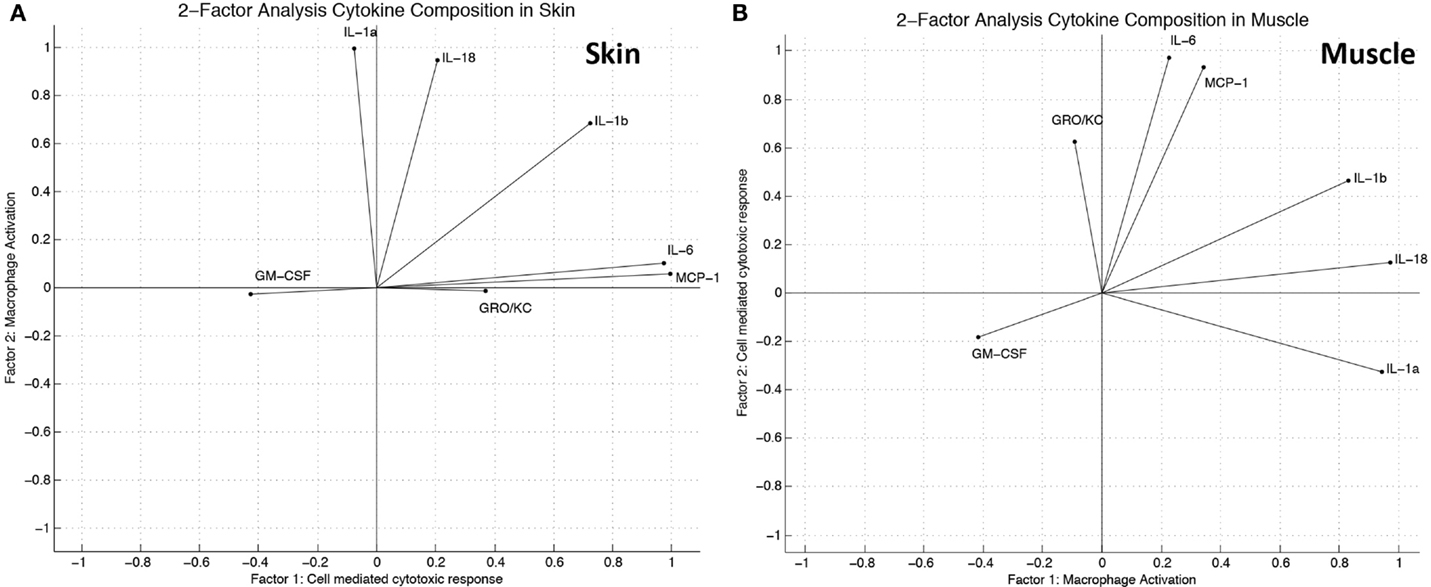
Figure 5. Factor analysis suggests distinct groups of inflammatory mediators induced in the “wound group” vs. the “cardiac arrest group” in skin and muscle. (A) Factor analysis (two factors) variable composition in skin. Modeled cytokine contribution to “cell-mediated cytotoxic response” and “macrophage activation” as calculated by linear combinations of provided latent factors. The direction of each vector indicates which factor the cytokine influences, while the length indicates the relative strength of influence as calculated by the model. (B) Factor analysis (two factors) variable composition in muscle. Modeled cytokine contribution to “macrophage activation” and “cell-mediated cytotoxic response” as calculated by linear combinations of provided latent factors.
Discussion
Inferring Inflammasome-Driven Networks from Data-Driven Modeling
In the present study, we sought to elucidate immune/inflammatory signaling patterns in the response to skin and muscle damages, in the presence or absence of cardiac arrest. We reasoned that this would represent an important step in understanding the potential mechanisms that drive immunological responses to tissue damage at the earliest stages in the settings of complex injury.
In the present study, we hypothesized that application of high-dimensional feature transformation methods, combined with biological knowledge from reports in the literature, can provide a deeper understanding of the immune signaling processes that underlie injury-associated inflammation. Based on the multiplexed analysis of inflammation biomarkers coupled with data-driven modeling, we suggest that tissue injury leads to the differential induction of IL-1α and IL-18 in skin vs. muscle. This inflammatory profile is associated with elevated caspase-1 immunoblotting in skin but not muscle. Cardiac arrest greatly elevates IL-1α, IL-18, and caspase-1 in the skin. Thus, inflammasome activity appears to be central to the type and severity of the inflammatory response to local tissue injury, and this activity appears to be augmented greatly in the presence of cardiac arrest.
We utilized PCA in the current study, because this method is capable of quantifying the amount of information contributed by individual cytokines to the observed inflammation (25, 47, 48). Because this method evaluates the contribution of new information derived from each inflammatory mediator to each of the inflammatory processes observed, PCA assigns the highest scoring to cytokines most correlated with a specific inflammatory response. As utilized herein, PCA seeks to find the linear combination of the original 14 inflammatory mediators that captures the most variance in the smallest number of synthesized variables. Thus, since PCA can show which combination of inflammatory mediators lies in a particular principal component, this method can suggest inflammatory networks which interact together to drive a particular facet of the overall response.
Although a well-established numerical analysis method, additional evidence to support the interpretation of PCA results is desirable and can be provided by FAN (49–51). PCA and FAN are related methods and can intuitively be understood as providing confirmatory analysis. While PCA is based on variance, FAN takes as the input the number of hypothesized factors driving the observed process and then seeks to find the optimal linear coefficients for each cytokine to project from the hypothesized factors back to the original values measured for each cytokine. In short, PCA reduces the dimensionality of a given dataset, while FAN expands that dimensionality. In both cases, the effect is to expose the orthogonal aspects of the data in order to gain a better perspective of what measured parameters are most responsible for driving the observed process, which in the present study is inflammation. When the two methods produce coefficient scores for the inflammatory mediators that are similar to each other, this can be interpreted as evidence in support of the hypothesis that inflammation is, to some degree, driven by those particular mediators. However, if the two methods produce coefficient scores that do not agree, it is likely that the number of hypothesized factors is insufficient to capture the complexity of the inflammatory process.
Using such dimensionality reduction methods, we have previously inferred principal characteristics or drivers of inflammation in mice subjected to surgical trauma alone vs. that same trauma in combination with hemorrhagic shock (25). More recently, we have utilized PCA to suggest a key module in a multicompartment mechanistic mathematical model of inflammation and organ pathophysiology in endotoxemic swine (48), to suggest key physiologic effects of peritoneal suction as a therapy for sepsis (32), to connect in vitro and in vivo outcomes in the inflammatory response to implanted biomaterials (28), as well as suggesting key changes in metabolism that occur in the setting of pulsatile perfusion of livers prior to transplantation (29). Importantly, we have recently used these methods to suggest a role for the inflammasome (including elevated IL-18 and caspase-1) in a rat model of chronic neuropathic pain (42).
To better understand the observed immune signaling activity, we investigated two classes of insult: tissue injury only and tissue injury with shock. In the first model, the inflammatory response to surgically excised wounds in skin and muscle tissue without any secondary contaminants reveals immune signaling patterns primarily associated with wound healing. In the model of injury with shock, changes in the patterns of the inflammatory response to surgically incised wounds under conditions of cardiac arrest represent the disruptive effect of a disruption in blood flow and the associated stresses have on wound-healing signaling. Changes in immune signaling under these circumstances are caused by metabolic stress and other factors accompanying shock.
In the present study, we observed elevations in a wide range of proinflammatory mediators, including IL-1α, IL-1β, IL-4, IL-6, IL-12, IL-18, MCP-1, and TNFα. These networks of inflammatory mediators, observed in both the “injury group” and the “cardiac arrest group” are indicative of highly activated immune cells, such as dendritic cells, mast cells, and neutrophils. Dendritic cells are known as sensitive and potent pathogen presentation cells, but they are also becoming recognized as important in wound detection and healing in a variety of contexts (52–54). The specific role of these immune cells in a tissue is affected by the cytokine milieu present (23, 55). Importantly, most of these mediators have been implicated in the response to trauma/hemorrhage, in prior studies, especially TNFα (56–59) and MCP-1 (31, 35).
Although the specific milieu of the “injury group” and “cardiac arrest group” animals appeared distinct, PCA and FAN suggested a central role for IL-1α and IL-18, leading us to hypothesize and confirm the concomitantly elevated expression of activated caspase-1 as evidence for strong inflammasome activation in these tissues. As cells are killed, are damaged, or are induced into apoptosis by the wounding process, they release endogenous damage-associated molecular pattern (DAMP) molecules, such as uric acid crystals and high-mobility group protein B1 (HMGB1), molecules that trigger inflammatory response mechanisms (60–64). One DAMP-induced mechanism of particular relevance to the inflammatory signaling profile observed in this study involves the activation of the NLRP3 inflammasome (65), which is one of several multiprotein complexes that play important roles in inflammation and cell death (66). Deficiency or overactivation of these cytoplasm-based protein assemblies has been implicated in inflammation-associated damage in a variety of disorders (67), and inflammasome activation is widely regarded as critical in the initiation of the innate immune response. NLRP3 responds to DAMPs and involves a spontaneous protein assembly that forms primarily in the cytoplasm of macrophages, monocytes, some keratinocytes (65), and mast cells (68). The NLRP3 inflammasome is known to respond to cytokines as well as DAMPs including HMGB1 and uric acid crystals (69). The inflammasome, in turn, produces and secretes bioactive proinflammatory cytokines. This process initiates an inflammation and wound-healing cycle that may moderate and conclude successfully with functional wound healing or may lead to runaway inflammation resulting in dysfunctional wound healing (68).
Although all classes of inflammasome produce proinflammatory cytokines, each assembly only forms in response to specific stimuli. In the present study, we found evidence for the activation of NLRP3 (cryopyrin, NALP3) inflammasome. This inflammasome forms specifically in response to the types of DAMPs released in wounding and shock, as well as pathogen-associated molecular patterns (PAMPs) (70, 71). This class of inflammasomes leads to the recruitment of caspase-1 and the cleavage of pro-IL-1β and pro-IL-18 to yield the bioactive, proinflammatory cytokines IL-1β and IL-18. These cytokines are able to induce degranulation in polymorphonuclear neutrophil (PMN) leukocytes (72). Importantly, caspase-1 activity appears to be essential for the innate immune response, given that caspase-1-deficient mice are resistant to developing shock in response to PAMPs, such as LPS (73).
NLRP3-depleted mice produce neither IL-1β nor IL-18 but are still able to express IL-1α, IL-12p40, and TNFα (74). In the present study, we found large quantities of IL-1α in wounded skin across all groups, suggesting that the observed immune response may in fact be comprised of several components that are driven by different activation and signaling pathways. Furthermore, we observed larger quantities of IL-18 vs. IL-1β, in line with reported requirement for prestimulation, for example with LPS, for the detection of large quantities of IL-1β (73). These findings are also in line with our recent studies in a rat model of chronic constriction injury and neuropathic pain (42).
In addition to inflammasome-derived cytokines, analysis of the biological functions of the cytokines identified by PCA suggested other potential mechanisms of inflammation induced by skin injury and cardiac arrest. For example, GM-CSF was highly expressed in skin in the “injury group” animals, while this cytokine was relatively absent in the “cardiac arrest group” animals. Since GM-CSF is an important regulator of macrophage and granulocyte populations and has been shown to play an important role in the onset and propagation of inflammation (75, 76), its presence indicates strong macrophage recruitment and host defense or inflammatory activity. Thus, we hypothesize that GM-CSF plays a key role in the phenotype of the skin of the “injury group,” a role which we hypothesize is supported by cytokines, such as TNFα and IL-4. Numerous reports on the healing properties of GM-CSF in the literature support our PCA-derived hypothesis for the role of GM-CSF in the “injury group” (77–82).
In muscle, IL-6, IL-18, and IL-1α shared both direction and strength of influence in both PCA and FAN models of muscle. Interestingly, we have suggested that MCP-1 in some way controls IL-6 expression (31), and it was notable that MCP-1 figured in both PCA and FAN. Additionally, GM-CSF and IL-1β were influential variables in PCA but were not highly influential in FAN. These findings suggest that the muscle response to injury, both with and without shock, is likely driven by a larger number of latent factors than in skin.
The high concentrations of MCP-1 seen in the “cardiac arrest group” may possibly be caused by the cellular metabolic stress associated with cardiac arrest, resulting in insufficient cellular perfusion that would then lead to vasodilation, shock, and widespread degranulation of mast cells (83, 84). This chemokine helps recruit monocytes, dendritic cells, and memory T cells to sites of inflammation. Perhaps more significantly, MCP-1 exerts an important role in the degranulation of basophils and mast cells, facilitating the release of serine proteases, histamine, serotonin, and proteoglycans (70). In turn, mast cell and neutrophil degranulation induced by chemokines, such as MCP-1, and cytokines, such as IL-18, releases TNFα, eosinophil chemotactic factor, histamine, and a number of other factors (85, 86). This release alerts other nearby cells of injury and is a part of the cascade that begins the immune response to the injury. This hypothesis is supported by evidence that the mast cells are required for optimal migration of dendritic cells, swelling, neutrophil infiltration, and other effects associated with wound healing (87). We have recently suggested, through combined in vitro, in silico, and clinical studies that liver-derived MCP-1 is a central mediator in dynamic networks of inflammation in trauma (31).
In the “cardiac arrest group,” it is likely that the release of TNFα as a proapoptotic cytokine was triggered, leading to higher concentrations of TNFα in the skin of the “cardiac arrest group.” However, the role of TNFα is varied and dependent on a wide range of factors, including receptor binding (TNF-R1 vs. TNF-R2), the local cytokine milieu, the presence of reactive oxygen species, as well as many additional factors (88, 89). It is therefore not surprising that TNFα is seen also seen in the “injury group” although not in as primary a role as in the “cardiac arrest group.”
TNFα also plays a potentially important role as a costimulator of IFNγ production with IL-12 (IL-12p70) (90–92). Levels of IFNγ in the skin of the “cardiac arrest group” suggest this as an important role of IL-12 in this context but is also capable of a variety of immunological capabilities, including stimulating proliferation in resting peripheral cells, promoting the generation of lymphokine-activated killer cells (LAK cells), and augmenting the cytolytic activity of natural killer cells (NK cells) (93–96).
Another prominent cytokine observed in our studies was IL-4, which is known to stimulate IgE B cell differentiation as well as alternative activation of macrophages into M2 repair cells (97), suggesting that IL-4 may be supporting a wound-healing pattern in the “injury group.” However, in the “cardiac arrest group,” significantly higher concentrations of IL-4, as well as the increased presence of additional cytokines that are associated with shock, suggest that in this context IL-4 may be acting as a promoter of IgE synthesis, contributing to the shock reaction and promoting widespread rapid degranulation of PMN leukocytes. Importantly, in the “injury group,” high levels of GM-CSF are seen along with moderate amounts of IL-4. This same combination of cytokines has been shown to enable the maturation of monocytes into dendritic cells in vitro (98).
Limitations
There are several challenges and limitations in this study. This study focuses on the early inflammatory phase following injury and therefore does not follow wounds through full healing. Although statistically significant differences in the expression levels of multiple inflammatory mediators were found between groups, the study size was limited to eight animals (four in the “injury group” and four in the “cardiac arrest group”). Additionally, a single time point proximal to the time of injury was studied. Future studies would ideally extend the findings reported here with larger study groups, sampled at multiple longitudinal time points. Our focus on the inflammasome was derived from an analysis of a limited number of inflammatory mediators. Thus, it is possible that (1) others, unmeasured inflammatory mediators play key roles in postinjury inflammation and (2) that our analysis methods were insufficient to inform our hypotheses. With regard to the former, we attempted to focus our analysis on mediators that have been studied previously in wound biology. With regard to the latter, we attempted to use corroborative methodologies (PCA and FAN) to gain confidence in our conclusions. In skin, the agreement between PCA and FAN models was high across all measured mediators, providing additional evidence that two factors are explanatory. Nonetheless, confirmation of this hypothesis requires studies in which the inflammasome, caspase-1, IL-1α, and/or IL-18 is antagonized.
Conclusion
The inflammasome appears to govern the early inflammatory response to tissue injury in the skin, releasing proinflammatory factors likely to initiate host defense and clearance of debris. Normal wound healing is characterized and regulated by characteristic immune signaling patterns, reflected in characteristic cytokine and chemokine profiles of skin and muscle (17, 99–102). The mediator profile characteristic of an early inflammatory response to tissue injury is modified extensively as a result of cellular stress induced by shock, likely interfering with eventual wound repair and regeneration. Moreover, the inflammatory network characteristics of skin and muscle injury responses (both with and without severe shock) are recognized as distinct from each other, as inferred from data-driven computational analyses. These findings support the use of feature transformation methods, in combination with literature-based analysis of the underlying biological functions, as a method to separate relevant biological processes from irrelevant homeostatic processes, or other background functions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grant P50-GM-53789. The authors would like to thank Dr. Qing Ye from Carnegie Mellon University’s NMR Center, Mr. Ed Gray, and Mr. Tim Starzl for their invaluable insight and suggestions on how to improve this work.
Abbreviations
BRD, band relative density; CTA, composite tissue allotransplantation; df, degrees of freedom; F, F-statistic (ratio of the between-group and within-group mean squares); FAN, confirmatory factor analysis; “injury only,” excisional wound group; LPS, lipopolysaccharide; MS, mean squares (the ratio SS/df); NMR, nuclear magnetic resonance; PCA, principal component analysis; PMN, polymorphonuclear neutrophil; “shock associated with cardiac arrest,” cardiac arrest group; SS, sum of squares.
References
1. Stadelmann WK, Digenis AG, Tobin GR. Physiology and healing dynamics of chronic cutaneous wounds. Am J Surg (1998) 176(2A Suppl):26S–38S. doi: 10.1016/S0002-9610(98)00183-4
2. Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med (1999) 341(10):738–46. doi:10.1056/NEJM199909023411006
3. Fahey TJ III, Sadaty A, Jones WG II, Barber A, Smoller B, Shires GT. Diabetes impairs the late inflammatory response to wound healing. J Surg Res (1991) 50(4):308–13. doi:10.1016/0022-4804(91)90196-S
4. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 10.7: cardiac arrest associated with trauma. Circulation (2005) 112(24_suppl):IV–146–IV–149. doi:10.1161/CIRCULATIONAHA.105.166569
5. Battistella FD, Nugent W, Owings JT, Anderson JT. Field triage of the pulseless trauma patient. Arch Surg (1999) 134(7):742–5. doi:10.1001/archsurg.134.7.742
6. Rosemurgy AS, Norris PA, Olson SM, Hurst JM, Albrink MH. Prehospital traumatic cardiac arrest: the cost of futility. J Trauma (1993) 35(3):468–73. doi:10.1097/00005373-199309000-00022
7. Cera SM, Mostafa G, Sing RF, Sarafin JL, Matthews BD, Heniford BT. Physiologic predictors of survival in post-traumatic arrest. Am Surg (2003) 69(2):140–4.
8. Scolletta S, Donadello K, Santonocito C, Franchi F, Taccone FS. Biomarkers as predictors of outcome after cardiac arrest. Expert Rev Clin Pharmacol (2012) 5(6):687–99. doi:10.1586/ecp.12.64
9. Adrie C, Laurent I, Monchi M, Cariou A, Dhainaou JF, Spaulding C. Postresuscitation disease after cardiac arrest: a sepsis-like syndrome? Curr Opin Crit Care (2004) 10(3):208–12. doi:10.1097/01.ccx.0000126090.06275.fe
10. Bro-Jeppesen J, Kjaergaard J, Wanscher M, Nielsen N, Friberg H, Bjerre M, et al. The inflammatory response after out-of-hospital cardiac arrest is not modified by targeted temperature management at 33 degrees C or 36 degrees C. Resuscitation (2014) 85(11):1480–7. doi:10.1016/j.resuscitation.2014.08.007
11. Segel GB, Halterman MW, Lichtman MA. The paradox of the neutrophil’s role in tissue injury. J Leukoc Biol (2011) 89(3):359–72. doi:10.1189/jlb.0910538
12. Knighton DR, Silver IA, Hunt TK. Regulation of wound-healing angiogenesis-effect of oxygen gradients and inspired oxygen concentration. Surgery (1981) 90(2):262–70.
13. Berghe GV, Zegher FD, Bouillon R. Acute and prolonged critical illness as different neuroendocrine paradigms. J Clin Endocrinol Metab (1998) 83(6):1827–34. doi:10.1210/jc.83.6.1827
14. Martin P. Wound healing – aiming for perfect skin regeneration. Science (1997) 276(5309):75–81. doi:10.1126/science.276.5309.75
15. Leoni G, Neumann PA, Sumagin R, Denning TL, Nusrat A. Wound repair: role of immune-epithelial interactions. Mucosal Immunol (2015) 8:959–68. doi:10.1038/mi.2015.63
16. Broughton G II, Janis JE, Attinger CE. The basic science of wound healing. Plast Reconstr Surg (2006) 117(7 Suppl):12S–34S. doi:10.1097/01.prs.0000225430.42531.c2
17. Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev (2003) 83(3):835–70. doi:10.1152/physrev.00032.2002
18. Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci (2004) 9:283–9. doi:10.2741/1184
19. Tuma MA, Stansbury LG, Stein DM, McQuillan KA, Scalea TM. Induced hypothermia after cardiac arrest in trauma patients: a case series. J Trauma (2011) 71(6):1524–7. doi:10.1097/TA.0b013e31823c5a06
20. Lundbye JB, Rai M, Ramu B, Hosseini-Khalili A, Li D, Slim HB, et al. Therapeutic hypothermia is associated with improved neurologic outcome and survival in cardiac arrest survivors of non-shockable rhythms. Resuscitation (2012) 83(2):202–7. doi:10.1016/j.resuscitation.2011.08.005
21. Sagalyn E, Band RA, Gaieski DF, Abella BS. Therapeutic hypothermia after cardiac arrest in clinical practice: review and compilation of recent experiences. Crit Care Med (2009) 37(7 Suppl):S223–6. doi:10.1097/CCM.0b013e3181aa5c7c
22. Oddo M, Schaller MD, Feihl F, Ribordy V, Liaudet L. From evidence to clinical practice: effective implementation of therapeutic hypothermia to improve patient outcome after cardiac arrest. Crit Care Med (2006) 34(7):1865–73. doi:10.1097/01.CCM.0000221922.08878.49
23. Nathan C, Sporn M. Cytokines in context. J Cell Biol (1991) 113(5):981–6. doi:10.1083/jcb.113.5.981
24. Donoho DL. High-dimensional data analysis: the curses and blessings of dimensionality. AMS Math Challenges Lecture (2000) 1–32.
25. Mi Q, Constantine G, Ziraldo C, Solovyev A, Torres A, Namas R, et al. A dynamic view of trauma/hemorrhage-induced inflammation in mice: principal drivers and networks. PLoS One (2011) 6(5):e19424. doi:10.1371/journal.pone.0019424
26. Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat Rev Mol Cell Biol (2006) 7(11):820–8. doi:10.1038/nrm2041
27. An G, Nieman G, Vodovotz Y. Computational and systems biology in trauma and sepsis: current state and future perspectives. Int J Burns Trauma (2012) 2(1):1–10.
28. Wolf MT, Vodovotz Y, Tottey S, Brown BN, Badylak SF. Predicting in vivo responses to biomaterials via combined in vitro and in silico analysis. Tissue Eng Part C Methods (2015) 21(2):148–59. doi:10.1089/ten.TEC.2014.0167
29. Fontes P, Lopez R, van der Plaats A, Vodovotz Y, Minervini M, Scott V, et al. Liver preservation with machine perfusion and a newly developed cell-free oxygen carrier solution under subnormothermic conditions. Am J Transplant (2015) 15(2):381–94. doi:10.1111/ajt.12991
30. Azhar N, Ziraldo C, Barclay D, Rudnick DA, Squires RH, Vodovotz Y, et al. Analysis of serum inflammatory mediators identifies unique dynamic networks associated with death and spontaneous survival in pediatric acute liver failure. PLoS One (2013) 8(11):e78202. doi:10.1371/journal.pone.0078202
31. Ziraldo C, Vodovotz Y, Namas RA, Almahmoud K, Tapias V, Mi Q, et al. Central role for MCP-1/CCL2 in injury-induced inflammation revealed by in vitro, in silico, and clinical studies. PLoS One (2013) 8(12):e79804. doi:10.1371/journal.pone.0079804
32. Emr B, Sadowsky D, Azhar N, Gatto LA, An G, Nieman GF, et al. Removal of inflammatory ascites is associated with dynamic modification of local and systemic inflammation along with prevention of acute lung injury: in vivo and in silico studies. Shock (2014) 41(4):317–23. doi:10.1097/SHK.0000000000000121
33. Zaaqoq AM, Namas R, Almahmoud K, Azhar N, Mi Q, Zamora R, et al. Inducible protein-10, a potential driver of neurally controlled interleukin-10 and morbidity in human blunt trauma. Crit Care Med (2014) 42(6):1487–97. doi:10.1097/CCM.0000000000000248
34. Wolfram D, Morandi EM, Eberhart N, Hautz T, Hackl H, Zelger B, et al. Differentiation between acute skin rejection in allotransplantation and T-cell mediated skin inflammation based on gene expression analysis. Biomed Res Int (2015) 2015:259160. doi:10.1155/2015/259160
35. Namas RA, et al. Temporal patterns of circulating inflammation biomarker networks differentiate susceptibility to nosocomial infection following blunt trauma in humans. Ann Surg (2014). doi:10.1097/SLA.0000000000001001
36. Almahmoud K, Namas RA, Zaaqoq AM, Abdul-Malak O, Namas R, Zamora R, et al. Pre-hospital hypotension is associated with altered inflammation dynamics and worse outcomes following blunt trauma in humans. Crit Care Med (2015) 43(7):1395–404. doi:10.1097/CCM.0000000000000964
37. Sadowsky D, Nieman G, Barclay D, Mi Q, Zamora R, Constantine G, et al. Impact of chemically-modified tetracycline 3 on intertwined physiological, biochemical, and inflammatory networks in porcine sepsis/ARDS. Int J Burns Trauma (2015) 5(1):22–35.
38. Dorsett-Martin WA. Rat models of skin wound healing: a review. Wound Repair Regen (2004) 12(6):591–9. doi:10.1111/j.1067-1927.2004.12601.x
39. Lindblad WJ. Considerations for selecting the correct animal model for dermal wound-healing studies. J Biomater Sci Polym Ed (2008) 19(8):1087–96. doi:10.1163/156856208784909390
40. Barclay D, Zamora R, Torres A, Namas R, Steed D, Vodovotz Y. A simple, rapid, and convenient Luminex-compatible method of tissue isolation. J Clin Lab Anal (2008) 22(4):278–81. doi:10.1002/jcla.20253
41. Wolfram D, Starzl R, Hackl H, Barclay D, Hautz T, Zelger B, et al. Profiling inflammatory cytokines mediating acute rejection after vascularized composite allotransplantation in a rat limb transplant model. PLoS One (2014) 9:e99926. doi:10.1371/journal.pone.0099926
42. Vasudeva K, Azhar N, Barclay D, Janjic JM, Pollock JA. In vivo and systems biology studies implicate interleukin-18 as a central mediator in chronic pain. J Neuroimmunol (2015) 283:43–9. doi:10.1016/j.jneuroim.2015.04.012
43. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi:10.1016/S1097-2765(02)00599-3
44. Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol (1999) 17:701–38. doi:10.1146/annurev.immunol.17.1.701
45. Tracey KJ, Cerami A. Tumor necrosis factor: an updated review of its biology. Crit Care Med (1993) 21(10 Suppl):S415–22. doi:10.1097/00003246-199310001-00002
47. Jolliffe IT. Principal component analysis. 2nd ed. Springer Series in Statistics. New York: Springer (2002).
48. Nieman G, Brown D, Sarkar J, Kubiak B, Ziraldo C, Dutta-Moscato J, et al. A two-compartment mathematical model of endotoxin-induced inflammatory and physiologic alterations in swine. Crit Care Med (2012) 40:1052–63. doi:10.1097/CCM.0b013e31823e986a
49. Brown TA. Confirmatory Factor Analysis for Applied Research 2nd ed. Guilford Publications (2015).
50. Thompson B. Exploratory and Confirmatory Factor Analysis: Understanding Concepts and Applications. Washington, DC: American Psychological Association (2004). 195 p.
51. Kim J-O, Mueller CW. Factor Analysis: Statistical Methods and Practical Issues. Vol. 14. Newbury Park, CA: SAGE (1978).
52. Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med (2010) 207(13):2921–30. doi:10.1084/jem.20101102
53. Trautmann A, Toksoy A, Engelhardt E, Bröcker EB, Gillitzer R. Mast cell involvement in normal human skin wound healing: expression of monocyte chemoattractant protein-1 is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol (2000) 190(1):100–6. doi:10.1002/(SICI)1096-9896(200001)190:1<100::AID-PATH496>3.0.CO;2-Q
54. Pruthi D, Munick ED, Fu Q, McGarr R, Moodie K. Critical role of dendritic cells in wound healing after myocardial infarction in CD11c-DTR-GFP mice. FASEB J (2009) 23.
55. Nathan C. Points of control in inflammation. Nature (2002) 420(6917):846–52. doi:10.1038/nature01320
56. Spielmann S, Kerner T, Ahlers O, Keh D, Gerlach M, Gerlach H. Early detection of increased tumour necrosis factor alpha (TNFalpha) and soluble TNF receptor protein plasma levels after trauma reveals associations with the clinical course. Acta Anaesthesiol Scand (2001) 45:364–70. doi:10.1034/j.1399-6576.2001.045003364.x
57. Sailhamer EA, Li Y, Smith EJ, Shuja F, Shults C, Liu B, et al. Acetylation: a novel method for modulation of the immune response following trauma/hemorrhage and inflammatory second hit in animals and humans. Surgery (2008) 144(2):204–16. doi:10.1016/j.surg.2008.03.034
58. Li S, Tao L, Jiao X, Liu H, Cao Y, Lopez B, et al. TNFalpha-initiated oxidative/nitrative stress mediates cardiomyocyte apoptosis in traumatic animals. Apoptosis (2007) 12(10):1795–802. doi:10.1007/s10495-007-0108-2
59. Foex BA. Systemic responses to trauma. Br Med Bull (1999) 55(4):726–43. doi:10.1258/0007142991902745
60. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol (2008) 8(4):279–89. doi:10.1038/nri2215
61. Straino S, Di Carlo A, Mangoni A, De Mori R, Guerra L, Maurelli R, et al. High-mobility group box 1 protein in human and murine skin: involvement in wound healing. J Invest Dermatol (2008) 128(6):1545–53. doi:10.1038/sj.jid.5701212
62. Ranzato E, Patrone M, Pedrazzi M, Burlando B. HMGb1 promotes scratch wound closure of HaCaT keratinocytes via ERK1/2 activation. Mol Cell Biochem (2009) 332(1–2):199–205. doi:10.1007/s11010-009-0192-4
63. Zampell JC, Yan A, Avraham T, Andrade V, Malliaris S, Aschen S, et al. Temporal and spatial patterns of endogenous danger signal expression after wound healing and in response to lymphedema. Am J Physiol Cell Physiol (2011) 300(5):C1107–21. doi:10.1152/ajpcell.00378.2010
64. Zhang Q, O’Hearn S, Kavalukas SL, Barbul A. Role of high mobility group box 1 (HMGB1) in wound healing. J Surg Res (2012) 176(1):343–7. doi:10.1016/j.jss.2011.06.069
65. Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest (2010) 120(6):1939–49. doi:10.1172/JCI40124
66. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature (1992) 356(6372):768–74. doi:10.1038/356768a0
67. Becker CE, O’Neill LA. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol (2007) 29(3):239–48. doi:10.1007/s00281-007-0081-4
68. Angele MK, Knöferl MW, Ayala A, Albina JE, Cioffi WG. Trauma-hemorrhage delays wound healing potentially by increasing pro-inflammatory cytokines at the wound site. Surgery (1999) 126(2):279–85. doi:10.1016/S0039-6060(99)70166-2
69. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440(7081):237–41. doi:10.1038/nature04516
70. Fujisawa T, Kato Y, Nagase H, Atsuta J, Terada A, Iguchi K, et al. Chemokines induce eosinophil degranulation through CCR-3. J Allergy Clin Immunol (2000) 106(3):507–13. doi:10.1067/mai.2000.108311
71. Chenais B, Tenu JP. Involvement of nitric oxide synthase in antiproliferative activity of macrophages: induction of the enzyme requires two different kinds of signal acting synergistically. Int J Immunopharmacol (1994) 16(5–6):401–6. doi:10.1016/0192-0561(94)90028-0
72. Leung BP, Culshaw S, Gracie JA, Hunter D, Canetti CA, Campbell C, et al. A role for IL-18 in neutrophil activation. J Immunol (2001) 167(5):2879–86. doi:10.4049/jimmunol.167.5.2879
73. Melnikov VY, Ecder T, Fantuzzi G, Siegmund B, Lucia MS, Dinarello CA, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest (2001) 107(9):1145–52. doi:10.1172/JCI12089
74. Walter K, Hölscher C, Tschopp J, Ehlers S. NALP3 is not necessary for early protection against experimental tuberculosis. Immunobiology (2010) 215(9–10):804–11. doi:10.1016/j.imbio.2010.05.015
75. Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol (2002) 23:403–8. doi:10.1016/S1471-4906(02)02260-3
76. Hamilton JA, Anderson GP. GM-CSF Biology. Growth Factors (2004) 22(4):225–31. doi:10.1080/08977190412331279881
77. Braunstein S, Kaplan G, Gottlieb AB, Schwartz M, Walsh G, Abalos RM, et al. GM-CSF activates regenerative epidermal growth and stimulates keratinocyte proliferation in human skin in vivo. J Invest Dermatol (1994) 103(4):601–4. doi:10.1111/1523-1747.ep12396936
78. Castrogiovanni P, Ventimiglia P, Imbesi R. rHuGM-CSF: a possible therapeutic treatment in resistant chronic wound healing: our first observations. Clin Ter (2010) 161(3):e101–4.
79. Zhang L, Chen J, Han C. A multicenter clinical trial of recombinant human GM-CSF hydrogel for the treatment of deep second-degree burns. Wound Repair Regen (2009) 17(5):685–9. doi:10.1111/j.1524-475X.2009.00526.x
80. Bernasconi E, Favre L, Maillard MH, Bachmann D, Pythoud C, Bouzourene H, et al. Granulocyte-macrophage colony-stimulating factor elicits bone marrow-derived cells that promote efficient colonic mucosal healing. Inflamm Bowel Dis (2010) 16(3):428–41. doi:10.1002/ibd.21072
81. Sutton TL, Pierpont YN, Robson MC, Payne WG. The use of growth factors and other humoral agents to accelerate and enhance burn wound healing. Eplasty (2011) 11:e41.
82. Hu X, Sun H, Han C, Wang X, Yu W. Topically applied rhGM-CSF for the wound healing: a systematic review. Burns (2011) 37(5):729–41. doi:10.1016/j.burns.2010.08.016
83. Sung FL, Zhu TY, Au-Yeung KK, Siow YL, O K. Enhanced MCP-1 expression during ischemia/reperfusion injury is mediated by oxidative stress and NF-kappaB. Kidney Int (2002) 62(4):1160–70. doi:10.1111/j.1523-1755.2002.kid577.x
84. Hillenbrand A, Knippschild U, Weiss M, Schrezenmeier H, Henne-Bruns D, Huber-Lang M, et al. Sepsis induced changes of adipokines and cytokines - septic patients compared to morbidly obese patients. BMC Surg (2010) 10:26. doi:10.1186/1471-2482-10-26
85. Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol (2010) 125(2):S73–80. doi:10.1016/j.jaci.2009.11.017
86. Yoshimoto T, Nakanishi K. Roles of IL-18 in basophils and mast cells. Allergol Int (2006) 55(2):105–13. doi:10.2332/allergolint.55.105
87. Suto H, Nakae S, Kakurai M, Sedgwick JD, Tsai M, Galli SJ. Mast cell-associated TNF promotes dendritic cell migration. J Immunol (2006) 176(7):4102–12. doi:10.4049/jimmunol.176.7.4102
88. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell (2005) 120(5):649–61. doi:10.1016/j.cell.2004.12.041
89. Grell M, Zimmermann G, Gottfried E, Chen CM, Grünwald U, Huang DC, et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J (1999) 18(11):3034–43. doi:10.1093/emboj/18.11.3034
90. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature (1995) 378(6552):88–91. doi:10.1038/378088a0
91. Tripp CS, Unanue WS. ER Interleukin 12 and tumor necrosis factor alpha are costimulators of interferon gamma production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc Natl Acad Sci U S A (1993) 90:3725–9. doi:10.1073/pnas.90.8.3725
92. Wherry JC, Schreiber RD, Unanue ER. Regulation of gamma interferon production by natural killer cells in scid mice: roles of tumor necrosis factor and bacterial stimuli. Infect Immun (1991) 59(5):1709–15.
93. Bertagnolli MM, Lin BY, Young D, Herrmann SH. IL-12 augments antigen-dependent proliferation of activated T lymphocytes. J Immunol (1992) 149(12):3778–83.
94. Chan SH, Perussia B, Gupta JW, Kobayashi M, Pospísil M, Young HA, et al. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med (1991) 173(4):869–79. doi:10.1084/jem.173.4.869
95. Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity (1996) 4(5):471–81. doi:10.1016/S1074-7613(00)80413-6
96. Chehimi J, Starr SE, Frank I, Rengaraju M, Jackson SJ, Llanes C, et al. Natural killer (NK) cell stimulatory factor increases the cytotoxic activity of NK cells from both healthy donors and human immunodeficiency virus-infected patients. J Exp Med (1992) 175(3):789–96. doi:10.1084/jem.175.3.789
97. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity (2005) 23(4):344–6. doi:10.1016/j.immuni.2005.10.001
98. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med (1994) 179(4):1109–18. doi:10.1084/jem.179.4.1109
99. Witte MB, Barbul A. General principles of wound healing. Surg Clin North Am (1997) 77(3):509–28. doi:10.1016/S0039-6109(05)70566-1
100. Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol (2001) 69(4):513–21.
101. McKay IA, Leigh IM. Epidermal cytokines and their roles in cutaneous wound healing. Br J Dermatol (1991) 124(6):513–8. doi:10.1111/j.1365-2133.1991.tb04942.x
Keywords: Inflammasome, inflammatory mediators, computational modeling, data driven modeling, cardiac arrest and trauma, immunoregulatory, localized tissue injury, hemorrhagic shock
Citation: Starzl R, Wolfram D, Zamora R, Jefferson B, Barclay D, Ho C, Gorantla V, Brandacher G, Schneeberger S, Andrew Lee WP, Carbonell J and Vodovotz Y (2015) Cardiac Arrest Disrupts Caspase-1 and Patterns of Inflammatory Mediators Differently in Skin and Muscle Following Localized Tissue Injury in Rats: Insights from Data-Driven Modeling. Front. Immunol. 6:587. doi: 10.3389/fimmu.2015.00587
Received: 01 May 2015; Accepted: 02 November 2015;
Published: 20 November 2015
Edited by:
Julia Arciero, Indiana University-Purdue University Indianapolis, USACopyright: © 2015 Starzl, Wolfram, Zamora, Jefferson, Barclay, Ho, Gorantla, Brandacher, Schneeberger, Andrew Lee, Carbonell and Vodovotz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ravi Starzl, cnN0YXJ6bEBjcy5jbXUuZWR1