 Katharina Andrea Schuett
Katharina Andrea Schuett Michael Lehrke†
Michael Lehrke† Nikolaus Marx
Nikolaus Marx Mathias Burgmaier
Mathias Burgmaier- Department of Internal Medicine I, University Hospital RWTH Aachen, Aachen, Germany
Cardiovascular disease is the leading cause of death in the Western world with an increase over the last few decades. Atherosclerosis with its different manifestations in the coronary artery tree, the cerebral, as well as peripheral arteries is the basis for cardiovascular events, such as myocardial infarction, stroke, and cardiovascular death. The pathophysiological understanding of the mechanisms that promote the development of vascular disease has changed over the last few decades, leading to the recognition that inflammation and inflammatory processes in the vessel wall are major contributors in atherogenesis. In addition, a subclinical inflammatory status, e.g., in patients with diabetes or the presence of a chronic inflammatory disease, such as rheumatoid arthritis, have been recognized as strong risk factors for cardiovascular disease. The present review will summarize the different inflammatory processes in the vessel wall leading to atherosclerosis and highlight the role of inflammation in diabetes and chronic inflammatory diseases for cardiovascular morbidity and mortality.
Introduction
Cardiovascular disease is the leading cause of death in the western world with an increase over the last few decades (1–4).
Atherosclerosis may result in myocardial infarction (MI), stroke, or peripheral artery disease according to its manifestations in the coronary artery tree, in cerebral arteries as well as peripheral arteries. The pathophysiological understanding of the mechanisms that promote the development of vascular disease has changed over the last few decades, leading to the recognition that inflammation and inflammatory processes in the vessel wall are major contributors in atherogenesis (5).
Atherogenesis
For decades, atherogenesis has been seen as a degenerative process in the vessel wall leading to the progressive occlusion of the artery up to a certain point where a few activated platelets are sufficient to occlude the vessel and cause cardiac events, such as acute MI. In the 1990s, we learned from our pathology colleagues that a majority of lesions causing an acute coronary syndrome show a stenosis <50%, and further pathological analysis revealed that most of these lesions are the so-called unstable lesions (6). These lesions are characterized by a high inflammatory burden of cells, such as monocytes and T cells, and a large necrotic lipid core as well as a very thin fibrous cap making the atherosclerotic lesion prone to rupture. At the same time, experimental data have shown that atherogenesis is an inflammatory process in the vessel wall in different phases and stages (7). Also not strictly defined endothelial dysfunction, fatty streak formation and the formation of advanced and potentially complicated lesions have been described as the main phases in lesion development (8). Over the last decade, our understanding of the inflammatory cells involved in these processes has grown. The characteristic cell type of an atherosclerotic lesion is the lipid-loaden macrophage which is attracted by certain chemokines to the vessel wall and then can contribute to the local inflammatory response by the expression and release of inflammatory mediators. The uptake of LDL in these mononuclear cells by scavenger receptors results in the characteristic foam cell. In addition, the accumulation of cholesterol within macrophages leads to crystal formation, and these intracellular microcrystals can activate the inflammasome of the cells subsequently resulting in the release of cytokines (9). Interestingly, experimental work from various groups around the world has shown that different subtypes of macrophages can be found in atherosclerotic lesions with the predominance of the M1-subpopulation which release proinflammatory cytokines (10). Additional cellular components of the innate immune response in the vessel wall are mast cells and to a certain extent polymorphonuclear leukocytes, but the latter have mainly been found in murine models, and the relevance for human atherosclerosis remains unclear (11). In addition to the innate immune response, clinical and experimental data support the recognition that the adaptive immune system is of critical importance in atherogenesis. As such, dendritic cells have been identified as a critical “link” between the innate and adaptive immune responses by initiating T-cell response. T cells in the atherosclerotic lesion are mainly CD4 cells which to a large extent release proinflammatory Th1 cytokines. These cytokines, such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α, do not only induce the expression of certain chemokines thus favoring the recruitment of additional leukocytes but also lead to the activation of other vascular cells [reviewed in Ref. (12)].
In addition, recent work has shown that so-called regulatory T cells are an important atheroprotective cell population in the vascular wall through their capacity to control effector T cells (13).
The recognition that inflammation plays an important role in atherogenesis has been paralleled by the clinical observation that elevated levels of high-sensitive C-reactive protein (hsCRP) is a useful biomarker to predict cardiovascular events in patients (14). Elevated levels of hsCRP are associated with an increased risk for MI and major cardiovascular events in a primary prevention population. In addition, a clinical trial with stratification of patients according to their CRP levels has shown that the implementation of a lipid lowering therapy by statins is of greatest benefit in those subjects that exhibited elevated levels of the inflammatory marker hsCRP (15).
Coagulation and Inflammation
Over the last two decades, experimental data have shown an interaction of inflammation with procoagulatory mechanisms in the vessel wall: inflammatory cells, such as macrophages and T lymphocytes, produce cytokines leading to the expression of tissue factor (TF) by endothelial cells, macrophages, and smooth muscle cells (SMCs), thus enhancing prothrombotic properties of the plaque (16). However, the interaction of inflammation and coagulation is not limited to the vessel wall. In obesity, which is frequently associated with cardiovascular disease and diabetes, adipose tissue is a major source for elevated inflammatory cytokines (17, 18). Macrophages and T lymphocytes produce IFN-γ, interleukin (IL)-6, monocyte chemotactic protein (MCP)-1, TNF-α, and PAI-1 (19), which are all able to impact on the coagulation system (Figure 1). Not only inflammatory cytokines but also the complement system interacts with coagulation. The complement system is an important immune surveillance system with over 30 components involved. It can be activated by three different pathways, the classical, the lectin, and the alternative pathway (20). Activation of any of these consequently results in activation of complement C3, the central component of the complement system. Subsequently, complement C5 is activated leading to the formation of the terminal complement complex (C5b, C6, C7, C8, and C9). The biological functions of the complement system are broad including, among others, innate immune responses and coagulation (21). In coagulation, complement affects both platelet function and fibrin clot structure and lysis.
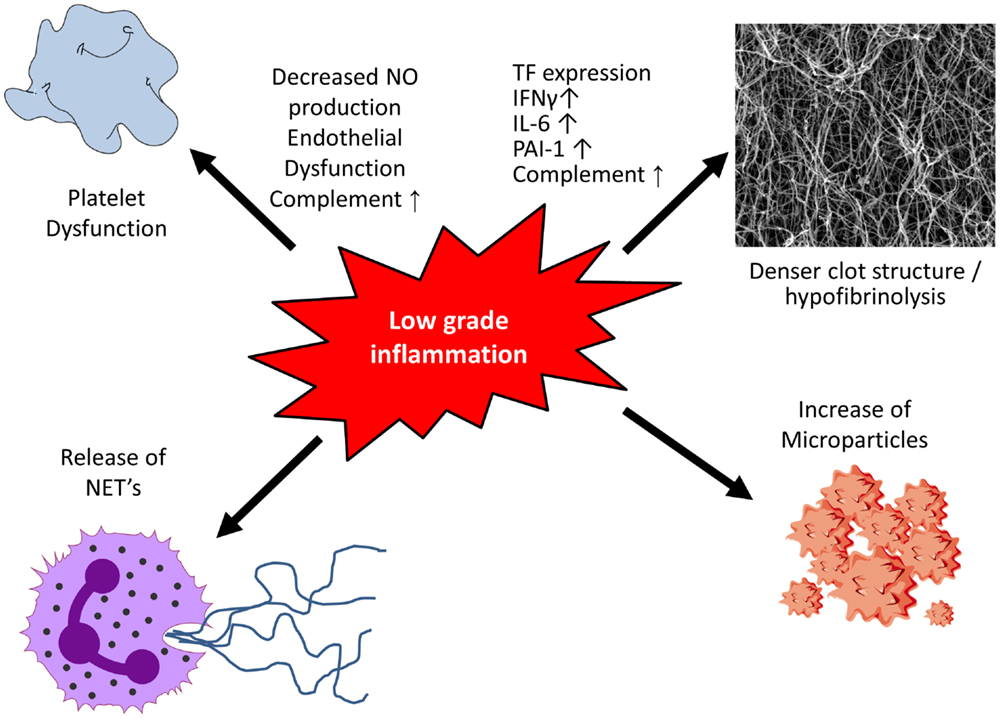
Figure 1. Low-grade inflammation and coagulation. Low-grade inflammation results in platelet dysfunction and the formation of a denser clot structure and hypofibrinolysis. Following activation or apoptosis various cell types release microparticles which contain procoagulants. Activation of neutrophils causes the release of neutrophil extracellular traps (NETs), which display procoagulant properties including platelet activation.
Primary Hemostasis
Following plaque rupture, thrombosis is initiated when thrombotic components of the plaque are exposed to circulating blood. Platelets are the first to respond and following their adhesion, platelets are activated leading to additional recruitment and aggregation with stabilization of the clot (22). In addition, inflammation can alter platelet function (Figure 1). Early changes include endothelial dysfunction with decreased nitric oxide (NO) production. NO is a key player in negative regulation of platelet activity, and decreased levels result in hyper-reactivity of platelets (23).
In addition to their primary role in hemostasis, platelets actively participate in inflammatory and immune processes. They carry a multiplicity of chemokines and cytokines, which are mostly found within the various platelet granules. Platelet activation and consequent degranulation result in the release of chemokines, including CXC-chemokine ligand 1 (CXCL1), platelet factor 4 (also known as CXCL4), CXCL5 and CXCL7, CC-chemokine ligand 3 [CCL3; also known as macrophage inflammatory protein 1 alpha (MIP-1α)], CCL5 [also known as regulated on activation, normal T Cell expressed and secreted (RANTES)], and CCL7, ILs (IL-1β, IL-7, and IL-8), prostaglandins, and the transmembrane protein CD154 (24, 25). CD154 interacts with CD40 on endothelial cells leading to an upregulation of cell adhesion molecules on endothelial cells, including intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1), and the release of CC-chemokine ligand-2 (CCL2), thereby facilitating leukocyte recruitment to the inflammatory site (26). In addition, activated platelets also release soluble CD154 likewise leading to an upregulation of vascular cell adhesion molecules and the release of IL-6. Through the expression of these chemokines, cytokines, CD154, and cell adhesion molecules, activated platelets promote neutrophil tethering and activation. Furthermore, they also activate monocytes and dendritic cells, particularly through the CD154–CD40 interaction (24). For an extended overview about this topic, see the review article of Semple et al. (24).
Complement and platelets interact on different levels. Platelets contain several complement receptors as well as complement regulatory molecules, and several components of the complement cascade have been recognized on the platelet surface. Complement activation can be initiated by several platelet agonists, such as adenosindiphosphate (ADP), epinephrine, arachidonic acid, thrombin, and exposure to shear stress (27). However, a study using complement C3 knock-out mice revealed C5 convertase activity of thrombin suggesting a platelet-independent mechanism of complement activation (28). The other way round, several complement components are known to activate platelets including the mannose-binding lectin (MBL)/MBL-associated serine protease (MASP) complex, binding of C1q to its receptor as well as the complement C3 cleavage products (iC3b, C3d, and C3dg) to their receptors CR2 and CR3 (only for iC3b) (27, 29–31). In vitro studies further suggest that activated platelets bind the anaphylatoxins C3a and C5a resulting in platelet aggregation (32, 33). In addition to classical complement receptors, P-selectin binds C3b resulting in the generation of C3a and the terminal complement complex C5b–9, thereby enhancing platelet response (34, 35). Furthermore, the anaphylatoxins C3a and C5a are generated which are known to have cytokine-like properties with enhanced leukocyte recruitment and support of the general inflammatory response (27). Under normal circumstances, complement activation is tightly regulated due to expression of complement regulatory molecules. On the platelet surface, factor H inactivates C3b and destabilizes the C3 convertase (36). Similarly, membrane cofactor (MCP or CD46) inactivates C3b, and decay-accelerating factor (DAF or CD55) interferes with C3 convertase. Furthermore, CD59 is known to destabilize the terminal complement complex (35).
Secondary Hemostasis
Fibrin clot structure plays a crucial role in determining predisposition to atherothrombotic events. Clots with compact structure and impaired fibrinolysis are associated with premature and more severe cardiovascular disease.
Inflammatory cytokines produced by macrophages and T lymphocytes, including IFN-γ, IL-6, MCP-1, and TNF-α, are able to influence both clot structure and lysis (Figure 1). By leading to increased production of acute phase products, such as fibrinogen and PAI-1, they alter clot structure and prolong fibrinolysis, thereby enhancing the prothrombotic milieu (19, 37). TF, the key initiator of the coagulation cascade, is produced by various cell types with monocytes being a major source. TF expression is very low under basal conditions (38) but increases upon stimulation with cytokines (i.e., TNF-α and IL-1β) resulting in activation of the coagulation system (39–41).
Complement interacts with the fluid phase of coagulation at different levels. Mannose-associated serine protease 1 (MASP-1) exhibits a thrombin-like profile by activating fibrinogen and FXIII. It thereby leads to the formation of a crosslinked fibrin clot although the catalytic efficiency compared with thrombin is greatly reduced (42). Furthermore, MASP-1 is able to induce prothrombin and thrombin activatable fibrinolysis inhibitor (TAFI), altogether leading to the formation of thinner fibrin fibers and longer lysis time (43). Similarly, MASP-2 cleaves prothrombin to thrombin (42, 44). The C1 inhibitor not only inhibits C1r/s, MASP-1, and MASP-2 but also inhibits FXIIa and FXIa (45). Shats-Tseytlina demonstrated already in 1994 that the activation of the complement cascade leads to the formation of a prothrombotic clot with thinner fibers. Using proteomics analysis, complements C1, C3, C4, C5a, and factor B were demonstrated in plasma clots (46). Of these, C3 binds with high affinity to fibrinogen leading to a prothrombotic clot structure and prolongation of clot lysis in vitro (47, 48). In addition to complement C3, C5a is known to induce TF and PAI-1 expressions (49, 50). Activation of the complement system and generation of the terminal C5b–9 complex further induce endothelial cells to produce TF and vWF thereby generating a prothrombotic phenotype (51).
Microparticles
Microparticles (MPs) are cell membrane-derived particles that can promote both coagulation and inflammation (Figure 1). Following activation or apoptosis, they are released from various cell types, including platelets, endothelial cells, red blood cells, and leukocytes. Depending on their origin they vary in size (0.2–1 μm) and their membrane composition of phospholipids and proteins. MPs can be detected in the circulation of healthy control individuals and are elevated in inflammatory situations, such as sepsis (52) and diabetes mellitus (53). MPs are directly able to modulate nitric oxide production from endothelial cells, induce cytokine release and prostacycline production as well as adherence of monocytes to the endothelium (54). The two major procoagulants found on the surface of MPs are phosphatidylserine and TF (55) thereby contributing to a prothrombotic state in various diseases. Furthermore, MPs can harbor and transport microRNA thereby impacting protein expression of target cells (56, 57).
Neutrophil Extracellular Traps
Activation of neutrophils causes the release of web-like structures of DNA, so-called neutrophil extracellular traps (NETs). Studies demonstrated NETs to display procoagulatory properties, including the induction of platelet adhesion, aggregation, and fibrin deposition on their surface. Histones, cationic proteins that are associated with DNA, can be actively secreted by activated inflammatory cells, such as neutrophils, contributing to the formation of NETs. In this context, they have been shown to promote platelet aggregation and thrombin formation through platelet-dependent mechanisms, including platelet toll-like receptor (TLR) 2 and TLR4 (58). Bosmann and colleagues demonstrated complement C5a to be able to trigger the appearance of histones by binding to its receptors C5aR and C5L2 (59), thereby contributing to an increased inflammatory and prothrombotic milieu. A more recent study in acute STEMI demonstrated that the interaction of thrombin-activated platelets with polymorphonuclear neutrophils at the site of plaque rupture results in local NET formation and delivery of active TF (60).
Atherosclerosis in Patients with Chronic Inflammatory Diseases
As highlighted above, atherosclerosis is characterized by an inflammatory process in the vessel wall (61). Thus, inflammatory biomarkers and cytokines, such as hsCRP, have been related with progressive cardiovascular disease as well as an increased risk for cardiovascular events (14, 62–65). On the other hand, patients with a chronic high state of inflammation, such as autoimmune diseases, including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), are at a high risk for cardiovascular morbidity and mortality, and the increased cardiovascular risk of these patients is related to the extent of inflammation (66) (Figure 2). However, the underlying pattern, pathophysiology, and phenotype of this increased cardiovascular risk in patients with chronic inflammation vary between diseases and are discussed below for RA and SLE.
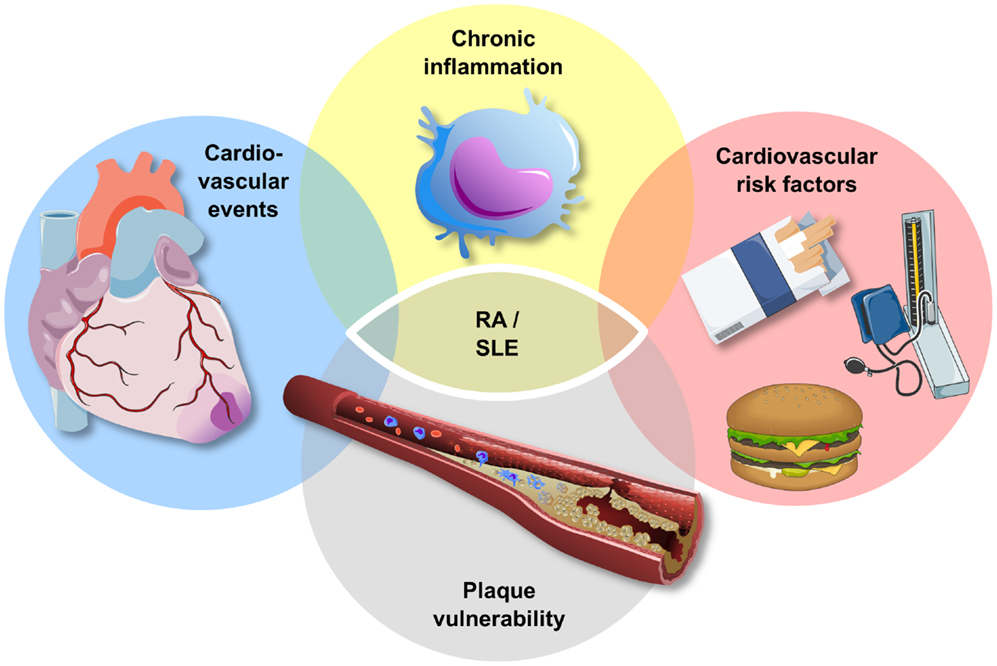
Figure 2. Atherosclerosis and chronic inflammatory disease. Patients with chronic inflammatory disease, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), exhibit an increased risk for traditional cardiovascular risk factors, plaque vulnerability, and cardiovascular events. The increase in cardiovascular events in this high-risk group cannot be explained entirely by the increase in cardiovascular risk factors.
Rheumatoid Arthritis
Patients with RA exhibit an increased cardiovascular risk with higher rates of sudden cardiac death and unrecognized MI compared with age- and sex-matched patients without RA (67). Furthermore, atherosclerosis starts early and is more diffuse over multiple vascular beds in this high-risk group (68). It could be demonstrated that patients with RA and diabetes share similar frequency and severity regarding preclinical atherosclerosis (69). An autopsy study of 41 patients with RA and 82 age- and sex-matched controls revealed a higher incidence of vulnerable plaques in the left anterior descending coronary artery and more plaque inflammation in patients with RA (70). Recently, a study by Karpouzas et al. analyzed plaque morphology using CT angiography and found a higher atherosclerotic burden in patients with RA. Interestingly, disease activity was associated with the presence of non-calcified and mixed, but not calcified plaques in this study, suggesting that disease activity is associated with plaque vulnerability (71). In light of these data, it is tempting to speculate that a more vulnerable plaque phenotype is responsible for the increased cardiovascular risk in patients with RA. Furthermore, these findings are in line with an increased prevalence of traditional cardiovascular risk factors, such as diabetes mellitus, hypertension, smoking, and obesity, in patients with RA [for review, see Ref. (72)]. However, several studies demonstrated that the cardiovascular risk in patients with RA following adjustment for classical cardiovascular risk factors remained high, suggesting that the increased risk in these patients cannot be fully explained by classical cardiovascular risk factors (67, 73). Vice versa, traditional cardiovascular risk assessment models are of limited value in patients with RA and should thus be used with caution in this high-risk group (74). Taken together, these data emphasize the link between systemic inflammation on the one hand and atherothrombosis on the other hand. However, patients with RA are not solely characterized by increased atherothrombosis but also by high rates of valvular heart disease, myocarditis, and non-ischemic cardiac failure, which all contribute to the increased cardiovascular risk in this high-risk group of patients (75).
The recognition of progressive vascular disease by the use of circulating biomarkers is a challenge in patients with a chronic high state of inflammation as conventional, inflammatory biomarkers may lack specificity in this high-risk group. Recently, we have been able to associate C-peptide, which is mainly regulated by insulin secretion and kidney function (76), to the presence of coronary artery calcification in patients with RA (77). Interestingly, the inflammatory biomarker YKL-40, which is thought to play a role in both atherogenesis and RA (62, 78– 80), was a predictor for the presence of coronary artery calcification in patients with RA in this study, supporting the link between inflammation and atherosclerosis in patients with RA (Figure 2).
In addition, others found striking similarities between the pathogenesis of both RA and atherosclerosis, including endothelial activation, inflammatory cell infiltration and activation, neovascularization, and collagen degradation via matrix metalloproteinases [for review, see Ref. (72)].
Systemic Lupus Erythematosus
Patients with SLE exhibit an increased risk for mortality (81, 82), which is in part attributed to disease activity and in part to increased cardiovascular deaths. This used to result in a bimodal mortality pattern and was described almost four decades ago (83). Presently, the mortality associated with disease activity has declined with advances in disease treatment, which results in increased life expectancy and a constant mortality rate over time (84). With these improvements in treating patients with SLE, cardiovascular and infectious complications are now the main direct cause of death among both early and late fatalities (84). Recently, a large retrospective cohort study in Taiwan compared the incidence of acute MI between patients with SLE compared to age- and sex-matched controls. The investigators found an adjusted hazard ratio of 5.11 for acute MI associated with SLE, which was even larger in the female population (hazard ratio 6.28) (85). Interestingly, patients with SLE were not only associated with a higher risk of acute MI but also with an increased mortality post-MI (85).
Similar to what is described above for patients with RA, patients with SLE exhibit an increased prevalence of traditional cardiovascular risk factors, including smoking, dyslipidemia, hypertension, and diabetes (86). However, adjustment for Framingham risk factors could not fully account for the increased cardiovascular risk in patients with SLE (87). These findings are in line with a recent meta-analysis including 17,187 patients, which found that both traditional and disease specific risk factors contribute to the risk in patients with SLE. Disease-specific risk factors included the presence of autoantibodies, neurological disorders, and to a lesser extent organ damage and SLE activity (88). Furthermore, antiphospholipid antibodies were related to mitral valve nodules and significant mitral regurgitation (89), suggesting that certain SLE-specific risk factors may also be associated with cardiovascular disease but not related to atherothrombosis.
Taken together, there appears to be a link between patients with a chronic high state of inflammation and progressive cardiovascular disease. Whereas RA and SLE demonstrate distinct immunological profiles and unique cytokine patterns [for review, see Ref. (90, 91, 75)], both share an increased cardiovascular mortality. Further studies are needed to evaluate the molecular mechanisms involved in the increased cardiovascular risk in patients with chronic inflammation and the interplay between chronic inflammation, cardiovascular risk factors, and progressive cardiovascular disease.
Inflammation in Patients with Diabetes Mellitus
The cardiovascular vulnerability of diabetic and prediabetic patients is predictable by a variety of inflammatory markers. These are indicative of an activated innate immune system featuring elevated circulating levels of IL-1, TNF-α, IL-6, and hsCRP (92). Among these, hsCRP has gained the most attention as a predictor of diabetes, cardiovascular disease, and mortality (14, 93). In addition, low-grade inflammation is linked to activation of the coagulation system featuring increased levels of fibrinogen and PAI-1 as predisposing factor for thrombus formation (94).
A variety of mechanisms have been proposed for the initiation of subclinical inflammation in metabolic disease. Among these, obesity leads to hypertrophic adipocytes, which are unable to cope with an overwhelming supply of fatty acids (95). The consequential release of free fatty acids into the circulation causes steatosis of various organs including liver and muscle (95). Limited lipid storage capacity of their organelles results in metabolic disturbance and oxidative stress, which has been termed lipid toxicity. The consequential cellular damage and apoptosis requires debris-clearing recruitment of macrophages. These eliminate cell fragments as a prerequisite for tissue regeneration. Low-grade inflammation accompanying this cellular turnover is beneficial and part of a healing process. Persistence of pathogenic stimuli with ongoing cellular damage, however, leads to chronic inflammation and disturbed tissue integrity (96). Initiation of fibrosis may lead to an irreversible scaring process as found for steatohepatitis with transition to liver cirrhosis.
Saturated fatty acids further serve as ligands for pattern recognition receptors including TLR2 and 4 (97, 98). These provide a primitive line of defense of the innate immune system able to recognize a variety of microbial fragments and cell debris. TLRs signal downstream to various inflammatory pathways, including NF-κB and MAP-kinase, with secretion of inflammatory cytokines, including TNF-α, IL-1, and IL-6. In addition, activation of NF-κB causes direct inhibition of the insulin receptor cascade as an alternative mechanism of inflammation-mediated insulin resistance (99). Genetic or pharmaceutical inhibition of NF-κB improves glucose metabolism in mice and men. Indeed treatment with salsalate – a prodrug of salicylic acid, which inhibits NF-κB at high doses – lowers fasting glucose in non-diabetic, obese individuals (99, 100). Salsalate, however, also slightly increases plasma LDL-cholesterol levels and was ineffective in improving flow-mediated dilatation in patients with diabetes, questioning its clinical utility (100). Another anti-inflammatory strategy in metabolic disease, which has advanced to the clinics, is blockade of the IL-1 receptor system. This improved glucose metabolism and insulin sensitivity in small clinical studies and was associated with a reduction of inflammatory serum parameters (101). Blockade of the IL-1 system is currently evaluated in a large cardiovascular endpoint study (CANTOS) in which the IL-1-directed antibody canakinumab is tested versus placebo in a randomized trial in 17,200 stable, post-MI patients with persistent high CRP levels (102). A dedicated sub-study further investigates the effect of IL-1 system blockade on glucose metabolism and insulin resistance with trial termination being expected in 2017. The efficacy of inflammation targeting therapies in metabolic disease is further investigated in the Cardiovascular Inflammation Reduction Trial (CIRT) using low-dose methotrexate (target dose 15–20 mg/week) treatment. This study randomizes 7000 prior MI patients with either type 2 diabetes or the metabolic syndrome with an average follow-up period of 3–5 years expecting termination in 2018 (103).
More recently, metabolic inflammation has further been attributed to a disturbed intestinal barrier function (104). Increased circulating concentrations of endotoxin are detected in patients with obesity and diabetes with direct associations to cardiovascular disease (105). A disturbed intestinal barrier enables the passive diffusion of bacterial fragments and lipopolysaccarides across the intestinal mucosa. Interestingly, this seems to be modulated by dietary factors with high nutritional fat intake promoting endotoxin absorption (106). Circulating endotoxin serves as a ligand for TLR2 and 4 with activation of the innate immune system. Chronic low-dose endotoxin application was consequently found to cause obesity and insulin resistance in rodent models while knockout of TLR4 prevented diet-induced obesity, glucose intolerance, and atherosclerosis (107–109). Furthermore, mice lacking the functional LPS receptor CD14 were resistant to diet-induced obesity and related disorders (110) while antibiotic eradication of the gut flora led to reduced metabolic inflammation, insulin resistance, and fat mass development (111). In consequence, germ-free mice were protected from high-fat diet-induced inflammation (112). Modulation of the intestinal microbiome as a source for endotoxin generation might therefore provide a new treatment strategy for diabetes and cardiovascular disease prevention.
These observations demonstrate a close interaction between the immune system and metabolism which is particularly present in patients with diabetes. Activation of the immune system does thereby not depend on an external stressor but is the consequence of overnutrition as an endogenous stressor. Concepts of anti-inflammatory therapies might prove effective in improving diabetes and cardiovascular outcome.
Conclusion
Over the last 30 years, inflammation has increasingly been recognized as an important pathophysiological link between vascular disease, atherothrombosis, metabolic disorders, and chronic autoimmune diseases. A plethora of mediators, such as cytokines, chemokines, and adipokines, released from various cells in different organs contribute to both local and remote stimulations of inflammatory cells, thus creating a network of interactions at different levels in the organism. Future therapeutic strategies need to be developed to examine how and by which tools this interaction can be targeted to modulate cardiovascular disease in high-risk patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med (1998) 339:229–34. doi: 10.1056/NEJM199807233390404
2. Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA (2002) 287:2570–81. doi:10.1001/jama.287.19.2570
3. Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, et al. Heart disease and stroke statistics – 2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation (2006) 113:e85–151. doi:10.1161/CIRCULATIONAHA.105.171600
4. Rugg SS, Bailey AL, Browning SR. Preventing cardiovascular disease in Kentucky: epidemiology, trends, and strategies for the future. J Ky Med Assoc (2008) 106:149–61; quiz 149, 162–3.
5. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation (2002) 105:1135–43. doi:10.1161/hc0902.104353
6. Falk E, Fernandez-Ortiz A. Role of thrombosis in atherosclerosis and its complications. Am J Cardiol (1995) 75:3B–11B. doi:10.1016/0002-9149(95)80003-B
8. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med (2005) 352:1685–95. doi:10.1056/NEJMra043430
9. Libby P, Hansson GK. Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res (2015) 116:307–11. doi:10.1161/CIRCRESAHA.116.301313
10. Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol (2009) 20:370–8. doi:10.1097/MOL.0b013e3283309848
11. Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol (2008) 8:802–15. doi:10.1038/nri2415
12. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity (2013) 38:1092–104. doi:10.1016/j.immuni.2013.06.009
13. Taleb S, Tedgui A, Mallat Z. Regulatory T-cell immunity and its relevance to atherosclerosis. J Intern Med (2008) 263:489–99. doi:10.1111/j.1365-2796.2008.01944.x
14. Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, et al. C-reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation (1999) 99:237–42.
15. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med (2008) 359:2195–207. doi:10.1056/NEJMoa0807646
16. Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med (2010) 38:S26–34. doi:10.1097/CCM.0b013e3181c98d21
17. Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes (2003) 52:1799–805. doi:10.2337/diabetes.52.7.1799
18. Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res (2005) 96:939–49. doi:10.1161/01.RES.0000163635.62927.34
19. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol (2005) 25:2062–8. doi:10.1161/01.ATV.0000183883.72263.13
20. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344:1058–66. doi:10.1056/NEJM200104053441406
21. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi:10.1038/ni.1923
22. Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J (2010) 74:597–607. doi:10.1253/circj.CJ-09-0982
23. Anderson TJ. Nitric oxide, atherosclerosis and the clinical relevance of endothelial dysfunction. Heart Fail Rev (2003) 8:71–86. doi:10.1023/A:1022199021949
24. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol (2011) 11:264–74. doi:10.1038/nri2956
25. Zhang W, Huang W, Jing F. Contribution of blood platelets to vascular pathology in Alzheimer’s disease. J Blood Med (2013) 4:141–7. doi:10.2147/JBM.S45071
26. Andre P, Nannizzi-Alaimo L, Prasad SK, Phillips DR. Platelet-derived CD40L: the switch-hitting player of cardiovascular disease. Circulation (2002) 106:896–9. doi:10.1161/01.CIR.0000028962.04520.01
27. Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol (2010) 47:2170–5. doi:10.1016/j.molimm.2010.05.009
28. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, Mcguire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med (2006) 12:682–7. doi:10.1038/nm1419
29. Cosgrove LJ, D’Apice AJ, Haddad A, Pedersen J, Mckenzie IF. CR3 receptor on platelets and its role in the prostaglandin metabolic pathway. Immunol Cell Biol (1987) 65:453–60. doi:10.1038/icb.1987.54
30. Nunez D, Charriaut-Marlangue C, Barel M, Benveniste J, Frade R. Activation of human platelets through gp140, the C3d/EBV receptor (CR2). Eur J Immunol (1987) 17:515–20. doi:10.1002/eji.1830170413
31. La Bonte LR, Pavlov VI, Tan YS, Takahashi K, Takahashi M, Banda NK, et al. Mannose-binding lectin-associated serine protease-1 is a significant contributor to coagulation in a murine model of occlusive thrombosis. J Immunol (2012) 188:885–91. doi:10.4049/jimmunol.1102916
32. Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med (1983) 158:603–15. doi:10.1084/jem.158.2.603
33. Martel C, Cointe S, Maurice P, Matar S, Ghitescu M, Théroux P, et al. Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PLoS One (2011) 6:e18812. doi:10.1371/journal.pone.0018812
34. Del Conde I, Crúz MA, Zhang H, López JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med (2005) 201:871–9. doi:10.1084/jem.20041497
35. Verschoor A, Langer HF. Crosstalk between platelets and the complement system in immune protection and disease. Thromb Haemost (2013) 110:910–9. doi:10.1160/TH13-02-0102
36. Mnjoyan Z, Li J, Afshar-Kharghan V. Factor H binds to platelet integrin alphaIIbbeta3. Platelets (2008) 19:512–9. doi:10.1080/09537100802238494
37. Alzahrani SH, Ajjan RA. Coagulation and fibrinolysis in diabetes. Diab Vasc Dis Res (2010) 7:260–73. doi:10.1177/1479164110383723
38. Breitenstein A, Tanner FC, Lüscher TF. Tissue factor and cardiovascular disease. Circ J (2010) 74:3–12. doi:10.1253/circj.CJ-09-0818
39. Napoleone E, Di Santo A, Lorenzet R. Monocytes upregulate endothelial cell expression of tissue factor: a role for cell-cell contact and cross-talk. Blood (1997) 89:541–9.
40. Steffel J, Hermann M, Greutert H, Gay S, Lüscher TF, Ruschitzka F, et al. Celecoxib decreases endothelial tissue factor expression through inhibition of c-Jun terminal NH2 kinase phosphorylation. Circulation (2005) 111:1685–9. doi:10.1161/01.CIR.0000160358.63804.C9
41. Steffel J, Lüscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation (2006) 113:722–31. doi:10.1161/CIRCULATIONAHA.105.567297
42. Gulla KC, Gupta K, Krarup A, Gal P, Schwaeble WJ, Sim RB, et al. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology (2010) 129:482–95. doi:10.1111/j.1365-2567.2009.03200.x
43. Hess K, Ajjan R, Phoenix F, Dobó J, Gál P, Schroeder V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS One (2012) 7:e35690. doi:10.1371/journal.pone.0035690
44. Krarup A, Wallis R, Presanis JS, Gál P, Sim RB. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One (2007) 2:e623. doi:10.1371/journal.pone.0000623
45. Wuillemin WA, Minnema M, Meijers JC, Roem D, Eerenberg AJ, Nuijens JH, et al. Inactivation of factor XIa in human plasma assessed by measuring factor XIa-protease inhibitor complexes: major role for C1-inhibitor. Blood (1995) 85:1517–26.
46. Distelmaier K, Adlbrecht C, Jakowitsch J, Winkler S, Dunkler D, Gerner C, et al. Local complement activation triggers neutrophil recruitment to the site of thrombus formation in acute myocardial infarction. Thromb Haemost (2009) 102:564–72. doi:10.1160/TH09-02-0103
47. Hess K, Alzahrani S, Mathai M, Schroeder V, Carter AM, Howell G, et al. A novel mechanism for hypofibrinolysis in diabetes: the role of Complement C3. Diabetologia (2012) 55:1103–13. doi:10.1007/s00125-011-2301-7
48. Howes JM, Richardson VR, Smith KA, Schroeder V, Somani R, Shore A, et al. Complement C3 is a novel plasma clot component with anti-fibrinolytic properties. Diab Vasc Dis Res (2012) 9:216–25. doi:10.1177/1479164111432788
49. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol (2006) 177:4794–802. doi:10.4049/jimmunol.177.7.4794
50. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol (2007) 28:184–92. doi:10.1016/j.it.2007.02.006
51. Tedesco F, Fischetti F, Pausa M, Dobrina A, Sim RB, Daha MR. Complement-endothelial cell interactions: pathophysiological implications. Mol Immunol (1999) 36:261–8. doi:10.1016/S0161-5890(99)90054-8
52. Zafrani L, Gerotziafas G, Byrnes C, Hu X, Perez J, Lévi C, et al. Calpastatin controls polymicrobial sepsis by limiting procoagulant microparticle release. Am J Respir Crit Care Med (2012) 185:744–55. doi:10.1164/rccm.201109-1686OC
53. Feng B, Chen Y, Luo Y, Chen M, Li X, Ni Y. Circulating level of microparticles and their correlation with arterial elasticity and endothelium-dependent dilation in patients with type 2 diabetes mellitus. Atherosclerosis (2010) 208:264–9. doi:10.1016/j.atherosclerosis.2009.06.037
54. Puddu P, Puddu GM, Cravero E, Muscari S, Muscari A. The involvement of circulating microparticles in inflammation, coagulation and cardiovascular diseases. Can J Cardiol (2010) 26:140–5. doi:10.1016/S0828-282X(10)70371-8
55. Diamant M, Tushuizen ME, Sturk A, Nieuwland R. Cellular microparticles: new players in the field of vascular disease? Eur J Clin Invest (2004) 34:392–401. doi:10.1111/j.1365-2362.2004.01355.x
56. Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA (2004) 10:1957–66. doi:10.1261/rna.7135204
57. Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia (2006) 20:847–56. doi:10.1038/sj.leu.2404132
58. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi:10.1182/blood-2011-03-343061
59. Bosmann M, Grailer JJ, Ruemmler R, Russkamp NF, Zetoune FS, Sarma JV, et al. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J (2013) 27:5010–21. doi:10.1096/fj.13-236380
60. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J (2015) 36(22):1405–14. doi:10.1093/eurheartj/ehv007
61. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med (1999) 340:115–26. doi:10.1056/NEJM199901143400207
62. Kastrup J, Johansen JS, Winkel P, Hansen JF, Hildebrandt P, Jensen GB, et al. High serum YKL-40 concentration is associated with cardiovascular and all-cause mortality in patients with stable coronary artery disease. Eur Heart J (2009) 30:1066–72. doi:10.1093/eurheartj/ehp049
63. Corrado E, Rizzo M, Coppola G, Fattouch K, Novo G, Marturana I, et al. An update on the role of markers of inflammation in atherosclerosis. J Atheroscler Thromb (2010) 17:1–11. doi:10.5551/jat.2600
64. Hochholzer W, Morrow DA, Giugliano RP. Novel biomarkers in cardiovascular disease: update 2010. Am Heart J (2010) 160:583–94. doi:10.1016/j.ahj.2010.06.010
65. Turkyilmaz AK, Devrimsel G, Kirbas A, Cicek Y, Karkucak M, Capkin E, et al. Relationship between pulse wave velocity and serum YKL-40 level in patients with early rheumatoid arthritis. Rheumatol Int (2013) 33:2751–6. doi:10.1007/s00296-013-2810-4
66. Dregan A, Charlton J, Chowienczyk P, Gulliford MC. Chronic inflammatory disorders and risk of type 2 diabetes mellitus, coronary heart disease, and stroke: a population-based cohort study. Circulation (2014) 130:837–44. doi:10.1161/CIRCULATIONAHA.114.009990
67. Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum (2005) 52:402–11. doi:10.1002/art.20878
68. Wang S, Yiu KH, Mok MY, Ooi GC, Khong PL, Mak KF, et al. Prevalence and extent of calcification over aorta, coronary and carotid arteries in patients with rheumatoid arthritis. J Intern Med (2009) 266:445–52. doi:10.1111/j.1365-2796.2009.02123.x
69. Stamatelopoulos KS, Kitas GD, Papamichael CM, Chryssohoou E, Kyrkou K, Georgiopoulos G, et al. Atherosclerosis in rheumatoid arthritis versus diabetes: a comparative study. Arterioscler Thromb Vasc Biol (2009) 29:1702–8. doi:10.1161/ATVBAHA.109.190108
70. Aubry MC, Maradit-Kremers H, Reinalda MS, Crowson CS, Edwards WD, Gabriel SE. Differences in atherosclerotic coronary heart disease between subjects with and without rheumatoid arthritis. J Rheumatol (2007) 34:937–42.
71. Karpouzas GA, Malpeso J, Choi TY, Li D, Munoz S, Budoff MJ. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann Rheum Dis (2014) 73:1797–804. doi:10.1136/annrheumdis-2013-203617
72. Skeoch S, Bruce IN. Atherosclerosis in rheumatoid arthritis: is it all about inflammation? Nat Rev Rheumatol (2015) 11(7):390–400. doi:10.1038/nrrheum.2015.40
73. del Rincon ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum (2001) 44:2737–45. doi:10.1002/1529-0131(200112)44:12<2737::AID-ART460>3.0.CO;2-#
74. Arts EE, Popa C, Den Broeder AA, Semb AG, Toms T, Kitas GD, et al. Performance of four current risk algorithms in predicting cardiovascular events in patients with early rheumatoid arthritis. Ann Rheum Dis (2015) 74:668–74. doi:10.1136/annrheumdis-2013-204024
75. Mason JC, Libby P. Cardiovascular disease in patients with chronic inflammation: mechanisms underlying premature cardiovascular events in rheumatologic conditions. Eur Heart J (2015) 36:482c–9c. doi:10.1093/eurheartj/ehu403
76. Cahill GF Jr. C-peptide: a new method of assessing pancreatic beta-cell function. N Engl J Med (1973) 288:1181–2. doi:10.1056/NEJM197305312882210
77. Burgmaier M, Hoppe S, Kruger T, Mahnken AH, Ketteler M, Reith S, et al. Serum levels of C-peptide are associated with coronary artery calcification in patients with rheumatoid arthritis. Rheumatol Int (2015) 35(9):1541–7. doi:10.1007/s00296-015-3244-y
78. Peltomaa R, Paimela L, Harvey S, Helve T, Leirisalo-Repo M. Increased level of YKL-40 in sera from patients with early rheumatoid arthritis: a new marker for disease activity. Rheumatol Int (2001) 20:192–6. doi:10.1007/s002960100115
79. Zheng JL, Lu L, Hu J, Zhang RY, Zhang Q, Chen QJ, et al. Increased serum YKL-40 and C-reactive protein levels are associated with angiographic lesion progression in patients with coronary artery disease. Atherosclerosis (2010) 210:590–5. doi:10.1016/j.atherosclerosis.2009.12.016
80. Bakker MF, Cavet G, Jacobs JW, Bijlsma JW, Haney DJ, Shen Y, et al. Performance of a multi-biomarker score measuring rheumatoid arthritis disease activity in the CAMERA tight control study. Ann Rheum Dis (2012) 71:1692–7. doi:10.1136/annrheumdis-2011-200963
81. Abu-Shakra M, Urowitz MB, Gladman DD, Gough J. Mortality studies in systemic lupus erythematosus. Results from a single center. I. Causes of death. J Rheumatol (1995) 22:1259–64.
82. Abu-Shakra M, Urowitz MB, Gladman DD, Gough J. Mortality studies in systemic lupus erythematosus. Results from a single center. II. Predictor variables for mortality. J Rheumatol (1995) 22:1265–70.
83. Urowitz MB, Bookman AA, Koehler BE, Gordon DA, Smythe HA, Ogryzlo MA. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med (1976) 60:221–5. doi:10.1016/0002-9343(76)90431-9
84. Nossent J, Cikes N, Kiss E, Marchesoni A, Nassonova V, Mosca M, et al. Current causes of death in systemic lupus erythematosus in Europe, 2000-2004: relation to disease activity and damage accrual. Lupus (2007) 16:309–17. doi:10.1177/0961203307077987
85. Lin CY, Shih CC, Yeh CC, Chou WH, Chen TL, Liao CC. Increased risk of acute myocardial infarction and mortality in patients with systemic lupus erythematosus: two nationwide retrospective cohort studies. Int J Cardiol (2014) 176:847–51. doi:10.1016/j.ijcard.2014.08.006
86. Petri M, Spence D, Bone LR, Hochberg MC. Coronary artery disease risk factors in the Johns Hopkins Lupus Cohort: prevalence, recognition by patients, and preventive practices. Medicine (Baltimore) (1992) 71:291–302. doi:10.1097/00005792-199209000-00004
87. Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, Du Berger R, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum (2001) 44:2331–7. doi:10.1002/1529-0131(200110)44:10<2331::AID-ART395>3.0.CO;2-I
88. Ballocca F, D’Ascenzo F, Moretti C, Omede P, Cerrato E, Barbero U, et al. Predictors of cardiovascular events in patients with systemic lupus erythematosus (SLE): a systematic review and meta-analysis. Eur J Prev Cardiol (2015) 22(11):1435–41. doi:10.1177/2047487314546826
89. Farzaneh-Far A, Roman MJ, Lockshin MD, Devereux RB, Paget SA, Crow MK, et al. Relationship of antiphospholipid antibodies to cardiovascular manifestations of systemic lupus erythematosus. Arthritis Rheum (2006) 54:3918–25. doi:10.1002/art.22265
90. Bartoloni Bocci E, Luccioli F, Angrisani C, Moscatelli S, Alunno A, Gerli R. Accelerated atherosclerosis in systemic lupus erythematosus and other connective tissue diseases. Expert Rev Clin Immunol (2007) 3:531–41. doi:10.1586/1744666X.3.4.531
91. Kahlenberg JM, Kaplan MJ. Mechanisms of premature atherosclerosis in rheumatoid arthritis and lupus. Annu Rev Med (2013) 64:249–63. doi:10.1146/annurev-med-060911-090007
92. Devaraj S, Dasu MR, Jialal I. Diabetes is a proinflammatory state: a translational perspective. Expert Rev Endocrinol Metab (2010) 5:19–28. doi:10.1586/eem.09.44
93. Ridker PM. Inflammatory biomarkers and risks of myocardial infarction, stroke, diabetes, and total mortality: implications for longevity. Nutr Rev (2007) 65:S253–9. doi:10.1111/j.1753-4887.2007.tb00372.x
94. Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab (2009) 94:3171–82. doi:10.1210/jc.2008-2534
95. Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome – an allostatic perspective. Biochim Biophys Acta (2010) 1801:338–49. doi:10.1016/j.bbalip.2009.12.006
96. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi:10.1146/annurev-immunol-031210-101322
97. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology (2007) 132:2169–80. doi:10.1053/j.gastro.2007.03.059
98. Huang S, Rutkowsky JM, Snodgrass RG, Ono-Moore KD, Schneider DA, Newman JW, et al. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res (2012) 53:2002–13. doi:10.1194/jlr.D029546
99. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med (2005) 11:183–90. doi:10.1038/nm1166
100. Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med (2010) 152:346–57. doi:10.7326/0003-4819-152-6-201003160-00004
101. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356:1517–26. doi:10.1056/NEJMoa065213
102. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J (2011) 162:597–605. doi:10.1016/j.ahj.2011.06.012
103. Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, Zaharris E, et al. Rationale and design of the cardiovascular inflammation reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J (2013) 166(199–207):e115. doi:10.1016/j.ahj.2013.03.018
104. Serino M, Luche E, Chabo C, Amar J, Burcelin R. Intestinal microflora and metabolic diseases. Diabetes Metab (2009) 35:262–72. doi:10.1016/j.diabet.2009.03.003
105. Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care (2011) 34:392–7. doi:10.2337/dc10-1676
106. Laugerette F, Vors C, Peretti N, Michalski MC. Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie (2011) 93:39–45. doi:10.1016/j.biochi.2010.04.016
107. Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A (2004) 101:10679–84. doi:10.1073/pnas.0403249101
108. Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes (2007) 56:1986–98. doi:10.2337/db06-1595
109. Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) (2008) 16:1248–55. doi:10.1038/oby.2008.210
110. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56:1761–72. doi:10.2337/db06-1491
111. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes (2008) 57:1470–81. doi:10.2337/db07-1403
Keywords: atherosclerosis, type 2 diabetes mellitus, systemic lupus erythematosus, rheumatoid arthritis, coagulation, inflammation, cardiovascular disease
Citation: Schuett KA, Lehrke M, Marx N and Burgmaier M (2015) High-Risk Cardiovascular Patients: Clinical Features, Comorbidities, and Interconnecting Mechanisms. Front. Immunol. 6:591. doi: 10.3389/fimmu.2015.00591
Received: 13 July 2015; Accepted: 03 November 2015;
Published: 23 November 2015
Edited by:
Mette M. M. Rosenkilde, University of Copenhagen, DenmarkCopyright: © 2015 Schuett, Lehrke, Marx and Burgmaier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathias Burgmaier, bWJ1cmdtYWllckB1a2FhY2hlbi5kZQ==
†Katharina Andrea Schuett and Michael Lehrke have contributed equally to this work.