 Silvia Gregori
Silvia Gregori Laura Passerini
Laura Passerini Maria-Grazia Roncarolo
Maria-Grazia Roncarolo- 1Division of Regenerative Medicine, Stem Cells and Gene Therapy, IRCCS San Raffaele Scientific Institute, San Raffaele Telethon Institute for Gene Therapy (TIGET), Milan, Italy
- 2Department of Pediatric Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Palo Alto, CA, USA
T regulatory cells (Tregs) are subsets of T lymphocytes specialized in modulating antigen-specific immune responses in vivo. Hence, Tregs represent an ideal therapeutic tool to control detrimental immune reactions. Based on solid pre-clinical results, investigators started testing the safety and efficacy of Treg-based therapies in humans. Despite promising results, a number of issues remain to be solved. We will discuss the results obtained from clinical trials and the challenges and risks we are facing in the further development of Treg-based therapies.
Introduction
T regulatory cells (Tregs) are a component of the immune system involved in modulating immune reactions and in inducing tolerance. Due to their potential as immune modulators, therapeutic application of Tregs to control undesirable immune responses and to promote tolerance has become an active field of investigation (1). Over the years, several types of Tregs have been identified, and the forkhead box P3 (FOXP3)-expressing Tregs (FOXP3+ Tregs) (2) and the T regulatory type 1 (Tr1) cells (3) are the best characterized (Figure 1).
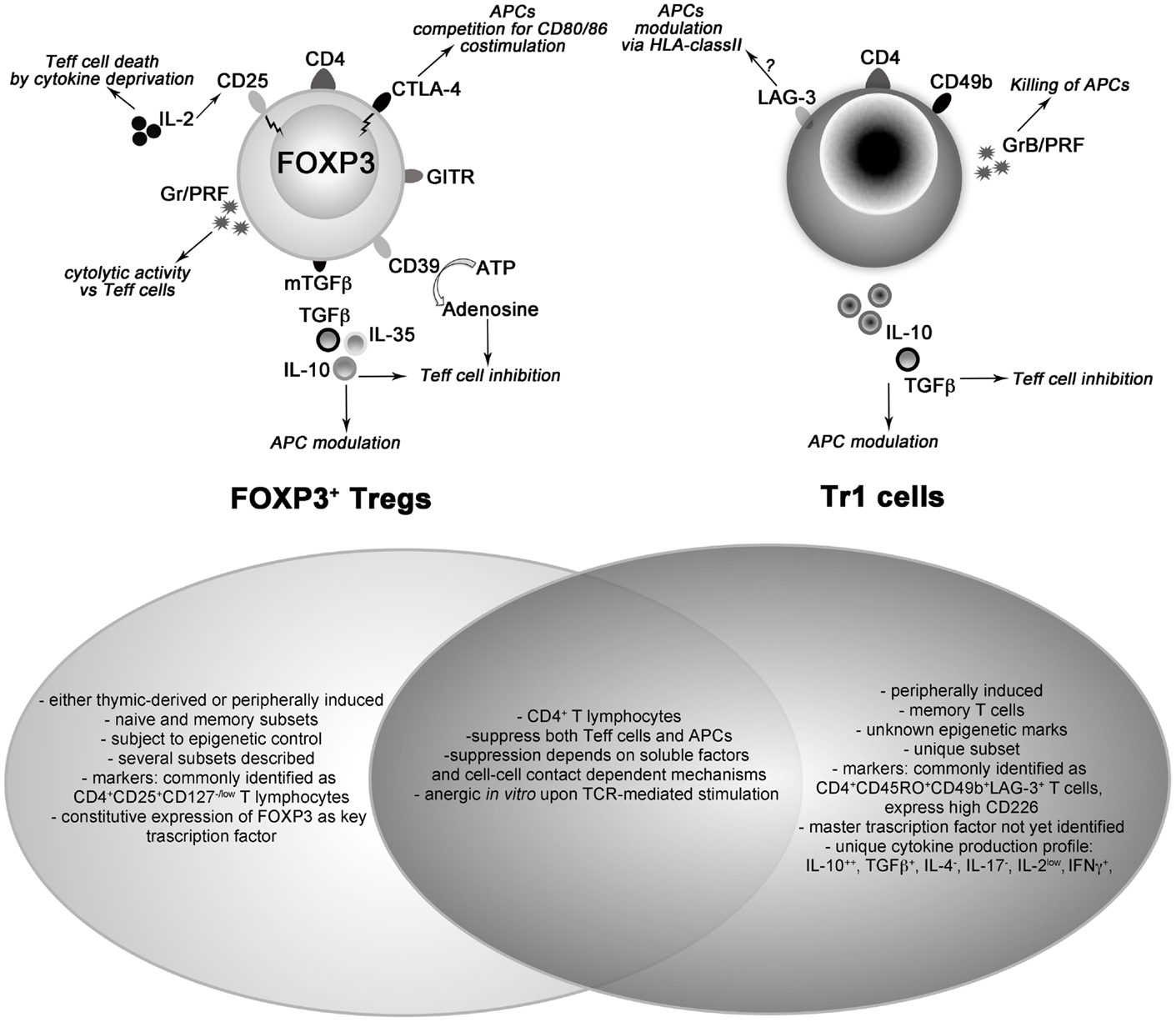
Figure 1. Schematic representation of cell surface and intracytoplasmic markers and mechanisms of action characterizing FOXP3-expressing (left cartoon) and type 1 (right cartoon) T regulatory cells. The shared and unique features of both cell types are listed in the frames. Ag, antigen; APC, antigen-presenting cell; ATP, adenosine triphosphate; CTLA-4, cytotoxic T-lymphocyte antigen 4; FOXP3, forkhead box protein 3; GITR, glucocorticoid-induced TNFR family related gene; Gr; Granzymes; LAG-3, lymphocyte-activation gene 3; PRF, perforin; Teff, effector T cell; TGFβ, transforming growth factor β; Tr1, type 1 T regulatory cell.
FOXP3+ Tregs can be either thymus-derived (tTregs), or induced in the periphery (pTregs) (4, 5). Regardless of their origin, both subsets are characterized by constitutive expression of the IL-2Rα-chain (CD25), in the absence of the IL-7Rα-chain (CD127), and of FOXP3 (6), making the two subsets indistinguishable based on their phenotype. High expression of Helios has been identified in FOXP3+ Tregs (7), and suggested to be specific for tTregs (8). However, this notion was later challenged by the demonstration that Helios is also expressed by non-tTregs (9, 10). To date, the most reliable feature unambiguously identifying tTregs is the epigenetic remodeling of a specific region in the FOXP3 locus, indicated as Treg-specific-demethylated-region (TSDR) (11). A more comprehensive CpG hypomethylation pattern of tTregs including several Treg-related genes has been described (12).
In addition to CD25, along the years, the expression of several molecules, i.e., CTLA-4 (13), GITR (14), CD39 (15), Galectin 10 (16), latency-associated-peptide (LAP) (17), and glycoproteinA-repetitions-predominant (GARP) (18) has been attributed to human FOXP3+ Tregs. The expression of the above-mentioned molecules is not exclusive to FOXP3+ Tregs, since they are often shared with activated conventional T cells.
CTLA-4, GITR, and CD39 are specifically associated with FOXP3+ Treg suppressive function, which is primarily dependent on contact with target cells. Additional mechanisms of suppression have been described for FOXP3+ Tregs, including release of IL-10 (19), TGF-β (20, 21), and IL-35 (22), direct killing of T effector (Teff) cells through the granzyme/perforin axis (23), modulation of antigen-presenting cells (APCs) stimulatory capacity via CTLA-4 (24), cytokine deprivation (25), and generation of immunosuppressive metabolites, such as extracellular adenosine (26) and intracellular cAMP (27). The variety of phenotypes and weapons discovered led from the original idea of FOXP3+ Tregs as homogeneous population to the modern view of a heterogeneous pool, including several specialized subtypes characterized by expression of specific cell surface markers such as ICOS (19), HLA-DR (28, 29), and CD45 isoforms (30, 31).
Tr1 cells are memory T lymphocytes expressing CD49b and LAG-3 (32). Tr1 cells, upon activation, secrete high levels of IL-10 and TGF-β, variable amounts of IL-5, GM-CSF, and IFN-γ, and minimal amounts of IL-2, IL-4, and IL-17 (3, 33, 34). Tr1 cells express CTLA-4, (35, 36), PD-1 (36), and ICOS (37). Similar to FOXP3+ Tregs, Tr1 cells can express CD39 and CD73 [Ref. (38–41) and (Gregori et al. unpublished data)]. Tr1 cells do not constitutively express FOXP3 (42), thus they are distinct from both tTregs and pTregs; however, upon activation, Tr1 cells can transiently up-regulate FOXP3, but its expression never reaches the levels of FOXP3+ Tregs (33, 43–45).
The main mechanism by which Tr1 cells control immune responses is the secretion of IL-10 and TGF-β. Importantly, to exert their suppressive function, Tr1 cells need to be activated via their TCR, but, once activated, they can mediate bystander suppressive activity against other antigen(Ag)s (3, 33). IL-10 and TGF-β directly inhibit T-cell responses by suppressing IL-2 and IFN-γ production and T-cell proliferation, and indirectly act on APCs by down-modulating costimulatory molecules, HLA-class-II, and pro-inflammatory cytokine production (34). In addition to the cytokine-mediated suppression, Tr1 cells inhibit T-cell responses by killing myeloid APCs via granzyme B (46). Tr1 cell-mediated cytotoxicity of myeloid APCs requires stable adhesion with target cells and activation via HLA-class-I molecules and CD112/CD155 expressed on target cells (46). New evidence suggests that Tr1 cells use additional modes of immune regulation to achieve tolerance: they can inhibit T-cell responses by cell-contact dependent mechanisms (36) and by metabolic disruption (33, 39, 41).
Results from pre-clinical murine and humanized models convinced investigators that Tregs can be used to control graft-versus-host disease (GvHD) as well as organ rejection, or to treat autoimmune diseases (47, 48). Good-manufacturing-practice (GMP)-grade protocols to isolate and expand human Tregs in vitro without losing their suppressive function and to generate human Ag-specific Tregs have been established allowing translation of Treg-based therapy to the clinical practice.
Completed and Ongoing Treg-Based Clinical Trials
Treg-based therapy has been used for the first time to prevent GvHD in patients undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT). Six independent trials, using either FOXP3+ Tregs or Tr1 cells, have been concluded, and all of them showed the feasibility and safety of Treg-based approaches (49–54) (Table 1). In five of these trials, either freshly isolated (51, 54, 55) or ex vivo expanded FOXP3+ Tregs (49, 50) were infused in patients undergoing allo-HSCT for onco-hematological diseases. Three of these trials also indicated the potential efficacy of the treatment. Brunstein et al. (50) reported a decreased incidence of grade II–IV GvHD as compared to historical controls when umbilical cord blood (UBC)-derived Tregs were injected, without increased risk of infections. Similarly, Di Ianni et al. (51) described few cases of low grade GvHD (2 out of 26 patients) and no development of chronic GvHD in patients injected with un-manipulated peripheral Tregs. More recently, it has been reported that in Treg-treated patients, the cumulative incidence of relapse was significantly lower than in historical controls (54). Previous trials based on the adoptive transfer of alloAgs-specific anergic T cells generated in vitro in the presence of Belatacept (CTLA-4-Ig) to prevent GvHD after allo-HSCT were performed (56, 57). Later, it was demonstrated that alloAgs-specific anergic T cells generated with CTLA-4-Ig contained a small fraction of FOXP3+ Tregs (58).
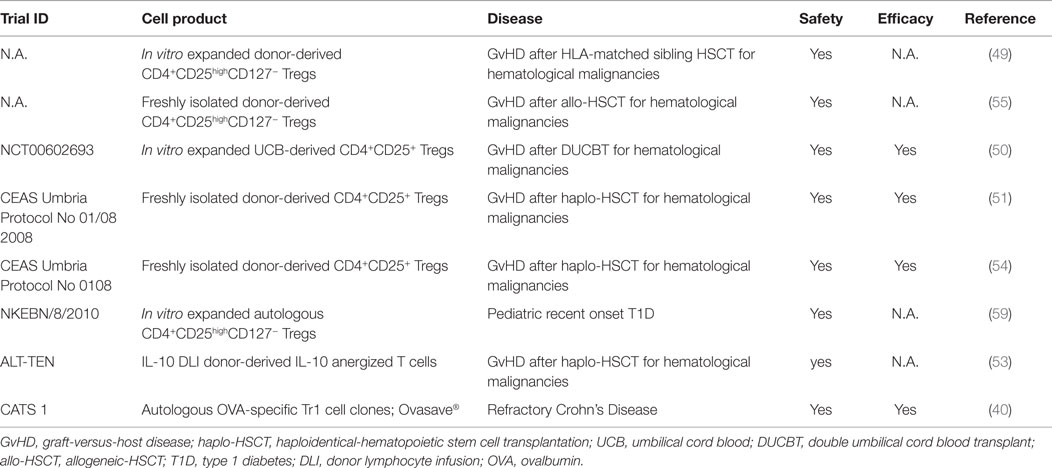
Table 1. Completed Treg-based clinical trials.
Our group has completed a phase-I clinical trial in which IL-10-anergized T cells (IL-10 DLI) containing Tr1 cells were injected in patients undergoing haploidentical-HSCT (53). Donor-derived IL-10-anergized T cells specific for host allo-Ags were generated in vitro through activation of T cells by host-derived APCs in the presence of exogenous IL-10 (60). An improved protocol for the generation of Tr1 cells, which foresees the use of tolerogenic dendritic cells (DC-10)(61), has been developed (60, 62). Although a small cohort of patients was treated, our results demonstrated that after infusion of IL-10 DLI no acute adverse events and only mild GvHD (grade II or III responsive to therapy) were observed. Furthermore, the treatment accelerated immune reconstitution after transplant and long-lasting disease remission (53).
The above-mentioned trials paved the way to a wider application of Tregs as advanced medical products for the treatment of autoimmunity in type 1 diabetes (T1D), inflammatory diseases, and rejection after solid organ transplantation. Ex vivo expanded CD4+CD25hiCD127− Tregs were administered to children with recent onset T1D in a phase-I trial (59) (Table 1). The procedure appeared to be safe, as no adverse reactions related to the treatment were reported. However, the few data available do not allow drawing conclusions on the clinical relevance of the procedure (59). The group of Bluestone is currently testing the safety of ex vivo expanded polyclonal CD4+CD25hiCD127low/− Tregs in a phase-I clinical trial (NCT01210664) in which increasing doses of Tregs will be injected in recent onset adult T1D patients (63). A phase-I/IIa clinical study in which Ag-specific Tr1 cell clones were used to treat patients with Crohn’s Disease has been recently reported. Overall, a response was observed in 40% of patients, with stronger effect in the group of patients who received the lowest Tr1 cell dose (40) (Table 1). The France-based company TxCell is currently heading a consortium dedicated to the clinical development of collagen-specific Tr1 cells (Col-Treg) to be tested in a first-in-man clinical study for severe and refractory autoimmune uveitis scheduled to start in 2016.1
The power of Tregs in inducing tolerance to allo-Ags after solid organ transplantation is currently under evaluation. In liver transplantation, several clinical trials are ongoing using polyclonal expanded Tregs with or without rapamycin (Treg trial, NCT01624077, ThRIL trial NCT02166177) or donor-specific expanded Tregs (darTreg: deLTA Trial NCT02188719, and ARTEMIS Trial NCT02474199). In addition, ex vivo expanded autologous polyclonal CD4+CD25+ Tregs are currently tested in the context of kidney transplantation (TRACT Trial, NCT 02145325 and TASK Trial NCT0288931). Moreover, an ambitious project in which the efficacy of different immune-regulatory cells, including polyclonal expanded Tregs with or without rapamycin (One Treg1 Trial, NCT02129881, ONE nTreg13 Trial NCT02371434), darTreg cells (DART Trial NCT02244801), and donor-specific T cells anergized in the presence of Belatacept (NCT02091232), and Tr1 cells induced with DC-10, will be compared in kidney transplant recipients (“The ONE study,” discussed in details below) is currently ongoing. Results of these trials will definitely address the safety of this approach and will also provide hints on their efficacy as therapeutic agents.
Open Issues in Treg-Based Immunotherapy
Despite the promising results obtained from the above-mentioned pilot clinical trials, many open questions remain on the best source and subtype of Tregs to be administered, the survival of these cells in the host, and their mechanisms of action.
The ONE Study2 is a large-scale, collaborative project funded by the Seventh Framework Programme (FP7) of the European Commission, envisioned to ascertain which immuno-modulatory cell type (among ex vivo isolated and in vitro expanded polyclonal or allo-specific FOXP3+ Tregs, Tr1 cells, and tolerogenic APCs) is best fit to induce tolerance to allo-Ags in patients receiving kidney transplants (64, 65). Results from this study will define which regulatory cell population is the most efficient in promoting graft acceptance and tolerance.
Recent work has led to the identification of specialized subsets of Tregs, which reside in peripheral tissues, including skin, intestinal mucosa, adipose tissue, autoimmune target tissues, and injured muscle (66). Although tissue-resident Tregs represent a small fraction of total Tregs, their peculiar phenotype and function confer the ability to regulate tissue-specific physiological and pathological processes. Therapies aimed at targeting tissue-specific Tregs may potentially allow the local control of the disease, without affecting systemic immunity. Although the clinical application of tissue-resident Tregs remains unexplored, the possibility of exploiting these subsets deserves to be investigated in the near future.
One pre-requisite for Treg-based therapies is their in vivo viability and persistence. In a clinical trial in allo-HSCT, upon in vivo infusion Tregs were no longer detected in the circulation after 2 weeks (50). Similarly, in T1D patients, in vitro expanded CD4+CD25+CD127− Tregs labeled with deuterium were found at high frequency in the peripheral blood 2 weeks after injection, then declined but they were still detectable at low frequency 6 months after therapy [Bluestone JA, unpublished data presented at FOCIS Annual Meeting 2015]. It is still unclear whether infused Tregs migrate to tissues or have limited in vivo survival because of in vitro expansion. In IL-10 DLI-treated patients, we found an expansion of circulating granzyme B/IL-10 and CD49b/LAG-3-expressing CD4+ T cells that progressively increased during follow up. The percentages of these cells were higher in the IL-10 DLI-treated long-term surviving patients (up to 8 years after haplo-HSCT), as compared to those in healthy subjects (53). These data support the hypothesis that IL-10 DLI infusion supports either Tr1 cell expansion, or the de novo induction of Tr1 cells.
Increasing evidence suggests that FOXP3-expressing Tregs are intrinsically plastic (67–69). Therefore, the risk of their in vivo conversion into Teff cells under inflammatory conditions, and consequent loss of their suppressive ability, cannot be ignored. To allow safe clinical application of Tregs, investigators are currently trying to address this issue. For example, rapamycin permits the in vitro expansion of FOXP3+ Tregs, while impairing the proliferation of contaminating Teff cells (70, 71). Importantly, rapamycin-expanded FOXP3+ Tregs maintain their regulatory phenotype, even upon exposure to a pro-inflammatory environment (72, 73). Clinical-grade Treg expansion protocols with rapamycin have been implemented for ongoing clinical trials under the umbrella of the European consortium “The ONE study” (65, 74). On the same line, in order to avoid infusion of Teff cell contaminants potentially allo-reactive, allo-anergization of T cells in the presence of costimulatory blockade with Belatacept has been proposed (58) and is currently being tested (NCT02091232).
One major concern for the use of immunotherapy with Tregs to control GvHD after allo-HSCT for hematological malignancies is the potential inhibition of the beneficial graft-versus-leukemia (GvL) effects. Results from one of the completed phase II trials showed that in CD25+ Treg-treated patients the cumulative incidence of relapse was significantly lower than in historical controls. The Authors proposed that the failure of human CD4+CD25+ Tregs to home to the bone marrow does not hamper the GvL activity of the donor conventional T cells (54). Although promising, these results are still preliminary and required further confirmation.
Up-Coming Challenges in Treg-Based Immunotherapy
As previously mentioned, increasing evidence suggests that FOXP3+ Tregs are a heterogeneous population, including several specialized subtypes, making it difficult to choose the “right” variety of cells for specific treatments. To overcome this limitation, we developed a novel and efficient method to generate homogeneous populations of human FOXP3-expressing Tregs by Lentiviral-Vector (LV)-mediated hFOXP3 gene transfer into conventional CD4+ T cells, hereafter indicated as CD4FOXP3 T cells. Constitutive over-expression of FOXP3 generates functional and stable FOXP3+ Treg-like cells, with potent in vitro and in vivo suppressive activity, reduced proliferative capacity and cytokine production (75, 76). CD4FOXP3 T cells generated from naïve CD4+ T cells have stable expression of FOXP3 in steady state and inflammatory conditions, whereas CD4FOXP3 T cells generated from memory cells show reduced percentage of FOXP3+ T cells upon activation, especially in the presence of inflammatory cytokines. The instability of FOXP3 expression in memory CD4FOXP3 T cells results in weaker suppressive function and increased proliferative capacity, confirming that acquisition of Treg functions is dependent on stable FOXP3 expression (76).
Despite recent advances in the establishment of protocols to efficiently generate Allo Ag-specific Tr1 cells in vitro, the resulting populations still contain contaminants that could potentially limit the in vivo efficacy of Tr1 cells (60, 61). The recent discovery of CD49b and LAG-3 as specific biomarkers of Tr1 cells that allow the isolation of Tr1 cells from in vitro Tr1-polarized populations (32) will open the possibility to select human Tr1 cells from mixed cultures. As an alternative to obtain a large and homogeneous population of Tr1 cells, the LV-mediated hIL-10 gene transfer has been used to convert conventional T cells into Tr1-like cells, termed CD4IL-10 (77). CD4IL-10 cells mirror the phenotype and function of Tr1 cells and suppress xeno-GvHD (77). These findings pave the way for adoptive cell therapy with FOXP3- or IL-10-engineered T cells in patients with autoimmune disorders and in patients undergoing allogeneic organ or HSC transplantation. Issues related to undesired effects of therapy with genetically modified cells, such as induction of general immunosuppression, impairment of immune reconstitution, and GvL activity in the context of allo-HSCT for hematological diseases are still under investigation.
In humanized pre-clinical models, allo-specific Tregs are more effective in preventing graft rejection as compared to polyclonal Tregs (78, 79). It is possible to select allo-specific Tregs from peripheral blood according to the expression of early activation markers and/or then in vitro expand them (78, 80). Moreover, a GMP-grade protocol to selectively expand human allo-specific Tregs using CD40L-activated B cells has also been established (79). As an alternative, ectopic expression of genes encoding for TCR with known specificity has been proposed. Forced expression of specific TCRs confers the desired specificity to human polyclonal Tregs. As a proof-of-concept, it has been shown that TCR or chimeric receptor specific for tumor Ags can be introduced in human polyclonal Tregs, conferring them the ability to potently suppress anti-tumor responses (81–83). It was also proposed to generate Ag-specific Tregs starting from conventional T cells engineered to over-express both hFOXP3 and TCR specific for a birch pollen allergen-derived peptide Betv1. The resulting T cells acquired a Treg phenotype and suppressed T-cell responses in an Ag-specific manner (84). Despite these data provided the proof-of-concept for such approaches, several questions regarding the potential clinical application of these engeneered T cells have to be addressed. Among others, one of the major concerns regards the need to eliminate endogenous TCRs to avoid double specificity and the risk of bystander undesired suppressive function. An interesting and promising approach to overcome this limitation is LV-mediated gene transfer of either hFOXP3 or hIL-10 in Ag-experienced T cells isolated from peripheral blood.
An additional crucial question for the success of Treg-based therapy, in particular in the context of solid organ transplantation, is how immunosuppressive treatments affect Treg survival and function. The impact of current immunosuppressive drugs on Tregs has been extensively reviewed in Ref. (48, 85). The general consensus is that calcineurin inhibitors are likely to be detrimental to Tregs, whereas drugs such as rapamycin or mycophenolate mofetil (MMF) preserve Tregs in vivo. However, indications will come from the results of “The ONE study” in which Tregs will be infused in patients receiving kidney transplantation and standard triple-therapy protocol (prednisolone, MMF, and tacrolimus) (65).
Finally, the heterogeneity of the parameters selected to monitor Treg activity in the recently completed trials hampers comparison of the results. To overcome this limitation, the EU COST Action “BM1305: action to focus and accelerate cell-based tolerance-inducing therapies3” has been funded to identify shared and disease specific biomarkers of tolerance in patients undergoing Treg-based therapies. This action is complementary to “The ONE study” and aims at defining general tolerance signatures and standardized immune monitoring protocols (65).
Concluding Remarks
The discovery that Tregs modulate immune responses led to the idea that they could be developed as a therapeutic tool to promote/restore tolerance to transplanted grafts and in inflammatory and autoimmune diseases. The recent clinical trials proved the safety of this approach and suggested a possible therapeutic effect. Thus far, the major challenges in the field were to expand hard-to-grow polyclonal Tregs to great purity, and to generate Ag-specific Tregs. Despite technical advances in the field, many questions relating to Treg-based therapies remain unanswered: Which cell type to be used? Which schedule of cell infusion? How long Tregs will survive in vivo? How long their effect will last? What is their mechanism of action? Do they interfere with GvL in the context of allo-HSCT? Moreover, reliable biomarkers of tolerance and standardized methods to evaluate the efficacy of Treg-based therapy are required to compare the outcome of present and future trials. To address these questions, close collaboration between groups in the field is required to allow the systematic comparison of Tregs and outcomes of cell therapy trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Rosa Bacchetta (Pediatric Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics Stanford School of Medicine Stanford) and Dr. Manuela Battaglia (Diabetes Research Institute IRCCS San Raffaele Scientific Institute, Milan, Italy) for their continuous helpful scientific discussion. This work was supported by grants to SG from the Italian Telethon Foundation (TGT11E02) and from the Italian Association for Cancer Research, IG 14105 – AIRC 2013; and to MGR from the Italian Association for Cancer Research, IG 10439 – AIRC 2010; IG 14555 – AIRC 2013 and by EU COST Action BM1305 http://www.afactt.eu.
Footnotes
References
1. Bluestone JA, Trotta E, Xu D. The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert Opin Ther Targets (2015) 19(8):1091–103. doi:10.1517/14728222.2015.1037282
2. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
3. Groux H, O’garra A, Bigler M, Rouleau M, Antonenko S, De Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389:737–42. doi:10.1038/39614
4. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10:490–500. doi:10.1038/nri2785
5. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14:307–8. doi:10.1038/ni.2554
6. Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J (2012) 26:2253–76. doi:10.1096/fj.11-193672
7. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity (2007) 27:786–800. doi:10.1016/j.immuni.2007.09.010
8. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184:3433–41. doi:10.4049/jimmunol.0904028
9. Akimova T, Beier UH, Wang L, Levine MH, Hancock WW. Helios expression is a marker of T cell activation and proliferation. PLoS One (2011) 6:e24226. doi:10.1371/journal.pone.0024226
10. Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol (2012) 188:976–80. doi:10.4049/jimmunol.1102964
11. Wieczorek G, Asemissen A, Model F, Turbachova I, Floess S, Liebenberg V, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res (2009) 69:599–608. doi:10.1158/0008-5472.CAN-08-2361
12. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37:785–99. doi:10.1016/j.immuni.2012.09.010
13. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192:303–10. doi:10.1084/jem.192.2.303
14. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity (2002) 16:311–23. doi:10.1016/S1074-7613(02)00280-7
15. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi:10.1182/blood-2006-12-064527
16. Kubach J, Lutter P, Bopp T, Stoll S, Becker C, Huter E, et al. Human CD4+CD25+ regulatory T cells: proteome analysis identifies galectin-10 as a novel marker essential for their anergy and suppressive function. Blood (2007) 110:1550–8. doi:10.1182/blood-2007-01-069229
17. Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood (2009) 113:5125–33. doi:10.1182/blood-2009-01-199950
18. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A (2009) 106:13445–50. doi:10.1073/pnas.0901944106
19. Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, et al. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity (2008) 28:870–80. doi:10.1016/j.immuni.2008.03.018
20. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol (2004) 172:834–42. doi:10.4049/jimmunol.172.2.834
21. Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol (2009) 39:3315–22. doi:10.1002/eji.200939684
22. Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting edge: human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol (2011) 186:6661–6. doi:10.4049/jimmunol.1100315
23. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21:589–601. doi:10.1016/j.immuni.2004.09.002
24. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi:10.1126/science.1160062
25. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8:1353–62. doi:10.1038/ni1536
26. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
27. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med (2007) 204:1303–10. doi:10.1084/jem.20062129
28. Baecher-Allan C, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol (2006) 176:4622–31. doi:10.4049/jimmunol.176.8.4622
29. Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113:4240–9. doi:10.1182/blood-2008-10-183251
30. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood (2006) 108:4260–7. doi:10.1182/blood-2006-06-027409
31. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30:899–911. doi:10.1016/j.immuni.2009.03.019
32. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med (2013) 19:739–46. doi:10.1038/nm.3179
33. Gregori S, Goudy KS, Roncarolo MG. The cellular and molecular mechanisms of immuno-suppression by human type 1 regulatory T cells. Front Immunol (2012) 3:30. doi:10.3389/fimmu.2012.00030
34. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi:10.1007/978-3-662-43492-5_3
35. Bacchetta R, Sartirana C, Levings MK, Bordignon C, Narula S, Roncarolo MG. Growth and expansion of human T regulatory type 1 cells are independent from TCR activation but require exogenous cytokines. Eur J Immunol (2002) 32:2237–45. doi:10.1002/1521-4141(200208)32:8<2237::AID-IMMU2237>3.0.CO;2-2
36. Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, et al. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J Exp Med (2004) 199:1567–75. doi:10.1084/jem.20032058
37. Haringer B, Lozza L, Steckel B, Geginat J. Identification and characterization of IL-10/IFN-gamma-producing effector-like T cells with regulatory function in human blood. J Exp Med (2009) 206:1009–17. doi:10.1084/jem.20082238
38. Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion and characteristics of human T regulatory type 1 cells in co-cultures simulating tumor microenvironment. Cancer Immunol Immunother (2007) 56:1429–42. doi:10.1007/s00262-007-0280-9
39. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, et al. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem (2010) 285:27571–80. doi:10.1074/jbc.M110.127100
40. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’ disease. Gastroenterology (2012) 143(1207–1217):e1201–2. doi:10.1053/j.gastro.2012.07.116
41. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med (2015) 21:638–46. doi:10.1038/nm.3868
42. Vieira PL, Christensen JR, Minaee S, O’neill EJ, Barrat FJ, Boonstra A, et al. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol (2004) 172:5986–93. doi:10.4049/jimmunol.172.10.5986
43. Levings MK, Gregori S, Tresoldi E, Cazzaniga S, Bonini C, Roncarolo MG. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood (2005) 105:1162–9. doi:10.1182/blood-2004-03-1211
44. Brun V, Bastian H, Neveu V, Foussat A. Clinical grade production of IL-10 producing regulatory Tr1 lymphocytes for cell therapy of chronic inflammatory diseases. Int Immunopharmacol (2009) 9:609–13. doi:10.1016/j.intimp.2009.01.032
45. Brun V, Neveu V, Pers YM, Fabre S, Quatannens B, Bastian H, et al. Isolation of functional autologous collagen-II specific IL-10 producing Tr1 cell clones from rheumatoid arthritis blood. Int Immunopharmacol (2011) 11:1074–8. doi:10.1016/j.intimp.2011.03.001
46. Magnani CF, Alberigo G, Bacchetta R, Serafini G, Andreani M, Roncarolo MG, et al. Killing of myeloid APCs via HLA class I. CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. Eur J Immunol (2011) 41:1652–62. doi:10.1002/eji.201041120
47. Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol (2007) 7:585–98. doi:10.1038/nri2138
48. Tang Q, Bluestone JA, Kang SM. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. J Mol Cell Biol (2012) 4:11–21. doi:10.1093/jmcb/mjr047
49. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (2009) 133:22–6. doi:10.1016/j.clim.2009.06.001
50. Brunstein CG, Miller JS, Cao Q, Mckenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi:10.1182/blood-2010-07-293795
51. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117:3921–8. doi:10.1182/blood-2010-10-311894
52. Edinger M, Hoffmann P. Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr Opin Immunol (2011) 23:679–84. doi:10.1016/j.coi.2011.06.006
53. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T cells. Front Immunol (2014) 5:16. doi:10.3389/fimmu.2014.00016
54. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T cell adoptive immunotherapy prevents acute leukemia relapse. Blood (2014) 124(4):638–44. doi:10.1182/blood-2014-03-564401
55. Edinger M. Treg cells in allogeneic stem cell transplantation. Keystone Symposia on Molecular and Cellular Biology Regulatory T cells (C5) 2009 Abstract 028 (2009).
56. Guinan EC, Boussiotis VA, Neuberg D, Brennan LL, Hirano N, Nadler LM, et al. Transplantation of anergic histoincompatible bone marrow allografts. N Engl J Med (1999) 340:1704–14. doi:10.1056/NEJM199906033402202
57. Davies JK, Gribben JG, Brennan LL, Yuk D, Nadler LM, Guinan EC. Outcome of alloanergized haploidentical bone marrow transplantation after ex vivo costimulatory blockade: results of 2 phase 1 studies. Blood (2008) 112:2232–41. doi:10.1182/blood-2008-03-143636
58. Davies JK, Nadler LM, Guinan EC. Expansion of allospecific regulatory T cells after anergized, mismatched bone marrow transplantation. Sci Transl Med (2009) 1(1):1ra3. doi:10.1126/scitranslmed.3000153
59. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care (2012) 35:1817–20. doi:10.2337/dc12-0038
60. Bacchetta R, Gregori S, Serafini G, Sartirana C, Schulz U, Zino E, et al. Molecular and functional characterization of allogantigen-specific anergic T cells suitable for cell therapy. Haematologica (2010) 95:2134–43. doi:10.3324/haematol.2010.025825
61. Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, et al. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood (2010) 116:935–44. doi:10.1182/blood-2009-07-234872
62. Petrelli A, Et BS, Mfarrej BG, Paganelli A, Spotti D, Caldara R, et al. Generation of donor-specific T regulatory type 1 cells from patients on dialysis for cell therapy after kidney transplantation. Transplantation (2015) 99(8):1582–9. doi:10.1097/TP.0000000000000751
63. Putnam A, Lares A, Lee M, Liu W, Herold KC, Gitelman S, et al. Results from 10 study participant treated with ex vivo expanded CD4+CD127low/-CD25+ polyclonal Tregs in a phase I clinical trial for the treatment of recent-onset type 1 diabetes. Clin Immunol (2013) 104:156.
64. Leslie M. Immunology. Regulatory T cell get their chance to shine. Science (2011) 332:1020–1. doi:10.1126/science.332.6033.1020
65. Hutchinson JA, Geissler EK. Now or never? The case for cell-based immunosuppression in kidney transplantation. Kidney Int (2015) 87:1116–24. doi:10.1038/ki.2015.50
66. Lu J, Meng H, Zhang A, Yang J, Zhang X. Phenotype and function of tissue-resident unconventional Foxp3-expressing CD4(+) regulatory T cells. Cell Immunol (2015) 297:53–9. doi:10.1016/j.cellimm.2015.06.005
67. Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol (2009) 21:281–5. doi:10.1016/j.coi.2009.05.007
68. Da Silva Martins M, Piccirillo CA. Functional stability of Foxp3+ regulatory T cells. Trends Mol Med (2012) 18:454–62. doi:10.1016/j.molmed.2012.06.001
69. MacDonald KG, Orban PC, Levings MK. T regulatory cell therapy in transplantation: stability, localization and functional specialization. Curr Opin Organ Transplant (2012) 17:343–8. doi:10.1097/MOT.0b013e328355aaaf
70. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105:4743–8. doi:10.1182/blood-2004-10-3932
71. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol (2006) 177:8338–47. doi:10.4049/jimmunol.177.12.8338
72. Tresoldi E, Dell’albani I, Stabilini A, Jofra T, Valle A, Gagliani N, et al. Stability of human rapamycin-expanded CD4(+)CD25(+) T regulatory cells. Haematologica (2011) 96:1357–65. doi:10.3324/haematol.2011.041483
73. Scotta C, Esposito M, Fazekasova H, Fanelli G, Edozie FC, Ali N, et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4(+)CD25(+)FOXP3(+) T regulatory cell subpopulations. Haematologica (2013) 98:1291–9. doi:10.3324/haematol.2012.074088
74. Van Der Net JB, Bushell A, Wood KJ, Harden PN. Regulatory T cells: first steps of clinical application in solid organ transplantation. Transpl Int (2015). doi:10.1111/tri.12608
75. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther (2008) 16:194–202. doi:10.1038/sj.mt.6300341
76. Passerini L, Mel ER, Sartirana C, Fousteri G, Bondanza A, Naldini L, et al. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med (2013) 5(215):215ra174. doi:10.1126/scitranslmed.3007320
77. Andolfi G, Fousteri G, Rossetti M, Magnani CF, Jofra T, Locafaro G, et al. Enforced IL-10 expression confers type 1 regulatory T cell (Tr1) phenotype and function to human CD4(+) T cells. Mol Ther (2012) 20:1778–90. doi:10.1038/mt.2012.71
78. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med (2011) 3(83):83ra42. doi:10.1126/scitranslmed.3002076
79. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant (2013) 13:3010–20. doi:10.1111/ajt.12433
80. Noyan F, Lee YS, Zimmermann K, Hardtke-Wolenski M, Taubert R, Warnecke G, et al. Isolation of human antigen-specific regulatory T cells with high suppressive function. Eur J Immunol (2014) 44:2592–602. doi:10.1002/eji.201344381
81. Hombach AA, Kofler D, Rappl G, Abken H. Redirecting human CD4+CD25+ regulatory T cells from the peripheral blood with pre-defined target specificity. Gene Ther (2009) 16:1088–96. doi:10.1038/gt.2009.75
82. Brusko TM, Koya RC, Zhu S, Lee MR, Putnam AL, Mcclymont SA, et al. Human antigen-specific regulatory T cells generated by T cell receptor gene transfer. PLoS One (2010) 5:e11726. doi:10.1371/journal.pone.0011726
83. Wan Q, Kozhaya L, Imberg K, Mercer F, Zhong S, Krogsgaard M, et al. Probing the effector and suppressive functions of human T cell subsets using antigen-specific engineered T cell receptors. PLoS One (2013) 8:e56302. doi:10.1371/journal.pone.0056302
84. Schmetterer KG, Haiderer D, Leb-Reichl VM, Neunkirchner A, Jahn-Schmid B, Kung HJ, et al. Bet v 1-specific T-cell receptor/forkhead box protein 3 transgenic T cells suppress Bet v 1-specific T-cell effector function in an activation-dependent manner. J Allergy Clin Immunol (2011) 127:.e231–3. doi:10.1016/j.jaci.2010.10.023
Keywords: T regulatory cells, T regulatory type 1 cells, tolerance, Treg-based therapy, IL-10, FOXP3, gene transfer
Citation: Gregori S, Passerini L and Roncarolo M-G (2015) Clinical Outlook for Type-1 and FOXP3+ T Regulatory Cell-Based Therapy. Front. Immunol. 6:593. doi: 10.3389/fimmu.2015.00593
Received: 31 August 2015; Accepted: 05 November 2015;
Published: 25 November 2015
Edited by:
Karina Pino-Lagos, Universidad de los Andes, ChileReviewed by:
Thomas Wekerle, Medical University of Vienna, AustriaGiovanna Lombardi, King’s College London, UK
Tobias Bopp, University Medical Center Mainz, Germany
Copyright: © 2015 Gregori, Passerini and Roncarolo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Gregori, Z3JlZ29yaS5zaWx2aWFAaHNyLml0;
Maria-Grazia Roncarolo, bWcxQHN0YW5mb3JkLmVkdQ==
†Silvia Gregori and Laura Passerini have contributed equally to this work.