 Cariad Chester
Cariad Chester Katherine Fritsch
Katherine Fritsch Holbrook E. Kohrt
Holbrook E. Kohrt- 1Division of Oncology, Department of Medicine, Stanford University, Stanford, CA, USA
- 2Institute for Immunity, Transplantation and Infection, Stanford University School of Medicine, Stanford, CA, USA
There is compelling clinical and experimental evidence to suggest that natural killer (NK) cells play a critical role in the recognition and eradication of tumors. Efforts at using NK cells as antitumor agents began over two decades ago, but recent advances in elucidating NK cell biology have accelerated the development of NK cell-targeting therapeutics. NK cell activation and the triggering of effector functions is governed by a complex set of activating and inhibitory receptors. In the early phases of cancer immune surveillance, NK cells directly identify and lyse cancer cells. Nascent transformed cells elicit NK cell activation and are eliminated. However, as tumors progress, cancerous cells develop immunosuppressive mechanisms that circumvent NK cell-mediated killing, allowing for tumor escape and proliferation. Therapeutic intervention aims to reverse tumor-induced NK cell suppression and sustain NK cells’ tumorlytic capacities. Here, we review tumor–NK cell interactions, discuss the mechanisms by which NK cells generate an antitumor immune response, and discuss NK cell-based therapeutic strategies targeting activating, inhibitory, and co-stimulatory receptors.
Introduction
The recent FDA approvals of the programmed cell death protein 1 (PD-1)-targeted checkpoint inhibitors pembrolizumab and nivolumab mark the latest successes in the rapidly expanding field of cancer immunotherapies. Immunotherapy represents a paradigm shift in cancer treatment; instead of targeting tumor cells, the goal of immunotherapy is to augment and expand the immune system’s intrinsic antitumor response. To date, diverse immunotherapeutic modalities have been accepted as viable strategies for eliminating cancerous cells. Cytokines, cancer vaccines, adoptive cell transfers, and especially checkpoint inhibitors constitute valuable elements in the immunotherapeutic armamentarium. However, a class of important immune-modulators is conspicuously absent: agents that utilize the power of innate immune cells to eradicate tumors. An important class of innate immune cells that play a critical role in mediating the antitumor immune response is the natural killer (NK) cell.
First described in 1975, NK cells were initially identified as a distinct sub-population of lymphocytes by their capacity to spontaneously lyse tumor cells (1). NK cells are now accepted to play an important role in both the adaptive and innate immune responses that govern infection, autoimmunity, and tumor immunosurveillance (2, 3). Human NK cells are phenotypically characterized by the expression of CD56 and the absence of CD3 and can be further subdivided into a CD56bright population and a CD56dim population. The CD56bright population produces immunoregulatory cytokines, including interferon-γ (IFN-γ), tumor necrosis factor-beta (TNF-B), tumor necrosis factor-α (TNF-α), granulocyte macrophage-colony stimulating factor (GMCSF), IL-10, and IL-13 (4). The CD56dim subset is the terminally differentiated successor of the CD56bright population and is primarily responsible for exerting cytolytic functions (5, 6). However, CD56dim NK cells can produce cytokines, specifically IFN-γ, after cell triggering via NKp46 of NKp30 activating receptors or after stimulation with combinations of IL-2, IL-12, and IL-15 (7).
The defining functional feature of NK cells remains their intrinsic ability to conduct “natural killing” of cellular targets without prior sensitization. The antitumor effect provided by natural killing has been observed in tumors of hematopoietic and non-hematopoietic origins and reported in diverse in vivo models and clinical series (8). NK cell infiltration into tumor tissue is associated with better disease prognosis in colorectal cancer, clear cell renal cell carcinoma, and lung carcinomas (9–11). Additionally, a 11-year prospective cohort study of Japanese inhabitants linked low peripheral-blood NK cell cytotoxicity with increased cancer risk (12). The combination of compelling preclinical evidence and early clinical success has established NK cell immunotherapy as a promising therapeutic strategy in cancer. Here, we review the current understanding of the NK cell mechanisms underpinning antitumor immunity and discuss immunomodulatory targets for augmenting NK cell-mediated tumor clearance.
Natural Killing
The initial hypothesis for the mechanism of NK cell-mediated killing postulated that the absence or altered expression of major histocompatibility complex (MHC) class I molecules would render target cells susceptible to NK cell attack (13). The “missing-self” hypothesis was the result of observations that NK cells can directly reject MHC class I-deficient tumors (14). Later in vivo experiments in murine and human systems confirmed that NK cytotoxicity was directly related to the absence of MHC class I expression on target cells (15, 16). However, the contemporary understanding of NK cell activation suggests that the transition of the NK cell from quiescence to activation is mediated by a network of activating and inhibitory receptors (17). While NK cells do express inhibitory receptors that detect the presence of MHC Class I molecules, it is the integration of multiple activating and inhibitory signals that determines if the NK cell becomes cytotoxic.
Natural killer cell cytotoxicity can be demonstrated in several related ways. The primary mechanism of cytotoxicity is based on granule exocytosis upon formation of an immunological synapse. NK cells contain preformed cytoplasmic granules that resemble secretory lysosomes and contain perforin and granzymes (18). Perforin is a membrane-disrupting protein that perforates the target cell membrane, while granzymes are a family of serine proteases that trigger cell apoptosis (19, 20). Upon activation, NK cells rapidly polarize the granules and reposition the microtubule organizing center toward the synapse with the target cell (21). The granule membrane then fuses with the plasma membrane, externalizes, and releases the cytotoxic granule contents, triggering target cell apoptosis (22).
NK cells can also contribute to target cell death indirectly by secreting pro-inflammatory cytokines. Two of the primary cytokines released by activated NK cells are IFN-γ and TNF-α. IFN-γ is a type II interferon that plays a critical role in promoting host resistance to microbial infection and protecting against tumor development (4). In the tumor microenvironment (TME), the IFN-γ released by NK cells stimulates CD4+ T cells to polarize toward a Th1 subset and accelerates the development of activated macrophages and cytotoxic, tumor-targeting CD8+ T cells (23). TNF-α is a multifunctional cytokine that can cause direct tumor necrosis by inflicting tumor-associated capillary injury, but also generates an adaptive immune response (24). TNF-α can enhance B cell proliferation and also promote monocyte and macrophage differentiation (25, 26). Together IFN-γ and TNF-α help to activate both innate and adaptive immune cells in the TME and generate a sustained antitumor immune response.
Antibody-Dependent Cell-Mediated Cytotoxicity
Another granule-mediated mechanism of NK cell targeted killing is antibody-dependent cell-mediated cytotoxicity (ADCC). ADCC is thought to play an important role in mediating the antitumor effects of many of the monoclonal antibody (mAb) therapies used today as standard of care treatments for both solid tumors and hematologic malignancies (27). In ADCC, the Fc receptor expressed by NK cells (FcγRIII or CD16) binds to the Fc portion of the therapeutic antibody, which in turn is bound to tumor-associated antigen (TAA) on the tumor surface. The effectiveness of ADCC depends on the FcγRIII ligation on the NK cell. Patients with a FcγRIIIa polymorphism, resulting in high-affinity binding of FcγRIII to IgG1, demonstrate enhanced clinical benefit. This effect has been seen in patients treated with rituximab, trastuzumab, and cetuximab (28–30). ADCC was initially described as the release of cytotoxic perforin and granzyme by NK cells following ligation of FcγRIII by IgG target cells. However, ADCC is now recognized as a multi-tiered process that involves a network of coordinated immune cells and an adaptive immune response (31). For example, FcγR ligation on NK cells can induce the secretion of pro-inflammatory cytokines like IFN-γ, which can accelerate dendritic cell (DC) maturation (32). Mature DCs enhance antigen presentation and train tumor-specific lymphocytes, producing an immunological memory response (33).
Death Receptor-Induced Apoptosis
Death receptor-induced apoptosis is a perforin-independent mechanism by which NK cells lyse target cells (34). This cytotoxic pathway relies on target cell expression of tumor necrosis factor (TNF) receptor superfamily members. The two main TNF receptors used in apoptotic induction are Fas (CD95) and TNF-related apoptosis-inducing ligand (TRAIL) (35). Fas is expressed on a wide variety of tissues, but Fas ligand (FasL) expression is restricted to activated NK cells and cytotoxic T lymphocytes (CTLs) (36). Fas cross-linking induces nuclear condensation, membrane blebbing, and caspase activation (37). The initial optimism surrounding the Fas–FasL pathway as a means of tumor control has decreased following the observations that Fas is downregulated in a variety of cancers during tumor progression (38).
TNF-related apoptosis-inducing ligand-mediated signaling is another death receptor-induced mechanism NK cells employ to kill target cells. TRAIL is constitutively expressed on some populations of NK cells and TRAIL-mediated signaling can induce spontaneous cytotoxicity against TRAIL-sensitive tumor cells (39, 40). Binding of TRAIL to its receptor, TRAILR1 or TRAILR2, results in receptor oligomerization on the cell membrane and triggering of a pro-apoptotic signal through activation of caspases (41). Preclinically, recombinant forms of TRAIL and agonistic anti-TRAIL receptor antibodies can have single-agent activity against TRAIL-sensitive tumor cells in vitro and in vivo (42). Recently, artificial nanoparticles coated with bioactive TRAIL demonstrated cytotoxicity against primary leukemic cells from a patient with acute lymphocytic leukemia (ALL) (43). However, despite preclinical successes, clinical trials of TRAIL-based therapies have demonstrated little efficacy and tumors rapidly develop resistance mechanisms to TRAIL (44). A better understanding of how tumors evade targeting and removal by NK cells is needed to overcome immunosuppression in the TME.
NK–Tumor Interactions
Despite the diverse repertoire of killing strategies utilized by NK cells, the tumor cell often avoids attack by direct and indirect mechanisms (45). Direct mechanisms consist of shedding soluble ligands for NK cell-activating receptors, upregulation of HLA molecules, and release of inhibitory cytokines. Indirect mechanisms consist of activation of inhibitory regulatory T cells (Tregs), DC killing, and phagocyte-derived inhibitory cytokines. These immunosuppressive mechanisms collectively create a TME where NK cell cytotoxic functions are inhibited. By stifling NK-mediated tumor eradication, the tumor escapes immunosurveillance and is able to grow and develop. Restoring and augmenting NK cell cytotoxic functions in the TME is an important step in overcoming immunosuppression and eliminating tumor. In an attempt to generate potent tumor-lysing NK cells, therapeutics are being developed that target NK cell activating, inhibitory, and co-stimulatory receptors (Figure 1).
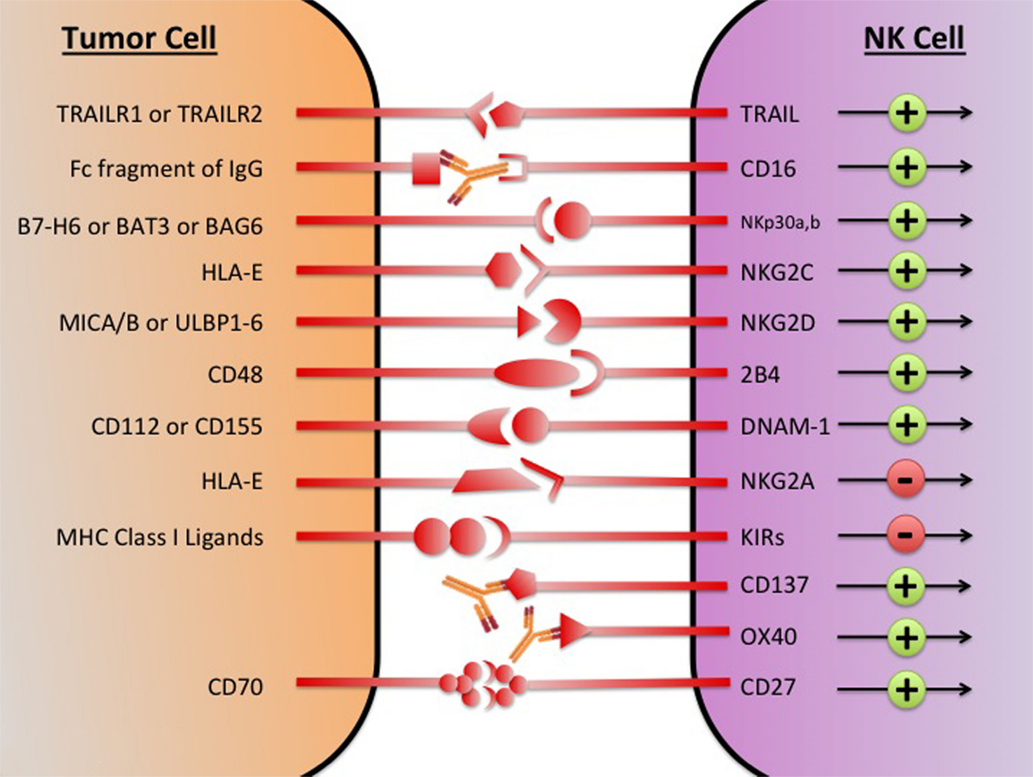
Figure 1. The major NK cell receptors that are potential immunotherapeutic targets. The transition of the NK cell from quiescence to activation is mediated by a network of activating and inhibitory receptors; it is the integration of the activating and inhibitory signals that determines if the NK cell becomes cytotoxic. Using immunotherapeutic agents to increase activation and decreases inhibitory signaling has the potential to generate NK cells with enhanced tumor lytic capacity. MICA/B, MHC class I chain-related proteins A and B; ULBP, UL16-binding protein; BAG, Bcl2-associated athanogene.
Activating Receptors
Activating receptors are a crucial element in regulating NK cell function. In the last decade, researchers have identified major signaling axes that control NK cell activation and suggested novel routes for therapeutic interventions (46). Some of the dominant activating receptors on NK cells are NKG2D, signaling lymphocytic activation molecule (SLAM) family molecule 2B4 (CD244), the DNAX accessory molecule (DNAM-1, CD226), and the NCRs: NKp30, NKp44, NKp46, and NKp80 (42). Recent work suggests that in NK cells, there is not a dominant receptor for activation, but instead receptors induce activation through combinatorial synergy (17). Only when multiple activating receptors are simultaneously engaged does the resulting signal surpass the requisite activation threshold and trigger cytokine secretion or direct cellular cytotoxicity. The requirement for activating receptor combinations helps prevent unrestrained activation of NK cells and provides flexibility in sensing and responding to environmental stimuli. What follows is a brief exploration of the dominant NK cell-activating receptors and summaries of attempts to target their tumorlytic capacity therapeutically.
NCRs
All NK cells express NKp30 and NKp46, whereas NKp44 is only expressed on activated NK cells (47, 48). The acquisition of NCR during NK cell maturation correlates with the acquisition of cytolytic activity against tumor target cells (49). Inversely, downregulation of NKp30, NKp44, and NKp46 correlates with low NK cytolytic activity (50). NKp80 is expressed by virtually all fresh NK cells and mAb-mediated cross-linking of NKp80 resulted in induction of cytolytic activity and Ca2+ mobilization (51).
The NKp30 activating receptor has emerged as a promising therapeutic target in multiple cancer histologies. Downregulation is observed in patients with cervical cancer and high-grade squamous intraepithelial lesions (52). In lymphoma and leukemia models, ligation of NKp30 has been shown to activate human NK cells, trigger degranulation, and increase cytotoxicity (53). In patients with gastrointestinal sarcoma, the NKp30 isoform predicts the clinical outcome; patients with the immunostimulatory NKp30a and NKp30b isoforms have increased survival relative to patients with the immunosuppressive NKp30c isoform (54). Recently, the expression of distinct forms of NKp30 has been linked to 10-year progression free survival in patients with high-risk neuroblastoma (NB) (55). In NB patients with metastatic disease, the percentage of CD3−CD56+ NK cells in the peripheral blood and bone marrow was significantly elevated relative to patients with localized tumors. Additionally, NKp30 expression in the bone marrow of patients with metastatic NB was lower than expression in patients with localized NB (55). The ligand for NKp30, B7-H6, was highly expressed in neuroblasts, and the serum soluble form of B7-H6 correlated with tumor load and disease dissemination. The authors conclude that NK cell modulating immunotherapeutics offer a promising strategy for treating NB patients and that antibodies neutralizing sB7-H6 serum molecules and antibodies targeting NKp30 are worth pursuing in future clinical development.
NKG2D
NKG2D, a homodimeric activating receptor and member of the C-type lectin superfamily, is expressed by all NK cells and subsets of T cells (56). NKG2D serves as a major recognition receptor for detection and elimination of infected and transformed cells (57). Ligands of the human NKG2D receptor are the MHC I-related molecules MICA/MICB, and the UL16-binding proteins (ULBP-1 to ULBP-6) (57). These ligands are rarely expressed in healthy tissues but induced by various cellular stresses, such as DNA damage, heat shock, or cellular transformation. Primary tumors frequently express NKG2D ligands: NK cell killing of both an urothelial tumor cell line and a bladder cancer cell appeared to be triggered by NK cell detection of the NKG2D ligands MICA/MICB (58, 59). However, tumors have also developed mechanisms for NK cell evasion despite NKG2D ligand expression. One such mechanism is the systemic release of NKG2D ligands by tumors in cancer patients (60, 61). The secreted NKG2D ligand was believed to cause downregulation of NK cell-expressed NKG2D, thus, depriving the NK cell of an activating signal and facilitating tumor escape. Recently, evidence has emerged that demonstrates an activating, antitumor role for soluble NKG2D ligands. The high-affinity MULT1 mouse NKG2D ligand can stimulate NK cells and enhance antitumor activity (62). The NKG2D pathway is integral to immune surveillance and an active area of immunotherapy research.
2B4 and DNAM-1
One of the best-characterized NK cell activation receptors is 2B4, a member of the SLAM receptor family. The first data to suggest a role for 2B4 in regulating NK cell activation demonstrated that ligation of 2B4 by 2B4-specific antibodies induced IFN-y production in vitro and triggered NK cell-mediated cytotoxicity (63). Following the identification of the natural ligand for 2B4, CD48, researchers reported that target cell expression of CD48 augmented NK cell-mediated cytotoxicity (64). Researchers also reported significantly greater cytotoxic effects if 2B4 ligation was accompanied by ligation of DNAM-1 (65). DNAM-1 is an Ig-like family glycoprotein expressed on most human NK cells, monocytes, and T lymphocytes (66). Early support for DNAM-1 controlling NK cell activation was provided by Lanier and colleagues using DX11, an anti-DNAM-1 mAb (67). Blockade via DX11 inhibited the cytotoxicity of NK cells against an array of different tumor cell lines. CD112 and CD155, two nectin family proteins regulated by cellular stress, were soon identified as the ligands for DNAM-1 (68). CD155 and CD112 are expressed in a wide range of both solid and hematologic tumors (69). In patients with NB, expression levels of CD155 and CD112 correlate with tumor cells susceptibility to NK cell-mediated lysis (70). However, tumors have developed mechanisms for downregulating NK cell DNAM-1 and effecting NK cell immunosuppression (71). In the design of future NK cell-based immunotherapies, mechanisms for preserving activation receptor surface expression need to be considered. Additionally, combinations of synergistic activating receptor pairs, like DNAM-1 and 2B4, need to be taken into account.
Checkpoint Blockade in NK Cells
Immune checkpoint blockade strategies have proven a powerful approach to cancer immunotherapy. By blocking the receptors that transmit inhibitory signals to effector immune cells, checkpoint blockade aims to reverse immune suppression and generate robust antitumor immune responses. The successes of ipilimumab (anti-CTLA-4 mAb) and nivolumab and pembrolizumab (anti-PD-1 mAbs) demonstrate the potential of this therapeutic strategy. Ipilimumab (Yervoy, BMS) was approved in 2011 for the treatment of unresectable or metastatic melanoma, and blocks the CTLA-4-mediated signaling in T cells (72). CTLA-4 is an inhibitory receptor that upon ligation sends a negative regulatory signal to the T-cell receptor (TCR), limiting T cell activation (73). Nivolumab (Opdivo, BMS) and pembrolizumab (Keytruda, Merck) target programmed cell death protein-1 (PD-1). PD-1 is upregulated on T cells following activation and ligation of PD-1 transmits a negative regulatory signal (74). Histologically, diverse tumors upregulate the ligands of PD-1, PD-L1, and PD-L2 to take advantage of this immunosuppressive signaling pathway (75). Analogously to negative regulators of T cell activity, NK cells express surface receptors that can be targeted in checkpoint blockade strategies.
Killer Cell Immunoglobulin-Like Receptor
Within the signaling pathways that govern NK lytic capacity, the killer cell immunoglobulin-like receptor (KIR) family is a dominant group of negative regulators. KIR receptors bind to the self-MHC class I ligands (HLA-A, -B, -C) and upon ligation transmit signals that abrogate the effects of activating receptors (76). The prevalence of MHC I on healthy cells provides an inhibitory signal that prevents NK cells from inducing autoimmune responses. However, in acute myeloid leukemia (AML) patients following haploidentical stem cell transplantation from KIR mismatched donors, the absence of KIR–HLA class I interactions resulted in potent NK cell-mediated antitumor efficacy and increased survival (77, 78). The antitumor effect can also be obtained without undergoing stem cell transplant; mAb therapy provides a viable route for blocking KIR–HLA interactions. Preventing HLA ligation to KIRs with an anti-KIR mAb has been shown to increase NK cell degranulation, IFN-γ secretion, and tumor cell lysis as well as increasing overall survival in murine cancer models (79).
The development of a candidate anti-KIR antibody had to overcome significant challenges. The KIR gene content varies substantially from individual to individual depending on the inherited KIR haplotype and the KIR family is composed of several structurally different proteins, necessitating an antibody that has cross-reactivity between different KIRs (80). Despite these challenges, the anti-KIR mAb lirilumab (Innate Pharma) has entered clinical trials. The initial phase I safety trial reported safety and potential efficacy in patients with AML (81). A second phase I trial confirmed the early reports of safety and durable KIR-blocking ability in patients with multiple myeloma (82). Recently, it has been reported that rituximab-mediated ADCC, a potent therapeutic mechanism of rituximab therapy, is reduced by KIR signaling (83). We have demonstrated that this KIR-mediated ADCC suppression can be overcome by combining rituximab with anti-KIR mAb therapy (84). Currently, multiple phase I and phase II clinical trials are ongoing, testing lirilumab (IPH2102/BMS-986015), as a monotherapy or in combination with other checkpoint inhibitors in patients with hematological and solid tumors (NCT01714739) and (NCT01750580).
NKG2A
In addition to KIRs, the CD94/NKG2A heterodimer is another target for NK cell checkpoint blockade. The natural ligand of CD94/NKG2A is HLA-E, a non-classical HLA class I molecule that is expressed on the cell surface of most leukocytes and on transformed cells, including virus-infected cells and tumor cells (85, 86). Ligation of CD94/NKG2A by HLA-E transmits inhibitory signaling that suppresses the effector functions of NK cells, resulting in decreased cytotoxicity and cytokine secretion. HLA-E and CD94/NKG2A expression has been reported in multiple tumor histologies and is associated with poor prognosis. In colorectal cancer patients, tumor expression of HLA-E is associated with shorter disease-free survival time (87). In patients with head and neck squamous cell cancers (HNSCC), 78 to 86% of tumors express HLA-E (88). In patients with non-small cell lung cancer, intratumoral NK cells display higher expression levels of NKG2A mRNA relative to non-tumor NK cells (89). In breast cancer patients, expression of NKG2A by tumor infiltrating NK cells increases with cancer progression and correlates with impaired NK cell functions (90). Similarly to blocking KIR-mediated interactions, blockade of CD94/NKG2A-mediated signaling has the potential to restore and preserve NK cell cytotoxicity, leading to antitumor responses. A phase I/II trial testing an anti-NKG2A antibody (IPH2201, Innate Pharma) in HNSCC patients is ongoing (NCT02331875).
Co-stimulatory Signaling via mAbs
Activating co-stimulatory pathways to potentiate antitumor immune responses is a promising approach for augmenting NK-mediated tumor clearance. Members of the tumor necrosis factor receptor superfamily (TNFRsf) include several co-stimulatory proteins with key roles in the regulation of the activation, proliferation, and apoptosis of lymphocytes, including NK cells.
CD137
First identified in 1989, CD137 (or 4-1BB) is a co-stimulatory receptor and member of the TNF receptor superfamily (91). CD137 is expressed on T cells and DCs and is upregulated on NK cells following FcγRIIIa ligation (92). In a variety of different tumor models, agonistic anti-CD137 mAbs have demonstrated the capacity to amplify antitumor immune responses and eliminate established tumors (93). Despite the broad expression of CD137 and its multiple contributions to immune dynamics, the therapeutic efficacy of anti-CD137 relies on functional NK cells. In preclinical models, the selective depletion of NK cells via the anti-AsialoGM1 or anti-NK1.1 antibodies completely abrogated the antitumor effect of anti-CD137 mAb therapy (94). Simultaneously, anti-CD137 agonistic antibodies increase NK cell proliferation, degranulation, and IFN-γ secretion, leading to enhanced ADCC of tumor cells (95). Because of the potential to enhance ADCC-mediated tumor clearance, anti-CD137 antibodies are being tested in combination treatment strategies with FDA-approved mAbs. We have previously demonstrated that antibodies targeting CD137 synergize with rituximab and trastuzumab to clear tumors in murine xenograft models of lymphoma and breast cancer (27, 96). Recently, we combined cetuximab and anti-CD137 antibody therapy to obtain complete tumor resolution and prolonged survival in xenograft models of EGFR-expressing cancer cells (97). In all three disease models and combination treatment regimens, expression of CD137 on NK cells increases significantly when NK cells encounter mAbs bound to tumor cells. We believe the synergy between anti-CD137 treatment and established mAbs demonstrates a promising therapeutic strategy and warrants future investigation.
Anti-CD137 mAb therapy has also entered clinical testing. The anti-CD137 antibody, urelumab, is currently in clinical trials with rituximab for patients with non-Hodgkin’s lymphoma (NCT01775631) and with cetuximab in patients with colorectal cancer or head and neck cancer (NCT02110082). In addition to urelumab, clinical trials of Pfizer’s anti-CD137 mAb, PF–05082566, are also ongoing (NCT01307267). A recent presentation of the preliminary findings reports that 27 patients with mixed tumor types have been treated with PF-05082566; disease stabilization was the best overall response, observed in 22% (6/27) patients (98).
OX40
OX40, also known as CD134 or TNFRSF4, is a co-stimulatory molecule expressed primarily by activated T cells, but also expressed on natural killer T (NKT) cells and NKs (99). In NK cells, OX40 ligation appears to induce an activating signal and IFN-γ production (100). Engagement of the OX40 receptor in vivo in tumor-bearing mice enhanced antitumor immunity, resulting in increased survival in four separate murine tumor models of diverse histology and immunogenicity (101). The initial phase I/II trial of an anti-OX40 mAb demonstrated tolerability and regression of at least one metastatic lesion in 12 out of 30 study patients (102). Immunologically, treatment with agonistic anti-OX40 increased the proliferation of NK cells as well as CD4+ T cells (103). Additional trials of anti-OX40 are ongoing, including combination therapies with rituximab in patients with CLL and NHL (NCT01775631), with stereotactic body radiation in patients with metastatic breast cancer (NCT01862900), and with tremelimumab, an anti-CTLA-4 antibody, in patients with solid tumors (NCT02205333).
CD27
In addition to its co-stimulatory role on T cells, the expression of CD27, or TNFRSF7, differentiates the NK cell compartment into two functionally distinct subsets. Circulating CD27+ NK have lower levels of perforin and granzyme B and demonstrate lower levels of cytotoxicity relative to CD27- NK cells (104). The absence of CD27 expression in combination with the expression of CD11b is an indicator of cytolytic effector cells within human NK cell subsets. The natural ligand for CD27, CD70, induces downregulation of CD27 in a process controlled by the common γ-chain cytokine IL-15 (105). Signaling via CD27–CD70 interactions have been shown to accelerate NK-mediated tumor clearance while simultaneously stimulating cytokine secretion by NK cells that elicits an adaptive immune response (106).
The potential for CD27 ligation to generate an antitumor response has been confirmed in preclinical models. In a xenograft models of lymphoma, administration of the humanized anti-CD27 antibody, 1F5, significantly prolonged survival (107). The fully human 1F5 cannot bind to mouse CD27, therefore, any observed antitumor activity is attributed to effector mechanisms such as direct inhibition/apoptosis via CD27 signaling in tumors or ADCC. In syngeneic colorectal and lymphoma models with little to no expression of CD27, treatment with the 1F5 mAb also elicited antitumor activity and increased survival (108). By testing an aglycosylated version of the 1F5 mAb, the researchers demonstrated that FcR engagement was required for the antitumor effects of 1F5 therapy. An anti-CD27 mAb (Varlilumab or CDX-1127, Celldex Therapeutics) is currently being tested in a phase I trial in patients with solid tumors and hematologic cancers (NCT01460134). Preliminary findings report that of the 19 treated patients, 3 had stable disease and 1 had a complete readmission (109).
Conclusion
In the future, immunotherapeutic agents that directly enhance NK cell-mediated tumor eradication will play a leading role in cancer treatment strategies. NK cells have novel mechanisms of participating in immune defense, making them uniquely appealing for cancer immunotherapy. Enhancing NK cell tumorlytic capacity is also a compelling combinatorial treatment strategy and would complement current standard of care treatments based on mAb therapy. The potential for NK-targeted agents to augment the antitumor effects of T cell checkpoint blockade is actively under consideration. As NK cell-based therapies move into the clinic, identifying prognostic biomarkers in the treatment populations will be crucial to the rational design of clinical studies. Concurrently, a greater effort must be made to profile the effects of novel immunotherapeutic agents, like checkpoint inhibitors, on NK cell function. The NK cell is now accepted as an integral part of the immunologic antitumor response. A number of promising NK-targeting therapeutics are in early-phase trials, and the results are eagerly awaited.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene (2008) 27:5932–43. doi: 10.1038/onc.2008.267
2. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi:10.1038/ni1582
3. Caligiuri MA. Human natural killer cells. Blood (2008) 112:461–9. doi:10.1182/blood-2007-09-077438
4. Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56bright subset. Blood (2001) 97:3146–51. doi:10.1182/blood.V97.10.3146
5. Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol (2007) 179:89–94. doi:10.4049/jimmunol.179.1.89
6. Romagnani C, Juelke K, Falco M, Morandi B, D’Agostino A, Costa R, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol (2007) 178:4947–55. doi:10.4049/jimmunol.178.8.4947
7. De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci U S A (2011) 108:728–32. doi:10.1073/pnas.1012356108
8. Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol (2007) 7:329–39. doi:10.1038/nri2073
9. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res (2011) 17:678–89. doi:10.1158/1078-0432.CCR-10-2173
10. Eckl J, Buchner A, Prinz PU, Riesenberg R, Siegert SI, Kammerer R, et al. Transcript signature predicts tissue NK cell content and defines renal cell carcinoma subgroups independent of TNM staging. J Mol Med (Berl) (2012) 90:55–66. doi:10.1007/s00109-011-0806-7
11. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res (2011) 71:5412–22. doi:10.1158/0008-5472.CAN-10-4179
12. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356:1795–9. doi:10.1016/S0140-6736(00)03231-1
13. Borrego F. The first molecular basis of the “missing self” hypothesis. J Immunol (2006) 177:5759–60. doi:10.4049/jimmunol.177.9.5759
14. Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. 1986. J Immunol (2005) 174:6566–9. doi:10.1038/319675a0
15. Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigen specificity of. Ly-49 (1992) 358:66–70. doi:10.1038/358066a0
16. Ljunggren HG, Kärre K. In search of the missing self: MHC molecules and NK cell recognition. Immunol Today (1990) 11:237–44. doi:10.1016/0167-5699(90)90097-S
17. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi:10.1146/annurev-immunol-020711-075005
18. Clark R, Griffiths GM. Lytic granules, secretory lysosomes and disease. Curr Opin Immunol (2003) 15:516–21. doi:10.1016/S0952-7915(03)00113-4
19. Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol (2003) 3:361–70. doi:10.1038/nri1083
20. Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, et al. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat Immunol (2011) 12:770–7. doi:10.1038/ni.2050
21. Mentlik AN, Sanborn KB, Holzbaur EL, Orange JS. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol Biol Cell (2010) 21:2241–56. doi:10.1091/mbc.E09-11-0930
22. Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev (2010) 235:105–16. doi:10.1111/j.0105-2896.2010.00908.x
23. Ikeda H, Old LJ, Schreiber RD. The roles of IFNγ in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev (2002) 13:95–109. doi:10.1016/S1359-6101(01)00038-7
24. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer (2009) 9:361–71. doi:10.1038/nrc2628
25. Higuchi M, Nagasawa K, Horiuchi T, Oike M, Ito Y, Yasukawa M, et al. Membrane tumor necrosis factor-alpha (TNF-alpha) expressed on HTLV-I-infected T cells mediates a costimulatory signal for B cell activation – characterization of membrane TNF-alpha. Clin Immunol Immunopathol (1997) 82:133–40. doi:10.1006/clin.1996.4291
26. Takeda K, Iwamoto S, Sugimoto H, Takuma T, Kawatani N, Noda M, et al. Identity of differentiation inducing factor and tumour necrosis factor. Nature (1986) 323:338–40. doi:10.1038/323338a0
27. Kohrt HE, Houot R, Marabelle A, Cho HJ, Osman K, Goldstein M, et al. Combination strategies to enhance antitumor ADCC. Immunotherapy (2012) 4:511–27. doi:10.2217/imt.12.38
28. Rodríguez J, Zarate R, Bandres E, Boni V, Hernández A, Sola JJ, et al. Fc gamma receptor polymorphisms as predictive markers of cetuximab efficacy in epidermal growth factor receptor downstream-mutated metastatic colorectal cancer. Eur J Cancer (2012) 48:1774–80. doi:10.1016/j.ejca.2012.01.007
29. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood (2002) 99:754–8. doi:10.1182/blood.V99.3.754
30. Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol (2008) 26:1789–96. doi:10.1200/JCO.2007.14.8957
31. Rajasekaran N, Chester C, Yonezawa A, Zhao X, et al. Enhancement of antibody-dependent cell mediated cytotoxicity: a new era in cancer treatment. Biologics (2015) 9:35–43. doi:10.2147/ITT.S61292
32. Márquez ME, Millet C, Stekman H, Conesa A, Deglesne PA, Toro F, et al. CD16 cross-linking induces increased expression of CD56 and production of IL-12 in peripheral NK cells. Cell Immunol (2010) 264:86–92. doi:10.1016/j.cellimm.2010.05.002
33. Walzer T, Dalod M, Vivier E, Zitvogel L. Natural killer cell-dendritic cell crosstalk in the initiation of immune responses. Expert Opin Biol Ther (2005) 5(Suppl 1):S49–59. doi:10.1517/14712598.5.1.S49
34. Berke G. The binding and lysis of target cells by cytotoxic lymphocytes: molecular and cellular aspects. Annu Rev Immunol (1994) 12:735–73. doi:10.1146/annurev.iy.12.040194.003511
35. Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med (1998) 188:2375–80. doi:10.1084/jem.188.12.2375
36. Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H, et al. Activation of NK cell cytotoxicity. Mol Immunol (2005) 42:501–10. doi:10.1016/j.molimm.2004.07.034
37. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26:239–57. doi:10.1038/bjc.1972.33
38. Reichmann E. The biological role of the Fas/FasL system during tumor formation and progression. Semin Cancer Biol (2002) 12:309–15. doi:10.1016/S1044-579X(02)00017-2
39. Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med (2001) 7:94–100. doi:10.1038/83416
40. Yagita H, Takeda K, Hayakawa Y, Smyth MJ, Okumura K. TRAIL and its receptors as targets for cancer therapy. Cancer Sci (2004) 95:777–83. doi:10.1111/j.1349-7006.2004.tb02181.x
41. Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol (2007) 39:1462–75. doi:10.1016/j.biocel.2007.02.007
42. Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer (2008) 8:782–98. doi:10.1038/nrc2465
43. De Miguel D, Gallego-Lleyda A, Galan-Malo P, Rodriguez-Vigil C, Marzo I, Anel A, et al. Immunotherapy with liposome-bound TRAIL overcomes partial protection to soluble TRAIL-induced apoptosis offered by down-regulation of Bim in leukemic cells. Clin Transl Oncol (2015) 17:657–67. doi:10.1007/s12094-015-1295-x
44. Dimberg LY, Anderson CK, Camidge R, Behbakht K, Thorburn A, Ford HL. On the TRAIL to successful cancer therapy? predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene (2013) 32:1341–50. doi:10.1038/onc.2012.164
45. Jacobs B, Ullrich E. The interaction of NK cells and dendritic cells in the tumor environment: how to enforce NK cell & DC action under immunosuppressive conditions? Curr Med Chem (2012) 19:1771–9. doi:10.2174/092986712800099857
46. Kaifu T, Escalière B, Gastinel LN, Vivier E, Baratin M. B7-H6/NKp30 interaction: a mechanism of alerting NK cells against tumors. Cell Mol Life Sci (2011) 68:3531–9. doi:10.1007/s00018-011-0802-7
47. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol (2009) 9:568–80. doi:10.1038/nri2604
48. Hudspeth K, Silva-Santos B, Mavilio D. Natural cytotoxicity receptors: broader expression patterns and functions in innate and adaptive immune cells. Front Immunol (2013) 4:69. doi:10.3389/fimmu.2013.00069
49. Sivori S, Cantoni C, Parolini S, Marcenaro E, Conte R, Moretta L, et al. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur J Immunol (2003) 33:3439–47. doi:10.1002/eji.200324533
50. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol (2001) 19:197–223. doi:10.1146/annurev.immunol.19.1.197
51. Vitale M, Falco M, Castriconi R, Parolini S, Zambello R, Semenzato G, et al. Identification of NKp80, a novel triggering molecule expressed by human NK cells. Eur J Immunol (2001) 31:233–42. doi:10.1002/1521-4141(200101)31:1<233::AID-IMMU233>3.3.CO;2-W
52. Garcia-Iglesias T, Del Toro-Arreola A, Albarran-Somoza B, Del Toro-Arreola S. Low NKp30, NKp46 and NKG2D expression and reduced cytotoxic activity on NK cells in cervical cancer and precursor lesions. BMC Cancer (2009) 9:186. doi:10.1186/1471-2407-9-186
53. Kellner C, Maurer T, Hallack D, Repp R, van de Winkel JG, Parren PW, et al. Mimicking an induced self phenotype by coating lymphomas with the NKp30 ligand B7-H6 promotes NK cell cytotoxicity. J Immunol (2012) 189:5037–46. doi:10.4049/jimmunol.1201321
54. Delahaye NF, Rusakiewicz S, Martins I, Ménard C, Roux S, Lyonnet L, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med (2011) 17:700–7. doi:10.1038/nm.2366
55. Semeraro M, Rusakiewicz S, Minard-Colin V, Delahaye NF, Enot D, Vély F, et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci Transl Med (2015) 7:283ra55. doi:10.1126/scitranslmed.aaa2327
56. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413. doi:10.1146/annurev-immunol-032712-095951
57. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res (2015) 3:575–82. doi:10.1158/2326-6066.CIR-15-0098
58. Higuchi T, Shimizu M, Owaki A, Takahashi M, Shinya E, Nishimura T, et al. A possible mechanism of intravesical BCG therapy for human bladder carcinoma: involvement of innate effector cells for the inhibition of tumor growth. Cancer Immunol Immunother (2009) 58:1245–55. doi:10.1007/s00262-008-0643-x
59. García-Cuesta EM, López-Cobo S, Álvarez-Maestro M, Esteso G, Romera-Cárdenas G, Rey M, et al. NKG2D is a key receptor for recognition of bladder cancer cells by IL-2-activated NK cells and BCG promotes NK cell activation. Front Immunol (2015) 6:284. doi:10.3389/fimmu.2015.00284
60. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419:734–8. doi:10.1038/nature01112
61. Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature (2007) 447:482–6. doi:10.1038/nature05768
62. Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science (2015) 348:136–9. doi:10.1126/science.1258867
63. Garni-Wagner BA, Purohit A, Mathew PA, Bennett M, Kumar V. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J Immunol (1993) 151:60–70.
64. Nakajima H, Cella M, Langen H, Friedlein A, Colonna M. Activating interactions in human NK cell recognition: the role of 2B4-CD48. Eur J Immunol (1999) 29:1676–83. doi:10.1002/(SICI)1521-4141(199905)29:05<1676::AID-IMMU1676>3.0.CO;2-Y
65. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107:159–66. doi:10.1182/blood-2005-04-1351
66. de Andrade LF, Smyth MJ, Martinet L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol Cell Biol (2014) 92:237–44. doi:10.1038/icb.2013.95
67. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity (1996) 4:573–81. doi:10.1016/S1074-7613(00)70060-4
68. Sakisaka T, Takai Y. Biology and pathology of nectins and nectin-like molecules. Curr Opin Cell Biol (2004) 16:513–21. doi:10.1016/j.ceb.2004.07.007
69. Tahara-Hanaoka S, Shibuya K, Kai H, Miyamoto A, Morikawa Y, Ohkochi N, et al. Tumor rejection by the poliovirus receptor family ligands of the DNAM-1 (CD226) receptor. Blood (2006) 107:1491–6. doi:10.1182/blood-2005-04-1684
70. Castriconi R, Dondero A, Corrias MV, Lanino E, Pende D, Moretta L, et al. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res (2004) 64:9180–4. doi:10.1158/0008-5472.CAN-04-2682
71. Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG, et al. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J Immunol (2009) 183:4921–30. doi:10.4049/jimmunol.0901226
72. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
73. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229:12–26. doi:10.1111/j.1600-065X.2009.00770.x
74. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
75. Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology (2012) 1:1223–5. doi:10.4161/onci.21335
76. Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol (2002) 20:217–51. doi:10.1146/annurev.immunol.20.092501.134942
77. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295:2097–100. doi:10.1126/science.1068440
78. Hsu KC, Keever-Taylor CA, Wilton A, Pinto C, Heller G, Arkun K, et al. Improved outcome in HLA-identical sibling hematopoietic stem cell transplantation for acute myelogenous leukemia (AML) predicted by KIR and HLA genotypes. Blood (2005) 105:4878–84. doi:10.1182/blood-2004-12-4825
79. Koh CY, Blazar BR, George T, Welniak LA, Capitini CM, Raziuddin A, et al. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood (2001) 97:3132–7. doi:10.1182/blood.V97.10.3132
80. Garcia CA, Robinson J, Guethlein LA, Parham P, Madrigal JA, Marsh SG. Human KIR sequences 2003. Immunogenetics (2003) 55:227–39. doi:10.1007/s00251-003-0572-y
81. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120:4317–23. doi:10.1182/blood-2012-06-437558
82. Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120:4324–33. doi:10.1182/blood-2012-06-438028
83. Terszowski G, Klein C, Stern M. KIR/HLA interactions negatively affect rituximab-but not GA101 (obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J Immunol (2014) 192:5618–24. doi:10.4049/jimmunol.1400288
84. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood (2014) 123:678–86. doi:10.1182/blood-2013-08-519199
85. Braud VM, Allan DS, O’Callaghan CA, Söderström K. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391:795–9. doi:10.1038/35869
86. Brooks AG, Borrego F, Posch PE, Patamawenu A, Scorzelli CJ, Ulbrecht M, et al. Specific recognition of HLA-E, but not classical, HLA class I molecules by soluble CD94/NKG2A and NK cells. J Immunol (1999) 162:305–13.
87. Levy EM, Bianchini M, Von Euw EM, Barrio MM, Bravo AI, Furman D, et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int J Oncol (2008) 32:633–41. doi:10.3892/ijo.32.3.633
88. Silva TG, Crispim JC, Miranda FA, Hassumi MK, de Mello JM, Simões RT, et al. Expression of the nonclassical HLA-G and HLA-E molecules in laryngeal lesions as biomarkers of tumor invasiveness. Histol Histopathol (2011) 26:1487–97. doi:10.1016/j.semcancer.2012.03.003
89. Gillard-Bocquet M, Caer C, Cagnard N, Crozet L, Perez M, Fridman WH, et al. Lung tumor microenvironment induces specific gene expression signature in intratumoral NK cells. Front Immunol (2013) 4:19. doi:10.3389/fimmu.2013.00019
90. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest (2011) 121:3609–22. doi:10.1172/JCI45816
91. Kwon BS, Weissman SM. cDNA sequences of two inducible T-cell genes. Proc Natl Acad Sci U S A (1989) 86:1963–7. doi:10.1073/pnas.86.6.1963
92. Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med (1997) 186:47–55. doi:10.1084/jem.186.1.47
93. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med (1997) 3:682–5. doi:10.1038/nm0697-682
94. Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol (1998) 190:167–72. doi:10.1006/cimm.1998.1396
95. Wilcox RA, Tamada K, Strome SE, Chen L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J Immunol (2002) 169:4230–6. doi:10.4049/jimmunol.169.8.4230
96. Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood (2011) 117:2423–32. doi:10.1182/blood-2010-08-301945
97. Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest (2014) 124:2668–82. doi:10.1172/JCI73014
98. Segal NH, Gopal AK, Bhatia S, Kohrt HE, Levy R, Pishvaian MJ, et al. A phase 1 study of PF-05082566 (anti-4-1BB) in patients with advanced cancer [abstract]. J Clin Oncol (2014) 32:3007. doi:10.1007/s00262-012-1237-1
99. Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol (2010) 28:57–78. doi:10.1146/annurev-immunol-030409-101243
100. Liu C, Lou Y, Lizée G, Qin H, Liu S, Rabinovich B, et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J Clin Invest (2008) 118:1165–75. doi:10.1172/JCI33583
101. Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol (2000) 164:2160–9. doi:10.4049/jimmunol.164.4.2160
102. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res (2013) 73:7189–98. doi:10.1158/0008-5472.CAN-12-4174
103. Kovacsovics-Bankowski M, Chisholm L, Vercellini J, Crittenden M, Lary S, et al. Phase I/II clinical trial of anti-OX40, radiation and cyclophosphamide in patients with prostate cancer: immunological analysis. J Immunother Cancer (2013) 1:1–1. doi:10.1186/2051-1426-1-S1-P255
104. Fu B, Tian Z, Wei H. Subsets of human natural killer cells and their regulatory effects. Immunology (2014) 141:483–9. doi:10.1111/imm.12224
105. Vossen MT, Matmati M, Hertoghs KM, Baars PA, Gent MR, Leclercq G, et al. CD27 defines phenotypically and functionally different human NK cell subsets. J Immunol (2008) 180:3739–45. doi:10.4049/jimmunol.180.6.3739
106. Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, et al. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol (2002) 3:83–90. doi:10.1038/ni746
107. Vitale LA, He LZ, Thomas LJ, Widger J, Weidlick J, Crocker A, et al. Development of a human monoclonal antibody for potential therapy of CD27-expressing lymphoma and leukemia. Clin Cancer Res (2012) 18:3812–21. doi:10.1158/1078-0432.CCR-11-3308
108. He LZ, Prostak N, Thomas LJ, Vitale L, Weidlick J, Crocker A, et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol (2013) 191:4174–83. doi:10.4049/jimmunol.1300409
109. Ansell SM, Northfelt DW, Flinn I, Burris HA, et al. Phase I evaluation of an agonist anti-CD27 human antibody (CDX-1127) in patients with advanced hematologic malignancies. ASCO Annual Meeting (2014). Available from: http://meetinglibrary.asco.org/content/132758-144
Keywords: natural killer cells, immunotherapy, adoptive cell therapy, monoclonal antibody, cancer vaccines, checkpoint blockade
Citation: Chester C, Fritsch K and Kohrt HE (2015) Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Front. Immunol. 6:601. doi: 10.3389/fimmu.2015.00601
Received: 02 May 2015; Accepted: 09 November 2015;
Published: 02 December 2015
Edited by:
Raquel Tarazona, University of Extremadura, SpainReviewed by:
Francisco Borrego, BioCruces Health Research Institute and Cruces University Hospital, SpainJohn T. Vaage, Oslo University Hospital, Rikshospitalet and University of Oslo, Norway
Copyright: © 2015 Chester, Fritsch and Kohrt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holbrook E. Kohrt, a29ocnRAc3RhbmZvcmQuZWR1