 Carin I. M. Dahlberg
Carin I. M. Dahlberg Dhifaf Sarhan
Dhifaf Sarhan Michael Chrobok
Michael Chrobok Adil D. Duru
Adil D. Duru Evren Alici
Evren Alici- 1Cell Therapies Institute, Nova Southeastern University, Fort Lauderdale, FL, USA
- 2Cell and Gene Therapy Group, Center for Hematology and Regenerative Medicine (HERM), Karolinska University Hospital Huddinge, NOVUM, Stockholm, Sweden
- 3Oncology-Pathology, Cancer Center Karolinska, Karolinska Institutet, Stockholm, Sweden
- 4Division of Hematology, Oncology and Transplantation, Masonic Cancer Research Center, University of Minnesota, Minnesota, MN, USA
- 5Hematology Center, Karolinska University Hospital Huddinge, Stockholm, Sweden
Natural killer (NK) cells were discovered 40 years ago, by their ability to recognize and kill tumor cells without the requirement of prior antigen exposure. Since then, NK cells have been seen as promising agents for cell-based cancer therapies. However, NK cells represent only a minor fraction of the human lymphocyte population. Their skewed phenotype and impaired functionality during cancer progression necessitates the development of clinical protocols to activate and expand to high numbers ex vivo to be able to infuse sufficient numbers of functional NK cells to the cancer patients. Initial NK cell-based clinical trials suggested that NK cell-infusion is safe and feasible with almost no NK cell-related toxicity, including graft-versus-host disease. Complete remission and increased disease-free survival is shown in a small number of patients with hematological malignances. Furthermore, successful adoptive NK cell-based therapies from haploidentical donors have been demonstrated. Disappointingly, only limited anti-tumor effects have been demonstrated following NK cell infusion in patients with solid tumors. While NK cells have great potential in targeting tumor cells, the efficiency of NK cell functions in the tumor microenvironment is yet unclear. The failure of immune surveillance may in part be due to sustained immunological pressure on tumor cells resulting in the development of tumor escape variants that are invisible to the immune system. Alternatively, this could be due to the complex network of immune-suppressive compartments in the tumor microenvironment, including myeloid-derived suppressor cells, tumor-associated macrophages, and regulatory T cells. Although the negative effect of the tumor microenvironment on NK cells can be transiently reverted by ex vivo expansion and long-term activation, the aforementioned NK cell/tumor microenvironment interactions upon reinfusion are not fully elucidated. Within this context, genetic modification of NK cells may provide new possibilities for developing effective cancer immunotherapies by improving NK cell responses and making them less susceptible to the tumor microenvironment. Within this review, we will discuss clinical trials using NK cells with a specific reflection on novel potential strategies, such as genetic modification of NK cells and complementary therapies aimed at improving the clinical outcome of NK cell-based immune therapies.
Introduction
Natural killer (NK) cells are lymphocytes of the innate immune system. They are cytokine producing and have cytotoxic ability to kill both viral infected and tumor cells. Tumor-killing lymphocytes were first reported in 1968 by Hellström et al. (1). Kiessling and colleagues, in parallel with Ronald Herberman’s research laboratory, defined a novel lymphocyte population named NK cells that are able to target tumor cells in 1975 (2–5). Unlike T cells and B cells, NK cell recognition is not governed by high-resolution antigen specificity. Target cell recognition is mediated by the signals delivered through several activating and inhibitory receptors. The balance between activating and inhibitory signals decides the response of NK cells. When there is a mismatch between an inhibitory subgroup of killer immunoglobulin-like receptors (KIRs) on NK cells and self-human leukocyte antigen (HLA) class I proteins on the surface of target cells the NK cells can get activated due to lack of inhibitory signals leading to lysis of the host cell. This mismatch mediates alloreactivity and is the strategy behind the missing-self concept (6). KIRs can be divided into two haplotypes; the A haplotype with predominantly inhibitory KIRs plus only one activating KIR and the B haplotype containing inhibitory and activating receptors (7). During NK cell education, KIRs go through a random sequential acquisition process where they get functionally competent after they encounter self-MHC class I molecules. Consequently, mature NK cell function is inhibited by self-MHC class I and KIR interaction (8). When a NK cell confronts a target cell without expression of self-MHC class I molecules, the inhibitory signals are not active and the NK cell gets activated.
The majority of NK cells, as well as certain T cell subpopulations, may express the receptor family NKG2. One of the ligands for most NKG2 receptors is HLA-E, which is expressed on all nucleated cells. NKG2-family consists of seven members: NKG2A, B, C, D, E, F, and H in which NKG2A and B are inhibitory receptors. NK cells also express activation receptors on the surface, such as natural cytotoxicity receptors (NCRs), DNAM-1, and receptor members of the 2B4 family. NCRs, including NKp30, NKp44, and NKp46, are one of the main and initial groups of NK cell-activating receptors identified and they recognize viral ligands, heat shock-associated proteins, or tumor antigens (9). NK cells can also get activated by crosslinking of Fc receptor CD16 to target cell leading to antibody-dependent cellular cytotoxicity (ADCC) and lysis of the target cell (10, 11).
Natural killer cells perform their cytotoxic activity through granzyme B- and perforin-mediated apoptosis or by expression of death receptor ligands such as FasL and TNF-related apoptosis-inducing ligand (TRAIL). While the release of cytolytic granules is one of the essential cytotoxic responses, perforin deficient NK cells can still kill tumor cells through Fas-mediated apoptosis (12). Moreover, TRAIL-TRAILR mediated cytotoxicity also plays an important role in eliminating the target cells. Various tumor cells express TRAIL death receptors, which could be upregulated by proteasome inhibitors such as bortezomib (13). Additionally, immunomodulatory drugs (IMiDs) such as lenalidomide upregulates TRAIL expression on NK cells that potentially enhance the TRAIL-mediated elimination of tumor cells (14, 15).
Natural killer cells are derived from hematopoietic stem cells (HSC) in the bone marrow. The differentiation from HSC can be divided into five stages based on surface markers [detailed review in Ref. (16)]. The stages can be identified by the following surface markers, CD34, CD117, CD94, and CD16 among the Lin− events, where stage 1 is CD34+CD117−CD94−CD16−. First at stage 2, the cells are able to respond to IL-15, which is necessary for NK cell development (17, 18). In the transition between stage 2 and 3, they lose their CD34 expression. At stage 4, the NK cells are CD56bright, produce IFNγ, and are capable of cytotoxic killing of K562 cells in vitro (19). NK cells in stage 5 are CD56dim and express CD16.
The majority of human NK cells are CD14−CD19−CD3−CD56+. While most of the CD56+ cells express lower levels of CD56 (~90% CD56dim), they are potent cytotoxic killers of target cells and secrete cytokines such as IFNγ. Approximately 10% of peripheral NK cells express high levels of CD56 (CD56bright), have low cytolytic activity, and have the capacity to produce high titers of immunoregulatory cytokines. The cell surface phenotypes of these two subpopulations also differ in respect to the receptors they express: the CD56bright population expresses the inhibitory receptor NKG2A that could also be expressed on CD56dim NK cells. While the CD56dim population expresses FcγRIIIa (CD16a) as well as the inhibitory receptors KIRs (20).
NK Cells in Cancer
Natural killer cells recognize tumor cells by the activating receptors like NCRs, which detect the altered expression of their ligands on the tumor cell surface. Additionally, downregulation or lack of MHC class I molecules on the cell surface of tumor cells can trigger NK cell activation since it diminishes the inhibitory signals transduced through KIR-MHC interactions. Moreover, since NK cells’ target recognition and activation are mainly through NCRs and missing-self, this engagement could induce upregulation of FasL on the NK cell surface leading to an alternative pathway inducing apoptosis in tumor cells. Nevertheless, both IL-2 stimulation and NK cell activation through NCRs also upregulate Fas on NK cells that may initiate regulation of the NK cell activation and expansion (21, 22).
Many tumors have gained methods to evade the surveillance by NK cells and other members of the immune system. For example, 16 of 18 patients with acute myeloid leukemia (AML) had reduced NCR surface expression compared to healthy donor NK cells, resulting in reduced cytotoxic capacity against target cells (23). Another way for tumor cells to escape recognition by NK cells is upregulation of the non-classical MHC class I molecule HLA-G, which dampens NK cell responses (24, 25). In numerous malignancies, there are also abnormalities found in the NK cell population. Examples of this include defective expression of activating receptors found in hepatocellular carcinoma (26), metastatic melanoma (27), AML (23), chronic lymphocytic leukemia (CLL) (28), and multiple myeloma (29, 30) or defective NK cell proliferation in metastatic renal cell carcinoma (31) and chronic myelogenous leukemia (CML) (32).
In renal cell carcinoma, infiltrating NK cells have, compared to peripheral blood NK cells, increased expression of NKG2A receptor contributing to decreased NK cell activity (33). NKG2D is a well-studied activating receptor on NK cells. Membrane-bound NKG2D ligand has a stimulatory effect on immunity, while soluble NKG2D ligands have the opposite effect on immune system leading to metastatic cancer progression (34). Patients with colorectal cancer have increased serum titers of the soluble NKG2D ligand, MHC class I chain-related protein A (sMICA), compared to healthy controls, leading to downmodulation of activating and cytokine receptors on the NK cells (35). A potential way to reduce the risk of soluble NKG2D ligand is to give the patients neutralizing antibody treatment. Clinical observations demonstrate that patients treated with cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) antibody blockade have reduced sMICA in a close correlation with increased titers of autoantibodies against MICA (36). Interestingly, a new report from Deng et al. shows that the soluble high-affinity ligand MUL1 causes NK cell activation and stimulates tumor rejection in mice, instead of inhibition of NK cells as earlier reported (37).
The potential benefits of NK cell-based cancer immunotherapy products have led to the design of in vitro methods aiming to cultivate NK cells in cGMP conditions. Some of these methods have already been tested in clinical trials, which will be discussed later in this review.
Clinical-Grade NK Cell Products
It is possible to activate NK cells and increase their anti-tumor activity through short-term cytokine exposure in vitro prior to adoptive transfer (38). However, to achieve clinically relevant numbers of NK cells, there also needs to be development of long-term NK cell expansion protocols (Table 1; Figure 1) (39–47). Yet, there are concerns when expanding NK cells in vitro, such as potential phenotypic changes, selective expansion, and reduced cytotoxic killing. When expanded in vitro with IL-2, there is a chance of CD3+ cell expansion as well (48, 49). Thus, there is still room for improvement to achieve optimum clinically relevant NK cell numbers, in vivo NK cell persistence and survival, and most importantly, anti-tumor activity. There are numerous parameters affecting the clinical-grade NK cell manufacturing such as source of the NK cells, cytokine stimulation, cell culture medium, and expansion platform. Here, in this section, we will address these parameters.
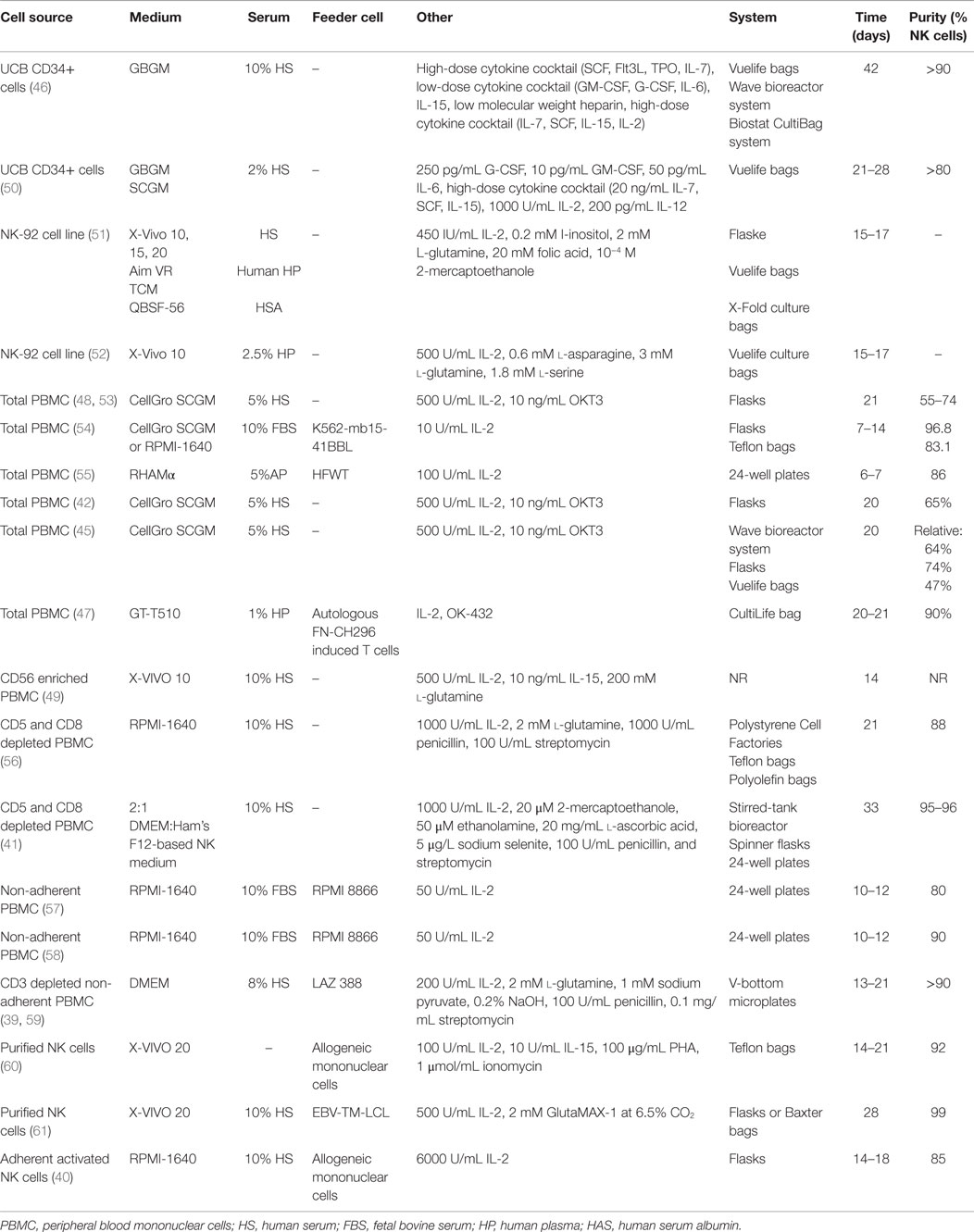
Table 1. Clinical-grade NK cell products.
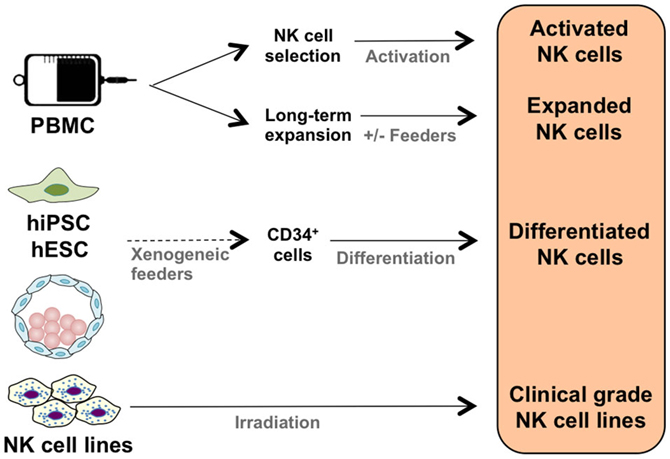
Figure 1. Clinical NK cell therapy products.
Source of the NK Cells
The majority of clinical NK cell products or pre-clinical research on efficient NK cell manufacturing platforms are making use of peripheral blood mononuclear cells (PBMC), umbilical cord blood (UCB), cell lines, and human embryonic stem cells (hESC), as well as induced pluripotent stem cells (iPSC) as a source of start material.
Peripheral Blood Mononuclear Cells
The majority of NK cell products are generated through utilization of PBMCs either by apheresis or ficoll separation under cGMP conditions. An advantage of using PBMCs is the ability to collect cells in a closed aseptic system. Although PBMC consists of 5–20% NK cells, it is not possible to achieve sufficient numbers of potent NK cells. Thus, various techniques to expand NK cells ex vivo have been developed. For example, we have designed a feeder-free NK cell expansion system where it is possible to expand and activate tumor-reactive NK cells in a clinically compatible manner (45). These cells have a high cytotoxic effect specifically against autologous and allogeneic tumors in vitro and in vivo (42, 45). We have also completed a first-in-man clinical trial using donor-derived ex vivo expanded NK cells in terminal cancer patients that had CLL, kidney cancer, colorectal cancer, and hepatocellular carcinoma with promising results (43). Having optimized the procedure for NK cell expansion in a closed-automated bioreactor using clinical-grade GMP-compliant components, we have initiated a first-in-man phase I/II clinical trial to expand and restore the function of patients’ own NK cells (45, 62). To our knowledge, this is the first advanced therapy investigational medicinal product trial performed using autologous NK cells in Sweden.
Sakamoto et al. have established another similar approach that generates large numbers of activated NK cells from peripheral blood without prior purification of the cells. The PBMCs are cultured with autologous plasma, IL-2, OK-432, and γ-irradiated autologous FN-CH296 stimulated T cells, reaching up to a median purity of 90.96% of NK cells at day 21 or 22. Many of the NK expansion protocols are based on enrichment of NK cells either prior to NK cell activation and expansion through cell selection or sorting in order to achieve pure cell therapy product and avoid unwanted side effects stemming from T cells especially in allogeneic NK cell transfusions.
One of the main methods of enriching the purity and the number of initial NK cells is the clinical-grade immuno-magnetic depletion of other lymphocyte subsets such as T cells and/or B cells as well as myeloid cells (60). Depletion of CD3+ cells followed by CD56+ cell enrichment can lead to highly pure NK cells which could be supplemented by CD19+ cell depletion before infusion in order to prevent passenger lymphocyte syndrome in allogeneic transplantation (63). Nguyen et al. have shown that a partial depletion of T cells could get a more beneficial clinical outcome compared to a complete T cell depletion after hematopoietic stem cell transplantation, suggesting that T cells may have a positive role in in vivo NK cell function (64).
Additionally, direct enrichment of CD56+ NK cells through immuno-magnetic selection is an option to achieve high purity initial NK cell product. Nevertheless, NK cells might require physical and cytokine-dependent communication with other cells such as monocytes (65) in order to activate and expand. Thus, it is essential to fine-tune the enrichment of NK cells by making use of feeder cells and/or optimizing the cytokine cocktail used in ex vivo NK cell expansion protocols.
Furthermore, using feeder cells and cell lines is another approach in expanding NK cells ex vivo since feeder cells can provide essential stimulatory signals for NK cells proliferation. Monocytes, irradiated PBMC, feeder cell lines, and engineered feeder cell lines are the most commonly used sources for stimulation of NK cell expansion through humoral signals and cell-to-cell contact. Example of feeder cells that have been used in clinical trials are irradiated autologous PBMCs (60, 66), irradiated Epstein–Barr virus-transformed lymphoblastoid cells (61), and K562 engineered cells expressing 4-1BB ligand (67) or membrane-bound IL-21 (68, 69) on cell surface.
Stem Cells
While PBMC is one of the major sources for achieving clinically relevant doses of tumor-reactive NK cells, HSC and potentially hESC as well as iPSC are likewise essential sources for achieving clinically relevant doses of NK cells.
One of the potential sources to accomplish clinically relevant doses of tumor-reactive NK cells is making use of HSC (CD34+) through differentiation and expansion of CD34+ cells isolated from bone marrow, peripheral blood, or UCB into functional NK cells. It was recently demonstrated that it is possible to expand activated, tumor cytotoxic and pure NK cells by differentiating UCB CD34+ HSC under cGMP condition (46). Furthermore, NK cells derived from CD34+ UCB cells lack expression of KIRs such as KIR2DL1 (CD158a), KIR2DL2/DL3 (CD158b), and NKB1, as well as diminished CD16 expression in the CD56dim population (70). Even though NK cells derived from UCB have reduced cytotoxicity, this could be restored by ex vivo cytokine stimulation such as IL-2, IL-12, and IL-15 (50, 71–73). Infusion of UCB-derived NK cells supplemented with IL-15 has shown to inhibit growth of human bone marrow resident leukemia cells in vivo (74). Recently, it was demonstrated that frozen UCB CD34+ cells differentiate into NK cells with better expansion than freshly isolated UCB CD34+ cells, and more importantly, UCB CD34+ cells gave more NK cell product than peripheral blood HSC without jeopardizing NK cell functionality (75). Thus, UCB CD34+ cells are one of the essential sources for manufacturing NK cell therapy protocols, providing an option to create NK cell biobanks.
Another potential source of NK cells is hESC and iPSC, with the advantage of potential usage of iPSCs in autologous settings with reduced risk of immune rejection. The first step is to generate CD34+ hematopoietic precursor cells from the hESCs and iPSCs and then differentiate these cells into NK cells, which could be efficiently achieved through growing hESCs and iPSCs on murine stromal cells (76, 77). Yet, the involvement of xenogeneic cells could limit the potential clinical usage of hESCs and iPSCs. Addressing this potential problem, Knorr et al. developed a two-stage culture method where hESCs and iPSCs are first differentiated to CD34+ hematopoietic cells by spin-EB system in xeno-free and serum-free conditions followed by stroma-free NK cell differentiation, which enables generation of cytotoxic NK cells without involvement of xenogeneic cells taking a step forward toward clinical-scale production (78). Since IL-2-activated NK cells are potent killers of both allogeneic and autologous iPSCs (79), it is possible to manufacture a pure NK cell therapy product. This sticks out as one of the advantages of using in vitro NK cell differentiation from iPSCs.
Cell Lines
Cell lines derived from NK cells with similar biological functions (NK-92, NKL, KYHG-1, and NKG) are potential candidates for NK cell-based products enabling design and development of off-the-shelf anti-cancer cell therapy products. Furthermore, it is more feasible to generate genetically modified NK cell lines expressing intracellular IL-2 for activation or cell surface molecules such as CD16, NCRs, and chimeric antigen receptors (CARs). To our knowledge, the NK-92 cell line is the most clinically studied one. The IL-2-dependent NK-92 cell line is cytotoxic to a wide range of malignant cells (80–83). It has also been used as a source of NK cells for cGMP-grade cellular therapy products (51) as well as in clinical trials (52, 84). The NK-92 cell line expresses several activating receptors but lacks most of the inhibitory KIRs, NKp44, and CD16 (80, 85). NK-92 cells require irradiation to prevent proliferation prior to being used effectively in immunotherapeutic approaches without compromising hematopoietic cell function. For example, recently, clinical-grade NK-92 cells have been manufactured and were safely used as anti-tumor therapy for patients with a variety of tumors (84) with promising results (52). As of today’s date, two phase I clinical trials (NCT00900809 and NCT00990717) are recruiting patients with hematological malignancies for treatment with NK-92 cells. The first clinical phase II study (NCT02465957) with NK-92 cells has recently been initiated.
KHYG-1 is the first NK cell line derived from NK leukemia and has higher cytotoxicity than NK-92 cell line (86). Likewise NK-92 cells, these cells can also be irradiated to inhibit proliferation and can still efficiently kill tumor targets. Furthermore, NKL cell line, which is the most biologically and functionally similar to primary NK cells, is more cytotoxic to certain tumor cells than NK-92 cell line and, additionally, it has the ADCC capacity whereas NK-92 cells lack CD16 expression. Thus, both KHYG-1 and NKL cell lines have the potential to be used as anti-cancer NK cell products.
Additionally, one of the advantages of using such master cell bank is an appealing opportunity in the manufacture of cellular therapy products since it is possible to establish a comprehensive standardization and characterization of the cell source. It is also possible to genetically modify these cell lines to exert more tumor specificity and cytotoxicity. For example, NK-92 cell lines are dependent on external IL-2 stimulation, which increases manufacturing costs as well as potentially reducing the long-term cytotoxic capacity of these cells unless they are supported by IL-2 infusions. Thus, constitutive expression of IL-2 in NK-92 cells through genetic modification leads to auto-activated and -proliferating cells, which reduces the manufacturing costs as well as potentially increases the in vivo tumor reactivity (87, 88).
Cytokines
Ex vivo manufacturing of NK cell-based products is dependent on extensive use of cytokines to stimulate, differentiate, activate, and expand NK cells in order to get clinically relevant doses and enhanced anti-tumor reactivity. Historically, one of the most popular cytokines in NK cell research is IL-2 since it was the first cytokine to be injected to patients to treat metastatic melanoma (89). Thirty years ago, Rosenberg et al. published the first report where they treated 25 metastatic cancer patients, who did not respond to standard therapy, with autologous lymphokine-activated killer (LAK) cells together with recombinant-derived IL-2. LAK cells are generated from mononuclear cells collected from IL-2 injected patients. In 11 patients, the cancer regression was observed with >50% of tumor volume (90). This adoptive immunotherapy was followed by a larger scale study, where 157 patients with advanced metastatic cancer were treated with successful results (91). In the same year, it was shown that it was the NK cells that mediated the cytotoxic activity in response to systemic administered recombinant IL-2 (92). These reports were followed by many years of IL-2 and NK cell research. In a dose-dependent manner, IL-2 is important for NK cell infiltration and killing of the tumor. For example, in the bone marrow, there are hypoxic regions leading to reduced NK cell killing of plasma cells in multiple myeloma. IL-2-activated NK cells ex vivo have increased NKG2D expression resulting in increased targeting of multiple myeloma upon infusion (93). Cytokine-activated NK cells in vitro are dependent on constant stimulation both in vitro and in vivo. Basse et al. reported that when no exogenous IL-2 is present the amount of injected NK cells found in tumors were very low (94). The half-life of IL-2 in serum is not more than 10 min, which makes the administration of IL-2-dependent cells difficult (95). By transducing NK cells to produce IL-2 prior to transplantation, the activated NK cells would have a constant source of IL-2 in vivo (87, 96). One of the disadvantages of using IL-2 to activate NK cell in vivo is the competition over IL-2 by regulatory T cells, which express high levels of the high-affinity receptor for IL-2, IL-2Rα (CD25). By treating patients with lympho-depleting agents (fludarabine and cyclophosphamide) followed by NK cell infusion and IL-2 fused with diphtheria toxin (IL-2DT), CD25+ cells are selectively depleted, leading to increased NK cell expansion and complete remission rate for patients with AML compared to regular IL-2 treatment (97). Overall, the majority of cGMP-grade NK cell therapy protocols include IL-2 as a main cytokine to stimulate NK cell activation and proliferation.
Another important cytokine is IL-15 which is required for both NK cell maturation and survival (98). IL-2 and IL-15 share the same receptor components: IL-2/15Rβ and common γ chain (also shared with IL-4, IL-7, IL-9, and IL-21). Recent advances in the production of cGMP quality cytokines enabled further optimization of cytokine supplementation during NK cell expansion. For example, use of IL-15 in combination with IL-2 has a synergetic effect on product viability and NK cell proliferation (66). This highlights the necessity of other cytokines to achieve NK cell product potency especially when it comes to the NK cell expansion protocols that are not using feeder cell support. Additionally, IL-21, primarily described in 2000 (99), has significant homology with IL-2 and IL-15. Compared to IL-2 and IL-15, IL-21 promotes maturation and survival but does not promote proliferation of NK cells alone. However, IL-21 does have synergetic effects with IL-2 and IL-15 (100). Interestingly, it has been suggested that IL-21 does not drive proliferation of regulatory T cells in vivo and might be a good candidate to substitute for IL-2 in CLL (101).
Other Factors
Besides NK cell source, feeder support, and cytokine stimulation, other parameters such as expansion platform, cell culture media, and serum supplementation are also very important in achieving clinically relevant cell numbers, viability, and tumor cytotoxicity. More specifically, we have recently investigated the importance of the culture vessels on the quality and efficacy of the NK cell product. Briefly, PBMCs from healthy donors and myeloma patients were cultured for 21 days using flasks, cell culture bags, and bioreactors. Even though we have achieved high yield in NK cell expansions in all systems, NK cells expanded in the bioreactor displayed significantly higher cytotoxic capacity. These results demonstrate that highly active NK cells can be produced in a closed, automated, large-scale bioreactor under feeder-free current GMP conditions facilitating adoptive immunotherapy clinical trials (45).
Additionally, cell culture media is another important factor to consider in the manufacturing of cellular therapy products. There are very few cGMP quality medias that work optimally for ex vivo NK cell expansion protocols. The most commonly preferred media in the generation of NK cell products are stem cell growth medium (SCGM; CellGenix, Freiburg, Germany), X-VIVO serum-free media (BioWhittaker, Verviers, Belgium), or AIM V (Life Technologies, Grand Island, NY, USA) (49, 102, 103). Generally, medium is supplemented by human AB serum or fetal bovine serum from certified sources.
Finally, there are numerous variables that may impact quality and quantity of NK cell products. Future pre-clinical research and results from more clinical trials will evaluate the contribution of each factor to the product purity, potency, and safety, as well as assist in acquiring NK cell products that can be manufactured reproducibly with the optimal safety and anti-tumor responses.
Clinical Use of NK Cell-Based Anticancer Products
Autologous NK Cells
Several clinical studies have been performed with adoptive autologous NK cells in an attempt to target tumors, such as breast cancer, lymphoma, glioma renal cell carcinoma, non-small cell lung cancer, and adenocarcinoma (Table 2) (39, 40, 55, 103–107). In general, autologous NK cell trials are safe with no toxic side effects (39, 40, 55, 105). For example, ex vivo activated autologous peripheral blood lymphocytes get enhanced cytolytic activity against heat shock protein 70 (Hsp70) membrane-positive tumors in vivo if pre-incubated with Hsp70 peptide and IL-2 (105, 107). However, some clinical trials with autologous NK cells have only partial effect on tumors, such as glioma (55). While other tumors, such as metastatic carcinoma or relapsed lymphoma, do not demonstrate any improvement (103, 104, 108). Moreover, a recent clinical trial used ex vivo FN-CH296 stimulated T cells and OK-432 expanded, autologous NK cells with enrolled patients diagnosed with rectal, esophageal, gastric, or colon cancer that was either recurrent or at metastatic disease stage. The NK cell therapy in these patients was well tolerated with no severe adverse events and the cytotoxicity of peripheral blood was elevated approximately twofold up to 4 weeks post the last transfer (47).
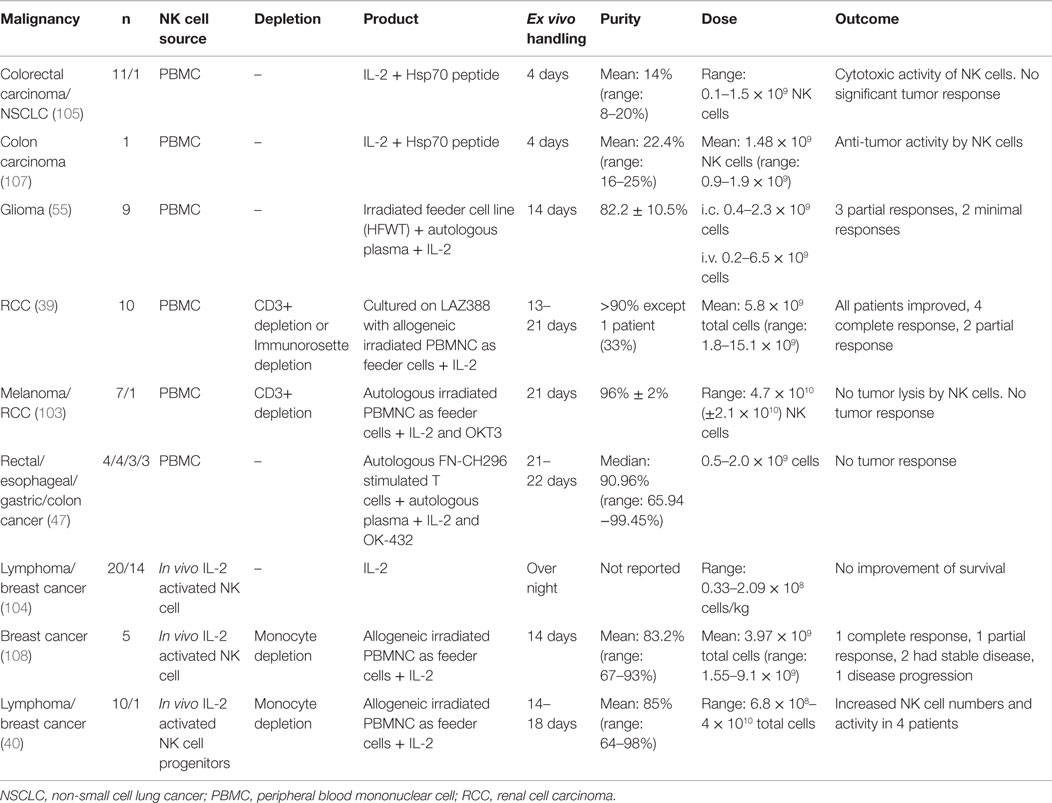
Table 2. Clinical trials with infusion of autologous NK cells.
Allogeneic NK Cells
Allogeneic NK cell products have been used in the treatment of a range of malignancies, such as leukemia, renal cell carcinoma, leukemia, colorectal cancer, hepatocellular cancer, lymphoma, and melanoma (Table 3) (38, 109–113). The major risk with allogeneic NK cell transplantation is the development of graft-versus-host disease (GvHD). Several precautions can be taken to reduce the risk of GvHD, for example, immunosuppression, infusion of CD3 depleted high purity NK cells and if available, selecting the donor that matches the host HLA (44, 114, 115).
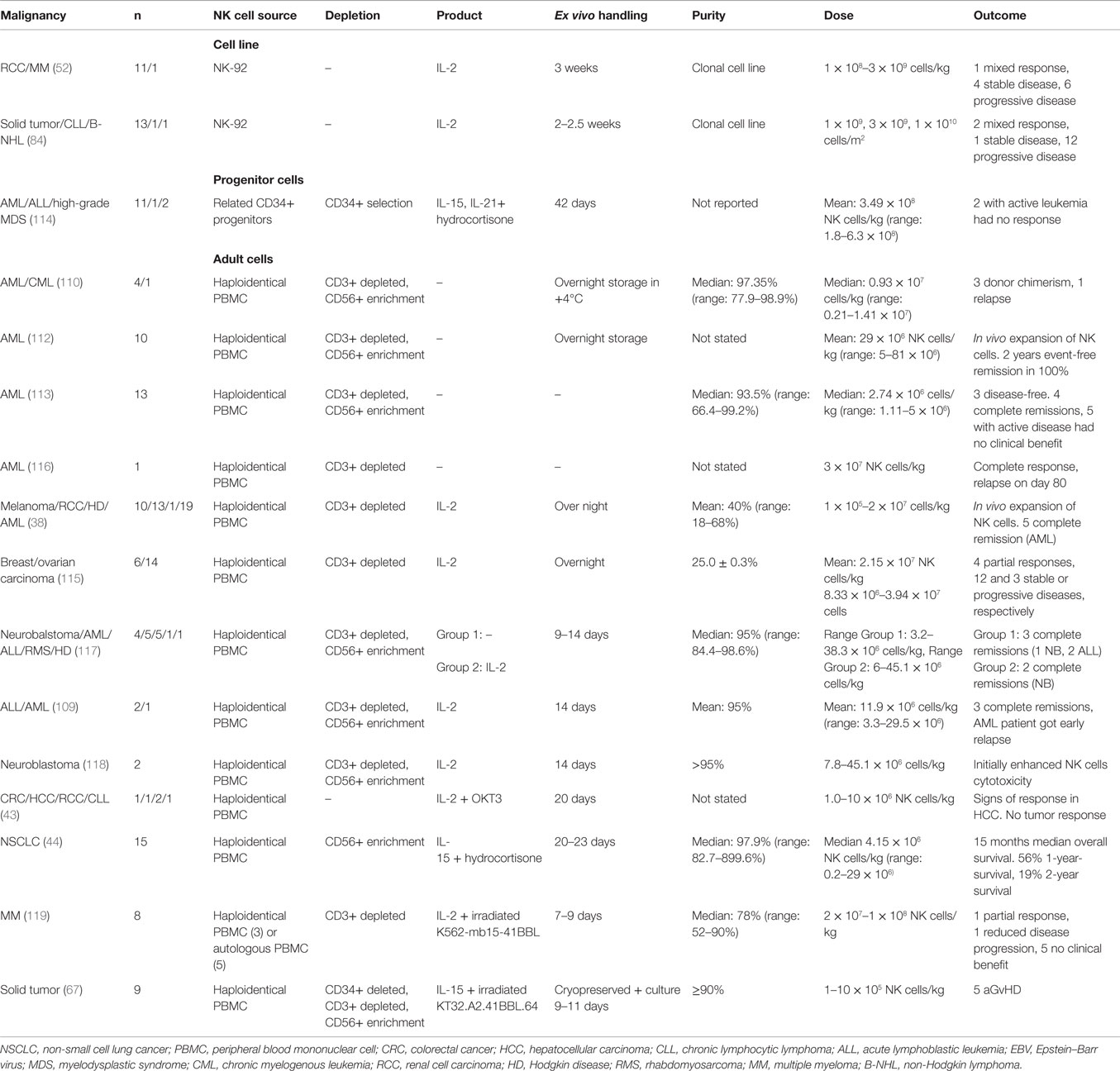
Table 3. Clinical trials with infusion of allogeneic NK cells.
In the first phase I clinical trial using the feeder-free ex vivo expansion platform, adoptive transfer of NK cells from HLA identical siblings into patients with leukemia or carcinoma was well tolerated and safe alongside in vivo NK cell expansion, with only some infusion-related complications (43).
If no HLA identical donor is available, host cells from a receptor–ligand-mismatched donor can be used. If the donor is HLA matched, it is preferentially better if the donor cells are KIR B haplotype. Also, to further improve the outcome, T cell depletion is performed (120). In haploidentical transplantation, at least one KIR ligand is not expressed on the host cells leading to reduced inhibition of donor NK cells. Less inhibited NK cells could lead to better prognosis and might be the best treatment for a good clinical outcome if GvHD can be avoided (38, 121). When haploidentical transplantation is performed, it is strictly necessary to make extensive T cell depletion to avoid GvHD. In most clinical trials, NK cells are collected from leukapheresis followed by a two-step purification procedure, with depletion of CD3+ T cells followed by enrichment of CD56+ cell (109, 110, 117, 118).
Completed clinical trials with haploidentical donors are safe with only a few reports of infusion-related complications such as dyspnea, nausea, hypertension, stroke, febrile reaction, and vomiting (38, 115). So far, allogeneic NK cell transplantations derived from PBMCs or CD34+ cells have shown promising results with engraftment, in vivo expansion of NK cells, complete remission, and a 100% 2-year event-free survival in one clinical trial by Rubnitz et al. (109, 112–114, 116).
Immune Suppression of NK Cells in the Tumor Microenvironment
Natural killer cells can recognize and kill tumor cells in vitro. However, their efficiency in targeting solid tumors has not yet been fully acknowledged in the clinical setting even though endogenous and adoptively transferred activated NK cells can be detected in various tumors (122, 123). Nevertheless, not all tumors are equally well infiltrated by NK cells, and many of the infiltrating cells are dysfunctional (124–127). The failure of immune surveillance may in part be due to sustained immunological selection pressure on tumor cells resulting in the development of tumor escape variants that are in fact invisible to the immune system (Figure 2). In addition, cytotoxic function of immune effector cells is also largely suppressed in the tumor microenvironment (128), which could be explained by suppressive tumor-secreted factors as well as suppressive immune compartments, such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAM), and regulatory T cells (Figure 2). One of the most studied immune-suppressive cell types associated with tumor progression is regulatory T cells (Treg), characterized by their expression of CD4, high CD25 (CD4+CD25+CD127low/neg) as well as the transcription factor forkhead box P3 (FoxP3) (129). The expansion of Treg population is promoted in different cancers and their accumulation correlates with impaired immune cell function and poor prognosis (130–135). In vitro, NK cells are suppressed by Treg cells in a cell contact-dependent manner where membrane-bound TGF-β is utilized to attenuate their cytotoxicity (136). In line with this, inverse correlation between NK cell activity and Treg cell expansion has been observed in patients with gastrointestinal stromal tumor (GIST) (136) as well as in hepatocellular carcinoma patients (137). Treg cells express the high-affinity IL-2 receptor alpha (CD25, IL-2Rα) and need IL-2 for their full function. Recent studies have indicated that NK cell proliferation, accumulation, and activation can be limited by Treg cells through hampering the availability of IL-2 released by activated CD4+ T cells (138, 139). Consequently, inadequate IL-2 levels in the tumor microenvironment limits the extent of NK cell-mediated tumor rejection.
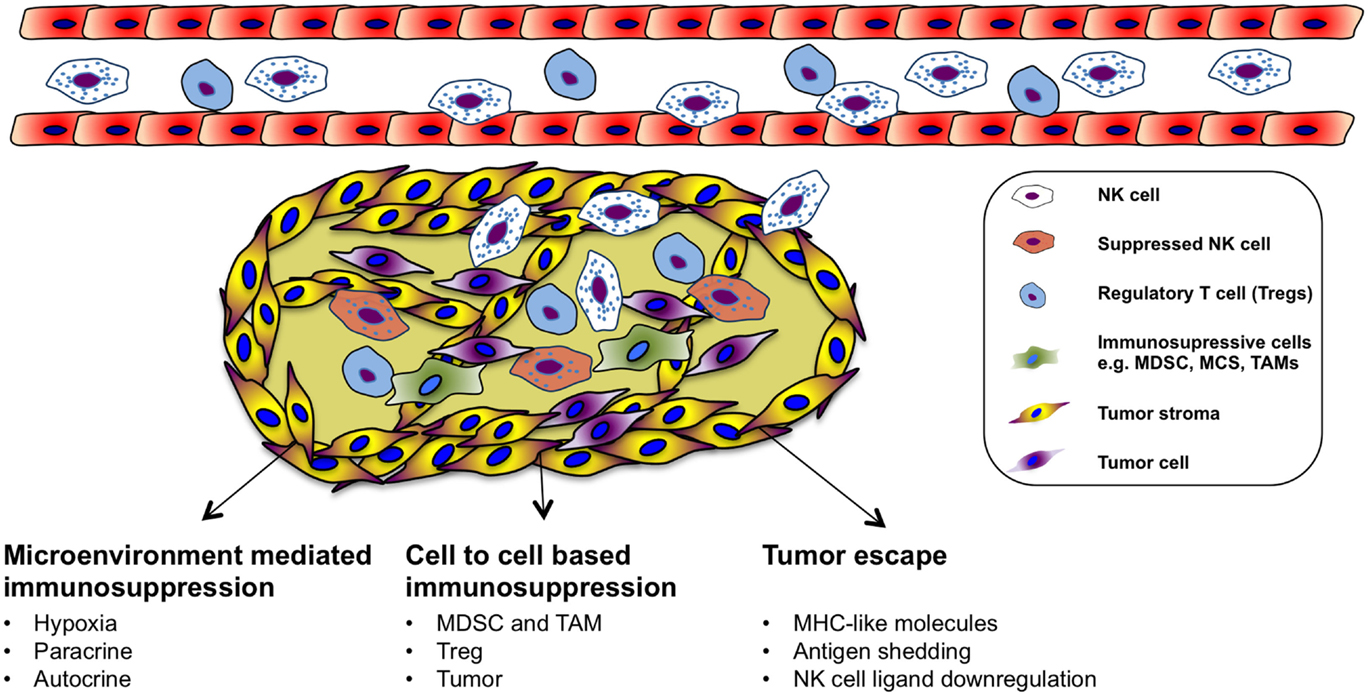
Figure 2. Immune evasion and immunosuppressive in the tumor microenvironment.
Another group of immunosuppressive cells in the tumor is the MDSCs. MDSCs are heterogeneous precursors of the myeloid cells, granulocytes, macrophages, and immature dendritic cells with immunosuppressive activity (140). Recently, MDSCs have been proposed as a key immunoregulator in various solid and hematologic malignancies (141, 142). MDSCs are divided into two groups that can originate from granulocytic (grMDSCs) and monocytic precursors (moMDSCs) (143). In human beings, distinct phenotypes of MDSCs are associated with different types of cancers (144–148). Their suppressive function is mediated by a few different mechanisms such as production of suppressive cytokines including IL-10 and TGF-β, depletion of arginine in the tumor or production of reactive oxygen species (ROS) (144, 149–151). Additionally, recent studies investigated the induction mechanism of MDSCs and how they suppress T cells in vitro (152–154). Furthermore, several studies have characterized cytokines that can induce MDSCs from healthy human PBMCs. We found that prostaglandin E2 treated healthy monocytes resemble patient-derived moMDSCs and suppress NK cell responses through TGF-β-dependent mechanism (155). In patients with hepatocellular carcinoma, NK cells were shown to be suppressed by monocytic MDSC in a cell contact-dependent manner, but did not rely on the arginase activity of MDSCs, which is a hallmark function of these cells; however, MDSC-mediated inhibition of NK cell function was revealed to be mainly dependent on the NKp30 on NK cells (146). Moreover, a negative correlation between increased CD33+-MDSC accumulation and functional loss of NK cells has been demonstrated in patients with myelodysplastic syndromes (156).
Macrophages are the dominant myeloid-derived population that is found in the tumor microenvironment. TAM has been identified as regulators of solid tumor development based on their capacity to enhance angiogenic, invasive, and metastatic programing of neoplastic tissue (157–160). TAMs could be found in several types of human cancer correlating with poor clinical outcome (161, 162). The immune-suppressive mechanisms applied by TAMs on NK cells in the tumor microenvironment can be different, such as recruitment of Treg, prostaglandin E2-mediated inactivation, and production of IL-10 (163–165). Furthermore, tumors are able to escape NK cells by releasing indoleamine 2,3-dioxygenase and prostaglandin E2, which inhibit the expression of activating receptors of NCRs and NKG2D (166). These molecules are also released by mesenchymal stem cells to inhibit NK cell function in the tumor microenvironment (167). There is a direct association between the surface density of NCRs (NKp46) and the intensity of anti-tumor cytolytic activity of the NK cells (168).
As mentioned earlier, the tumor microenvironment plays a significant role in suppressing NK cell responses against cancer. Therefore, therapies aim to target immunosuppressive cell populations are emerging (169–174). In the next section, some of the alternative ways aiming to enhance tumor-specific targeting and NK cell survival in order to overcome immunosuppressive effect of the tumor microenvironment on NK cells and to improve intra-tumoral NK cell responses will be discussed.
Future Perspectives
Genetically Modified NK Cells
In the last decade, several NK cell based anti-cancer products have been taken to clinical trial stage with promising clinical outcomes. However, in order to manufacture more efficient NK cell therapy products, it is essential to develop novel potential strategies such as genetic modification of NK cells (Figure 3). Although NK cells are inherently resistant to retroviral infections (96, 175–177), our group has significantly enhanced retroviral and lentiviral gene delivery to NK cells through enhanced proliferation and targeting intracellular viral defense mechanism by small molecule inhibitors (96). Therefore, it is easier to design genetically modified NK cells expressing cytokine transgenes, silenced inhibitory receptors, overexpressing activating receptors, or retargeting NK cells by expression of CARs on the cell surface. By genetically modifying NK cells to produce cytokines such as IL-2 or IL-15, their survival capacity and proliferation increase and their activation and anti-tumor activity in vivo are enhanced (83, 87, 88, 178, 179). To enhance the specificity for the target cells, NK cells can be modified to recognize antigens specifically expressed on the tumor cells.
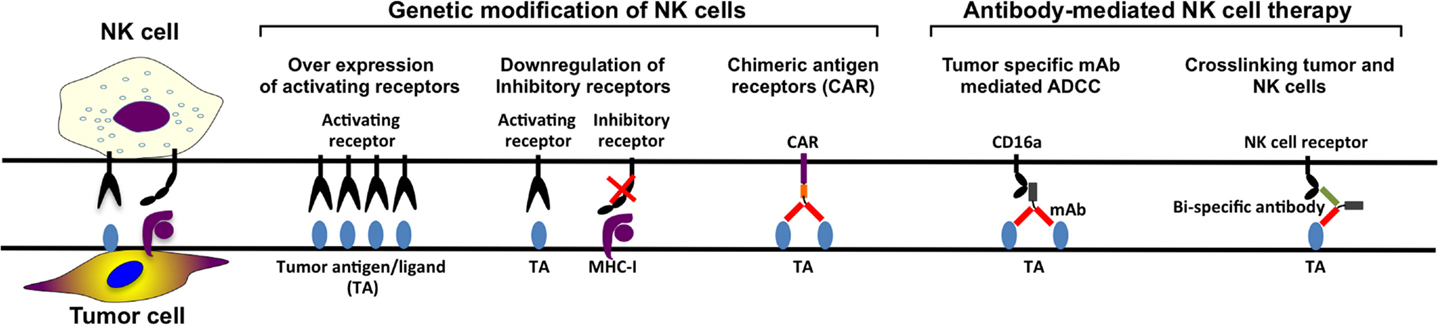
Figure 3. NK cell therapy approaches.
Furthermore, another approach aiming to enhance tumor specificity is to make use of ADCC. The constant region of the tumor-specific monoclonal antibodies (mAbs) targeting the tumor cells can engage to the FcγRIIIa receptor (CD16a) on the NK cell, activating the NK cell. However, NK-92 cell line cannot perform ADCC since they lack CD16a expression (80, 85). This defect on NK-92 cells can be reverted by the introduction of CD16a through genetic modification so that they are able to perform ADCC in antibody combination treatments (180). Finally, CAR-modified NK cell lines can also function as tumor-specific standardized and characterized NK cell-based therapy products. Most of the NK cell lines require further in vivo characterization with a potential to become standard NK cell-based products for certain tumors.
Monoclonal Antibodies
When the antigen-binding fraction (Fab) of the antibody binds to the tumor target cell and the constant region (Fc) of the antibody binds to CD16 on the NK cells, NK cells get activated and ADCC is triggered. Several different mAbs have been developed for targeting specific tumor antigens, such as anti-CD20 (retuximab), anti-Her2 (trastuzumab), anti-CD52 (alemtuzumab), anti-EGFR (certuximab), and anti-CD38 (daratumumab) (181). Daratumumab treatment of patients with relapsed myeloma has mild infusion-related reactivity, complete or very good partial responses with reduced bone marrow plasma cell levels (182). mAbs bind to the target tumor cell plus engaging CD16 on NK cells and other cell types resulting in killing of tumor cell by ADCC both in vivo and in vitro [reviewed in Ref. (183)]. New generations of mAbs have been developed to increase ADCC and complement-dependent cytotoxicity. Second-generation anti-CD20 mAbs, such as veltuzumab (hA20) (184, 185) and ofatumumab (HuMax-CD20) (186–193), have the advantage of being humanized or of fully human origin. Both veltuzumab and ofatumumab had promising preliminary outcomes in various studies (184, 186, 187, 189, 190, 193). The benefit of third-generation anti-CD20 mAbs, ublituximab (TG-1101), ocaratuzumab (AME-133) (194, 195), and obinutuzumab (GA-101) (196–200), is that they are both humanized and that their Fc regions have been modified for increased binding affinity to CD16a. So far, the most studied third-generation anti-CD20 mAbs is obinutuzumab. The overall response rate for obinutuzumab is 44.6%, which is higher than the overall response rate for rituximab treatment which is 33.7% (200). In the same study, the progression-free survival did not promote obinutuzumab over rituximab. By increased affinity between CD16a and mAb better NK cell cytolysis can be induced by ADCC. Ublituximab, ocaratuzumab, or obinutuzumab-treated NK cells from CLL patients or healthy donors have more efficient ADCC compared to same cells treated with first- or second-generation anti-CD20 mAb in vitro (201–203).
Monoclonal antibody therapies in combination with already existing treatments can potentially enhance NK cell activity in anti-tumor therapy. The completely human IgG4 anti-KIR antibody, IPH-2102, has been tested in several clinical trials for hematological diseases both as single treatment and as combination (204, 205). Some clinical trials for combination treatment of advanced solid tumors with anti-KIR antibodies are done as well, for example, in combination with anti-CTLA antibody or anti-PD1 antibody (NCT01750580 and NCT01714739, respectively). Thus, use of mAbs enhancing ADCC and stimulation of NK cells as well as blocking NK cell inhibition could potentially improve outcome of clinical anti-cancer NK cell products (Figure 3).
Bi- and Trispecific Antibodies
Likewise designing CARs through tumor-specific mAbs can be used to engineer bi- and trispecific antibodies crosslinking CD16 with tumor-specific mAbs in order to enhance NK cell tumor reactivity (Figure 3). Briefly, the design of bi- and trispecific antibodies, fusing the Fab region of the antibody targeting the tumor cell antigen, such as CD19, CD20, and CD33, in combination with another Fab region recognizing CD16 on NK cell leads to stimulation of the NK cells followed by tumor cell killing. This technology makes it possible to select the amount of NK cells that should be activated as well as it is possible to add more Fab regions targeting other tumor-associated antigens. These Fab regions can be exchanged to other tumor-associated antigen-recognizing antibody parts, as long as the part crosslinking CD16 on the NK cell is present (206, 207).
Chimeric Antigen Receptors (CARs)
Design of CARs using antigen-specific variable part of these tumor antigen antibodies fused with intracellular lymphocyte stimulatory molecules (CD3ξ, CD28, 4-1BB) enables high-affinity specific recognition of tumor antigens and tumors. CAR modifications of T cells have been studied extensively and have led to several phase I and phase II clinical trials (208–211). NK cells are less explored and so far only two clinical trials using CAR NK cells have been approved. The first study (NCT00995137) at St. Jude Children’s Research Hospital is completed and was a phase I clinical trial with 14 relapsed or refractory B-lineage ALL patients below 18 years. Haploidentical NK cells were expanded by co-culture with irradiated K562 cell line expressing IL-15 and 4-1BB ligand on the surface to be transduced with a signaling receptor binding CD19 (anti-CD19 CAR). The second study (NCT01974479) is a phase II pilot study, which is still recruiting refractory B-lineage ALL patients in all ages. NK cells are expanded by co-culture with K562 cells as the previous trial, together with IL-2 before transduction with the same construct. The patients will also receive IL-2 after NK cell administration to support NK cell viability and expansion. Although CAR T cell studies have been extremely promising, CARs designed for T cell therapies are still suboptimal for NK cells. Thus, it is essential to further optimize the construct design, especially the intracellular stimulatory adapter molecules, in order to trigger most efficient NK cell responses.
Immunomodulatory Drugs (IMiDs)
Immunomodulatory drugs (IMiDs) such as thalidomide, lenalidomide, and pomalidomide, can stimulate both NK cells and T cells, potentially resulting in better targeting cancer cells (212). Lenalidomide upregulates TRAIL molecules on NK cells and enhances anti-tumor activity (14, 15). So far, several different malignancies, both solid and hematological, have been treated with IMiDs. A large part of the nearly 100 clinical trials with IMiDs that has been reported with results to clinicaltrials.gov is treatment of myeloma, lymphoma, and leukemia. IMiDs can be used as combination treatment, such as lenalidomide in combination with IPH-2102, anti-inhibitory KIR antibody therapy (205). Lenalidomide expands and activates the NK cells, while anti-inhibitory KIR antibody (IPH2101) promotes NK cell recognition and lysis of tumor cells. This combination could give a better therapeutic outcome.
Combination Treatments
It is possible that NK cell products cannot fully eliminate tumor cells due to several immunosuppressive effects of tumor microenvironment as well as reduced in vivo expansion and cytotoxicity. These obstacles could be overcome by combination treatments using NK cell therapy products together with other drugs either directly targeting tumor cells or modulating cytotoxic activity of NK cells. As mentioned earlier, use of mAbs and IMiDs together with appropriate NK cell products could enhance tumor targeting and elimination. Another way to enhance NK cell-mediated killing is to combine drug therapy with NK cell stimulating cytokines such as IL-2, IL-12, IL-15, and IL-21 (213).
Furthermore, chemotherapy in combination with NK cell infusions is an alternative way to overcome tumor-induced dysfunctions. NK cells from haploidentical donor require combination treatments with the intense chemotherapy drugs high-dose fludarabin and cyclophosphamide (Hi-Cy/Flu) plus daily infusion of IL-2 to be able to expand in vivo (38). Total body irradiation could help to create immunological space for expanding NK cells in addition to chemotherapy after short-term ex vivo activation of NK cells (214).
Conclusion
In this review, we have summarized current NK cell-based therapy strategies as well as some of the challenges that need to be addressed. Even though NK cell-based therapies represent one of the most promising strategies to combat cancer, to our knowledge, no clinical trial has clearly demonstrated a significant benefit in patients with malignancies. This is in part due to the lack of prospective large-scale clinical trials and partly due to a lack of consensus in which NK cell product preparation would show the best effect. Further comparative clinical studies are definitely warranted; however, the design of such clinical trials is challenging due to the advanced therapy regulations in major countries such as European Union member states and the United States of America. Although cell therapy clinical trials are reaching a log-linear expansion, the number of NK cell-based therapies is not aligned with this increase. Nevertheless, there is a lot of promise in early clinical and pre-clinical data that cannot be omitted. In the near future, different NK cell-based products will reach multicenter clinical trial stage and we will start to see efficacy data.
Separately, NK cell-based therapies are in theory complementary to many different upfront, maintenance, and late-line therapies. Further studies clarifying the complementary efficacies and synergies have to be initiated to conclusively state if there is any place for these intriguing cells in search for an effective treatment of cancer.
Author Contributions
All the authors performed the review of the literature, wrote, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by generous grants from the Swedish Cancer Fund (Cancerfonden) (Reference number 141022), Swedish Cancer Society in Stockholm (Cancerföreningen/Radiumhemmets forskningsfonder), VINNOVA (Project number 2010-00501), and State of Florida Department of Health. The authors acknowledge the linguistic comments from Kim Kusser at Nova Southeastern University and Elizabeth Henry Alici.
References
1. Hellström I, Hellström KE, Pierce GE, Yang JP. Cellular and humoral immunity to different types of human neoplasms. Nature (1968) 220:1352–4. doi:10.1038/2201352a0
2. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer (1975) 16:230–9.
3. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16:216–29.
4. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol (1975) 5:117–21. doi:10.1002/eji.1830050208
5. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5:112–7. doi:10.1002/eji.1830050208
6. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319:675–8. doi:10.1038/319675a0
7. Pyo CW, Guethlein LA, Vu Q, Wang R, Abi-Rached L, Norman PJ, et al. Different patterns of evolution in the centromeric and telomeric regions of group A and B haplotypes of the human killer cell Ig-like receptor locus. PLoS One (2010) 5:e15115. doi:10.1371/journal.pone.0015115
8. Fauriat C, Ivarsson MA, Ljunggren HG, Malmberg KJ, Michaelsson J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood (2010) 115:1166–74. doi:10.1182/blood-2009-09-245746
9. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol (2013) 34:182–91. doi:10.1016/j.it.2013.01.003
10. Titus JA, Perez P, Kaubisch A, Garrido MA, Segal DM. Human K/natural killer cells targeted with hetero-cross-linked antibodies specifically lyse tumor cells in vitro and prevent tumor growth in vivo. J Immunol (1987) 139:3153–8.
11. Garrido MA, Perez P, Titus JA, Valdayo MJ, Winkler DF, Barbieri SA, et al. Targeted cytotoxic cells in human peripheral blood lymphocytes. J Immunol (1990) 144:2891–8.
12. Wallin RP, Screpanti V, Michaelsson J, Grandien A, Ljunggren HG. Regulation of perforin-independent NK cell-mediated cytotoxicity. Eur J Immunol (2003) 33:2727–35. doi:10.1002/eji.200324070
13. De Wilt LH, Kroon J, Jansen G, De Jong S, Peters GJ, Kruyt FA. Bortezomib and TRAIL: a perfect match for apoptotic elimination of tumour cells? Crit Rev Oncol Hematol (2013) 85:363–72. doi:10.1016/j.critrevonc.2012.08.001
14. Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res (2008) 14:4650–7. doi:10.1158/1078-0432.CCR-07-4405
15. Jungkunz-Stier I, Zekl M, Stuhmer T, Einsele H, Seggewiss-Bernhardt R. Modulation of natural killer cell effector functions through lenalidomide/dasatinib and their combined effects against multiple myeloma cells. Leuk Lymphoma (2014) 55:168–76. doi:10.3109/10428194.2013.794270
16. Freud AG, Yu J, Caligiuri MA. Human natural killer cell development in secondary lymphoid tissues. Semin Immunol (2014) 26:132–7. doi:10.1016/j.smim.2014.02.008
17. Suzuki H, Duncan GS, Takimoto H, Mak TW. Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor beta chain. J Exp Med (1997) 185:499–505. doi:10.1084/jem.185.3.499
18. Carotta S, Pang SH, Nutt SL, Belz GT. Identification of the earliest NK-cell precursor in the mouse BM. Blood (2011) 117:5449–52. doi:10.1182/blood-2010-11-318956
19. Freud AG, Yokohama A, Becknell B, Lee MT, Mao HC, Ferketich AK, et al. Evidence for discrete stages of human natural killer cell differentiation in vivo. J Exp Med (2006) 203:1033–43. doi:10.1084/jem.20052507
20. Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst (2014) 106:dju200. doi:10.1093/jnci/dju200
21. Medvedev AE, Johnsen AC, Haux J, Steinkjer B, Egeberg K, Lynch DH, et al. Regulation of Fas and Fas-ligand expression in NK cells by cytokines and the involvement of Fas-ligand in NK/LAK cell-mediated cytotoxicity. Cytokine (1997) 9:394–404. doi:10.1006/cyto.1996.0181
22. Poggi A, Massaro AM, Negrini S, Contini P, Zocchi MR. Tumor-induced apoptosis of human IL-2-activated NK cells: role of natural cytotoxicity receptors. J Immunol (2005) 174:2653–60. doi:10.4049/jimmunol.174.5.2653
23. Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood (2002) 99:3661–7. doi:10.1182/blood.V99.10.3661
24. Rouas-Freiss N, Moreau P, Ferrone S, Carosella ED. HLA-G proteins in cancer: do they provide tumor cells with an escape mechanism? Cancer Res (2005) 65:10139–44. doi:10.1158/0008-5472.CAN-05-0097
25. Urosevic M, Dummer R. Human leukocyte antigen-G and cancer immunoediting. Cancer Res (2008) 68:627–30. doi:10.1158/0008-5472.CAN-07-2704
26. Jinushi M, Takehara T, Tatsumi T, Hiramatsu N, Sakamori R, Yamaguchi S, et al. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol (2005) 43:1013–20. doi:10.1016/j.jhep.2005.05.026
27. Konjevic G, Mirjacic Martinovic K, Vuletic A, Jovic V, Jurisic V, Babovic N, et al. Low expression of CD161 and NKG2D activating NK receptor is associated with impaired NK cell cytotoxicity in metastatic melanoma patients. Clin Exp Metastasis (2007) 24:1–11. doi:10.1007/s10585-006-9043-9
28. Veuillen C, Aurran-Schleinitz T, Castellano R, Rey J, Mallet F, Orlanducci F, et al. Primary B-CLL resistance to NK cell cytotoxicity can be overcome in vitro and in vivo by priming NK cells and monoclonal antibody therapy. J Clin Immunol (2012) 32:632–46. doi:10.1007/s10875-011-9624-5
29. Fauriat C, Mallet F, Olive D, Costello RT. Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia (2006) 20:732–3. doi:10.1038/sj.leu.2404096
30. El-Sherbiny YM, Meade JL, Holmes TD, Mcgonagle D, Mackie SL, Morgan AW, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res (2007) 67:8444–9. doi:10.1158/0008-5472.CAN-06-4230
31. Gati A, Da Rocha S, Guerra N, Escudier B, Moretta A, Chouaib S, et al. Analysis of the natural killer mediated immune response in metastatic renal cell carcinoma patients. Int J Cancer (2004) 109:393–401. doi:10.1002/ijc.11730
32. Pierson BA, Miller JS. CD56+bright and CD56+dim natural killer cells in patients with chronic myelogenous leukemia progressively decrease in number, respond less to stimuli that recruit clonogenic natural killer cells, and exhibit decreased proliferation on a per cell basis. Blood (1996) 88:2279–87.
33. Schleypen JS, Von Geldern M, Weiss EH, Kotzias N, Rohrmann K, Schendel DJ, et al. Renal cell carcinoma-infiltrating natural killer cells express differential repertoires of activating and inhibitory receptors and are inhibited by specific HLA class I allotypes. Int J Cancer (2003) 106:905–12. doi:10.1002/ijc.11321
34. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest (2013) 123:4410–22. doi:10.1172/JCI69369
35. Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O’Reilly RJ, Dupont B, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol (2003) 171:6891–9. doi:10.4049/jimmunol.171.12.6891
36. Jinushi M, Hodi FS, Dranoff G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc Natl Acad Sci U S A (2006) 103:9190–5. doi:10.1073/pnas.0603503103
37. Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science (2015) 348:136–9. doi:10.1126/science.1258867
38. Miller JS, Soignier Y, Panoskaltsis-Mortari A, Mcnearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi:10.1182/blood-2004-07-2974
39. Escudier B, Farace F, Angevin E, Charpentier F, Nitenberg G, Triebel F, et al. Immunotherapy with interleukin-2 (IL2) and lymphokine-activated natural killer cells: improvement of clinical responses in metastatic renal cell carcinoma patients previously treated with IL2. Eur J Cancer (1994) 30A:1078–83. doi:10.1016/0959-8049(94)90460-X
40. Lister J, Rybka WB, Donnenberg AD, Demagalhaes-Silverman M, Pincus SM, Bloom EJ, et al. Autologous peripheral blood stem cell transplantation and adoptive immunotherapy with activated natural killer cells in the immediate post-transplant period. Clin Cancer Res (1995) 1:607–14.
41. Pierson BA, Europa AF, Hu WS, Miller JS. Production of human natural killer cells for adoptive immunotherapy using a computer-controlled stirred-tank bioreactor. J Hematother (1996) 5:475–83. doi:10.1089/scd.1.1996.5.475
42. Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood (2008) 111:3155–62. doi:10.1182/blood-2007-09-110312
43. Barkholt L, Alici E, Conrad R, Sutlu T, Gilljam M, Stellan B, et al. Safety analysis of ex vivo-expanded NK and NK-like T cells administered to cancer patients: a phase I clinical study. Immunotherapy (2009) 1:753–64. doi:10.2217/imt.09.47
44. Iliopoulou EG, Kountourakis P, Karamouzis MV, Doufexis D, Ardavanis A, Baxevanis CN, et al. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol Immunother (2010) 59:1781–9. doi:10.1007/s00262-010-0904-3
45. Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, et al. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy (2010) 12:1044–55. doi:10.3109/14653249.2010.504770
46. Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, De Witte T, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One (2011) 6:e20740. doi:10.1371/journal.pone.0020740
47. Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med (2015) 13:277. doi:10.1186/s12967-015-0632-8
48. Carlens S, Gilljam M, Chambers BJ, Aschan J, Guven H, Ljunggren HG, et al. A new method for in vitro expansion of cytotoxic human CD3-CD56+ natural killer cells. Hum Immunol (2001) 62:1092–8. doi:10.1016/S0198-8859(01)00313-5
49. Klingemann HG, Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy (2004) 6:15–22. doi:10.1080/14653240310004548
50. Cany J, Van Der Waart AB, Spanholtz J, Tordoir M, Jansen JH, Van Der Voort R, et al. Combined IL-15 and IL-12 drives the generation of CD34-derived natural killer cells with superior maturation and alloreactivity potential following adoptive transfer. Oncoimmunology (2015) 4:e1017701. doi:10.1080/2162402X.2015.1017701
51. Tam YK, Martinson JA, Doligosa K, Klingemann HG. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy (2003) 5:259–72. doi:10.1080/14653240310001523
52. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy (2008) 10:625–32. doi:10.1080/14653240802301872
53. Guven H, Gilljam M, Chambers BJ, Ljunggren HG, Christensson B, Kimby E, et al. Expansion of natural killer (NK) and natural killer-like T (NKT)-cell populations derived from patients with B-chronic lymphocytic leukemia (B-CLL): a potential source for cellular immunotherapy. Leukemia (2003) 17:1973–80. doi:10.1038/sj.leu.2403083
54. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res (2009) 69:4010–7. doi:10.1158/0008-5472.CAN-08-3712
55. Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res (2004) 24:1861–71.
56. Miller JS, Klingsporn S, Lund J, Perry EH, Verfaillie C, Mcglave P. Large scale ex vivo expansion and activation of human natural killer cells for autologous therapy. Bone Marrow Transplant (1994) 14:555–62.
57. Torelli GF, Guarini A, Palmieri G, Breccia M, Vitale A, Santoni A, et al. Expansion of cytotoxic effectors with lytic activity against autologous blasts from acute myeloid leukaemia patients in complete haematological remission. Br J Haematol (2002) 116:299–307. doi:10.1046/j.1365-2141.2002.03277.x
58. Torelli GF, Guarini A, Maggio R, Alfieri C, Vitale A, Foa R. Expansion of natural killer cells with lytic activity against autologous blasts from adult and pediatric acute lymphoid leukemia patients in complete hematologic remission. Haematologica (2005) 90:785–92.
59. Hercend T, Farace F, Baume D, Charpentier F, Droz JP, Triebel F, et al. Immunotherapy with lymphokine-activated natural killer cells and recombinant interleukin-2: a feasibility trial in metastatic renal cell carcinoma. J Biol Response Mod (1990) 9:546–55.
60. Luhm J, Brand JM, Koritke P, Hoppner M, Kirchner H, Frohn C. Large-scale generation of natural killer lymphocytes for clinical application. J Hematother Stem Cell Res (2002) 11:651–7. doi:10.1089/15258160260194794
61. Berg M, Lundqvist A, Mccoy P Jr., Samsel L, Fan Y, Tawab A, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy (2009) 11:341–55. doi:10.1080/14653240902807034
62. Sutlu T, Alici E. Ex vivo expansion of natural killer cells: a question of function. Cytotherapy (2011) 13:767–8. doi:10.3109/14653249.2011.563295
63. Skeate R, Singh C, Cooley S, Geller M, Northouse J, Welbig J, et al. Hemolytic anemia due to passenger lymphocyte syndrome in solid malignancy patients treated with allogeneic natural killer cell products. Transfusion (2013) 53:419–23. doi:10.1111/j.1537-2995.2012.03942.x
64. Nguyen S, Kuentz M, Vernant JP, Dhedin N, Bories D, Debre P, et al. Involvement of mature donor T cells in the NK cell reconstitution after haploidentical hematopoietic stem-cell transplantation. Leukemia (2008) 22:344–52. doi:10.1038/sj.leu.2405041
65. Miller JS, Oelkers S, Verfaillie C, Mcglave P. Role of monocytes in the expansion of human activated natural killer cells. Blood (1992) 80:2221–9.
66. Siegler U, Meyer-Monard S, Jorger S, Stern M, Tichelli A, Gratwohl A, et al. Good manufacturing practice-compliant cell sorting and large-scale expansion of single KIR-positive alloreactive human natural killer cells for multiple infusions to leukemia patients. Cytotherapy (2010) 12:750–63. doi:10.3109/14653241003786155
67. Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood (2015) 125:784–92. doi:10.1182/blood-2014-07-592881
68. Somanchi SS, Senyukov VV, Denman CJ, Lee DA. Expansion, purification, and functional assessment of human peripheral blood NK cells. J Vis Exp (2011): doi:10.3791/2540
69. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One (2012) 7:e30264. doi:10.1371/journal.pone.0030264
70. Kalberer CP, Siegler U, Wodnar-Filipowicz A. Human NK cell development in NOD/SCID mice receiving grafts of cord blood CD34+ cells. Blood (2003) 102:127–35. doi:10.1182/blood-2002-07-2024
71. Tanaka H, Kai S, Yamaguchi M, Misawa M, Fujimori Y, Yamamoto M, et al. Analysis of natural killer (NK) cell activity and adhesion molecules on NK cells from umbilical cord blood. Eur J Haematol (2003) 71:29–38. doi:10.1034/j.1600-0609.2003.00081.x
72. Xing D, Ramsay AG, Gribben JG, Decker WK, Burks JK, Munsell M, et al. Cord blood natural killer cells exhibit impaired lytic immunological synapse formation that is reversed with IL-2 ex vivo expansion. J Immunother (2010) 33:684–96. doi:10.1097/CJI.0b013e3181e475e9
73. Lehmann D, Spanholtz J, Sturtzel C, Tordoir M, Schlechta B, Groenewegen D, et al. IL-12 directs further maturation of ex vivo differentiated NK cells with improved therapeutic potential. PLoS One (2014) 9:e87131. doi:10.1371/journal.pone.0087131
74. Cany J, Van Der Waart AB, Tordoir M, Franssen GM, Hangalapura BN, De Vries J, et al. Natural killer cells generated from cord blood hematopoietic progenitor cells efficiently target bone marrow-residing human leukemia cells in NOD/SCID/IL2Rg(null) mice. PLoS One (2013) 8:e64384. doi:10.1371/journal.pone.0064384
75. Luevano M, Domogala A, Blundell M, Jackson N, Pedroza-Pacheco I, Derniame S, et al. Frozen cord blood hematopoietic stem cells differentiate into higher numbers of functional natural killer cells in vitro than mobilized hematopoietic stem cells or freshly isolated cord blood hematopoietic stem cells. PLoS One (2014) 9:e87086. doi:10.1371/journal.pone.0087086
76. Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol (2005) 175:5095–103. doi:10.4049/jimmunol.175.8.5095
77. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood (2009) 113:6094–101. doi:10.1182/blood-2008-06-165225
78. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med (2013) 2:274–83. doi:10.5966/sctm.2012-0084
79. Kruse V, Hamann C, Monecke S, Cyganek L, Elsner L, Hubscher D, et al. Human induced pluripotent stem cells are targets for allogeneic and autologous natural killer (NK) cells and killing is partly mediated by the activating NK receptor DNAM-1. PLoS One (2015) 10:e0125544. doi:10.1371/journal.pone.0125544
80. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia (1994) 8:652–8.
81. Klingemann HG, Miyagawa B. Purging of malignant cells from blood after short ex vivo incubation with NK-92 cells. Blood (1996) 87:4913–4.
82. Yan Y, Steinherz P, Klingemann HG, Dennig D, Childs BH, Mcguirk J, et al. Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res (1998) 4:2859–68.
83. Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther (1999) 10:1359–73. doi:10.1089/10430349950018030
84. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15:1563–70. doi:10.1016/j.jcyt.2013.06.017
85. Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematother Stem Cell Res (2001) 10:369–83. doi:10.1089/152581601750288975
86. Suck G, Branch DR, Smyth MJ, Miller RG, Vergidis J, Fahim S, et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol (2005) 33:1160–71. doi:10.1016/j.exphem.2005.06.024
87. Nagashima S, Mailliard R, Kashii Y, Reichert TE, Herberman RB, Robbins P, et al. Stable transduction of the interleukin-2 gene into human natural killer cell lines and their phenotypic and functional characterization in vitro and in vivo. Blood (1998) 91:3850–61.
88. Konstantinidis KV, Alici E, Aints A, Christensson B, Ljunggren HG, Dilber MS. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp Hematol (2005) 33:159–64. doi:10.1016/j.exphem.2004.11.003
89. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am (2000) 1(6 Suppl):S11–4.
90. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med (1985) 313:1485–92. doi:10.1056/NEJM198512053132327
91. Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. N Engl J Med (1987) 316:889–97. doi:10.1056/NEJM198704093161501
92. Phillips JH, Gemlo BT, Myers WW, Rayner AA, Lanier LL. In vivo and in vitro activation of natural killer cells in advanced cancer patients undergoing combined recombinant interleukin-2 and LAK cell therapy. J Clin Oncol (1987) 5:1933–41.
93. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, Van Gelder M, Bos GM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS One (2013) 8:e64835. doi:10.1371/journal.pone.0064835
94. Basse PH, Goldfarb RH, Herberman RB, Hokland ME. Accumulation of adoptively transferred A-NK cells in murine metastases: kinetics and role of interleukin-2. In vivo (1994) 8:17–24.
95. Donohue JH, Rosenberg SA. The fate of interleukin-2 after in vivo administration. J Immunol (1983) 130:2203–8.
96. Sutlu T, Nystrom S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther (2012) 23:1090–100. doi:10.1089/hum.2012.080
97. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, Mckenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood (2014) 123:3855–63. doi:10.1182/blood-2013-10-532531
98. Cooper MA, Bush JE, Fehniger TA, Vandeusen JB, Waite RE, Liu Y, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood (2002) 100:3633–8. doi:10.1182/blood-2001-12-0293
99. Ozaki K, Kikly K, Michalovich D, Young PR, Leonard WJ. Cloning of a type I cytokine receptor most related to the IL-2 receptor beta chain. Proc Natl Acad Sci U S A (2000) 97:11439–44. doi:10.1073/pnas.200360997
100. De Rham C, Ferrari-Lacraz S, Jendly S, Schneiter G, Dayer JM, Villard J. The proinflammatory cytokines IL-2, IL-15 and IL-21 modulate the repertoire of mature human natural killer cell receptors. Arthritis Res Ther (2007) 9:R125. doi:10.1186/ar2336
101. Gowda A, Ramanunni A, Cheney C, Rozewski D, Kindsvogel W, Lehman A, et al. Differential effects of IL-2 and IL-21 on expansion of the CD4+ CD25+ Foxp3+ T regulatory cells with redundant roles in natural killer cell mediated antibody dependent cellular cytotoxicity in chronic lymphocytic leukemia. MAbs (2010) 2:35–41. doi:10.4161/mabs.2.1.10561
102. Mckenna DH Jr., Sumstad D, Bostrom N, Kadidlo DM, Fautsch S, Mcnearney S, et al. Good manufacturing practices production of natural killer cells for immunotherapy: a six-year single-institution experience. Transfusion (2007) 47:520–8. doi:10.1111/j.1537-2995.2006.01145.x
103. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res (2011) 17:6287–97. doi:10.1158/1078-0432.CCR-11-1347
104. Burns LJ, Weisdorf DJ, Defor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant (2003) 32:177–86. doi:10.1038/sj.bmt.1704086
105. Krause SW, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, et al. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: a clinical phase I trial. Clin Cancer Res (2004) 10:3699–707. doi:10.1158/1078-0432.CCR-03-0683
106. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res (2006) 12:6079–86. doi:10.1158/1078-0432.CCR-06-0114
107. Milani V, Stangl S, Issels R, Gehrmann M, Wagner B, Hube K, et al. Anti-tumor activity of patient-derived NK cells after cell-based immunotherapy – a case report. J Transl Med (2009) 7:50. doi:10.1186/1479-5876-7-50
108. Demagalhaes-Silverman M, Donnenberg A, Lembersky B, Elder E, Lister J, Rybka W, et al. Posttransplant adoptive immunotherapy with activated natural killer cells in patients with metastatic breast cancer. J Immunother (2000) 23:154–60. doi:10.1097/00002371-200001000-00018
109. Koehl U, Sorensen J, Esser R, Zimmermann S, Gruttner HP, Tonn T, et al. IL-2 activated NK cell immunotherapy of three children after haploidentical stem cell transplantation. Blood Cells Mol Dis (2004) 33:261–6. doi:10.1016/j.bcmd.2004.08.013
110. Passweg JR, Tichelli A, Meyer-Monard S, Heim D, Stern M, Kuhne T, et al. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia (2004) 18:1835–8. doi:10.1038/sj.leu.2403524
111. Rizzieri DA, Storms R, Chen DF, Long G, Yang Y, Nikcevich DA, et al. Natural killer cell-enriched donor lymphocyte infusions from A 3-6/6 HLA matched family member following nonmyeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2010) 16:1107–14. doi:10.1016/j.bbmt.2010.02.018
112. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol (2010) 28:955–9. doi:10.1200/JCO.2009.24.4590
113. Curti A, Ruggeri L, D’addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118:3273–9. doi:10.1182/blood-2011-01-329508
114. Yoon SR, Lee YS, Yang SH, Ahn KH, Lee JH, Lee JH, et al. Generation of donor natural killer cells from CD34(+) progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant (2010) 45:1038–46. doi:10.1038/bmt.2009.304
115. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13:98–107. doi:10.3109/14653249.2010.515582
116. Nguyen S, Beziat V, Norol F, Uzunov M, Trebeden-Negre H, Azar N, et al. Infusion of allogeneic natural killer cells in a patient with acute myeloid leukemia in relapse after haploidentical hematopoietic stem cell transplantation. Transfusion (2011) 51:1769–78. doi:10.1111/j.1537-2995.2010.03058.x
117. Brehm C, Huenecke S, Quaiser A, Esser R, Bremm M, Kloess S, et al. IL-2 stimulated but not unstimulated NK cells induce selective disappearance of peripheral blood cells: concomitant results to a phase I/II study. PLoS One (2011) 6:e27351. doi:10.1371/journal.pone.0027351
118. Huenecke S, Zimmermann SY, Kloess S, Esser R, Brinkmann A, Tramsen L, et al. IL-2-driven regulation of NK cell receptors with regard to the distribution of CD16+ and CD16- subpopulations and in vivo influence after haploidentical NK cell infusion. J Immunother (2010) 33:200–10. doi:10.1097/CJI.0b013e3181bb46f7
119. Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother (2015) 38:24–36. doi:10.1097/CJI.0000000000000059
120. Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood (1999) 94:333–9.
121. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295:2097–100. doi:10.1126/science.1068440
122. Kumano M, Hara I, Furukawa J, Oniki S, Nagai H, Miyake H, et al. Interleukin-21 activates cytotoxic T lymphocytes and natural killer cells to generate antitumor response in mouse renal cell carcinoma. J Urol (2007) 178:1504–9. doi:10.1016/j.juro.2007.05.115
123. Rocha FG, Calvo FB, Chaves KC, Peron JP, Marques RF, De Borba TR, et al. Endostatin- and interleukin-2-expressing retroviral bicistronic vector for gene therapy of metastatic renal cell carcinoma. J Gene Med (2011) 13:148–57. doi:10.1002/jgm.1547
124. Watanabe M, Kono K, Kawaguchi Y, Mizukami Y, Mimura K, Maruyama T, et al. NK cell dysfunction with down-regulated CD16 and up-regulated CD56 molecules in patients with esophageal squamous cell carcinoma. Dis Esophagus (2010) 23:675–81. doi:10.1111/j.1442-2050.2010.01073.x
125. Izawa S, Kono K, Mimura K, Kawaguchi Y, Watanabe M, Maruyama T, et al. H(2)O(2) production within tumor microenvironment inversely correlated with infiltration of CD56(dim) NK cells in gastric and esophageal cancer: possible mechanisms of NK cell dysfunction. Cancer Immunol Immunother (2011) 60:1801–10. doi:10.1007/s00262-011-1082-7
126. Koo KC, Shim DH, Yang CM, Lee SB, Kim SM, Shin TY, et al. Reduction of the CD16(-)CD56bright NK cell subset precedes NK cell dysfunction in prostate cancer. PLoS One (2013) 8:e78049. doi:10.1371/journal.pone.0078049
127. Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol (2015) 12(3):292–302. doi:10.1038/cmi.2014.91
128. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris JH, Noman MZ, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol (2015) 6:482. doi:10.3389/fimmu.2015.00482
129. Hartigan-O’Connor DJ, Poon C, Sinclair E, Mccune JM. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J Immunol Methods (2007) 319:41–52. doi:10.1016/j.jim.2006.10.008
130. Berendt MJ, North RJ. T-cell-mediated suppression of anti-tumor immunity. An explanation for progressive growth of an immunogenic tumor. J Exp Med (1980) 151:69–80.
131. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res (2001) 61:4766–72.
132. Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res (2003) 9:4404–8.
133. Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res (2005) 65:2457–64. doi:10.1158/0008-5472.CAN-04-3232
134. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology (2007) 132:2328–39. doi:10.1053/j.gastro.2007.03.102
135. Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology (2009) 137:1270–9. doi:10.1053/j.gastro.2009.06.053
136. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med (2005) 202:1075–85. doi:10.1084/jem.20051511
137. Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol (2008) 129:428–37. doi:10.1016/j.clim.2008.08.012
138. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8:1353–62. doi:10.1038/ni1536
139. Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med (2013) 210:1167–78. doi:10.1084/jem.20122462
140. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67:425. doi:10.1158/0008-5472.CAN-06-3037
141. Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother (2013) 62:1421–30. doi:10.1007/s00262-013-1447-1
142. Gorgun GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood (2013) 121:2975–87. doi:10.1182/blood-2012-08-448548
143. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181:5791–802. doi:10.4049/jimmunol.181.8.5791
144. Loercher AE, Nash MA, Kavanagh JJ, Platsoucas CD, Freedman RS. Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J Immunol (1999) 163:6251–60.
145. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203:2691–702. doi:10.1084/jem.20061104
146. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (2009) 50:799–807. doi:10.1002/hep.23054
147. Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res (2010) 70:4335–45. doi:10.1158/0008-5472.CAN-09-3767
148. Lin Y, Gustafson MP, Bulur PA, Gastineau DA, Witzig TE, Dietz AB. Immunosuppressive CD14+HLA-DR(low)/- monocytes in B-cell non-Hodgkin lymphoma. Blood (2011) 117:872–81. doi:10.1182/blood-2010-05-283820
149. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol (2003) 24:302–6. doi:10.1016/S1471-4906(03)00132-7
150. Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol (2003) 74:186–96. doi:10.1189/jlb.0103010
151. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol (2007) 25:2546–53. doi:10.1200/JCO.2006.08.5829
152. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol (2010) 185:2273–84. doi:10.4049/jimmunol.1000901
153. Kohanbash G, Mckaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res (2013) 73:6413–23. doi:10.1158/0008-5472.CAN-12-4124
154. Mao Y, Poschke I, Wennerberg E, Pico De Coana Y, Egyhazi Brage S, Schultz I, et al. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res (2013) 73:3877–87. doi:10.1158/0008-5472.CAN-12-4115
155. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res (2014) 20:4096–106. doi:10.1158/1078-0432.CCR-14-0635
156. Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood (2014) 123:3016–26. doi:10.1182/blood-2013-10-533398
157. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol (2009) 9:259–70. doi:10.1038/nri2528
158. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi:10.1016/j.cell.2010.03.014
159. Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int (2013) 2013:187204. doi:10.1155/2013/187204
160. Wyler L, Napoli CU, Ingold B, Sulser T, Heikenwalder M, Schraml P, et al. Brain metastasis in renal cancer patients: metastatic pattern, tumour-associated macrophages and chemokine/chemoreceptor expression. Br J Cancer (2014) 110:686–94. doi:10.1038/bjc.2013.755
161. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362:875–85. doi:10.1056/NEJMoa0905680
162. Campbell MJ, Tonlaar NY, Garwood ER, Huo D, Moore DH, Khramtsov AI, et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res Treat (2011) 128:703–11. doi:10.1007/s10549-010-1154-y
163. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS One (2011) 6:e19495. doi:10.1371/journal.pone.0019495
164. Chen C, Qu QX, Shen Y, Mu CY, Zhu YB, Zhang XG, et al. Induced expression of B7-H4 on the surface of lung cancer cell by the tumor-associated macrophages: a potential mechanism of immune escape. Cancer Lett (2012) 317:99–105. doi:10.1016/j.canlet.2011.11.017
165. Helm O, Held-Feindt J, Grage-Griebenow E, Reiling N, Ungefroren H, Vogel I, et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int J Cancer (2014) 135(4):843–61. doi:10.1002/ijc.28736
166. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res (2012) 72:1407–15. doi:10.1158/0008-5472.CAN-11-2544
167. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood (2008) 111:1327–33. doi:10.1182/blood-2007-02-074997
168. Sivori S, Pende D, Bottino C, Marcenaro E, Pessino A, Biassoni R, et al. NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol (1999) 29:1656–66. doi:10.1002/(SICI)1521-4141(199905)29:05<1656::AID-IMMU1656>3.0.CO;2-1
169. Bournazou E, Bromberg J. Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT (2013) 2:e23828. doi:10.4161/jkst.23828
170. Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res (2013) 73:4965–77. doi:10.1158/0008-5472.CAN-13-0661
171. Samples J, Willis M, Klauber-Demore N. Targeting angiogenesis and the tumor microenvironment. Surg Oncol Clin N Am (2013) 22:629–39. doi:10.1016/j.soc.2013.06.002
172. Giordano G, Febbraro A, Venditti M, Campidoglio S, Olivieri N, Raieta K, et al. Targeting angiogenesis and tumor microenvironment in metastatic colorectal cancer: role of aflibercept. Gastroenterol Res Pract (2014) 2014:526178. doi:10.1155/2014/526178
173. Nelson D, Fisher S, Robinson B. The “Trojan Horse” approach to tumor immunotherapy: targeting the tumor microenvironment. J Immunol Res (2014) 2014:789069. doi:10.1155/2014/789069
174. Omidi Y, Barar J. Targeting tumor microenvironment: crossing tumor interstitial fluid by multifunctional nanomedicines. Bioimpacts (2014) 4:55–67. doi:10.5681/bi.2014.021
175. Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol (2008) 8:259–68. doi:10.1038/nri2276
176. Alici E, Sutlu T, Sirac Dilber M. Retroviral gene transfer into primary human natural killer cells. Methods Mol Biol (2009) 506:127–37. doi:10.1007/978-1-59745-409-4_10
177. Brandstadter JD, Yang Y. Natural killer cell responses to viral infection. J Innate Immun (2011) 3:274–9. doi:10.1159/000324176
178. Zhang J, Sun R, Wei H, Zhang J, Tian Z. Characterization of interleukin-15 gene-modified human natural killer cells: implications for adoptive cellular immunotherapy. Haematologica (2004) 89:338–47.
179. Jiang W, Zhang J, Tian Z. Functional characterization of interleukin-15 gene transduction into the human natural killer cell line NKL. Cytotherapy (2008) 10:265–74. doi:10.1080/14653240801965156
180. Clemenceau B, Vivien R, Pellat C, Foss M, Thibault G, Vie H. The human natural killer cytotoxic cell line NK-92, once armed with a murine CD16 receptor, represents a convenient cellular tool for the screening of mouse mAbs according to their ADCC potential. MAbs (2013) 5:587–94. doi:10.4161/mabs.25077
181. Mentlik James A, Cohen AD, Campbell KS. Combination immune therapies to enhance anti-tumor responses by NK cells. Front Immunol (2013) 4:481. doi:10.3389/fimmu.2013.00481
182. Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med (2015) 373(13):1207–19. doi:10.1056/NEJMoa1506348
183. Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science (2013) 341:1192–8. doi:10.1126/science.1241145
184. Morschhauser F, Leonard JP, Fayad L, Coiffier B, Petillon MO, Coleman M, et al. Humanized anti-CD20 antibody, veltuzumab, in refractory/recurrent non-Hodgkin’s lymphoma: phase I/II results. J Clin Oncol (2009) 27:3346–53. doi:10.1200/JCO.2008.19.9117
185. Kalaycio ME, Negrea OG, Allen SL, Rai KR, Abbasi RM, Horne H, et al. Subcutaneous injections of low doses of humanized anti-CD20 veltuzumab: a phase I study in chronic lymphocytic leukemia. Leuk Lymphoma (2015) 19:1–9. doi:10.3109/10428194.2015.1085531
186. Wierda WG, Padmanabhan S, Chan GW, Gupta IV, Lisby S, Osterborg A, et al. Ofatumumab is active in patients with fludarabine-refractory CLL irrespective of prior rituximab: results from the phase 2 international study. Blood (2011) 118:5126–9. doi:10.1182/blood-2011-04-348656
187. Czuczman MS, Fayad L, Delwail V, Cartron G, Jacobsen E, Kuliczkowski K, et al. Ofatumumab monotherapy in rituximab-refractory follicular lymphoma: results from a multicenter study. Blood (2012) 119:3698–704. doi:10.1182/blood-2011-09-378323
188. Coiffier B, Radford J, Bosly A, Martinelli G, Verhoef G, Barca G, et al. A multicentre, phase II trial of ofatumumab monotherapy in relapsed/progressive diffuse large B-cell lymphoma. Br J Haematol (2013) 163:334–42. doi:10.1111/bjh.12537
189. Ogawa Y, Ogura M, Suzuki T, Ando K, Uchida T, Shirasugi Y, et al. A phase I/II study of ofatumumab (GSK1841157) in Japanese and Korean patients with relapsed or refractory B-cell chronic lymphocytic leukemia. Int J Hematol (2013) 98:164–70. doi:10.1007/s12185-013-1393-x
190. Ogura M, Hatake K, Tobinai K, Uchida T, Suzuki T, Terui Y, et al. Phase I study of ofatumumab, a human anti-CD20 antibody, in Japanese patients with relapsed or refractory chronic lymphocytic leukemia and small lymphocytic lymphoma. Jpn J Clin Oncol (2013) 43:466–75. doi:10.1093/jjco/hyt022
191. Furtado M, Dyer MJ, Johnson R, Berrow M, Rule S. Ofatumumab monotherapy in relapsed/refractory mantle cell lymphoma – a phase II trial. Br J Haematol (2014) 165:575–8. doi:10.1111/bjh.12769
192. Moreno C, Montillo M, Panayiotidis P, Dimou M, Bloor A, Dupuis J, et al. Ofatumumab in poor-prognosis chronic lymphocytic leukemia: a phase IV, non-interventional, observational study from the European research initiative on chronic lymphocytic leukemia. Haematologica (2015) 100:511–6. doi:10.3324/haematol.2014.118158
193. Osterborg A, Wierda WG, Mayer J, Hess G, Hillmen P, Schetelig J, et al. Ofatumumab retreatment and maintenance in fludarabine-refractory chronic lymphocytic leukaemia patients. Br J Haematol (2015) 170:40–9. doi:10.1111/bjh.13380
194. Forero-Torres A, De Vos S, Pohlman BL, Pashkevich M, Cronier DM, Dang NH, et al. Results of a phase 1 study of AME-133v (LY2469298), an Fc-engineered humanized monoclonal anti-CD20 antibody, in FcgammaRIIIa-genotyped patients with previously treated follicular lymphoma. Clin Cancer Res (2012) 18:1395–403. doi:10.1158/1078-0432.CCR-11-0850
195. Ganjoo KN, De Vos S, Pohlman BL, Flinn IW, Forero-Torres A, Enas NH, et al. Phase 1/2 study of ocaratuzumab, an Fc-engineered humanized anti-CD20 monoclonal antibody, in low-affinity FcgammaRIIIa patients with previously treated follicular lymphoma. Leuk Lymphoma (2015) 56:42–8. doi:10.3109/10428194.2014.911859
196. Salles G, Morschhauser F, Lamy T, Milpied N, Thieblemont C, Tilly H, et al. Phase 1 study results of the type II glycoengineered humanized anti-CD20 monoclonal antibody obinutuzumab (GA101) in B-cell lymphoma patients. Blood (2012) 119:5126–32. doi:10.1182/blood-2012-01-404368
197. Morschhauser FA, Cartron G, Thieblemont C, Solal-Celigny P, Haioun C, Bouabdallah R, et al. Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large b-cell lymphoma or mantle-cell lymphoma: results from the phase II GAUGUIN study. J Clin Oncol (2013) 31:2912–9. doi:10.1200/JCO.2012.46.9585
198. Ogura M, Tobinai K, Hatake K, Uchida T, Suzuki T, Kobayashi Y, et al. Phase I study of obinutuzumab (GA101) in Japanese patients with relapsed or refractory B-cell non-Hodgkin lymphoma. Cancer Sci (2013) 104:105–10. doi:10.1111/cas.12040
199. Cartron G, De Guibert S, Dilhuydy MS, Morschhauser F, Leblond V, Dupuis J, et al. Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: final data from the phase 1/2 GAUGUIN study. Blood (2014) 124:2196–202. doi:10.1182/blood-2014-07-586610
200. Sehn LH, Goy A, Offner FC, Martinelli G, Caballero MD, Gadeberg O, et al. Randomized phase II trial comparing obinutuzumab (GA101) with rituximab in patients with relapsed CD20+ indolent B-cell non-Hodgkin lymphoma: final analysis of the GAUSS study. J Clin Oncol (2015) 33(30):3467–74. doi:10.1200/JCO.2014.59.2139
201. Le Garff-Tavernier M, Decocq J, De Romeuf C, Parizot C, Dutertre CA, Chapiro E, et al. Analysis of CD16+CD56dim NK cells from CLL patients: evidence supporting a therapeutic strategy with optimized anti-CD20 monoclonal antibodies. Leukemia (2011) 25:101–9. doi:10.1038/leu.2010.240
202. Kern DJ, James BR, Blackwell S, Gassner C, Klein C, Weiner GJ. GA101 induces NK-cell activation and antibody-dependent cellular cytotoxicity more effectively than rituximab when complement is present. Leuk Lymphoma (2013) 54:2500–5. doi:10.3109/10428194.2013.781169
203. Cheney CM, Stephens DM, Mo X, Rafiq S, Butchar J, Flynn JM, et al. Ocaratuzumab, an Fc-engineered antibody demonstrates enhanced antibody-dependent cell-mediated cytotoxicity in chronic lymphocytic leukemia. MAbs (2014) 6:749–55. doi:10.4161/mabs.28282
204. Alici E. IPH-2101, a fully human anti-NK-cell inhibitory receptor mAb for the potential treatment of hematological cancers. Curr Opin Mol Ther (2010) 12:724–33.
205. Benson DM Jr, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res (2015) 21(18):4055–61. doi:10.1158/1078-0432.CCR-15-0304
206. Gleason MK, Verneris MR, Todhunter DA, Zhang B, Mccullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther (2012) 11:2674–84. doi:10.1158/1535-7163.MCT-12-0692
207. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res (2013) 19:3844–55. doi:10.1158/1078-0432.CCR-13-0505
208. Jensen MC, Popplewell L, Cooper LJ, Digiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant (2010) 16:1245–56. doi:10.1016/j.bbmt.2010.03.014
209. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18:843–51. doi:10.1038/mt.2010.24
210. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest (2011) 121:1822–6. doi:10.1172/JCI46110
211. Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood (2013) 122:4129–39. doi:10.1182/blood-2013-08-519413
212. Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene (2013) 32:4191–202. doi:10.1038/onc.2012.599
213. Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo) (2014) 2014:205796. doi:10.1155/2014/205796
Keywords: natural killer cells, adoptive cell therapy, immunotherapy, cancer, clinical trials, expansion, tumor microenvironment, genetic modifications
Citation: Dahlberg CIM, Sarhan D, Chrobok M, Duru AD and Alici E (2015) Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 6:605. doi: 10.3389/fimmu.2015.00605
Received: 02 September 2015; Accepted: 13 November 2015;
Published: 30 November 2015
Edited by:
Francisco Borrego, Cruces University Hospital, SpainReviewed by:
Subramaniam Malarkannan, Medical College of Wisconsin, USAVincent Vieillard, Institut National de la Santé et de la Recherche Scientifique (INSERM), France
Copyright: © 2015 Dahlberg, Sarhan, Chrobok, Duru and Alici. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evren Alici, ZXZyZW4uYWxpY2lAa2kuc2U=