 Rui Li1
Rui Li1 Ayman Rezk1
Ayman Rezk1 Luke M. Healy1Gillian Muirhead1Alexandre Prat2
Luke M. Healy1Gillian Muirhead1Alexandre Prat2 Jennifer L. Gommerman3
Jennifer L. Gommerman3 Amit Bar-Or1,4*MSSRF Canadian B cells in MS Team†
Amit Bar-Or1,4*MSSRF Canadian B cells in MS Team†
- 1Neuroimmunology Unit, Montreal Neurological Institute and Hospital, McGill University, Montreal, QC, Canada
- 2Neuroimmunology Unit, Department of Neuroscience, Centre de Recherche du CHUM (CRCHUM), Université de Montreal, Montreal, QC, Canada
- 3Department of Immunology, University of Toronto, Toronto, ON, Canada
- 4Experimental Therapeutics Program, Montreal Neurological Institute and Hospital, McGill University, Montreal, QC, Canada
Important antibody-independent pathogenic roles of B cells are emerging in autoimmune diseases, including multiple sclerosis (MS). The contrasting results of different treatments targeting B cells in patients (in spite of predictions of therapeutic benefits from animal models) call for a better understanding of the multiple roles that distinct human B cell responses likely play in MS. In recent years, both murine and human B cells have been identified with distinct functional properties related to their expression of particular cytokines. These have included regulatory (Breg) B cells (secreting interleukin (IL)-10 or IL-35) and pro-inflammatory B cells (secreting tumor necrosis factor α, LTα, IL-6, and granulocyte macrophage colony-stimulating factor). Better understanding of human cytokine-defined B cell responses is necessary in both health and diseases, such as MS. Investigation of their surface phenotype, distinct functions, and the mechanisms of regulation (both cell intrinsic and cell extrinsic) may help develop effective treatments that are more selective and safe. In this review, we focus on mechanisms by which cytokine-defined B cells contribute to the peripheral immune cascades that are thought to underlie MS relapses, and the impact of B cell-directed therapies on these mechanisms.
Introduction
In addition to their potential to differentiate into antibody producing plasma cells, B cells can efficiently present antigens to T cells and modulate local immune responses through secretion of soluble products, such as pro-inflammatory or anti-inflammatory cytokines (Figure 1) (1). Historically, B-cell implication in multiple sclerosis (MS) pathogenesis was based on the common finding of abnormally increased immunoglobulin levels in the cerebrospinal fluid (CSF) of patients as well as antibody deposition noted in brain lesions (2–5). However, the success of B-cell-depleting therapy to limit new MS relapses without obviously impacting abnormal CSF antibody levels underscores antibody-independent contributions of B cells to relapsing disease activity (6–11). In this review, we focus on implication of pro-inflammatory or anti-inflammatory cytokine-defined B cell responses in MS and the impact of B-cell-directed therapies on their functions.
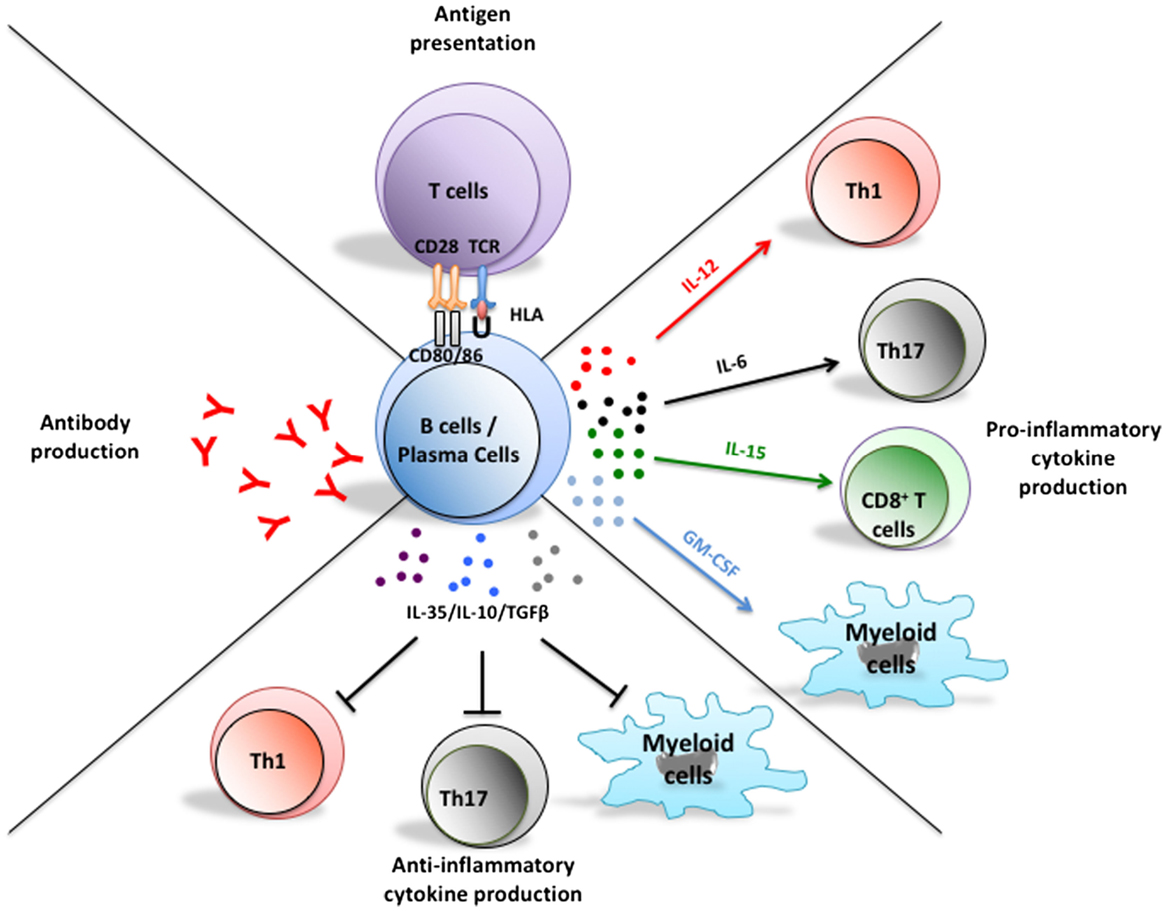
Figure 1. Multifaceted functions of B cells and their implications in the pathogenesis of MS. In addition to their potential to differentiate into antibody-secreting plasmablasts and plasma cells, B cells can present antigen to T cells, as well as up- or down-regulate local immune responses through elaboration of pro- or anti-inflammatory cytokine, respectively. Plasma cells can also secrete pro- and anti-inflammatory cytokines that could modulate T cell and myeloid-cell responses. Abnormal B cell responses of potential relevance to MS include aberrant production of autoreactive antibodies, exaggerated activation of T cells through antigen presentation, and induction of pro-inflammatory T cell and myeloid-cell responses through abnormal secretion of pro-inflammatory B cell cytokines and/or insufficient secretion of B cell regulatory cytokines.
Cytokine-Defined B Cell Responses in MS and EAE
Several functionally distinct cytokine-defined B cell responses have been implicated in the pathophysiology of MS and its commonly used animal model, experimental autoimmune encephalomyelitis (EAE). Nonetheless, translation of B-cell-related findings from mouse to human has not always been straightforward. Characterizing and defining the roles of distinct human B cell subsets in health and disease are important requisites for rational development of more selective and effective B cell-targeting therapies.
Interleukin-10-Producing B Cells
Interleukin (IL)-10 is a cytokine with pleiotropic effects in immunoregulation and inflammation (12). In mice, knock-out (KO) of IL-10 selectively from B cells results in more severe EAE (13), and adoptive transfer of in vitro-induced IL-10-producing B cells suppresses EAE in an IL-10-dependent manner (14–16). Inducing EAE in IL-10 reporter mice implicated the draining lymph nodes (rather than spleen or spinal cord) as the sites where IL-10+ B cells regulate disease-relevant immune responses (17). The IL-10+ B cells in this study exhibited plasma cell/plasmablast markers, consistent with an earlier report showing that CD138+ plasma cells are able to produce IL-10 (17), thus also highlighting the previously unappreciated antibody-independent functions of plasma cells. Although IL-10 production from B cells can be induced by both innate [toll-like receptors (TLRs)] and adaptive (cognate interaction) stimuli (14, 16, 18), the targets of regulation of innate- and adaptive-induced IL-10-producing B cells may differ depending on context. For example, innate signal-induced IL-10-producing B cells are able to down-regulate pathogenic T-cell responses indirectly through dendritic cells (14), whereas adaptive signal-induced IL-10-producing B cells directly down-regulate antigen-specific T-cell responses (15, 16).
In humans, both naïve and memory B cells are capable of producing IL-10 in a context-dependent manner (19–22). Human CD27− (naïve) B cells, but not CD27+ (memory) B cells, are able to produce IL-10 upon CD40-ligand stimulation (11, 23–25), a response found to be abnormally deficient in B cells of MS patients (24). By contrast, IL-10+ B10 cells are induced within the CD27+ memory pool by stimulation through TLR4 and TLR9 and can suppress tumor necrosis factor α (TNFα) production by monocytes through an IL-10-dependent mechanism. Unexpectedly, B10 cells were reportedly increased in several human autoimmune diseases, including MS, upon stimulation (21). A better understanding of these cells, including defining surface markers and master transcriptional regulators, could facilitate future cell-based therapies for MS.
IL-35 Producing B Cells
Interleukin-35 is an anti-inflammatory cytokine of the IL-12 family (26). Although the EBI3 subunit of IL-35 was first identified in EBV-infected B cells (27), functions of IL-35 were initially described in regulatory T cells (28–30). More recently, IL-35-producing B cells were found to play important roles in recovery from EAE and experimental autoimmune uveitis (31, 32). In these contexts, IL-35-producing B cells inhibited pro-inflammatory immune responses either directly through IL-35 (31) or indirectly through induction of IL-10-producing B cells (32). These IL-35-producing B cells also exhibited plasma cell phenotypic markers (31). Besides IL-10 and IL-35, B cells can also produce Transforming-growth factor β or Granzyme B that may down-regulate immune responses (33–39); their relevance to MS (or EAE) is yet to be determined.
Tumor Necrosis Factor α and Lymphotoxin-α Producing B Cells
Tumor necrosis factor α and Lymphotoxin-α (LTα) are actively involved in promoting pro-inflammatory immune responses to protect against pathogen invasion (40). In addition, TNFα is also known to play a pathogenic role in several autoimmune diseases, including rheumatoid arthritis (41) and inflammatory bowel disease (42), in which TNFα-blocking therapies have been successful (41). In MS, however, TNFα blockade increased disease activity (43) highlighting the challenge of broadly targeting individual cytokines (versus targeting particular cytokine-expressing cells). Stimulation through CD40 and the B-cell receptor (BCR) significantly increases TNFα and LTα secretion from human B cells, compared to either stimulation alone (19). B cells of MS patients produce abnormally higher levels of both TNFα and LTα upon such dual stimulation (11, 23, 24). A microRNA (miR)-132:SIRT1 axis controls expression of TNFα and LTα by human B cells (23). Abnormally increased expression of miR-132 by MS B cells inhibited their SIRT1 expression, resulting in enhanced pro-inflammatory cytokine production. In vitro addition of the SIRT1-agonist resveratrol normalized the exaggerated pro-inflammatory cytokine expression of MS B cells (23).
IL-6 Producing B Cells
Interleukin-6, a cytokine with both pro-inflammatory and anti-inflammatory properties, can be produced by both immune and non-immune cells (44). IL-6 can induce Th17-cell differentiation from naïve T cells (45) and inhibit regulatory T cells (46–48). By contrast, IL-6 may induce IL-10-producing regulatory B cells and myeloid cells (18, 49). B cells of MS patients secrete abnormally high levels of IL-6 (50) and IL-6 knock-out selectively from B cells resulted in decreased Th17 responses and diminished EAE severity (50, 51). How B cell-derived IL-6 is regulated, and whether B-cell IL-6 also contributes to Th17 differentiation and regulatory T-cell dysfunction in MS, remains unknown.
IL-15 Producing B Cells
Interleukin-15 belongs to the four α-helix bundle family of cytokines and can be produced by multiple cell types (52). IL-15 knock-out mice develop more severe EAE (53), in part attributed to IL-15’s ability to inhibit pathogenic Th17-cell differentiation (54), and to induce regulatory CD8+ CD122+ T cells (55). In patients with MS, however, IL-15 is abnormally increased in both serum and CSF (56, 57), where it may have disease-promoting (rather than disease-inhibiting) potential (58, 59). B cells from MS patients reportedly produce more IL-15 than controls, and activation of B cells through CD40 and the BCR induces IL-15 secretion that enhanced both the migratory capacity of CD8+ T cells across a model of the blood–brain barrier and CD8+ T cell cytotoxicity toward oligodentrocytes (59).
Granulocyte Macrophage Colony-Stimulating Factor-Producing B Cells
Granulocyte macrophage colony-stimulating factor (GM-CSF) is an important growth factor for myeloid lineage cell development and function, which is secreted by both immune and non-immune cells during infection and autoimmune disease (60). GM-CSF KO is resistant to active EAE induction (61), and GM-CSF KO Th17 cells fail to induce passive EAE (62–64). Since GM-CSF-producing T cells are reportedly increased in the circulation of MS patients (65–67), T cells have been thought to be the main source of GM-CSF of relevance to MS and EAE (65–68). A murine B-cell population generated from B1a cells, termed “innate response activator (IRA)” B cells (69), was described to produce GM-CSF and found to play a GM-CSF-mediated protective role during infections (69, 70), as well as a GM-CSF-mediated pathogenic role in atherosclerosis (71). In contrast to the murine IRA cells, a recently described human GM-CSF producing B cell subset belonged to the memory pool, and co-expressed high levels of TNFα and IL-6 (72). The human GM-CSF-producing B cells enhanced myeloid-cell pro-inflammatory responses in a GM-CSF-dependent manner and were abnormally increased in MS patients. B cell depletion in patients with MS resulted in a B cell–GM-CSF-dependent decrease of pro-inflammatory myeloid-cell responses, highlighting the potential pathogenic role of this B cell population in vivo and revealing a novel disease-implicated axis involving B cell:myeloid-cell interactions (72).
B Cell-Targeting Therapies and Effects in MS
The use of B cell-depleting agents in MS was initially driven by the long-standing recognition of abnormal antibody presence in both the CSF and brain lesions of MS patients (2–4, 73). Therapies directed against B cells include agents that impact their survival (rituximab, ocrelizumab, ofatumumab, alemtuzumab, and atacicept), and their trafficking to the CNS (natalizumab and fingolimod). In this section, we will highlight the mechanisms of action of these and other MS-related therapies that may impact B cells, with a focus on how such therapies may influence MS disease-relevant cytokine-defined B cells responses.
Anti-CD20 Monoclonal Antibodies
CD20 is a transmembrane protein with incompletely understood function, expressed on immature, transitional, naïve, and memory B cells, but not on stem cells, pro-B cells, and plasma cells (74). Rituximab, ocrelizumab, and ofatumumab are anti-CD20 monoclonal antibodies that induce B cell lysis via different combinations of antibody-dependent cell cytotoxicity, complement-dependent cytotoxicity, or apoptosis (75, 76). In MS, anti-CD20 antibodies rapidly and significantly reduced the number of new gadolinium-enhancing brain lesions and significantly reduced relapse rates (6–10, 77). Treatment reduced circulating B cell counts by >90% of baseline values, while serum and CSF immunoglobulin G levels remained largely unchanged (77–79), pointing to an important antibody-independent contribution of B cells to MS relapsing disease activity. An attractive hypothesis that has emerged is that pro-inflammatory B cells in untreated patients abnormally activate disease-relevant responses of other immune cells – hence removal of such B cells diminishes disease activity. In support of this view, anti-CD20-mediated B-cell depletion decreases both Th1 and Th17 T cell responses (11, 50) and pro-inflammatory myeloid-cell responses (that in turn could drive Th1 and Th17 responses) in the periphery of treated patients (72). In addition to cognate interactions in which B cells may serve as efficient antigen-presenting cells (APC) to activate T cells that recognize the same antigen, abnormal B-cell secretion of pro-inflammatory cytokines (including IL-6, TNFα, LTα, and GM-CSF) has now been implicated in abnormal T-cell and myeloid-cell responses of MS patients and may involve “bystander activation” (i.e., not be predicated by cognate antigen-specific interactions). Rituximab treatment could also diminish T cells within the CSF (79), providing further support that, when present, B cells may contribute to disease activity by enhancing peripheral T-cell activation and trafficking, and/or by CNS resident B cells promoting chemotaxis of T cells into the CNS. Alternative mechanisms proposed include an increased frequency of circulating regulatory T cells following B cell depletion (80) and in addition to depleting circulating B cells, anti-CD20 treatment also removes a small population of CD20dim T cells (81, 82). Initial studies of this T cell subset point to their potential to produce pro-inflammatory cytokines (81), though their significance in relation to MS disease activity warrants further investigation.
Elegant work using somatic mutation analysis of the Ig gene in B cells derived from both CNS and peripheral compartments of the same MS patients indicates that bi-directional trafficking of B cells occurs between the CNS and periphery and that much of the activation and maturation that results in clonal enrichment of B cells within the CNS may actually occur in the periphery (presumably through cognate–antigen interactions with T cells) (83–86). Hence, efforts to deplete or modulate the profile and functions of B cells in peripheral compartments of MS patients may meaningfully influence the profile and activities of B cells within the CNS, even if the treatment itself does not efficiently penetrate the CNS (as is generally the case for monoclonal antibodies). There have also been efforts to selectively eliminate B cells in the CNS using intrathecally administered rituximab (87). However, a complication in interpreting this result has been the finding that even small doses of rituximab infused into the CSF results in rapid and substantial peripheral B cell depletion (87). Data regarding effects of anti-CD20-mediated peripheral B-cell depletion on inflammation within the CNS compartment remain limited. Early work suggested that rituximab may be more effective at depleting CSF B cells in patients with relapsing compared to progressive forms of MS (77, 78), possibly due to differences in Blood-brain barrier permeability. In the earlier OLYMPUS trial (88), rituximab failed to limit progression of disability in PPMS patients compared to placebo treatment, though post hoc sub-group analysis suggested that patients who had gadolinium-enhancing lesions at baseline, and particularly younger patients, did benefit (88). The ORATORIO study, focusing on earlier disease, and using ocrelizumab, demonstrated that anti-CD20 therapy could limit disease progression in PPMS patients (89). The mechanisms underlying this benefit of B cell depletion in patients with progressive MS remain to be elucidated (see Michel et al., in this issue).
In addition to the decreased MS disease activity observed following B cell depletion with anti-CD20 antibodies, there is a suggestion that the benefit of B cell depletion may persist in at least some patients even as reconstitution of B cells occurs (6, 7). This would imply that the re-emerging B cells differ importantly from the B cells present prior to depletion. Indeed, the B cells that reconstitute following anti-CD20 depletion have been shown to be largely naïve B cells which, when activated, express more IL-10 and less pro-inflammatory cytokines, including TNFα, IL-6, and GM-CSF, compared to pre-treatment B cells (72).
Atacicept
B-cell activating factor of the TNF family (BAFF) and a proliferation-inducing ligand (APRIL) are expressed by a variety of immune and non-immune cells (90, 91). Both cytokines signal through transmembrane activator and cyclophilin ligand interactor (TACI) and B cell maturation antigen (BCMA), while only BAFF binds to BAFF-R (90, 91). Both play important roles in the survival, maturation, and function of B cells and plasma cells (92–94). BAFF can also promote differentiation and expansion of Th17 cells in models of infectious and autoimmune diseases (95). BAFF and APRIL levels are reportedly elevated in MS patients (96, 97), where they are highly expressed by peripheral blood monocytes and T cells. BAFF is also abnormally expressed by astrocytes within MS lesions (77–79, 98). Atacicept, a soluble, recombinant fusion protein containing the extracellular ligand-binding portion of TACI receptor and a modified Fc portion of human IgG, prevents binding of BAFF and APRIL to their receptors (99). Atacicept thereby limits survival of mature and activated B cells as well as antibody-secreting plasma cells but does not appear to target pro- or memory B cells (100, 101). Treatment with atacicept reduces circulating B cell counts (by 60–70%) and substantially reduces serum IgM and IgA (but to a lesser extent IgG) levels (100, 102–104). While emerging as beneficial in systemic lupus erythematosus, development of atacicept in MS was halted due to increased relapsing disease activity (100, 103–105). Why atacicept induced rather than limited new MS disease activity remains unknown but may reflect differential effects on functionally distinct B cell or plasma cell responses. Indeed, BAFF can induce IL-10-producing B cells and suppress the generation of IL-15+ B cells (106). Hence, the dysregulated cytokine balance in B cells of untreated MS patients may actually be aggravated by atacicept, leading to aberrant responses of disease-relevant immune cells, such as pathogenic T cells and myeloid cells.
Approved MS Therapies That may Impact B Cell Responses
While most approved MS therapies were developed based on their presumed ability to target T cells, many of them are now understood to also impact B cells in potentially disease-relevant ways (Table 1). For example, interferon (IFN)-β decreases the frequency of CD80-expressing B cells in treated MS patients, which could in turn limit peripheral T cell activation (107). IFN-β also enhances the numbers of circulating transitional B cells (108) and, such as glatiramer acetate (GA), may result in an anti-inflammatory shift of B cell cytokine responses (109, 110). Both treatment with IFN-β and GA unexpectedly increased serum BAFF (111, 112), which would not only support certain aspects of B cells and plasma cells but may also shape the balance between cytokine-defined B cells, as described above. While natalizumab modifies frequencies of circulating B cells and plasma cells (Table 1) its effects, if any, on B cell cytokine-response profiles are largely unknown (113–115). It is noteworthy that in vivo treatment with natalizumab not only limits trafficking but may also modify T cell activation, which may reflect a role for VLA-4 during T cell interaction with APC, including B cells (113). Treatment with fingolimod (FTY720), an S1P receptor targeting agent, not only reduces the overall number of circulating B cells but can also modulate the cytokine-response profile of B cells in treated patients (116–120). Indeed, the proportion of peripheral blood memory B cells was reduced in fingolimod-treated patients, while the proportion of regulatory and transitional B cells was increased (117–120). The shift in circulating B-cell subsets was paralleled by changes in B-cell cytokine production, with reduced TNFα and enhanced IL-10 expression in B cells from fingolimod-treated patients (117, 120). Fingolimod treatment may also enhance migration capacity of regulatory B cells based on in vitro modeling of blood–brain barrier trafficking (120). Mitoxantrone, a chemotherapeutic agent that targets type II topoisomerase (121), preferentially reduces memory B cell counts and leads to a profile of circulating B cells with a less pro-inflammatory profile (24). Alemtuzumab, a humanized monoclonal antibody-targeting CD52, rapidly depletes multiple immune cell subsets (including B cells) followed by distinct kinetics of reconstitution (122). B cells reconstitute faster than T cells (within 3–6 months post-treatment), with naïve B cells dominating the re-emerging B cell pool. Such a treatment effect would be expected to induce a favorable shift in the balance between pro- and anti-inflammatory B cell responses (122, 123). Further studies are required to elucidate potential MS-relevant effects of dimethyl-fumarate (BG12) and teriflunomide on B-cell profiles, including cytokine responses (124, 125).
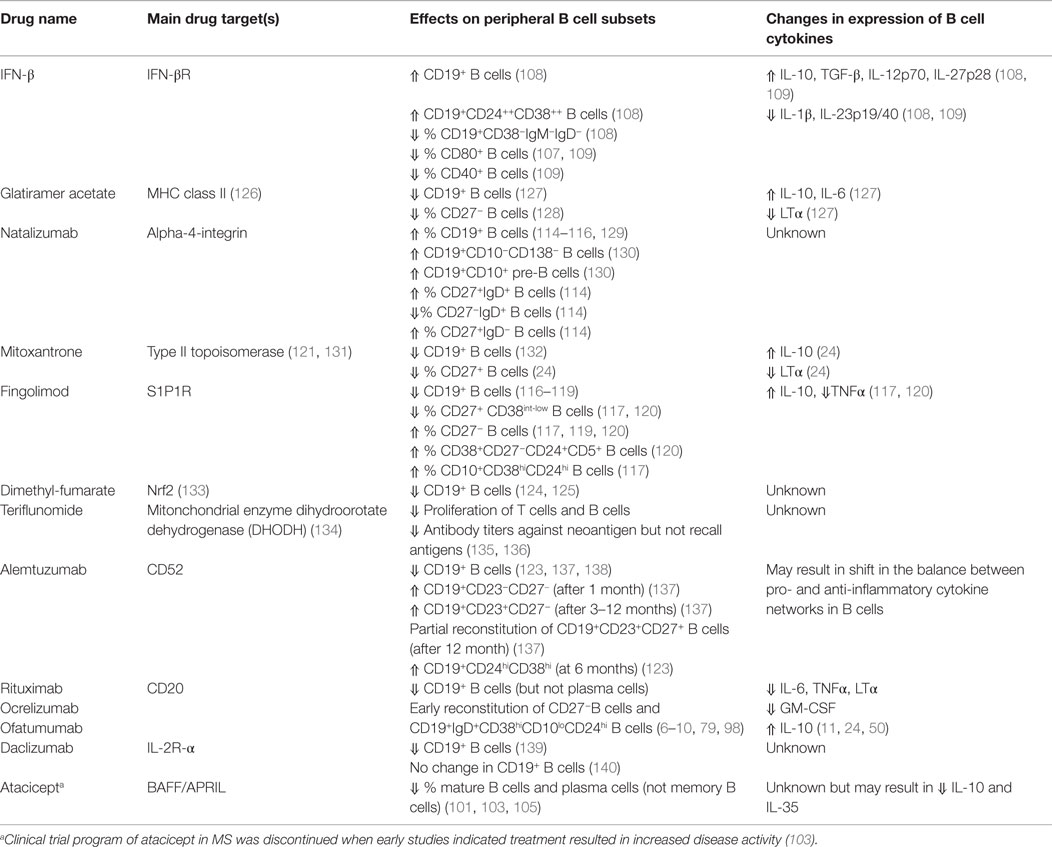
Table 1. Selected therapies approved for (or under investigation for) multiple sclerosis, and their in vivo effects on the profiles and cytokine responses of B cells.
Conclusion
The success of anti-CD20 therapy in MS establishes that B cells contribute to relapsing disease activity. Though unwelcome, the observation that treatment with atacicept (Table 1) exacerbates MS, serves to reinforce the concept that targeting B cells can change the face of CNS disease activity, while also underscoring the importance of elucidating the functional heterogeneity that exists within the B cell pool. Emerging studies indicate that responses of cytokine-secreting B cells in the periphery may influence new MS disease activity, potentially through aberrant peripheral activation of other immune cells. B cells may play additional roles in propagating disease activity within the CNS (see Michel et al., in this issue). Success of B cell targeting therapies may lie in restoring and maintaining a favorable balance between pro- and anti-inflammatory B cell activities in patients.
Conflict of Interest Statement
Rui Li, Ayman Rezk, Luke M. Healy, Gillian Muirhead, Alexandre Prat, and Jennifer L. Gommerman declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Dr. Bar-Or has participated as a speaker in meetings sponsored by and received consulting fees and/or grant support from Biogen Idec, Diogenix, Genentech, Sanofi-Genzyme, GlaxoSmithKline, Novartis, Ono Pharma, Teva Neuroscience, Receptos Inc, Roche, and Merck/EMD Serono.
The Handling Editor, Jorge Ivan Alvarez, declares that, despite having been supervised by author Alexandre Prat during post-doctoral work, the review process was handled objectively.
Funding
Supported by a Collaborative Team grant from the Research Foundation of the MS Society of Canada (RF-MSSC) to the “Canadian B cells in MS Team” (AB-O, AP, and JG).
References
1. Shen P, Fillatreau S. Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol (2015) 15(7):441–51. doi: 10.1038/nri3857
2. Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol (1998) 43(4):465–71. doi:10.1002/ana.410430409
3. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med (1999) 5(2):170–5. doi:10.1038/5532
4. Raine CS, Cannella B, Hauser SL, Genain CP. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: a case for antigen-specific antibody mediation. Ann Neurol (1999) 46(2):144–60. doi:10.1002/1531-8249(199908)46:2<144::AID-ANA3>3.0.CO;2-K
5. Walsh MJ, Tourtellotte WW, Roman J, Dreyer W. Immunoglobulin G, A, and M – clonal restriction in multiple sclerosis cerebrospinal fluid and serum – analysis by two-dimensional electrophoresis. Clin Immunol Immunopathol (1985) 35(3):313–27. doi:10.1016/0090-1229(85)90092-3
6. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med (2008) 358(7):676–88. doi:10.1056/NEJMoa0706383
7. Bar-Or A, Calabresi PA, Arnold D, Markowitz C, Shafer S, Kasper LH, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol (2008) 63(3):395–400. doi:10.1002/ana.21363
8. Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet (2011) 378(9805):1779–87. doi:10.1016/S0140-6736(11)61649-8
9. Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology (2014) 82(7):573–81. doi:10.1212/WNL.0000000000000125
10. Bar-Or A, Grove RA, Austin DJ, Tolson JM, VanMeter SA, Lewis E, et al. The MIRROR Study: a randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study to investigate the safety and MRI efficacy of subcutaneous ofatumumab in subjects with relapsing-remitting multiple sclerosis (RRMS). Neurology (2014) 82(10 Suppl):S23.006.
11. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol (2010) 67(4):452–61. doi:10.1002/ana.21939
12. Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10(3):170–81. doi:10.1038/nri2711
13. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3(10):944–50. doi:10.1038/ni833
14. Lampropoulou V, Hoehlig K, Roch T, Neves P, Calderon Gomez E, Sweenie CH, et al. TLR-activated B cells suppress T cell-mediated autoimmunity. J Immunol (2008) 180(7):4763–73. doi:10.4049/jimmunol.180.7.4763
15. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest (2008) 118(10):3420–30. doi:10.1172/JCI36030
16. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491(7423):264–8. doi:10.1038/nature11501
17. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41(6):1040–51. doi:10.1016/j.immuni.2014.10.016
18. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory B cells are induced by gut microbiota-driven interleukin-1beta and interleukin-6 production. Nat Med (2014) 20(11):1334–9. doi:10.1038/nm.3680
19. Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol (2004) 172(6):3422–7. doi:10.4049/jimmunol.172.6.3422
20. Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity (2010) 32(1):129–40. doi:10.1016/j.immuni.2009.11.009
21. Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood (2011) 117(2):530–41. doi:10.1182/blood-2010-07-294249
22. Rieger A, Bar-Or A. B-cell-derived interleukin-10 in autoimmune disease: regulating the regulators. Nat Rev Immunol (2008) 8(6):486–7. doi:10.1038/nri2315-c1
23. Miyazaki Y, Li R, Rezk A, Misirliyan H, Moore C, Farooqi N, et al. A novel microRNA-132-surtuin-1 axis underlies aberrant B-cell cytokine regulation in patients with relapsing-remitting multiple sclerosis. PLoS One (2014) 9(8):e105421. doi:10.1371/journal.pone.0105421
24. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol (2007) 178(10):6092–9. doi:10.4049/jimmunol.178.10.6092
25. Correale J, Farez M. Association between parasite infection and immune responses in multiple sclerosis. Ann Neurol (2007) 61(2):97–108. doi:10.1002/ana.21067
26. Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol (2012) 13(8):722–8. doi:10.1038/ni.2366
27. Devergne O, Hummel M, Koeppen H, Le Beau MM, Nathanson EC, Kieff E, et al. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J Virol (1996) 70(2):1143–53.
28. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450(7169):566–9. doi:10.1038/nature06306
29. Seyerl M, Kirchberger S, Majdic O, Seipelt J, Jindra C, Schrauf C, et al. Human rhinoviruses induce IL-35-producing Treg via induction of B7-H1 (CD274) and sialoadhesin (CD169) on DC. Eur J Immunol (2010) 40(2):321–9. doi:10.1002/eji.200939527
30. Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting edge: human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol (2011) 186(12):6661–6. doi:10.4049/jimmunol.1100315
31. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature (2014) 507(7492):366–70. doi:10.1038/nature12979
32. Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med (2014) 20(6):633–41. doi:10.1038/nm.3554
33. Caver TE, O’Sullivan FX, Gold LI, Gresham HD. Intracellular demonstration of active TGFbeta1 in B cells and plasma cells of autoimmune mice. IgG-bound TGFbeta1 suppresses neutrophil function and host defense against Staphylococcus aureus infection. J Clin Invest (1996) 98(11):2496–506. doi:10.1172/JCI119068
34. Parekh VV, Prasad DVR, Banerjee PP, Joshi BN, Kumar A, Mishra GC. B cells activated by lipopolysaccharide, but not by anti-Ig and anti-CD40 antibody, induce anergy in CD8+ T cells: role of TGF-1. J Immunol (2003) 170(12):5897–911. doi:10.4049/jimmunol.170.12.5897
35. Lee KM, Stott RT, Zhao G, SooHoo J, Xiong W, Lian MM, et al. TGF-beta-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol (2014) 44(6):1728–36. doi:10.1002/eji.201344062
36. Jahrsdorfer B, Blackwell SE, Wooldridge JE, Huang J, Andreski MW, Jacobus LS, et al. B-chronic lymphocytic leukemia cells and other B cells can produce granzyme B and gain cytotoxic potential after interleukin-21-based activation. Blood (2006) 108(8):2712–9. doi:10.1182/blood-2006-03-014001
37. Hagn M, Schwesinger E, Ebel V, Sontheimer K, Maier J, Beyer T, et al. Human B cells secrete granzyme B when recognizing viral antigens in the context of the acute phase cytokine IL-21. J Immunol (2009) 183(3):1838–45. doi:10.4049/jimmunol.0901066
38. Hagn M, Ebel V, Sontheimer K, Schwesinger E, Lunov O, Beyer T, et al. CD5+ B cells from individuals with systemic lupus erythematosus express granzyme B. Eur J Immunol (2010) 40(7):2060–9. doi:10.1002/eji.200940113
39. Hagn M, Sontheimer K, Dahlke K, Brueggemann S, Kaltenmeier C, Beyer T, et al. Human B cells differentiate into granzyme B-secreting cytotoxic B lymphocytes upon incomplete T-cell help. Immunol Cell Biol (2012) 90(4):457–67. doi:10.1038/icb.2011.64
40. Bradley JR. TNF-mediated inflammatory disease. J Pathol (2008) 214(2):149–60. doi:10.1002/path.2287
41. Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol (2001) 19:163–96. doi:10.1146/annurev.immunol.19.1.163
42. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14(5):329–42. doi:10.1038/nri3661
43. Arnason BGW, Jacobs G, Hanlon M, Harding B, Noronha ABC, Auty A, et al. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology (1999) 53(3):457–65. doi:10.1212/WNL.53.3.457
44. Kishimoto T. Interleukin-6: from basic science to medicine – 40 years in immunology. Annu Rev Immunol (2005) 23:1–21. doi:10.1146/annurev.immunol.23.021704.115806
45. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
46. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol (2010) 40(7):1830–5. doi:10.1002/eji.201040391
47. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A (2008) 105(47):18460–5. doi:10.1073/pnas.0809850105
48. Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, et al. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med (2013) 5(170):170ra15. doi:10.1126/scitranslmed.3004970
49. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol (2014) 15(5):423–30. doi:10.1038/ni.2865
50. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med (2012) 209(5):1001–10. doi:10.1084/jem.20111675
51. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med (2013) 210(13):2921–37. doi:10.1084/jem.20130699
52. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol (2006) 24:657–79. doi:10.1146/annurev.immunol.24.021605.090727
53. Gomez-Nicola D, Spagnolo A, Guaza C, Nieto-Sampedro M. Aggravated experimental autoimmune encephalomyelitis in IL-15 knockout mice. Exp Neurol (2010) 222(2):235–42. doi:10.1016/j.expneurol.2009.12.034
54. Pandiyan P, Yang XP, Saravanamuthu SS, Zheng L, Ishihara S, O’Shea JJ, et al. The role of IL-15 in activating STAT5 and fine-tuning IL-17A production in CD4 T lymphocytes. J Immunol (2012) 189(9):4237–46. doi:10.4049/jimmunol.1201476
55. Yu P, Bamford RN, Waldmann TA. IL-15-dependent CD8+ CD122+ T cells ameliorate experimental autoimmune encephalomyelitis by modulating IL-17 production by CD4+ T cells. Eur J Immunol (2014) 44(11):3330–41. doi:10.1002/eji.201444675
56. Blanco-Jerez C, Plaza JF, Masjuan J, Orensanz LM, Alvarez-Cermeno JC. Increased levels of IL-15 mRNA in relapsing-remitting multiple sclerosis attacks. J Neuroimmunol (2002) 128(1–2):90–4. doi:10.1016/S0165-5728(02)00146-7
57. Rentzos M, Cambouri C, Rombos A, Nikolaou C, Anagnostouli M, Tsoutsou A, et al. IL-15 is elevated in serum and cerebrospinal fluid of patients with multiple sclerosis. J Neurol Sci (2006) 241(1–2):25–9. doi:10.1016/j.jns.2005.10.003
58. Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N. Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J Immunol (2010) 185(10):5693–703. doi:10.4049/jimmunol.1002188
59. Schneider R, Mohebiany AN, Ifergan I, Beauseigle D, Duquette P, Prat A, et al. B cell-derived IL-15 enhances CD8 T cell cytotoxicity and is increased in multiple sclerosis patients. J Immunol (2011) 187(8):4119–28. doi:10.4049/jimmunol.1100885
60. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol (2008) 8(7):533–44. doi:10.1038/nri2356
61. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med (2001) 194(7):873–82. doi:10.1084/jem.194.7.873
62. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol (2007) 178(1):39–48. doi:10.4049/jimmunol.178.1.39
63. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol (2011) 12(6):560–7. doi:10.1038/ni.2027
64. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi:10.1038/ni.2031
65. Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun (2014) 5:5056. doi:10.1038/ncomms6056
66. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med (2014) 6(241):241ra80. doi:10.1126/scitranslmed.3008706
67. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-beta therapy. J Immunol (2015) 194(11):5085–93. doi:10.4049/jimmunol.1403243
68. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature (2013) 496(7446):518–22. doi:10.1038/nature11868
69. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science (2012) 335(6068):597–601. doi:10.1126/science.1215173
70. Weber GF, Chousterman BG, Hilgendorf I, Robbins CS, Theurl I, Gerhardt LM, et al. Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J Exp Med (2014) 211(6):1243–56. doi:10.1084/jem.20131471
71. Hilgendorf I, Theurl I, Gerhardt LM, Robbins CS, Weber GF, Gonen A, et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation (2014) 129(16):1677–87. doi:10.1161/CIRCULATIONAHA.113.006381
72. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med (2015) 7(310):310ra166. doi:10.1126/scitranslmed.aab4176
73. Kostulas VK, Link H, Lefvert AK. Oligoclonal IgG bands in cerebrospinal fluid. Principles for demonstration and interpretation based on findings in 1114 neurological patients. Arch Neurol (1987) 44(10):1041–4. doi:10.1001/archneur.1987.00520220043014
74. Scott SD. Rituximab: a new therapeutic monoclonal antibody for non-Hodgkin’s lymphoma. Cancer Pract (1998) 6(3):195–7. doi:10.1046/j.1523-5394.1998.006003195.x
75. Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant (2006) 6(5 Pt 1):859–66. doi:10.1111/j.1600-6143.2006.01288.x
76. Boross P, Leusen JH. Mechanisms of action of CD20 antibodies. Am J Cancer Res (2012) 2(6):676–90.
77. Stuve O, Cepok S, Elias B, Saleh A, Hartung HP, Hemmer B, et al. Clinical stabilization and effective B-lymphocyte depletion in the cerebrospinal fluid and peripheral blood of a patient with fulminant relapsing-remitting multiple sclerosis. Arch Neurol (2005) 62(10):1620–3. doi:10.1001/archneur.62.10.1620
78. Monson NL, Cravens PD, Frohman EM, Hawker K, Racke MK. Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch Neurol (2005) 62(2):258–64. doi:10.1001/archneur.62.2.258
79. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol (2006) 180(1–2):63–70. doi:10.1016/j.jneuroim.2006.06.029
80. Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood (2008) 112(4):1147–50. doi:10.1182/blood-2007-12-129262
81. Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol (2014) 193(2):580–6. doi:10.4049/jimmunol.1400118
82. Wilk E, Witte T, Marquardt N, Horvath T, Kalippke K, Scholz K, et al. Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum (2009) 60(12):3563–71. doi:10.1002/art.24998
83. Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol (2014) 75(2):266–76. doi:10.1002/ana.24088
84. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med (2014) 6(248):248ra106. doi:10.1126/scitranslmed.3008930
85. Stern JNH, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med (2014) 6(248):248ra107. doi:10.1126/scitranslmed.3008879
86. von Budingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest (2012) 122(12):4533–43. doi:10.1172/JCI63842
87. Studer V, Rossi S, Motta C, Buttari F, Centonze D. Peripheral B cell depletion and central proinflammatory cytokine reduction following repeated intrathecal administration of rituximab in progressive multiple sclerosis. J Neuroimmunol (2014) 276(1–2):229–31. doi:10.1016/j.jneuroim.2014.08.617
88. Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol (2009) 66(4):460–71. doi:10.1002/ana.21867
89. Montalban X, Hemmer B, Rammohan B, Giovannoni G, de Seze J, Bar-Or A. Efficacy and safety of ocrelizumab in primary progressive multiple sclerosis – results of the placebo-controlled, double-blind, phase III ORATORIO Study. Mult Scler J (2015) 21(S11):780–808. doi:10.1177/1352458515604791
90. Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol (2003) 21:231–64. doi:10.1146/annurev.immunol.21.120601.141152
91. Novak AJ, Darce JR, Arendt BK, Harder B, Henderson K, Kindsvogel W, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood (2004) 103(2):689–94. doi:10.1182/blood-2003-06-2043
92. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. doi:10.1084/jem.20031330
93. Dillon SR, Gross JA, Ansell SM, Novak AJ. An APRIL to remember: novel TNF ligands as therapeutic targets. Nat Rev Drug Discov (2006) 5(3):235–46. doi:10.1038/nrd1982
94. Bossen C, Schneider P. BAFF APRIL and their receptors: structure, function and signaling. Semin Immunol (2006) 18(5):263–75. doi:10.1016/j.smim.2006.04.006
95. Lai Kwan Lam Q, King Hung Ko O, Zheng BJ, Lu L. Local BAFF gene silencing suppresses Th17-cell generation and ameliorates autoimmune arthritis. Proc Natl Acad Sci U S A (2008) 105(39):14993–8. doi:10.1073/pnas.0806044105
96. Thangarajh M, Gomes A, Masterman T, Hillert J, Hjelmstrom P. Expression of B-cell-activating factor of the TNF family (BAFF) and its receptors in multiple sclerosis. J Neuroimmunol (2004) 152(1–2):183–90. doi:10.1016/j.jneuroim.2004.03.017
97. Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med (2005) 201(2):195–200. doi:10.1084/jem.20041674
98. Piccio L, Naismith RT, Trinkaus K, Klein RS, Parks BJ, Lyons JA, et al. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol (2010) 67(6):707–14. doi:10.1001/archneurol.2010.99
99. Lehmann-Horn K, Schleich E, Hertzenberg D, Hapfelmeier A, Kumpfel T, von Bubnoff N, et al. Anti-CD20 B-cell depletion enhances monocyte reactivity in neuroimmunological disorders. J Neuroinflammation (2011) 8:146. doi:10.1186/1742-2094-8-146
100. Dall’Era M, Chakravarty E, Wallace D, Genovese M, Weisman M, Kavanaugh A, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum (2007) 56(12):4142–50. doi:10.1002/art.23047
101. Hartung HP, Kieseier BC. Atacicept: targeting B cells in multiple sclerosis. Ther Adv Neurol Disord (2010) 3(4):205–16. doi:10.1177/1756285610371146
102. van Vollenhoven RF, Kinnman N, Vincent E, Wax S, Bathon J. Atacicept in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase II, randomized, placebo-controlled trial. Arthritis Rheum (2011) 63(7):1782–92. doi:10.1002/art.30372
103. Kappos L, Hartung HP, Freedman MS, Boyko A, Radu EW, Mikol DD, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol (2014) 13(4):353–63. doi:10.1016/S1474-4422(14)70028-6
104. Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis (2014) 74(11):2006–15. doi:10.1136/annrheumdis-2013-205067
105. Sergott RC, Bennett JL, Rieckmann P, Montalban X, Mikol D, Freudensprung U, et al. ATON: results from a Phase II randomized trial of the B-cell-targeting agent atacicept in patients with optic neuritis. J Neurol Sci (2015) 351(1–2):174–8. doi:10.1016/j.jns.2015.02.019
106. Ma N, Xing C, Xiao H, He Y, Han G, Chen G, et al. BAFF suppresses IL-15 expression in B cells. J Immunol (2014) 192(9):4192–201. doi:10.4049/jimmunol.1302132
107. Genc K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest (1997) 99(11):2664–71. doi:10.1172/JCI119455
108. Schubert RD, Hu Y, Kumar G, Szeto S, Abraham P, Winderl J, et al. IFN-beta treatment requires B cells for efficacy in neuroautoimmunity. J Immunol (2015) 194(5):2110–6. doi:10.4049/jimmunol.1402029
109. Ramgolam VS, Sha Y, Marcus KL, Choudhary N, Troiani L, Chopra M, et al. B cells as a therapeutic target for IFN-beta in relapsing-remitting multiple sclerosis. J Immunol (2011) 186(7):4518–26. doi:10.4049/jimmunol.1000271
110. Ireland SJ, Guzman AA, O’Brien DE, Hughes S, Greenberg B, Flores A, et al. The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol (2014) 71(11):1421–8. doi:10.1001/jamaneurol.2014.1472
111. Hedegaard CJ, Sellebjerg F, Krakauer M, Hesse D, Bendtzen K, Nielsen CH. Interferon-beta increases systemic BAFF levels in multiple sclerosis without increasing autoantibody production. Mult Scler (2011) 17(5):567–77. doi:10.1177/1352458510393771
112. Begum-Haque S, Sharma A, Christy M, Lentini T, Ochoa-Reparaz J, Fayed IF, et al. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol (2010) 219(1–2):47–53. doi:10.1016/j.jneuroim.2009.11.016
113. Niino M, Bodner C, Simard ML, Alatab S, Gano D, Kim HJ, et al. Natalizumab effects on immune cell responses in multiple sclerosis. Ann Neurol (2006) 59(5):748–54. doi:10.1002/ana.20859
114. Planas R, Jelcic I, Schippling S, Martin R, Sospedra M. Natalizumab treatment perturbs memory- and marginal zone-like B-cell homing in secondary lymphoid organs in multiple sclerosis. Eur J Immunol (2012) 42(3):790–8. doi:10.1002/eji.201142108
115. Warnke C, Stettner M, Lehmensiek V, Dehmel T, Mausberg AK, von Geldern G, et al. Natalizumab exerts a suppressive effect on surrogates of B cell function in blood and CSF. Mult Scler (2015) 21(8):1036–44. doi:10.1177/1352458514556296
116. Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kumpfel T, Buck D, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology (2011) 76(14):1214–21. doi:10.1212/WNL.0b013e3182143564
117. Miyazaki Y, Niino M, Fukazawa T, Takahashi E, Nonaka T, Amino I, et al. Suppressed pro-inflammatory properties of circulating B cells in patients with multiple sclerosis treated with fingolimod, based on altered proportions of B-cell subpopulations. Clin Immunol (2014) 151(2):127–35. doi:10.1016/j.clim.2014.02.001
118. Nakamura M, Matsuoka T, Chihara N, Miyake S, Sato W, Araki M, et al. Differential effects of fingolimod on B-cell populations in multiple sclerosis. Mult Scler (2014) 20(10):1371–80. doi:10.1177/1352458514523496
119. Claes N, Dhaeze T, Fraussen J, Broux B, Van Wijmeersch B, Stinissen P, et al. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One (2014) 9(10):e111115. doi:10.1371/journal.pone.0111115
120. Grutzke B, Hucke S, Gross CC, Herold MV, Posevitz-Fejfar A, Wildemann BT, et al. Fingolimod treatment promotes regulatory phenotype and function of B cells. Ann Clin Transl Neurol (2015) 2(2):119–30. doi:10.1002/acn3.155
121. Smith IE, Stuart-Harris R, Pavlidis N, Bozek T. Mitoxantrone (novantrone) as single agent and in combination chemotherapy in the treatment of advanced breast cancer. Cancer Treat Rev (1983) 10(Suppl B):37–40. doi:10.1016/0305-7372(83)90020-8
122. Hill-Cawthorne GA, Button T, Tuohy O, Jones JL, May K, Somerfield J, et al. Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry (2012) 83(3):298–304. doi:10.1136/jnnp-2011-300826
123. Heidt S, Hester J, Shankar S, Friend PJ, Wood KJ. B cell repopulation after alemtuzumab induction-transient increase in transitional B cells and long-term dominance of naive B cells. Am J Transplant (2012) 12(7):1784–92. doi:10.1111/j.1600-6143.2012.04012.x
124. Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BA, Zamvil SS. Reduction of CD8(+) T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm (2015) 2(3):e76. doi:10.1212/NXI.0000000000000076
125. Berkovich R, Weiner LP. Effects of dimethyl fumarate on lymphocyte subsets. Mult Scler Relat Disord (2015) 4(4):339–41. doi:10.1016/j.msard.2015.06.002
126. Fridkis-Hareli M, Teitelbaum D, Gurevich E, Pecht I, Brautbar C, Kwon OJ, et al. Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells – specificity and promiscuity. Proc Natl Acad Sci U S A (1994) 91(11):4872–6. doi:10.1073/pnas.91.11.4872
127. Carrieri PB, Carbone F, Perna F, Bruzzese D, La Rocca C, Galgani M, et al. Longitudinal assessment of immuno-metabolic parameters in multiple sclerosis patients during treatment with glatiramer acetate. Metabolism (2015) 64(9):1112–21. doi:10.1016/j.metabol.2015.05.001
128. Rovituso D, Heller S, Schroeter M, Kleinschnitz C, Kuerten S. B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients. J Neuroimmunol (2014) 272(1–2):86–90. doi:10.1016/j.jneuroim.2014.04.008
129. Mattoscio M, Nicholas R, Sormani MP, Malik O, Lee JS, Waldman AD, et al. Hematopoietic mobilization: potential biomarker of response to natalizumab in multiple sclerosis. Neurology (2015) 84(14):1473–82. doi:10.1212/WNL.0000000000001454
130. Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology (2008) 71(17):1350–4. doi:10.1212/01.wnl.0000327671.91357.96
131. Crespi MD, Ivanier SE, Genovese J, Baldi A. Mitoxantrone affects topoisomerase activities in human breast cancer cells. Biochem Biophys Res Commun (1986) 136(2):521–8. doi:10.1016/0006-291X(86)90471-7
132. Chanvillard C, Millward JM, Lozano M, Hamann I, Paul F, Zipp F, et al. Mitoxantrone induces natural killer cell maturation in patients with secondary progressive multiple sclerosis. PLoS One (2012) 7(6):e39625. doi:10.1371/journal.pone.0039625
133. Fox RJ, Kita M, Cohan SL, Henson LJ, Zambrano J, Scannevin RH, et al. BG-12 (dimethyl fumarate): a review of mechanism of action, efficacy, and safety. Curr Med Res Opin (2014) 30(2):251–62. doi:10.1185/03007995.2013.849236
134. Bar-Or A, Pachner A, Menguy-Vacheron F, Kaplan J, Wiendl H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs (2014) 74(6):659–74. doi:10.1007/s40265-014-0212-x
135. Bar-Or A, Freedman MS, Kremenchutzky M, Menguy-Vacheron F, Bauer D, Jodl S, et al. Teriflunomide effect on immune response to influenza vaccine in patients with multiple sclerosis. Neurology (2013) 81(6):552–8. doi:10.1212/WNL.0b013e31829e6fbf
136. Bar-Or A, Wiendl H, Miller B, Benamor M, Truffinet P, Church M, et al. Randomized study of teriflunomide effects on immune responses to neoantigen and recall antigens. Neurol Neuroimmunol Neuroinflamm (2015) 2(2):e70. doi:10.1212/NXI.0000000000000070
137. Thompson SA, Jones JL, Cox AL, Compston DA, Coles AJ. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol (2010) 30(1):99–105. doi:10.1007/s10875-009-9327-3
138. Cox AL, Thompson SA, Jones JL, Robertson VH, Hale G, Waldmann H, et al. Lymphocyte homeostasis following therapeutic lymphocyte depletion in multiple sclerosis. Eur J Immunol (2005) 35(11):3332–42. doi:10.1002/eji.200535075
139. Lin YC, Winokur P, Blake A, Wu T, Romm E, Bielekova B. Daclizumab reverses intrathecal immune cell abnormalities in multiple sclerosis. Ann Clin Transl Neurol (2015) 2(5):445–55. doi:10.1002/acn3.181
Keywords: multiple sclerosis, B-lymphocytes, cytokine-defined responses, immune modulation, B-cell depletion, B cell modulation
Citation: Li R, Rezk A, Healy LM, Muirhead G, Prat A, Gommerman JL, Bar-Or A and MSSRF Canadian B cells in MS Team (2016) Cytokine-Defined B Cell Responses as Therapeutic Targets in Multiple Sclerosis. Front. Immunol. 6:626. doi: 10.3389/fimmu.2015.00626
Received: 31 August 2015; Accepted: 30 November 2015;
Published: 08 January 2016
Edited by:
Jorge Ivan Alvarez, University of Pennsylvania, USAReviewed by:
Anne Spurkland, University of Oslo, NorwayMichael Karl Racke, The Ohio State University, USA
Copyright: © 2016 Li, Rezk, Healy, Muirhead, Prat, Gommerman, Bar-Or and MSSRF Canadian B cells in MS Team. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Bar-Or, YW1pdC5iYXItb3JAbWNnaWxsLmNh
†The “MSSRF Canadian B cells in MS Team” represents the Multiple Sclerosis of Canada Research Foundation supported ‘Canadian B cells in MS Team’. Co-principle investigators of the Canadian B cells in MS Team are Drs. Amit Bar-Or, Alexandre Prat and Jennifer Gommerman.