 Hong Zan
Hong Zan Paolo Casali
Paolo Casali- Department of Microbiology and Immunology, University of Texas School of Medicine, UT Health Science Center, San Antonio, TX, USA
Epigenetic modifications, such as histone post-translational modifications, DNA methylation, and alteration of gene expression by non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), are heritable changes that are independent from the genomic DNA sequence. These regulate gene activities and, therefore, cellular functions. Epigenetic modifications act in concert with transcription factors and play critical roles in B cell development and differentiation, thereby modulating antibody responses to foreign- and self-antigens. Upon antigen encounter by mature B cells in the periphery, alterations of these lymphocytes epigenetic landscape are induced by the same stimuli that drive the antibody response. Such alterations instruct B cells to undergo immunoglobulin (Ig) class switch DNA recombination (CSR) and somatic hypermutation (SHM), as well as differentiation to memory B cells or long-lived plasma cells for the immune memory. Inducible histone modifications, together with DNA methylation and miRNAs modulate the transcriptome, particularly the expression of activation-induced cytidine deaminase, which is essential for CSR and SHM, and factors central to plasma cell differentiation, such as B lymphocyte-induced maturation protein-1. These inducible B cell-intrinsic epigenetic marks guide the maturation of antibody responses. Combinatorial histone modifications also function as histone codes to target CSR and, possibly, SHM machinery to the Ig loci by recruiting specific adaptors that can stabilize CSR/SHM factors. In addition, lncRNAs, such as recently reported lncRNA-CSR and an lncRNA generated through transcription of the S region that form G-quadruplex structures, are also important for CSR targeting. Epigenetic dysregulation in B cells, including the aberrant expression of non-coding RNAs and alterations of histone modifications and DNA methylation, can result in aberrant antibody responses to foreign antigens, such as those on microbial pathogens, and generation of pathogenic autoantibodies, IgE in allergic reactions, as well as B cell neoplasia. Epigenetic marks would be attractive targets for new therapeutics for autoimmune and allergic diseases, and B cell malignancies.
Introduction
Epigenetic changes brought about by genetic susceptibility and/or environmental exposure can modulate gene expression and alter cellular functions without altering genomic sequences (1). Epigenetic modifications and factors, such as DNA methylation, histone post-translational modifications and non-coding RNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), comprise the epigenome and interact with genetic programs to regulate immune responses. Immunoglobulin (Ig) class switch DNA recombination (CSR) and somatic hypermutation (SHM) are critical for the production of protective antibodies against microbial pathogens, IgE in allergic responses, as well as pathogenic autoantibodies in autoimmune diseases. CSR recombines S region DNA located upstream of constant heavy-chain (CH) region exons, thereby encoding new Ig CH regions that endow antibodies new biological effector functions (2). SHM introduces mostly point mutations in variable regions of Ig heavy and light chains, thereby providing the structural substrate for antigen-mediated selection of B cell mutants with higher affinity B cell receptors (BCRs) (3–5). CSR and SHM occur mainly in germinal center and require activation-induced cytidine deaminase (AID, encoded by AICDA in humans and Aicda in mice), which is expressed in a differentiation stage-specific fashion in B cells (2–4). Class switched and hypermutated B cells further differentiate into long-lived memory B cells, which can react quickly to a recurrent antigenic challenge, or antibody-secreting plasma cells in a fashion critically dependent on B lymphocyte-induced maturation protein 1 (Blimp-1, encoded by PRDM1 in humans and Prdm1 in mice) (6, 7). Epigenetic modifications and factors influence gene expression and modulate critical B cell processes, such as CSR, SHM, and differentiation to memory B cells or plasma cells, thereby informing the antibody response (4, 8–10). Epigenetic dysregulation can result in aberrant antibody responses to exogenous antigens or self-antigens, such as chromatin, histones, and double-strand DNA in lupus.
B cell development and differentiation occur in two sequential stages. The initial, antigen-independent stage occurs in the bone marrow and involves recombination activating gene (RAG)1/RAG2-dependent V-(D)-J DNA rearrangement, which produces clonally unique Ig variable regions that specifically bind antigen. This stage generates mature, immunocompetent B cells that can bind to a unique antigen. The B cells move into the periphery and complete further, antigen-independent maturation into immunocompetent naïve mature B cells. In the periphery lymphoid organs, B cell undergoes the antigen-dependent stage of development or differentiation, upon activation by antigen binding and co-stimulation (5). In this stage, resting naïve mature B cells are induced to undergo cell proliferation, CSR, as well as SHM-mediated antibody affinity maturation, and differentiate into memory B cells, or short- or long-lived antibody-secreting plasma cells (6, 7). Multiple epigenetic changes are associated with each B cell development and differentiation stage. Resting, naïve B cells undergo VHDJH-Cμ transcription, which initiates at the VH promoter and runs through the intronic Sμ region and Cμ/Cδ exon clusters. This encodes the surface BCR, which comprises Igμ and Igδ heavy chain genes. These resting B cells display low levels of overall histone acetylation and genome-wide DNA hypermethylation, therefore most regions within the Ig heavy chain (IgH) locus are in a closed chromatin state (11), enriched in repressive histone post-translational modifications (e.g., H3K9me3 and H3K27me3) but lacking of activated histone modifications (12, 13). In B cells, epigenetic marks, such as DNA methylation, histone modifications, and miRNAs, are induced by the same stimuli that drive the antibody response, and modulate the transcriptome, especially the expression of AID, which is essential for SHM and CSR, and factors critical for plasma cell differentiation, such as Blimp-1 (4). By functioning as histone codes, combinatorial histone modifications also play a role in the targeting of the CSR and, possibly, SHM machinery to the Ig loci through recruiting specific scaffold proteins that stabilize CSR/SHM factors (8). These inducible B cell-intrinsic epigenetic marks control transcription programs that distinguish individual stages of B cell differentiation and underpin the molecular changes that are necessary for antibody response.
In this review, we provide a conceptual framework to understand how epigenetic modifications/factors modulate CSR and SHM, and the generation of plasma cells and memory B cells, with focus on AID-dependent peripheral B cell differentiation into memory B cells and long-lived plasma cells (but not differentiation of naïve B cells to short-lived plasma cells). We also highlight our current understanding of epigenetic modulations of CSR, SHM, and plasma cell differentiation by histone deacetylases (HDACs) inhibitors (HDIs). Finally, we summarize recent discoveries that indicate the importance of B cell epigenetic dysregulation in autoimmunity and B cell neoplasia.
Epigenetic Regulation of AID Induction
Somatic hypermutation and CSR are initiated by transcription through V(D)J and the donor/acceptor S regions that will undergo recombination, respectively, and are mediated by AID, a 198 amino acid protein, which is structurally and functionally similar to apolipoprotein B RNA-editing cytidine deaminases (APOBEC enzymes) (2, 3). AID shares a conserved catalytic domain with other members of the APOBEC family of cytosine or cytidine deaminases (3). It deaminates deoxycytidines (dCs) into deoxyuracils (dUs) yielding dU:dG mismatches. These mismatches can be repaired by an error-prone DNA repair pathway, which introduce somatic mutations, or processed by uracil DNA glycosylase (Ung), which is recruited to and stabilized on S regions by the scaffold functions of 14-3-3 adaptors, the translesion DNA synthesis (TLS) polymerase Rev1 and replication protein A (RPA), or elements of the mismatch repair (MMR) machinery, such as Msh2 and Msh6, which trigger DNA repair processes leading to introduction of point mutations (SHM) or double-strand DNA breaks (DSBs) (CSR) (2).
As a potent mutator, AID can effectively introduce mutations in not only Ig loci but also a variety of non-Ig genes, thereby causing genome instability in both B cells and non-B cells, including non-lymphoid cells, and contributing to tumorgenesis (3). Tight regulation of AID expression and function is necessary to maintain genomic stability in both B cells and non-B cells, and to avoid damages, such as chromosomal translocations, resulting from its dysregulation (14–18). This is achieved through fine control of transcription, post-transcription and post-translation regulation, nuclear/cytoplasmic distribution, stability, and activity (3) (Figure 1). AID express at a very low level (mostly undetectable) in naïve B cells, it is greatly induced in B cells undergoing SHM/CSR, and repressed in memory B cells and plasma cells to preserve the specificity, affinity, and isotype of antibody and BCR. Aicda transcription is under the control of multiple elements, particularly Homeobox protein C4 (HoxC4) (Figure 1). HoxC4 is a highly conserved helix-loop-helix homeodomain-containing transcription factor. As we have shown, HoxC4 directly binds to the Aicda promoter through an evolutionarily conserved 5′-ATTT-3′ site embedded within a conserved binding site for POU domain-containing transcription factors Oct1 and Oct2 (5′-ATTTGAAT-3′) (19). Sp1/Sp3 and NF-κB also bind the same promoter core and synergize with HoxC4 for Aicda induction (19, 20).
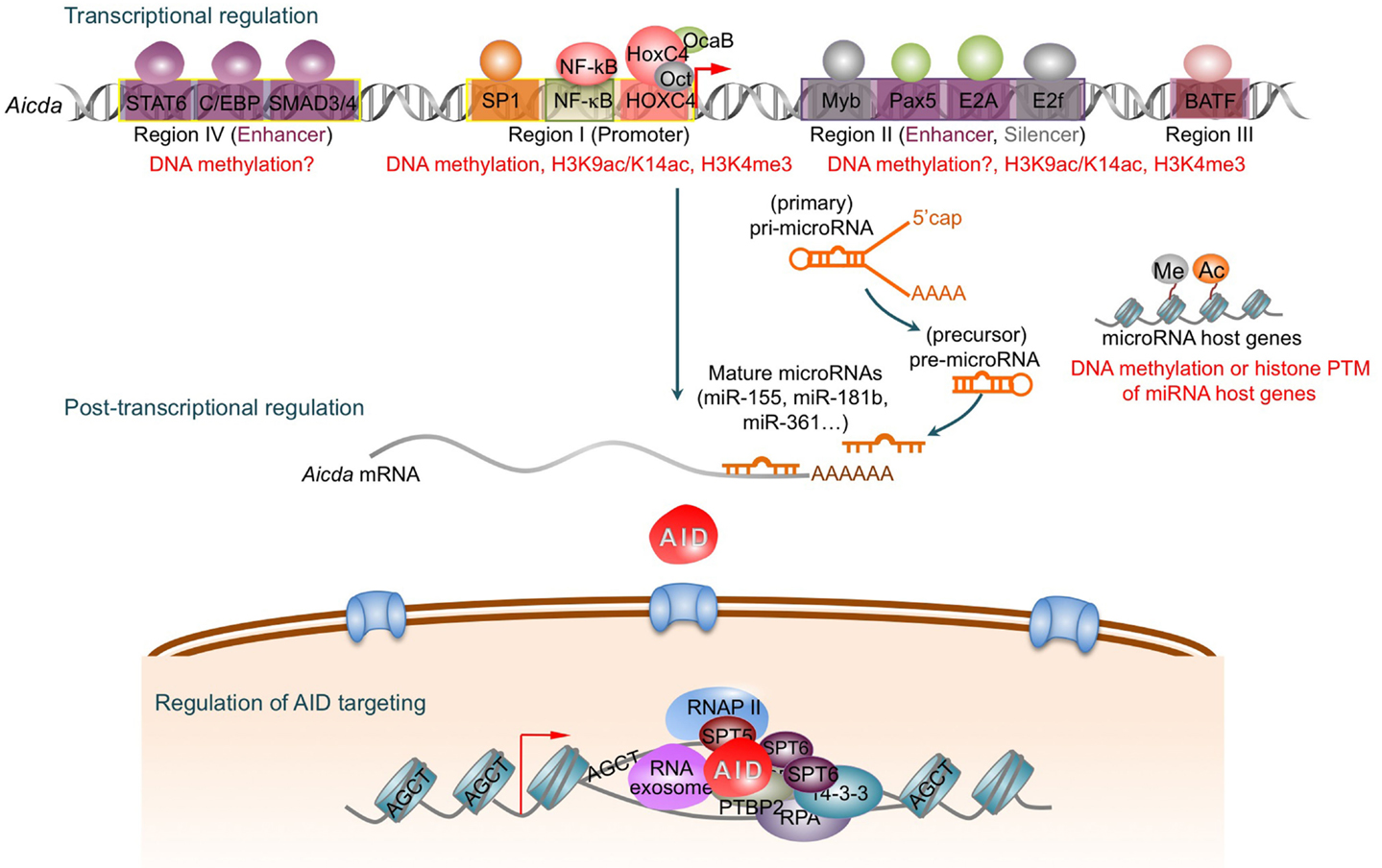
Figure 1. AID is tightly regulated at the transcriptional, post-transcriptional, and post-translational levels. Four distinct DNA regions (regions I–IV) of the AID gene (Aicda) locus possess binding sites for multiple transcription factors to regulate Aicda expression. Region I functions as promoters containing the binding sites for HoxC4/Oct and NF-κB/Sp1/Sp3, which can be induced by activating the Aicda promoter. In resting naive and memory B cells, and non-B cells, silencer elements in region II bind the repressor proteins E2f and c-Myb to counter the activity of the transcriptional activators. Stimulation of B cells with the primary inducing stimuli and cytokines that promote CSR induce activation signals through region IV enhancer in collaboration with the intronic enhancer in region II can overcome the effect of the region II silencer. After transcription, the Aicda mRNA can be negatively regulated by miR-155, miR-181b, and miR-361, which specifically bind to the conserved target sites on the 3′ UTR of Aicda mRNA. Nuclear AID is either degraded or exported back to the cytoplasm. Only a small proportion of AID molecules are targeted onto DNA at Ig or non-Ig loci by its co-factors. AID preferentially deaminates single-strand DNA, which emerges from transcription by RNA Pol II and depends on histone modifications in the transcribed locus. AID can be recruited to the open DNA before or during transcription. In CSR, AID recruitment to S regions occurs with interaction of RNA Pol II and AID-interacting factors, such as Spt5, Spt6, PTBP2, RNA exosome, lncRNAs, and 14-3-3 adaptor proteins, which would form a macromolecular complex. AID is enriched and stabilized on the targeted DNA by 14-3-3 adaptor proteins, which access the same S regions as the transcription machinery owing to their open chromatin state. These 14-3-3 adaptors are recruited and/or stabilized through interactions with 5′-AGCT-3′ repeats and possibly by H3K9acS10ph. The RNA exosome also interacts with AID and allows AID to deaminate both the transcribed and the non-transcribed DNA strand in the S regions undergoing transcription. AID deaminate dCs into dUs to yield dU:dG mismatches. Resolution of these lesions can lead to different physiological or pathological outcomes.
Regulation of AID Expression by DNA Modifications and Histone Modifications
The expression of Aicda is modulated by changes of Aicda epigenetic status. Suppression of Aicda expression in naïve B cells is mediated by promoter DNA hypermethylation (21). In naive B cells, in which AID is not expressed, histone H3 acetylation occurs in the Aicda gene at low levels comparable to the overall H3 acetylation in the genome and neighboring genes, such as Mfap5, which is not significantly expressed in B cells (22). Upon activation of B cells, DNA of the Aicda gene is demethylated and the locus becomes enriched in H3K4me3 and H3K9ac/K14ac. B cell activation by LPS plus IL-4, which induces Aicda transcription, greatly increases H3 acetylation at the Aicda locus, particularly in the Aicda regulatory regions (22). These epigenetic modifications, together with induction of nuclear factor NF-κB, homeobox protein HoxC4 and other transcription factors, activate Aicda transcription. Transcription elongation depends on the H3K36me3 posttranslational modicification, an intragenic mark of gene activation in the Aicda gene body, as suggested by AID down-regulation following depletion of the H3K36me3 methyltransferase Setd2 (3, 23). Post-CSR/SHM down-regulation of Aicda transcription probably results from remethylation of the Aicda DNA.
Regulation of AID Expression by miRNA
In addition to DNA methylation of the Aicda promoter and histone acetylation of the Aicda locus, selected miRNAs provide a more important mechanism of modulation of AID expression. miRNAs are small (~21–23 nt), evolutionarily conserved non-coding RNAs derived from much larger primary transcripts encoded by their “host genes.” miRNAs bind to complementary sequences within the 3′ untranslated region (3′ UTR) of their target mRNAs and negatively regulate protein expression at the post-transcriptional level. miRNA can recognize its target mRNA through the short (as few as 6–8 nt) “seed sequence” at the 5′ end of the miRNA. miRNA repress gene expression by accelerating mRNA degradation and inhibiting mRNA translation. A given miRNA can potentially target different mRNAs, and a given mRNA can be targeted by multiple miRNAs. The mammalian genome encodes hundreds of miRNAs that collectively impact the expression of about one-third of all genes. miRNAs are transcribed from intergenic genomic DNA by RNA polymerase (Pol) II or Pol III as primary transcripts (pri-miRNAs) that are then cleaved by the nuclear ribonuclease Drosha to generate an about 70 bp long characteristic stem-loop structure, known as a pre-miRNA, and exported to the cytoplasm (24). The pre-miRNA is further processed by cytoplasmic enzyme Dicer into mature miRNA (24), which forms complexes with ribonucleoproteins (RNPs) and expresses regulatory effects (25, 26).
microRNAs control various biological processes by fine-tuning gene expression at the post-transcriptional level. They can modulate a wide range of physiological and pathological processes by regulating cellular function at every aspect, from proliferation and apoptosis to differentiation. miRNAs are present in all human and mouse cells, in which they each modulate the expression of a few to hundreds of target genes. miRNAs likely play important roles in B cell development and peripheral differentiation, as well as T cell stage-specific differentiation and autoimmunity. Naïve B cells, germinal center B cells, memory B cells, and plasma cells show distinct miRNA expression profiles (27, 28). Deletion of Dicer, which is critical for miRNA maturation, in activated B cells resulted in defective generation of germinal center B cells, memory B cells, and plasma cells (29). Indeed, Dicer-deficient B cells showed impaired biogenesis of many miRNAs, including miR-155 and miR-125b, which regulate expression of genes that modulate B cell germinal center reaction and plasma cell differentiation (30, 31). A single miRNA, such as miR-155, can target multiple genes, including Myd88, Pu.1, and Aicda, and regulate sequential stages of B cell differentiation (30, 32–34). By contrast, multiple miRNAs, such as miR-15a and miR-16, can cooperatively repress one critical gene in germinal center B cells, Bcl2 (27).
miR-155, miR-181b, and miR-361 modulate AID expression by binding to the evolutionarily conserved target sites in the 3′ UTR of AICDA/Aicda mRNA, thereby reducing both AICDA/Aicda mRNA and AID protein levels (33–37). These miRNAs could suppress AID expression in naïve B cells and in B cells that completed SHM/CSR. miR-155 is the most abundant miRNA that has been shown to silence AID expression. miR-155 is processed from an RNA sequence encoded by miR-155 host gene (miR155HG). This was originally identified as a gene transcriptionally activated by promoter insertion at a common retroviral integration site in B-cell lymphomas (Bic, B-cell integration cluster). Bic RNA is a spliced and polyadenylated but non-protein-coding RNA that accumulates in lymphoma cells (32) and is induced along with AID in B cells activated by CSR-inducing stimuli (33, 34). The sequences of pre-miRNA-155 and mature miR-155 are highly conserved across more than 22 different organisms, including mammals, amphibians, birds, reptiles, sea squirts, and sea lampreys (38).
miR-155 and miR-361 are directly repressed by BCL-6, a transcriptional repressor required for germinal center formation. miR-361 is embedded in the CHM gene, which encodes a subunit of a Rab geranylgeranyl transferase and is known for its genetic inactivation in choroideremia (37). BCL-6 display a coordinated activity in sustaining high levels of AID expression in germinal center B cells undergoing CSR and SHM. By direct repressing miR-155 and miR-361, BCL-6 upregulates the expression of the target genes of these miRNAs, including AICDA and other elements involved in the maintenance of the germinal center B cell centroblast phenotype (37). The specific effect of miR-155 in the regulation of AID expression was demonstrated by the findings that disruption of the miR-155 binding site in the 3′ UTR of Aicda mRNA in B cells led to an increase in Aicda mRNA and AID protein by increasing the half-life of Aicda mRNA, resulting in increased CSR and c-Myc/IgH translocations (33, 34). Nevertheless, the role of miR-155 in regulating B cell function and antibody response is much more than modulating the expression of AID. miR-155 is expressed at a high level in germinal center B cells and plays an important role in germinal center formation and subsequent antibody response following antigen challenge (30, 32, 39). miR-155 deficiency in B cells resulted in reduced extra follicular and germinal center responses, decreased numbers of IgG1+ plasma cells and memory B cells, and failed production of high-affinity IgG1 antibodies (30). In B6/Faslpr/lpr mice, deficiency of miR-155 results in a reduced autoantibody production and autoimmunity (40). This is likely stemmed from dysregulation of a variety of genes in multiple immune cells, including derepressed expression of SHIP-1 in B cells, which lead to mitigation of B-cell activation, proliferation and autoantibody production (40).
The 3′ UTR of Aicda mRNA contains multiple putative binding sites for miR-181b, which is predominantly expressed in lymphoid cells (10, 41). miR-181b is expressed at the highest levels in resting B cells and is downregulated upon B cell activation by CSR-inducing stimuli (34, 42). Expression of miR-181b in B cells leads to down-regulation of AID at both the transcript and protein levels. It has been suggested that miR-181b and miR-155 have non-overlapping functions in controlling AID expression. miR-181b may inhibit premature AID activity but allows proper AID transcriptional activation at early time points, while miR-155 could narrow AID function at a later stage of activation (34). By controlling AID expression, miR-155 and miR-181b protect resting B cells and non-B cells from AID-mediated mutagenesis. Accordingly, in Burkitt’s lymphoma B cells, deficiency of miR-155 expression is associated with high levels of somatic mutations and inter-chromosomal translocations (43).
Epigenetic Regulation of AID Targeting in CSR and SHM
One fundamental question for B cell biology remains to be answered is how CSR and SHM machineries are targeted to the Ig locus. For CSR to take place, the IgH genes are subjected to transcriptional activation, RNA splicing, AID-mediated cytidine deamination, as well as DNA cleavage, repair, and recombination. Each of these events is likely associated with, and possibly regulated by specific changes in chromatin structure. Histone modifications in S and V(D)J regions are critical for targeting of the CSR and SHM machinery, respectively (Table 1). Chromatin structure that impacts on and likely regulate most aspects of gene expression, also contributes to the regulation of CSR and SHM. In B cells poised to undergo CSR, the IgH genes are in an “accessible” chromatin conformation before CSR (44). Upon induction of germ-line transcription, histones H3 and H4 have been shown to be acetylated at the IH exon promoters and S regions (45, 46).
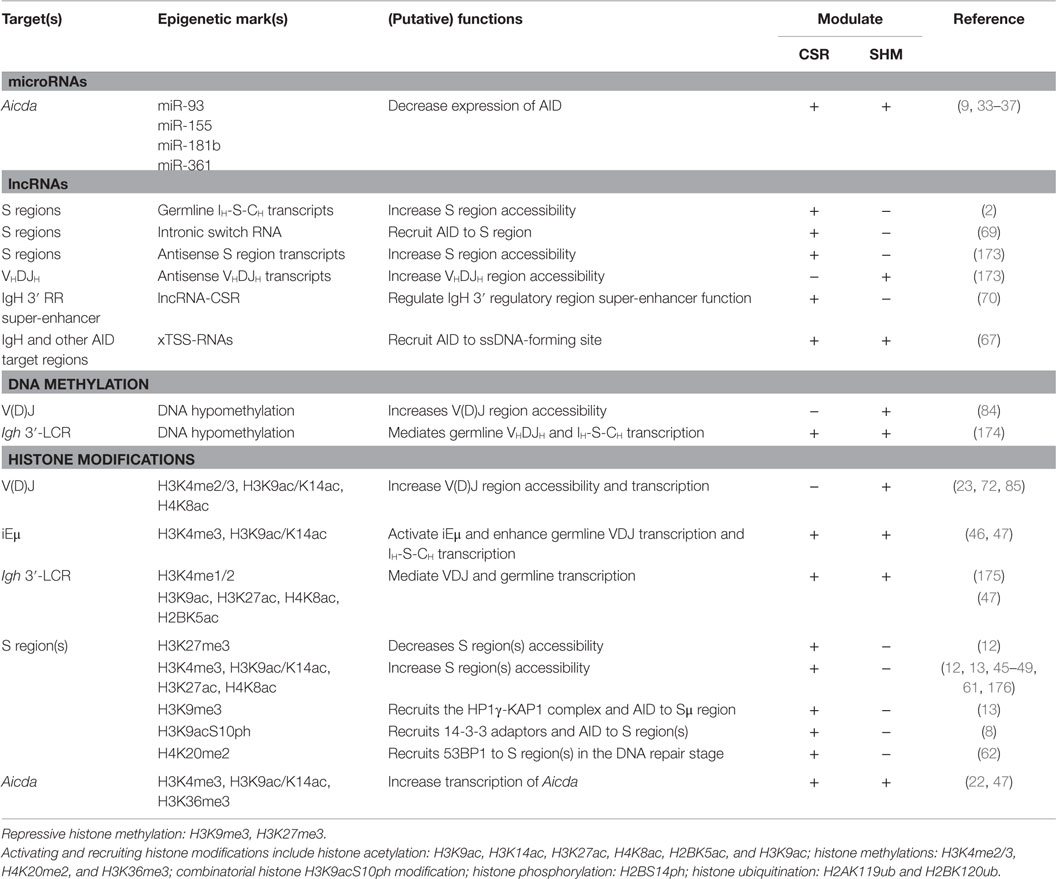
Table 1. Epigenetic marks/factors, and their functions in CSR and SHM.
Epigenetic Targeting of the CSR Machinery
Histone Modifications in CSR Targeting
Histone post-translational modifications are important for targeting of the CSR machinery to the upstream donor and the downstream acceptor S regions that are involved in CSR (Table 1). The significant levels of germline Iμ-Sμ-Cμ transcription and activating histone marks, such as H3K4me3, H3K36me3, H2BK5ac, H3K9ac/K14ac, H3K27ac, and H4K8ac, are observed in the donor Sμ region even in resting naïve B cells, suggesting that Sμ is in a constitutively open state and poised for switch recombination (8). CSR induction requires both primary and secondary (CSR-inducing) stimuli. Primary stimuli, such as engagement of B cell CD40 by CD154 expressed on activated T cells, and T-independent dual toll-like receptor (TLR) and BCR engagement, induce B cell activation, proliferation, and differentiation. In conjunction with primary stimuli, secondary stimuli, which comprise cytokines (e.g., IL-4, TGF-β, and IFN-γ), direct class switching to IgG, IgA, or IgE by selecting the acceptor S region, through activation of S region histone modifications and inducing specific germline IH-S-CH transcription (46–49). Primary stimuli induce histone-modifying enzymes and trigger chromatin decondensation in downstream S regions by removing repressive H3K9me3 and H3K27me3. These allow histone-modifying enzymes to ride on the RNA Pol II to reach S regions during germline IH-S-CH transcription elongation (46, 47, 50–52). Upon RNA Pol II stalling, caused by complex secondary DNA structures, such as cruciform-like structures and R-loop, in S regions (2, 53), histone-modifying enzymes catalyze histone modifications in these regions. DNA transcription together with modified histones opens the chromatin in S regions, thereby allowing for access of CSR factors. The role of histone-modifying enzymes in CSR is further emphasized by the reduced S region histone modifications and CSR in B cells knockdown H3K4 methyltransferase Set1 (48), or H3K9 acetyltransferases Pcaf and Gcn5 (8).
In the donor and acceptor S regions, activating histone modifications, such as H3K9ac and H3K4me3, are enriched at levels much higher than those in the associated IH promoter and CH regions (46, 47). These histone marks are read by CSR factors, including 14-3-3 adaptors, which, such as AID and histone-modifying enzymes, are induced by primary CSR stimuli (54). 14-3-3 adaptors directly interact with AID and target it to the upstream and downstream S regions that undergo recombination (2), thereby transducing the epigenetic code. 14-3-3 adaptors specifically bind to the combinatorial histone H3K9acS10ph modification in S regions and 5′-AGCT-3′ (2, 54) repeats. These are characteristic motifs of all IgH locus S regions, but not IH promoters, CH regions or other genome areas (54). Due to their high affinity for 5′-AGCT-3′ repeats, 14-3-3 can potentially bind all S regions. Nevertheless, these adaptors are recruited only to the S regions that undergo recombination (54). This is due to the open chromatin state of such regions as well as specific binding of 14-3-3 to H3K9acS10ph (8). The specificity of 14-3-3 adaptors for H3K9acS10ph and 5′-AGCT-3′ repeats are evocative of RAG1/RAG2 complex specificity for H3K4me3 and V, D and J gene recombination signal sequences (RSSs) (55).
H3K4 methylation, particularly H3K4me3, plays a critical role in AID-mediated DNA cleavage in S regions during CSR (48). The formation of H3K4me3 at AID target loci is dependent on the histone chaperone complex, facilitates chromatin transcription (FACT), a chromatin-modifying complex during RNA processing (48, 56). We have shown that specific H3K4 methyltransferases and H3K9 acetyltransferases can be induced by TLR or CD40 signaling and catalyze histone H3K4me3 and H3K9ac/K14ac modifications. These are decorated S regions, regardless of whether they are targets of CSR (8). Conversely, the combinatorial histone H3K9acS10ph modification specifically marks the S regions set to recombine and directly recruits 14-3-3 adaptors for AID stabilization (8). 14-3-3 adaptors, which possess no enzymatic activity, function as histone code readers to recruit/stabilize downstream effector molecules, which per se cannot read histone codes, consistent with the “histone code hypothesis” (57, 58). Inhibition of the enzymatic activity of Gcn5/Pcaf histone acetyltransferases leads to decreased H3K9acS10ph, 14-3-3 recruitment and AID stabilization in S regions, and CSR.
H3K9me3 is also present, although at a relatively low level, in Sμ but not downstream S regions. In the Sμ region, H3K9me3 recruits the KAP1-HP1γ complex to stabilize AID (13). Accordingly, CSR to IgA can be impaired by deletion of the H3K9 methyltransferase Suv39 (59). Histone modification readers that function as scaffolds, such as 14-3-3 adaptors, translesion DNA synthesis polymerase Rev1 (60) and RPA (61), act as core for the assembly of macromolecular complexes on S region DNA, to stabilize AID and/or Ung for generation of DNA lesions. Histone modifications are also recognized by DNA repair factors, such as p53-binding protein 1 (53BP1), which may functions as a scaffold to recruit/stabilize additional DNA repair factors for CSR. Abrogation of histone methyltransferase MMSET expression impairs H4K20me2 enrichment and 53BP1 recruitment in S regions, and results in reduced CSR (62).
Suppressor of Ty6 (Spt6), a RNA Pol II-interacting histone H3-H4 chaperone, also plays a role in the regulation of H3K4me3 for SHM and CSR (23). Depletion of Spt6 impaired H3K4me3 and AID-mediated DSBs in the S regions in CH12 B cells, which can otherwise be induced to express AID and CSR to IgA (23). In addition, knockdown Spt6 in human Burkitt’s lymphoma BL2 cells overexpressing mutant AID (JP8Bdel) that lacks C-terminal 16 aa residues, which are critical for CSR but not to SHM (63), abolished SHM and H3K4me3 in Ig VH region and non-Ig AID target genes (23). Thus, activating histone modifications are induced in S regions and create an open chromatin state, which allows for the access of the CSR machinery. Epigenetic specification of CSR targeting entails reading of histone codes by scaffold proteins, which orchestrate the assembly of macromolecular complexes in the sequential DNA lesion and repair stages.
Long Non-Coding RNA and CSR Targeting
Long non-coding RNAs are evolutionarily conserved non-coding RNA molecules that are longer than 200 nt and located within the intergenic loci or regions overlapping antisense transcripts of protein coding genes (64–66). Their expression can be tissue- and cell-type specific. lncRNAs are involved in numerous cellular functions, such as transcriptional regulation, RNA processing, RNA modification and epigenetic silencing. lncRNAs have been recently shown to play an important role in the targeting of the CSR machinery (Table 1). They target AID to divergently transcribed loci in B cells (67). In B cells undergoing CSR, the RNA exosome, a cellular RNA-processing/degradation complex is required for optimal CSR (68). The RNA exosome associates with AID, accumulates on S regions in an AID-dependent fashion. Both the cellular RNA exosome complex and a recombinant RNA exosome core complex inform robust transcription-dependent DNA deamination by AID in both strands of transcribed SHM substrates in vitro. In B cells, deficiency of Exosc3 or Exosc10, the essential subunits of the RNA exosome complex, impairs CSR and SHM (67, 69). Many novel RNA exosome substrate lncRNAs have been identified by transcriptome analysis of Exosc3- or Exosc10-deficient B cells. RNA exosome-regulated, antisense-transcribed regions accumulate single-strand DNA structures containing RNA-DNA hybrids and recruit AID in B cell. RNA exosome regulation of lncRNA has been suggested to recruit AID to single-strand DNA-forming sites of antisense and divergent transcription in the B cell genome (67). In addition, a recent study has shown that an lncRNA generated by S region transcription followed by lariat debranching can fold into G-quadruplex structures, which can be directly bound by AID, thereby targeting of AID to S region DNA (70).
Epigenetic Targeting of the SHM Machinery
Activation-induced cytidine deaminase initiates SHM by deaminating cytosine residues in Ig V(D)J genes. It also introduces DNA damages, including point-mutations, in non-Ig loci at a lower frequency. How AID is recruited to the target sites is not fully understood. AID has been suggested to target a specific microenvironment rather than a defined set of genes (71). The SHM machinery is targeted to the V(D)J region through unique targeting sequences, transcription, and possibly, DNA demethylation and histone modifications (Table 1) (72). Like CSR, SHM requires not only the expression of AID, but also transcription in the target regions. Alterations in chromatin structure at IgH may also play an important role in promoting and/or stabilizing AID targeting (73). AID targets a distinct set of hotspots, which are concentrated in genes that are highly transcribed but frequently stalled genes (74). AID associates with RNA Pol II (75). RNA Pol II transcription, as well as several chromatin alterations could facilitate the access of AID to Ig loci and give use to single-stranded DNA, a preferential substrate for AID deamination. However, transcription alone is insufficient to recruit AID activity (74). AID targets are predominantly grouped within topological complex, highly transcribed superenhancers and regulatory clusters, which are enriched in chromatin modifications associated with active enhancers (such as H3K27Ac), they are also and marks of active transcription (such as H3K36me3), indicating that these features are universal mediators of AID recruitment (71, 74, 76). In both human and mouse B cells, there is a strong overlap between hypermutated genes and superenhancer domains (71).
The phosphorylated histone H2B (H2BSer14P) correlates tightly with SHM and CSR. In Ig V(D)J and S regions, H2B phosphorylation requires AID and may be mediated by the histone kinase Mst1 (72, 77). It has been suggested that SHM and CSR trigger distinct DNA damage responses and identify a novel histone modification pattern for SHM consisting of H2B (Ser14P) in the absence of γH2AX (72, 77). The non-Ig AID targets share important characteristics with Ig genes, namely, repetitive sequences that can form non-B DNA structures upon efficient transcription, and the accumulation of chromatin H3K4me3 histone marks (78).
The FACT complex may also promote SHM (56). FACT, a histone chaperone-type elongation factor, was originally discovered for its biochemical activity to promote transcription elongation of RNAPII on the nucleosomal DNA template (79). It has been suggested to remove nucleosomal histones and deposit them at the RNAPII of transcription site, and this allows the RNA polymerase to be proceed beyond the nucleosomes (80). FACT is important for inducing H3K4me3, which can be recognized by a protein complex with DNA cleaving activity and accumulates at SHM-targeted genomic regions (48). Furthermore, FACT and histone variant H3.3, a hallmark of replication-independent histone turnover, are enriched at the heavy and light chain V(D)J regions, the light chain Jκ5 region and the Sμ region 5′ flanking sequence (48). The importance of the chromatin histone-exchanging dynamics in SHM target regions, especially Ig genes, is emphasized by high abundant FACT and H3.3 deposition in the most efficient targets of SHM (56, 81). Histone post-translational modifications would also mediate recruitment of DNA repair factors, such as error-prone TLS DNA polymerases, in SHM at the DNA repair stage. H2AK119 ubiquitination (ub) and H2BK120ub are enriched in VHDJH but not CH regions and colocalize with DNA Pol η, an important TLS polymerase for SHM, in AID nuclear foci (82). Pol η is likely recruited to those loci by H2AK119ub and H2BK120ub, as well as by monoubiquitinated PCNA scaffold, through its ubiquitin-binding domain (83). As mentioned before, AID-dependent histone H2BSer14P mark in the V(D)J region (72) may also contributes to recruitment of DNA repair factors. The role of DNA hypomethylation in SHM has been suggested by the finding that only the hypomethylated allele is hypermutated in B cells carrying two nearly identical pre-rearranged Igκ transgenic alleles, even though transcription of both alleles are comparable (84).
It is possible that DNA demethylation facilitates SHM targeting by promoting histone modifications H3K4me3, H3K9ac/K14ac and H4K8ac, which are enriched in the V(D)J region and associated with an open chromatin state (23, 72, 85). Both H3K4me3 and H4K8ac are involved in SHM. Decreased H3K4me3 in VHDJH regions in human BL2 cells, a Burkitt’s lymphoma cell line that can be induced to undergo SHM, upon knockdown of histone chaperone Spt6 is associated with reduced VHDJH mutations (23). In BL2 cells, H4K8ac increases concomitantly with VHDJH mutations upon treatment with HDI trichostatin A (TSA) (85). Persistent H4K8ac in VλJλ region required E2A, whose inactivation result in decreased mutations (72, 86).
Some cis-acting regulatory regions, such as Ig enhancer and Ig enhancer-like sequences, are important for targeting SHM to Ig loci (87, 88). In chicken, mouse and human B cells that Ig locus enhancers and enhancer-like elements function as core diversification activator (DIVAC) sequences that work together to target SHM (88). In chicken DT40 B cells, short mammalian Igλ and IgH enhancer fragments can increase mutation rates by more than 20-fold (88). lncRNAs, which are likely to regulate many biological functions, have been recently shown to be link to enhancer activity. lncRNAs are expressed in a lineage-specific fashion and function through RNA–protein, RNA–DNA, or RNA–RNA target interactions. They are induced to modulate innate and adaptive immunity (66). Many regulatory lncRNAs can be categorized as DNA accessibility modulators, and likely play a role in SHM targeting, especially in conjunction with the function of Ig enhancers (67). Thus, activating histone modifications, DNA hypomethylation, and possibly lncRNAs increase V(D)J region chromatin accessibility to the SHM machinery, including AID and error-prone DNA repair factors, which can be stabilized by modified histones.
Epigenetic Regulation of Plasma Cell Differentiation
B cell differentiation is initiated by extracellular stimuli that bind to cellular receptors and trigger a signaling cascade resulting in the induction of transcription factors that reprogram B cells to secrete antibodies. The function of transcription factors is controlled by the accessibility to DNA through epigenetic modifications. Little is known about how the epigenetic mechanisms direct B cell differentiation into antibody-secreting plasma cells (6). Plasma cells are terminally differentiated elements in the B cell lineage that mostly have undergone SHM and CSR. These cells do not proliferate, but secrete large volumes (107 molecules/h) of clone-specific antibodies. Plasma cells are derived from either germinal center or memory B cells. Although some memory B cells are IgM+, most memory B cells are class-switched and express mutated V(D)J gene segments. Upon reactivation by specific antigen, memory B cells differentiate into antibody-secreting plasma cells to mediate an anamnestic humoral response (89). Reactivated memory B cells can also reenter into the germinal center reaction and undergo further CSR and/or SHM before differentiating into plasma cells or reverting back to memory B cells (90, 91).
Plasma cells display a transcriptional signature that is distinct from B cells (92). Their changes in gene expression correlated with the acquisition of permissive histone modifications, including H3K4me1 and H3K4me4, which are enriched in active promoters and distal enhancers and play an important role in B cell development (92, 93). Upregulation of Blimp-1, a transcriptional repressor, is central to plasma differentiation (6). Blimp-1 down-regulates the expression of Bcl6, Pax5, and Spib, all of which inhibit B cell differentiation into plasma cells by binding to the promoters of these genes (6), and, possibly, deacetylating their promoters. Indeed, Pax5, Spib, and perhaps Bcl6 promoters display decreased histone acetylation in plasma cells (94, 95). Furthermore, Blimp-1 can interact with HDACs that remove acetyl groups on a histone (94). In addition, in plasma cells, Blimp-1 down-regulates c-Myc expression through a similar epigenetic mechanism (94), thereby maintaining the terminal differentiation state of these cells (6). Finally, Blimp-1 can interact with H3K9 methyltransferase G9a and likely recruits this enzyme to the Pax5 and Spib promoters, thereby increasing H3K9me3 and repressing activation of these promoters (95, 96). Thus, epigenetic induction of Blimp-1 and Blimp-1-mediated epigenetic inhibition of target genes drives plasma cell differentiation, and possibly maintains plasma cell identity. Down-regulation of Pax-5 and Pax-5-driven Bcl-6 lead to derepression of the Prdm1 promoter from Bcl-6-mediated epigenetic silencing. This is associated with increased histone acetylation in the Prdm1 promoter, likely resulting from release of Bcl-6-bound HDACs (97, 98). Reduction of Blimp-1 in a plasmacytoid cell line by enforced expression of Bcl-6 resulted in re-expression of B cell markers, including CD19 (98).
Prdm1 mRNA contains a long (>2,000 nt) 3′ UTR, which can be potentially targeted by multiple miRNAs, including miR-9, miR-23b, miR-30, miR-125b, miR-127, and let-7 (10, 27, 28, 31, 99–102). Overexpression of miR-125b in B cells impairs expression of Blimp-1 and inhibits B cell differentiation into plasma cells (31). In addition, miR-125b can downregulate IFN regulatory factor (Irf)-4, which reciprocally regulates Blimp-1 and is required for the generation of plasma cells (27, 31, 103). Furthermore, X-box binding protein (Xbp)-1 that governs late events of plasma cell differentiation can be downregulated by miR-127 (102).
Epigenetic Regulation of Memory B Cell Differentiation
B cell memory is a hallmark of adaptive immunity. Memory B cells are antigen-experienced quiescent B cells, which can be generated in response to both T-dependent antigens (usually proteins) and T-independent antigens (usually carbohydrates) (7). B cells quickly react to a second challenge with the same antigen, thereby providing humoral immune protection. While inherit epigenetic information from their active B cell precursors, memory B cells acquire new epigenetic marks, which make these resting B cells poised to quickly respond to experienced antigen. Expression of memory B cell hallmark genes, such as CD27 (in humans) and Cd38 (in mice), is likely mediated by histone modifications induced during B cell activation (4). Genes that control B cell identity and function, including Pax5 and Spib, are also expressed in memory B cells, likely reflecting the epigenetic state that originated in naïve B cells and led them to memory B cells differentiation. Post-recombined S–S regions in the IgH locus show constitutive (upstream Sμ portion) or induced (downstream Sγ, Sϵ, or Sα portion) histone modifications, which could be transferred to memory B cells and result in comparable epigenetic landscapes in class-switched memory B cells (4). The functional distinction between memory B cells and their naïve counterparts could at least partially result from the epigenetic alterations.
Quiescent and activated B cells display different histone marks (104). In resting cells, histone lysine methylation was reduced as compared to activated cells (105). Enhancer of zeste homolog 2 (Ezh2) catalyzes H3K27me3, which is enriched at transcription start sites of repressed genes, through its SET domain. EZH2 is highly expressed in human germinal center B cells. Inactivation of Ezh2 in mouse germinal center B cells resulted in a profound reduction of germinal center reactions, memory B cell formation, and antibody response (106). Ezh2 protected germinal center B cells against AID mutagenesis and facilitated cell cycle progression. Repression of Blimp-1 and Irf4 expression in germinal center B cells is also necessary to limit plasma cell differentiation (106). In Ezh2fl/flCγ1-Cre mice, the B cell differentiation stage-specific Ezh2 deficiency resulted in profound impairment of germinal center reactions and memory B cell formation, suggesting that the methyltransferase activity of Ezh2 is essential for not only germinal center B cell functions but also generation of memory B cells (106). Furthermore, it has been recently shown that histone acetyltransferase monocytic leukemia zinc finger protein (MOZ), which specifically targets H3K9 and plays a role in stem cell self-renewal, regulates B cell memory formation, controlling memory compartment composition (104). This activity of MOZ is B cell-intrinsic and is required for establishing the germinal center gene expression program. B cell stage-specific deletion of MOZ alters fate decisions in both primary and secondary antibody responses. The lack of MOZ affected the functional outcome of antibody responses, with an increase in secondary germinal centers and a corresponding decrease in secondary high-affinity antibody-secreting cell formation (104).
The differentiation of naïve B cells to germinal center B cells and then to plasma cells or memory B cells would also be associated with changes in DNA methylation. Several genes can be silenced by DNA methylation catalyzed by DNA methyltransferases (DNMTs), such as DNMT3a, which is highly expressed in memory B cells (107). In memory B cells, different expression, as compare to naïve B cells, of the immune activation related elements is likely concomitant with distinctive DNA methylation (108). This supports the concept that the memory B cell epigenome is poised to facilitate a more rapid and robust activation response than that of its naive counterparts.
As demonstrated by concomitant miRNA and mRNA profiling, miRNAs play a regulatory role at every stage of the B cell peripheral differentiation process (28). Selected miRNAs, including miR-125b and let-7, negatively regulate Prdm1 (10, 27, 28, 101). Down-regulation of miR-15a and miR-16, which target Bcl2, likely contributes to memory B cell survival (27, 109) and re-expression of Krüppel-like factors (KLFs), which can bind HDACs, mediates memory B cell quiescence (110). miR-223 is enriched in human memory B cells and down-regulates the expression of LMO2, a key transcription factor in B cell differentiation (28). In addition to function as a negative regulator for CSR and SHM by silencing AID, miR-155 also plays an important role in memory B cell responses. miR-155 deficiency greatly reduced memory B cells (111). Several lncRNAs have been shown to be preferentially expressed in human memory B cells, but not in naïve B cells or B1 cells (112), suggesting that lncRNA also play a role in memory B cell differentiation. Thus, DNA methylation, histone modifications and non-coding RNAs, especially miRNAs, control gene expression programs that lead to B cell differentiation into plasma cells and memory B cells, and maintain the identity of these differentiated cells.
Epigenetic Modulation of Aid and Blimp-1 Expression, CSR, SHM, and Plasma Cell Differentiation by HDAC Inhibitors
Histone deacetylases are a class of enzymes that remove the acetyl groups from the lysine residues on a histone leading to the formation of a condensed and transcriptionally silenced chromatin. HDIs block this action and can result in histone hyperacetylation, thereby affecting gene expression. HDIs have been shown to alter gene expression by altering chromatin accessibility (113–115). In immune cells, these epigenetic modifiers exert modulatory effects even at moderate concentrations. By using well-characterized short-chain fatty acid (SCFA) HDIs, valproic acid (VPA) (116), and butyrate (117), we have shown that HDIs regulate intrinsic B cell functions that are critical in shaping effective antibody and autoantibody responses. Our findings were further supported by a recent publication showing that HDIs Panobinostat (a novel broad-spectrum HDI) and Vorinostat (suberanilohydroxamic acid or SAHA) significantly impair antibody and autoantibody responses (118). VPA is an FDA-approved drug, widely used as an anticonvulsant and a mood-stabilizer. It selectively inhibits class I HDACs, particularly, HDAC1 and HDAC2, and, less effectively, class IIa HDACs, of the four HDAC classes identified in mammals (116, 119). Butyrate is a major metabolite in the digestive tract, arising from bacterial fermentation of dietary fibers, mainly “resistant” starch (120, 121), and it is widely available as a dietary supplement. Butyrate modulates gene expression by selectively inhibiting HDAC1 and HDAC3, and, less effectively, other members of class I and class IIa HDACs (117). SCFA HDIs have been suggested to display significant selectivity for different HDACs (122). HDAC activity is mostly associated with multiprotein complexes, the role and composition of which are often cell type-specific. HDAC-associated proteins would specify the selectivity of HDI, which display different affinities for different HDAC/co-factor complexes. HDIs with diverse chemical properties target different HDACs and HDAC/co-factor complexes, thereby regulating gene expression in a locus- and cell type-specific fashion (122). In B cells, HDIs would modulate miRNAs selectively, possibly as a result of HDACs existing in unique contexts of HDAC/co-factor complexes, as occurring in these lymphocytes, particularly when in an activated state (10).
Although HDIs may also indirectly modulate antibody responses or diminish autoimmunity by affecting elements other than B cells, such as innate immune cells (123) and T cells (Treg, TH1, and TH17 cells), or inhibit proinflammatory cytokines (115, 124–126), HDIs would directly regulate B cell genes that are central to the peripheral differentiation of these lymphocytes and the maturation of antibody and autoantibody responses (Figure 2) (10). Silencing of AICDA/Aicda and PRDM1/Prdm1 (and XBP1/Xbp1) by HDIs has been found to be intrinsic to B cells and independent of other cellular elements, as shown by our in vitro experiments using purified human and mouse B cells, as well as our in vivo studies of the T-dependent response to NP-CGG and the T-independent response to NP-LPS. In both in vivo and in vitro, HDI-mediated down-regulation of AICDA/Aicda and PRDM1/Prdm1 expression was associated with a concomitant increase of the respective B cell targeting miRNAs (miR-155, miR-181b, and miR-361 for AICDA/Aicda; miR-23b, miR-30a, and miR-125b for PRDM1/Prdm1), in a tight dose-dependent fashion (10). HDI-induced down-regulation of XBP1/Xbp1 could be secondary to decreased Blimp-1 expression.
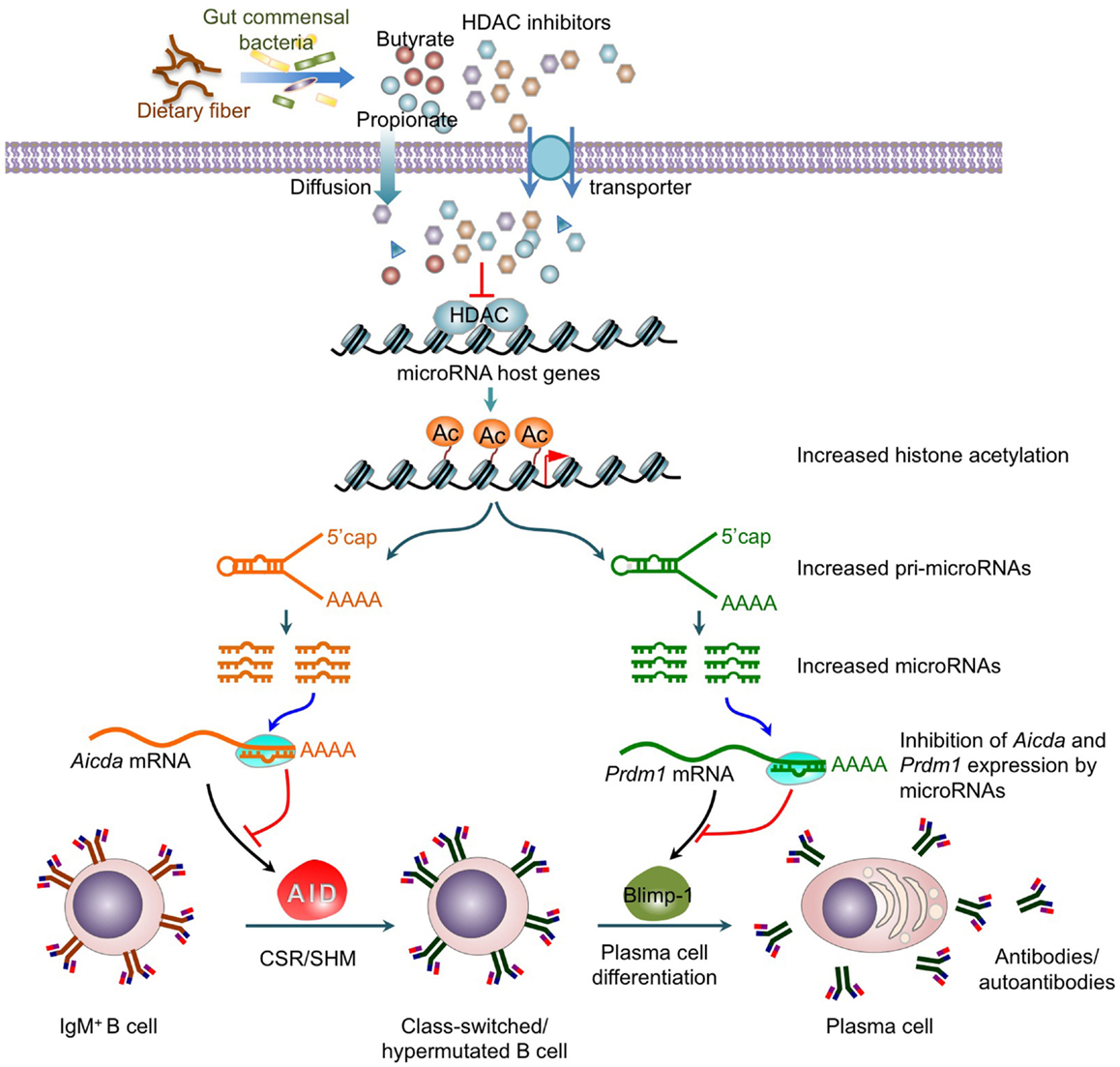
Figure 2. Histone deacetylase inhibitors upregulate selected B cell miRNAs that silence AID and Blimp-1 expression to epigenetically modulate CSR, SHM, plasma cell differentiation and antibody/autoantibody responses. HDAC inhibitors, including short-chain fatty acid butyrate and propionate produced by gut commensal bacteria through fermentation of dietary fiber, epigenetically modify CSR and SHM by upregulating miRNAs, which silence AICDA/Aicda mRNA and PRDM1/Prdm1 mRNA. The upregulation of miRNA expression results from an increase in the histone acetylation of the host genes of these miRNAs. This leads to down-regulation of AID and Blimp-1 expression, and the dampening of CSR, SHM, and plasma cell differentiation.
The selectivity of HDI-mediated silencing of AICDA/Aicda and PRDMI/Prdm1 in B cells that were induced to undergo CSR and plasma cell differentiation was demonstrated by genome-wide mRNA-Seq and further emphasized by the unchanged expression of Ung, Irf4, HoxC4, Rev1, and Bcl6, as well as the unchanged expression of miR-19a/b, miR-20a, and miR-25, which are not known to regulate AICDA/Aicda or PRDMI/Prdm1 (10, 99). This, however, cannot rule out the possibility that HDI regulated other B cell factors (e.g., NF-κB, Id2/3, or Pax5), which contributed to the reduction of AID or Blimp-1. Nevertheless, relief of the HDI-mediated repression of luciferase activity under the control of Aicda and Prdm1 mRNA 3′ UTRs bearing mutated miR-155, miR-181b, miR-23b, miR-30a, and miR-125b target sites demonstrated that miRNAs are indeed direct effectors of the HDI-mediated repression of such selected genes in B cells (10).
Potential Role for Gut Microbiota-Derived Short-Chain Fatty Acid HDAC Inhibitors in the Modulation of Antibody Response
At ant time, the human body carries 1013–1014 microorganisms, a number 10-fold more than the total number of human cells in the body. Human gastrointestinal tract microbiota composed of up to 1,000–1,150 bacterial species, which play an important role in nutritional, metabolic and physiological processes that are crucial for the maintenance of human health. Gut commensal bacteria are critical regulators of health and disease by protecting against pathogen while also maintaining immune tolerance to allergens (127–131). Commensal bacteria may modulate host immunity through metabolite-dependent mechanisms (129, 131). SCFAs, such as acetic acid, propionic acid, and butyric acid, which are generated in the colon by commensal bacteria through digestion of dietary fiber, are among the most abundant of these dietary metabolites. They are important for gut motility and colonocyte development. SCFAs function through binding to host cell surface receptors, such as GPR41, GPR43, and GPR109A, and through their HDI activity (132). It has been suggested that SCFAs produced in the gut could distribute systemically and shape the immunological environment in the respiratory system, thereby influencing the severity of allergic inflammation (132).
Mice fed a low-fiber diet displayed decreased serum levels of SCFAs and increased IgE-mediated allergic inflammation in the lung, while a high-fiber diet increased levels of SCFAs and were protected against allergic airway disease (132). Butyrate and propionate, which are potent HDIs, modulate the function of intestinal macrophages and naive T cells to promote epigenetic changes that regulate the expression of genes responsible for differentiation into regulatory T cells and IL-10-producing T cells (121, 132, 133). Our recent findings that butyrate modulates AID expression and CSR to IgG, IgA, and IgE, as well as plasma cell differentiation through its direct HDI activity on B cells (10), indicates that this SCFA can play an important role in modulating antibody responses of gut lymphoid organs. A diverse microbial population, which would produce an appropriate amount of SCFA HDIs, particularly, butyrate, is required to maintain a baseline immune-regulatory state, including IgG, IgA, and IgE levels. Elevated serum IgE and CSR to IgE in B cells at mucosal sites in the absence of microbial colonization in germ-free mice and in mice with low-diversity gut microbiota (134) further emphasize the important role for gut commensal bacteria-produced butyrate in modulating IgE production. Altered composition and decreased bacterial diversity of gut microbiota would lead to changes in absolute and relative levels of SCFA HDIs and, therefore, changes in systemic IgG, IgA and IgE levels and specificities, which contribute to altered immunity and increased susceptibility to immune-mediated diseases.
B Cell Epigenetic Dysregulation in Autoimmunity and Lymphomagenesis
Epigenetic factors also play an important role in the pathogenesis of B cell-related immune disorders, such as autoimmunity, allergic states and B cell malignancies, by integrating the effects of genetic makeup and the environment, two major disease-causing factors. Epigenetic dysregulation would compound genetic susceptibility in the generation of autoantibodies and autoimmunity, as suggested by alterations of histone modifications and DNA methylation in patients with lupus, autoimmunity in mice with miRNA dysregulation (9), and low penetrance (25–45%) of lupus in monozygotic twins (135). The development of allergic diseases has been associated with environmental conditions, such as diet, drugs, toxins, sex hormones, and microbiota, all of which can impact the epigenetic profile (136, 137). Aberrant AID expression that can result from epigenetic dysregulation can lead to autoantibody-mediated autoimmunity, IgE-mediated allergic responses and tumorigenesis (Figure 3).
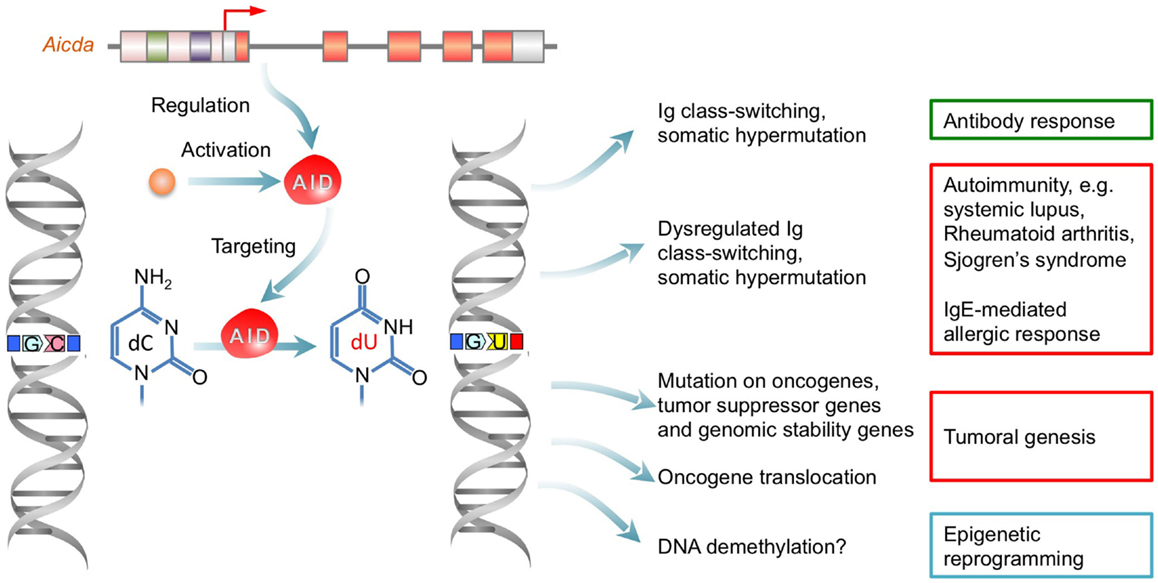
Figure 3. Dysregulation of AID result in autoantibody-mediated autoimmunity, IgE-mediated allergic response and tumoral genesis. AID initiates CSR and SHM by deaminating dCs into dUs yielding dU:dG mismatches, which lead to point-mutations in Ig V(D)J regions and DSBs in S regions. Aberrant AID expression result from epigenetic dysregulation can lead to a dysregulated antibody/autoantibody response and AID-mediated DNA mutagenesis, which can cause autoimmunity, allergic response, or tumoral genesis.
Epigenetic Changes and Autoimmunity
Systemic autoimmune diseases, such as SLE, rheumatoid arthritis, systemic sclerosis, and dermatomyositis, are associated with overall DNA hypomethylation (1, 138, 139). Autoreactive B cells in lupus patients are characterized by lack of ability to induce DNA methylation that extends their survival (140). In mice, prolonged treatment with DNA methylation inhibitors, or adaptive transfer of B cells treated with DNMT inhibitors, resulted in autoantibody production and lupus-like disease (141, 142). The role of DNA methylation in autoimmune diseases is further emphasized by the discordance of SLE in monozygotic twins. B cells isolated from SLE patients displayed profound defects in DNA methylation, as compared to those from healthy monozygotic siblings (135). The decreased DNA methylation in autoreactive B cells likely resulted from reduced DNMT1 and DNMT3b expression; it could also result from active DNA demethylation mediated by AID-mediated cytosine deamination (143). Indeed, AID is upregulated in B cells of lupus patients or lupus-prone mice (3, 9, 144). The contribution of aberrant histone modifications to lupus development has been strongly suggested by the increased histone acetylation and reduced autoantibody production in lupus-prone mice treated with HDIs (1, 10, 145).
Dysregulation of miRNAs has been associated with autoimminty. miRNAs are aberrantly expressed in different cell types and tissues in patients with autoimmune disease. B cell-specific deletion of Dicer, which is critical for miRNA maturation, resulted in a distorted BCR repertoire with increased of autoreactivity, suggesting a role for miRNAs in preventing the generation of self-reactive antibodies (9, 146, 147). In lupus-prone MRL/Faslpr/lpr mice, miR-150 is downregulated in spleen B cells, as compared to that in MRL/Fas+/+ mice (148), possibly as a result of decreased acetylation and transcription of the miR-150 host gene due to defective HDAC activity. Indeed, specific knock-in dominant negative mutant of histone acetyltransferase p300 in B cells resulted in production of class-switched anti-double-stranded DNA autoantibodies and development of lupus-like symptoms (149). Conversely, elevated expression of some miRNAs can also contribute to the development of autoimmunity. In lupus mice, expression miR-21 is upregulated in B cells, silencing of this miRNA ameliorates autoimmune splenomegaly (150). Constitutively expression of miR-17 ~ 92 in transgenic mice results in an increased numbers of germinal center B cells in the spleen and peripheral lymph nodes that may lead to autoimmune response (151).
Epigenetics Changes and Lymphomagenesis
At all stages of cancer development, inappropriate epigenetic marks interact with genetic alterations to promote neoplastic transformation and tumor cell progression (152, 153). The epigenome of B cell lymphomas is characterized by global changes in DNA methylation and histone modification patterns, which varies with chromosomal regions, local gene density, as well as DNA and histone modification status of neighboring genes (Table 2) (154, 155). Aberrant DNA hypomethylation of promoters can lead to increased transcription of genes with oncogenic potential, and aberrant DNA hypermethylation can lead to decreased transcription of genes with tumor suppressor function. For example, mantle B cell lymphomas display DNA hypomethylation in promoters of genes that are involved in pathways controlling cell cycle or apoptosis, such as Cdk5, and aberrant hypermethylation in the promoter of tumor suppressor genes, such as Cdkn2b (156). Hypermethylation is likely associated with increased expression of DNMTs, such as DNMT3b, which is upregulated in diffuse lager B cell lymphomas (157). Accordingly, Dnmt3b transgenic mice develop mediastinal B cell lymphomas, which display significantly altered methylation patterns (157). Aberrant hypomethylation could involve AID, which can mediate active DNA demethylation (158) and is upregulated in human B cell non-Hodgkin lymphomas (NHL) (159).

Table 2. B cell epigenetic dysregulation and lymphomagenesis.
Active DNA demethylation can also be initiated by the 10–11 translocation (Tet) family of proteins Tet1, Tet2, and Tet3, which catalyze the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), a critical step for ultimate removal of a methyl mark (160, 161). Reduced expression of Tet proteins that lead to decreased 5hmC has been shown to associate with tumor development, suggests an important role of Tet proteins and 5hmC in normal cellular function (162). Tet1 is required for maintaining the normal abundance and distribution of 5hmC, which prevented hypermethylation of DNA in B cells (163). It is important for regulation of the B cell lineage and of genes encoding molecules involved in chromosome maintenance and DNA repair. Tet1 may function as a tumor suppressor of B cell malignancy. Deletion of Tet1 in mice promotes the development of B cell lymphoma (163).
Altered expression or mutation of the histone-modifying enzymes promotes aberrant gene expression, which is responsible for many tumor changes (164). Aberrant germinal center development is common in many B cell malignancies. EZH2, a histone methyltransferase component of polycomb repression complex (PRC)2 that catalyzes H3K27me3 and promotes tumor growth, is highly expressed in germinal center B cells and is often constitutively activated in germinal center-derive NHLs. EZH2 prevents apoptosis caused by DNA damage, including that generated by AID, facilitated cell cycle progression, and silenced Blimp-1, which can function as tumor suppressor (106). Inhibition of EZH2 in NHL cells induces Blimp-1, which impairs tumor growth. Overexpression of EZH2 is associated with B lymphomagenesis (165). Somatic mutations at Y641 and A677 residues within the catalytic domain of EZH2 have been found in diffuse large B cell lymphoma and follicular lymphoma (166). These mutations promote EZH2 activity and increase H3K27me3 levels in those cells (165). Furthermore, other histone-modifying enzymes, such as histone acetyltransferases CBP and p300, histone methyltransferases MLL2, and histone demethylases UTX and JMJD2C, are frequently mutated in B cell lymphomas (157). Mutations in these enzymes are likely mediated by AID, which is highly expressed in those neoplastic B cells, and result in aberrant patterns of histone modifications and disruption of chromatin structure, ultimately leading to dysregulated gene transcription programs.
Dysregulated miRNA expression also contributes to B lymphomagenesis (28, 167). The miR-17 ~ 92 cluster, which consists of six miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92-1) that target the tumor repressor genes Bim and Pten, is often increased in human lymphomas (151, 168). Accordingly, ectopic expression of the miR-17 ~ 92 cluster in lymphocytes leads to development of lymphoproliferative disease (151, 168). Conversely, expression of miR-15 and miR-16-1, which target anti-apoptotic oncogene Bcl-2, is downregulated in chronic lymphocytic leukemia B cells (169, 170). Knockout miR-15a and miR-16-1 in B cells results in clonal lymphoproliferative disorders (171). B cell lymphomas often express significant amounts of mRNA isoforms with shorter 3′ UTRs, which lack miRNA-binding sites, thereby escaping miRNA-directed silencing (172). For example, a short Cyclin D1 mRNA isoform that lacks part of the 3′ UTR is expressed in a subset of mantle cell lymphomas (172). These cells have increased expression of Cyclin D1, an important regulator of cell cycle progression, and B cell proliferation. Thus, by allowing selected oncogenes to escape regulation by their modulatory miRNAs, the shortening of 3′ UTRs may provide an important mechanism in B cell neoplastic transformation.
Concluding Remarks
Epigenetic changes are critical in shaping B cell differentiation functions, such as CSR, SHM, generation of plasma cells as well as memory B cells, for the production of class-switched and high affinity antibodies. In addition, a growing body of evidence implicates the involvement of epigenetic mechanisms in immune programing and development of allergic and autoimmune diseases. Nevertheless, important questions on the nature and role of such epigenetic changes remain to be answered. These include the mechanisms by which histone posttranslational modifications and non-coding RNAs target the CSR and SHM machineries, particularly the selective recruitment of AID, to the Ig locus and how the dynamics of these epigenetic modifications orchestrate AID-mediated DNA lesion and DNA repair processes. High-affinity antibodies are generated in germinal centers through SHM and selection of higher affinity B cell submutants for survival and expansion. These B cells will then undergo differentiation to plasma cells or memory B cells. Much needs be understood on the role of epigenetic modifications in the selection of germinal center B cells and in what determines whether a germinal B cell becomes a memory B cell or a plasma cell. These processes would involve unique epigenetic and transcriptional changes. Furthermore, altered DNA methylation, histone methylation and acetylation, and miRNA expression, resulting in immune imbalance, have been shown to be associated with the onset and progression of allergic and autoimmune diseases, as well as B cell lymphomagenesis. Thus, knowledge of the epigenetic profiles associated with B cell development and peripheral differentiation, and molecular mechanisms that cause and result from disease-associated epigenetic patterns in B cells is required to understand the pathophysiology of allergic and autoimmune diseases, as well as B cell malignancies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize that owing to space limitations only a fraction of the relevant literature was cited in this review article. This work was supported by NIH grants AI 079705 and AI 105813, and the Alliance for Lupus Research Target Identification in Lupus Grant ALR 295955 (to PC) and an Arthritis National Research Foundation grant (to HZ).
References
1. Hedrich CM, Tsokos GC. Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol Med (2011) 17:714–24. doi:10.1016/j.molmed.2011.07.005
2. Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol (2012) 12:517–31. doi:10.1038/nri3216
3. Zan H, Casali P. Regulation of Aicda expression and AID activity. Autoimmunity (2013) 46:81–99. doi:10.3109/08916934.2012.749244
4. Li G, Zan H, Xu Z, Casali P. Epigenetics of the antibody response. Trends Immunol (2013) 34:460–70. doi:10.1016/j.it.2013.03.006
5. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol (2015) 15:137–48. doi:10.1038/nri3804
6. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol (2015) 15:160–71. doi:10.1038/nri3795
7. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol (2015) 15:149–59. doi:10.1038/nri3802
8. Li G, Pone EJ, Lan T, Tran DC, Mai T, Zan H, et al. Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14-3-3 adaptors and AID to specify antibody class-switch DNA recombination. Cell Rep (2013) 5:702–14. doi:10.1016/j.celrep.2013.09.031
9. Zan H, Tat C, Casali P. microRNAs in lupus. Autoimmunity (2014) 47:272–85. doi:10.3109/08916934.2014.915955
10. White CA, Pone EJ, Lam T, Tat C, Hayama KL, Li G, et al. Histone deacetylase inhibitors upregulate B cell microRNAs that silence AID and Blimp-1 expression for epigenetic modulation of antibody and autoantibody responses. J Immunol (2014) 193:5933–50. doi:10.4049/jimmunol.1401702
11. Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood (2011) 118:3559–69. doi:10.1182/blood-2011-06-357996
12. Chowdhury M, Forouhi O, Dayal S, McCloskey N, Gould HJ, Felsenfeld G, et al. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc Natl Acad Sci U S A (2008) 105:15872–7. doi:10.1073/pnas.0808462105
13. Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med (2011) 208:1649–60. doi:10.1084/jem.20110118
14. Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell (2008) 135:1028–38. doi:10.1016/j.cell.2008.09.062
15. Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet (2008) 40:108–12. doi:10.1038/ng.2007.35
16. Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell (2009) 36:631–41. doi:10.1016/j.molcel.2009.11.007
17. Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, et al. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol (2010) 11:820–6. doi:10.1038/ni.1909
18. Chandra V, Bortnick A, Murre C. AID targeting: old mysteries and new challenges. Trends Immunol (2015) 36(9):527–35. doi:10.1016/j.it.2015.07.003
19. Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, et al. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol (2009) 10:540–50. doi:10.1038/ni.1725
20. White CA, Seth Hawkins J, Pone EJ, Yu ES, Al-Qahtani A, Mai T, et al. AID dysregulation in lupus-prone MRL/Fas(lpr/lpr) mice increases class switch DNA recombination and promotes interchromosomal c-Myc/IgH loci translocations: modulation by HoxC4. Autoimmunity (2011) 44:585–98. doi:10.3109/08916934.2011.577128
21. Fujimura S, Matsui T, Kuwahara K, Maeda K, Sakaguchi N. Germinal center B-cell-associated DNA hypomethylation at transcriptional regions of the AID gene. Mol Immunol (2008) 45:1712–9. doi:10.1016/j.molimm.2007.09.023
22. Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, et al. Regulation of AID expression in the immune response. J Exp Med (2007) 204:1145–56. doi:10.1084/jem.20061952
23. Begum NA, Stanlie A, Nakata M, Akiyama H, Honjo T. The histone chaperone Spt6 is required for activation-induced cytidine deaminase target determination through H3K4me3 regulation. J Biol Chem (2012) 287:32415–29. doi:10.1074/jbc.M112.351569
24. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol (2014) 15:509–24. doi:10.1038/nrm3838
25. Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions-beyond repression of gene expression. Nat Rev Genet (2014) 15:599–612. doi:10.1038/nrg3765
26. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell (2014) 157:77–94. doi:10.1016/j.cell.2014.03.008
27. Malumbres R, Sarosiek KA, Cubedo E, Ruiz JW, Jiang X, Gascoyne RD, et al. Differentiation stage-specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas. Blood (2009) 113:3754–64. doi:10.1182/blood-2008-10-184077
28. Zhang J, Jima DD, Jacobs C, Fischer R, Gottwein E, Huang G, et al. Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood (2009) 113:4586–94. doi:10.1182/blood-2008-09-178186
29. Xu S, Guo K, Zeng Q, Huo J, Lam KP. The RNase III enzyme Dicer is essential for germinal center B-cell formation. Blood (2012) 119:767–76. doi:10.1182/blood-2011-05-355412
30. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science (2007) 316:604–8. doi:10.1126/science.1141229
31. Gururajan M, Haga CL, Das S, Leu CM, Hodson D, Josson S, et al. microRNA 125b inhibition of B cell differentiation in germinal centers. Int Immunol (2010) 22:583–92. doi:10.1093/intimm/dxq042
32. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity (2007) 27:847–59. doi:10.1016/j.immuni.2007.10.009
33. Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. microRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity (2008) 28:621–9. doi:10.1016/j.immuni.2008.03.015
34. Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, et al. microRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity (2008) 28:630–8. doi:10.1016/j.immuni.2008.04.002
35. de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med (2008) 205:2199–206. doi:10.1084/jem.20080579
36. Borchert GM, Holton NW, Larson ED. Repression of human activation induced cytidine deaminase by miR-93 and miR-155. BMC Cancer (2011) 11:347. doi:10.1186/1471-2407-11-347
37. Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, et al. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med (2012) 209:2455–65. doi:10.1084/jem.20121387
38. Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: a typical multifunctional microRNA. Biochim Biophys Acta (2009) 1792:497–505. doi:10.1016/j.bbadis.2009.02.013
39. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science (2007) 316:608–11. doi:10.1126/science.1139253
40. Thai TH, Patterson HC, Pham DH, Kis-Toth K, Kaminski DA, Tsokos GC. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Fas(lpr) mouse. Proc Natl Acad Sci U S A (2013) 110:20194–9. doi:10.1073/pnas.1317632110
41. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell (2007) 129:1401–14. doi:10.1016/j.cell.2007.04.040
42. Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol (2011) 32:194–201. doi:10.1016/j.it.2011.03.003
43. Kluiver J, van den Berg A, de Jong D, Blokzijl T, Harms G, Bouwman E, et al. Regulation of pri-microRNA BIC transcription and processing in Burkitt lymphoma. Oncogene (2007) 26:3769–76. doi:10.1038/sj.onc.1210147
44. Fear DJ, McCloskey N, O’Connor B, Felsenfeld G, Gould HJ. Transcription of Ig germline genes in single human B cells and the role of cytokines in isotype determination. J Immunol (2004) 173:4529–38. doi:10.4049/jimmunol.173.7.4529
45. Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, et al. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science (2003) 302:2137–40. doi:10.1126/science.1092481
46. Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med (2009) 206:1817–30. doi:10.1084/jem.20081678
47. Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science (2010) 329:917–23. doi:10.1126/science.1187942
48. Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A (2010) 107:22190–5. doi:10.1073/pnas.1016923108
49. Kuang FL, Luo Z, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc Natl Acad Sci U S A (2009) 106:5288–93. doi:10.1073/pnas.0901368106
50. Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, et al. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med (2009) 206:1237–44. doi:10.1084/jem.20082514
51. Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell (2010) 143:122–33. doi:10.1016/j.cell.2010.09.017
52. Storck S, Aoufouchi S, Weill JC, Reynaud CA. AID and partners: for better and (not) for worse. Curr Opin Immunol (2011) 23:337–44. doi:10.1016/j.coi.2011.02.002
53. Kenter AL. AID targeting is dependent on RNA polymerase II pausing. Semin Immunol (2012) 24:281–6. doi:10.1016/j.smim.2012.06.001
54. Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, et al. 14-3-3 adaptor proteins recruit AID to 5’-AGCT-3’-rich switch regions for class switch recombination. Nat Struct Mol Biol (2010) 17:1124–35. doi:10.1038/nsmb.1884
55. Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet (2011) 45:167–202. doi:10.1146/annurev-genet-110410-132552
56. Aida M, Hamad N, Stanlie A, Begum NA, Honjo T. Accumulation of the FACT complex, as well as histone H3.3, serves as a target marker for somatic hypermutation. Proc Natl Acad Sci U S A (2013) 110:7784–9. doi:10.1073/pnas.1305859110
57. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet (2008) 40:897–903. doi:10.1038/ng.154
58. Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem (2011) 80:473–99. doi:10.1146/annurev-biochem-061809-175347
59. Bradley SP, Kaminski DA, Peters AH, Jenuwein T, Stavnezer J. The histone methyltransferase Suv39h1 increases class switch recombination specifically to IgA. J Immunol (2006) 177:1179–88. doi:10.4049/jimmunol.177.2.1179
60. Zan H, White CA, Thomas LM, Mai T, Li G, Xu Z, et al. Rev1 recruits ung to switch regions and enhances du glycosylation for immunoglobulin class switch DNA recombination. Cell Rep (2012) 2:1220–32. doi:10.1016/j.celrep.2012.09.029
61. Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol (2011) 12:62–9. doi:10.1038/ni.1964
62. Pei H, Wu X, Liu T, Yu K, Jelinek DF, Lou Z. The histone methyltransferase MMSET regulates class switch recombination. J Immunol (2013) 190:756–63. doi:10.4049/jimmunol.1201811
63. Nagaoka H, Ito S, Muramatsu M, Nakata M, Honjo T. DNA cleavage in immunoglobulin somatic hypermutation depends on de novo protein synthesis but not on uracil DNA glycosylase. Proc Natl Acad Sci U S A (2005) 102:2022–7. doi:10.1073/pnas.0409491102
64. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell (2011) 43:904–14. doi:10.1016/j.molcel.2011.08.018
65. Lee JT. Epigenetic regulation by long noncoding RNAs. Science (2012) 338:1435–9. doi:10.1126/science.1231776
66. Satpathy AT, Chang HY. Long noncoding RNA in hematopoiesis and immunity. Immunity (2015) 42:792–804. doi:10.1016/j.immuni.2015.05.004
67. Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, et al. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature (2014) 514:389–93. doi:10.1038/nature13580
68. Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell (2011) 144:353–63. doi:10.1016/j.cell.2011.01.001
69. Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, Chaudhuri J. Non-coding RNA Generated following Lariat Debranching Mediates Targeting of AID to DNA. Cell (2015) 161:762–73. doi:10.1016/j.cell.2015.03.020
70. Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell (2015) 161:774–89. doi:10.1016/j.cell.2015.04.034
71. Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell (2014) 159:1524–37. doi:10.1016/j.cell.2014.11.013
72. Odegard VH, Kim ST, Anderson SM, Shlomchik MJ, Schatz DG. Histone modifications associated with somatic hypermutation. Immunity (2005) 23:101–10. doi:10.1016/j.immuni.2005.05.007
73. Daniel JA, Nussenzweig A. The AID-induced DNA damage response in chromatin. Mol Cell (2013) 50:309–21. doi:10.1016/j.molcel.2013.04.017
74. Wang Q, Oliveira T, Jankovic M, Silva IT, Hakim O, Yao K, et al. Epigenetic targeting of activation-induced cytidine deaminase. Proc Natl Acad Sci U S A (2014) 111:18667–72. doi:10.1073/pnas.1420575111
75. Willmann KL, Milosevic S, Pauklin S, Schmitz KM, Rangam G, Simon MT, et al. A role for the RNA pol II-associated PAF complex in AID-induced immune diversification. J Exp Med (2012) 209:2099–111. doi:10.1084/jem.20112145
76. Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell (2014) 159:1538–48. doi:10.1016/j.cell.2014.11.014
77. Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol (2006) 6:573–83. doi:10.1038/nri1896
78. Kato L, Begum NA, Burroughs AM, Doi T, Kawai J, Daub CO, et al. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes. Proc Natl Acad Sci U S A (2012) 109:2479–84. doi:10.1073/pnas.1120791109
79. Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell (1998) 92:105–16. doi:10.1016/S0092-8674(00)80903-4
80. Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science (2003) 301:1090–3. doi:10.1126/science.1085703
81. Aida M, Honjo T. FACT and H3.3: new markers for the somatic hypermutation. Cell Cycle (2013) 12:2923–4. doi:10.4161/cc.26178
82. Borchert GM, Holton NW, Edwards KA, Vogel LA, Larson ED. Histone H2A and H2B are monoubiquitinated at AID-targeted loci. PLoS One (2010) 5:e11641. doi:10.1371/journal.pone.0011641
83. Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell (2004) 14:491–500. doi:10.1016/S1097-2765(04)00259-X
84. Fraenkel S, Mostoslavsky R, Novobrantseva TI, Pelanda R, Chaudhuri J, Esposito G, et al. Allelic ‘choice’ governs somatic hypermutation in vivo at the immunoglobulin kappa-chain locus. Nat Immunol (2007) 8:715–22. doi:10.1038/ni1476
85. Woo CJ, Martin A, Scharff MD. Induction of somatic hypermutation is associated with modifications in immunoglobulin variable region chromatin. Immunity (2003) 19:479–89. doi:10.1016/S1074-7613(03)00261-9
86. Kitao H, Kimura M, Yamamoto K, Seo H, Namikoshi K, Agata Y, et al. Regulation of histone H4 acetylation by transcription factor E2A in Ig gene conversion. Int Immunol (2008) 20:277–84. doi:10.1093/intimm/dxm140
87. Blagodatski A, Batrak V, Schmidl S, Schoetz U, Caldwell RB, Arakawa H, et al. A cis-acting diversification activator both necessary and sufficient for AID-mediated hypermutation. PLoS Genet (2009) 5:e1000332. doi:10.1371/journal.pgen.1000332
88. Buerstedde JM, Alinikula J, Arakawa H, McDonald JJ, Schatz DG. Targeting of somatic hypermutation by immunoglobulin enhancer and enhancer-like sequences. PLoS Biol (2014) 12:e1001831. doi:10.1371/journal.pbio.1001831
89. McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat Rev Immunol (2012) 12:24–34. doi:10.1038/nri3128
90. Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol (2009) 10:1292–9. doi:10.1038/ni.1814
91. McHeyzer-Williams LJ, Milpied PJ, Okitsu SL, McHeyzer-Williams MG. Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat Immunol (2015) 16:296–305. doi:10.1038/ni.3095
92. Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol (2015) 16:663–73. doi:10.1038/ni.3154
93. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell (2010) 38:576–89. doi:10.1016/j.molcel.2010.05.004
94. Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol (2000) 20:2592–603. doi:10.1128/MCB.20.7.2592-2603.2000
95. Su ST, Ying HY, Chiu YK, Lin FR, Chen MY, Lin KI. Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation. Mol Cell Biol (2009) 29:1421–31. doi:10.1128/MCB.01158-08
96. Gyory I, Wu J, Fejer G, Seto E, Wright KL. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol (2004) 5:299–308. doi:10.1038/ni1046
97. Lemercier C, Brocard MP, Puvion-Dutilleul F, Kao HY, Albagli O, Khochbin S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J Biol Chem (2002) 277:22045–52. doi:10.1074/jbc.M201736200
98. Fujita N, Jaye DL, Geigerman C, Akyildiz A, Mooney MR, Boss JM, et al. MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell (2004) 119:75–86. doi:10.1016/j.cell.2004.09.014
99. Shen T, Nasrollahi H, Zan H, Casali P. Genome-wide analysis of B cells reveals selective modulation of microRNAs and mRNAs by histone deacetylase inhibitor. Front Immunol (2015) 6:627. doi:10.3389/fimmu.2015.00627
100. West JA, Viswanathan SR, Yabuuchi A, Cunniff K, Takeuchi A, Park IH, et al. A role for Lin28 in primordial germ-cell development and germ-cell malignancy. Nature (2009) 460:909–13. doi:10.1038/nature08210
101. Nie K, Zhang T, Allawi H, Gomez M, Liu Y, Chadburn A, et al. Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: a potential role of the microRNA let-7. Am J Pathol (2010) 177:1470–9. doi:10.2353/ajpath.2010.091291
102. Leucci E, Onnis A, Cocco M, De Falco G, Imperatore F, Giuseppina A, et al. B-cell differentiation in EBV-positive Burkitt lymphoma is impaired at posttranscriptional level by miRNA-altered expression. Int J Cancer (2010) 126:1316–26. doi:10.1002/ijc.24655
103. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity (2006) 25:225–36. doi:10.1016/j.immuni.2006.07.009
104. Good-Jacobson KL. Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front Immunol (2014) 5:596. doi:10.3389/fimmu.2014.00596
105. Baxter J, Sauer S, Peters A, John R, Williams R, Caparros ML, et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J (2004) 23:4462–72. doi:10.1038/sj.emboj.7600414
106. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest (2013) 123:5009–22. doi:10.1172/JCI70626
107. Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, Mathis D. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci U S A (2006) 103:3304–9. doi:10.1073/pnas.0511137103
108. Lai AY, Mav D, Shah R, Grimm SA, Phadke D, Hatzi K, et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res (2013) 23:2030–41. doi:10.1101/gr.155473.113
109. Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J Jr, Miljkovic V, et al. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci U S A (2003) 100:2639–44. doi:10.1073/pnas.0437996100
110. Cao Z, Sun X, Icli B, Wara AK, Feinberg MW. Role of Kruppel-like factors in leukocyte development, function, and disease. Blood (2010) 116:4404–14. doi:10.1182/blood-2010-05-285353
111. Calame K. microRNA-155 function in B Cells. Immunity (2007) 27:825–7. doi:10.1016/j.immuni.2007.11.010
112. Ranzani V, Rossetti G, Panzeri I, Arrigoni A, Bonnal RJ, Curti S, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol (2015) 16:318–25. doi:10.1038/ni.3093
113. Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, et al. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med (2003) 198:1427–37. doi:10.1084/jem.20030802
114. Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics (2012) 4:5. doi:10.1186/1868-7083-4-5
115. Reilly CM, Regna N, Mishra N. HDAC inhibition in lupus models. Mol Med (2011) 17:417–25. doi:10.2119/molmed.2011.00055
116. Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J (2001) 20:6969–78. doi:10.1093/emboj/20.24.6969
118. Waibel M, Christiansen AJ, Hibbs ML, Shortt J, Jones SA, Simpson I, et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat Commun (2015) 6:6838. doi:10.1038/ncomms7838
119. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem (2001) 276:36734–41. doi:10.1074/jbc.M101287200
120. Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev (2001) 81:1031–64.
121. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi:10.1038/nature12721
122. Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol (2011) 29:255–65. doi:10.1038/nbt.1759
123. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, et al. Histone deacetylase inhibitors impair innate immune responses to toll-like receptor agonists and to infection. Blood (2011) 117:1205–17. doi:10.1182/blood-2010-05-284711
124. Dowdell KC, Pesnicak L, Hoffmann V, Steadman K, Remaley AT, Cohen JI, et al. Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, diminishes lymphoproliferation in the Fas -deficient MRL/lpr(-/-) murine model of autoimmune lymphoproliferative syndrome (ALPS). Exp Hematol (2009) 37:487–94. doi:10.1016/j.exphem.2008.12.002
125. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol (2009) 10:92–100. doi:10.1038/ni.1673
126. Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood (2012) 119:2443–51. doi:10.1182/blood-2011-10-292003
127. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science (2012) 336:1268–73. doi:10.1126/science.1223490
128. Ivanov II, Honda K. Intestinal commensal microbes as immune modulators. Cell Host Microbe (2012) 12:496–508. doi:10.1016/j.chom.2012.09.009
129. Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol (2013) 14:676–84. doi:10.1038/ni.2640
130. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell (2014) 157:121–41. doi:10.1016/j.cell.2014.03.011
131. Thorburn AN, Macia L, Mackay CR. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity (2014) 40:833–42. doi:10.1016/j.immuni.2014.05.014
132. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med (2014) 20:159–66. doi:10.1038/nm.3444
133. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi:10.1038/nature12726
134. Cahenzli J, Koller Y, Wyss M, Geuking MB, McCoy KD. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe (2013) 14:559–70. doi:10.1016/j.chom.2013.10.004
135. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res (2010) 20:170–9. doi:10.1101/gr.100289.109
136. Kauffmann F, Demenais F. Gene-environment interactions in asthma and allergic diseases: challenges and perspectives. J Allergy Clin Immunol (2012) 130:1229–40. doi:10.1016/j.jaci.2012.10.038
137. Blumenthal MN. Genetic, epigenetic, and environmental factors in asthma and allergy. Ann Allergy Asthma Immunol (2012) 108:69–73. doi:10.1016/j.anai.2011.12.003
138. Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat Rev Rheumatol (2011) 7:263–71. doi:10.1038/nrrheum.2011.16
139. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol (2010) 28:1057–68. doi:10.1038/nbt.1685
140. Garaud S, Le Dantec C, Jousse-Joulin S, Hanrotel-Saliou C, Saraux A, Mageed RA, et al. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J Immunol (2009) 182:5623–32. doi:10.4049/jimmunol.0802412
141. Strickland FM, Richardson BC. Epigenetics in human autoimmunity. Autoimmunity (2008) 41:278–86. doi:10.1080/08916930802024616
142. Mazari L, Ouarzane M, Zouali M. Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc Natl Acad Sci U S A (2007) 104:6317–22. doi:10.1073/pnas.0610434104
143. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol (2010) 11:607–20. doi:10.1038/nrm2950
144. Zan H, Zhang J, Ardeshna S, Xu Z, Park SR, Casali P. Lupus-prone MRL/faslpr/lpr mice display increased AID expression and extensive DNA lesions, comprising deletions and insertions, in the immunoglobulin locus: concurrent upregulation of somatic hypermutation and class switch DNA recombination. Autoimmunity (2009) 42:89–103. doi:10.1080/08916930802629554
145. Pieterse E, Hofstra J, Berden J, Herrmann M, Dieker J, van der Vlag J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin Exp Immunol (2015) 179:68–74. doi:10.1111/cei.12359
146. Belver L, de Yébenes VG, Ramiro AR. microRNAs prevent the generation of autoreactive antibodies. Immunity (2010) 33:713–22. doi:10.1016/j.immuni.2010.11.010
147. Ceribelli A, Satoh M, Chan EK. microRNAs and autoimmunity. Curr Opin Immunol (2012) 24:686–91. doi:10.1016/j.coi.2012.07.011
148. Dai R, Zhang Y, Khan D, Heid B, Caudell D, Crasta O, et al. Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS One (2010) 5:e14302. doi:10.1371/journal.pone.0014302
149. Forster N, Gallinat S, Jablonska J, Weiss S, Elsässer HP, Lutz W. p300 protein acetyltransferase activity suppresses systemic lupus erythematosus-like autoimmune disease in mice. J Immunol (2007) 178:6941–8. doi:10.4049/jimmunol.178.11.6941
150. Garchow BG, Bartulos Encinas O, Leung YT, Tsao PY, Eisenberg RA, Caricchio R, et al. Silencing of microRNA-21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med (2011) 3:605–15. doi:10.1002/emmm.201100171
151. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol (2008) 9:405–14. doi:10.1038/ni1575
153. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet (2011) 12:861–74. doi:10.1038/nrg3074
154. Jones PA, Baylin SB. The epigenomics of cancer. Cell (2007) 128:683–92. doi:10.1016/j.cell.2007.01.029
155. De S, Shaknovich R, Riester M, Elemento O, Geng H, Kormaksson M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet (2013) 9:e1003137. doi:10.1371/journal.pgen.1003137
156. Leshchenko VV, Kuo PY, Shaknovich R, Yang DT, Gellen T, Petrich A, et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood (2010) 116:1025–34. doi:10.1182/blood-2009-12-257485
157. Shaknovich R, Melnick A. Epigenetics and B-cell lymphoma. Curr Opin Hematol (2011) 18:293–9. doi:10.1097/MOH.0b013e32834788cf
158. Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev (2010) 24:2107–14. doi:10.1101/gad.1963010
159. Greeve J, Philipsen A, Krause K, Klapper W, Heidorn K, Castle BE, et al. Expression of activation-induced cytidine deaminase in human B-cell non-Hodgkin lymphomas. Blood (2003) 101:3574–80. doi:10.1182/blood-2002-08-2424
160. Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem (2014) 83:585–614. doi:10.1146/annurev-biochem-060713-035513
161. Schubeler D. Function and information content of DNA methylation. Nature (2015) 517:321–6. doi:10.1038/nature14192
162. Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet (2014) 30:464–74. doi:10.1016/j.tig.2014.07.005
163. Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol (2015) 16:653–62. doi:10.1038/ni.3148
164. Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med (2011) 17:330–9. doi:10.1038/nm.2305
165. Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood (2010) 116:5247–55. doi:10.1182/blood-2010-04-280149
166. Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood (2011) 117:2451–9. doi:10.1182/blood-2010-11-321208
167. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. microRNA expression profiles classify human cancers. Nature (2005) 435:834–8. doi:10.1038/nature03702
168. Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. microRNAs: new regulators of immune cell development and function. Nat Immunol (2008) 9:839–45. doi:10.1038/ni.f.209
169. Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med (2008) 14:1271–7. doi:10.1038/nm.1880
170. Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ (2010) 17:215–20. doi:10.1038/cdd.2009.69
171. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell (2010) 17:28–40. doi:10.1016/j.ccr.2009.11.019
172. Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell (2003) 3:185–97. doi:10.1016/S1535-6108(03)00028-X
173. Perlot T, Alt FW. Cis-regulatory elements and epigenetic changes control genomic rearrangements of the IgH locus. Adv Immunol (2008) 99:1–32. doi:10.1016/S0065-2776(08)00601-9
174. Giambra V, Volpi S, Emelyanov AV, Pflugh D, Bothwell AL, Norio P, et al. Pax5 and linker histone H1 coordinate DNA methylation and histone modifications in the 3’ regulatory region of the immunoglobulin heavy chain locus. Mol Cell Biol (2008) 28:6123–33. doi:10.1128/MCB.00233-08
175. Garrett FE, Emelyanov AV, Sepulveda MA, Flanagan P, Volpi S, Li F, et al. Chromatin architecture near a potential 3’ end of the IgH locus involves modular regulation of histone modifications during B-Cell development and in vivo occupancy at CTCF sites. Mol Cell Biol (2005) 25:1511–25. doi:10.1128/MCB.25.4.1511-1525.2005
176. Wang L, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. J Exp Med (2006) 203:215–26. doi:10.1084/jem.20051774
177. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature (2011) 471:189–95. doi:10.1038/nature09730
178. Xu Y. DNA damage: a trigger of innate immunity but a requirement for adaptive immune homeostasis. Nat Rev Immunol (2006) 6:261–70. doi:10.1038/nri1804
179. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet (2010) 42:181–5. doi:10.1038/ng.518
180. Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet (2014) 10:e1004566. doi:10.1371/journal.pgen.1004566
181. Pasqualucci L. The genetic basis of diffuse large B-cell lymphoma. Curr Opin Hematol (2013) 20:336–44. doi:10.1097/MOH.0b013e3283623d7f
182. Chambwe N, Kormaksson M, Geng H, De S, Michor F, Johnson NA, et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood (2014) 123:1699–708. doi:10.1182/blood-2013-07-509885
183. Shaknovich R, Geng H, Johnson NA, Tsikitas L, Cerchietti L, Greally JM, et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood (2010) 116:e81–9. doi:10.1182/blood-2010-05-285320
184. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A (2005) 102:13944–9. doi:10.1073/pnas.0506654102
185. Lawrie CH, Soneji S, Marafioti T, Cooper CD, Palazzo S, Paterson JC, et al. microRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer (2007) 121:1156–61. doi:10.1002/ijc.22800
186. Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, et al. microRNA-17~92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J (2013) 32:2377–91. doi:10.1038/emboj.2013.178
Keywords: AID, B cell, Blimp-1, class switch DNA recombination, epigenetics, histone post-translational modification, memory B cell, microRNA, plasma cell differentiation, somatic hypermutation
Citation: Zan H and Casali P (2015) Epigenetics of Peripheral B-Cell Differentiation and the Antibody Response. Front. Immunol. 6:631. doi: 10.3389/fimmu.2015.00631
Received: 31 August 2015; Accepted: 30 November 2015;
Published: 14 December 2015
Edited by:
Harry W. Schroeder, University of Alabama at Birmingham, USAReviewed by:
Wenxia Song, University of Maryland, USARonald B. Corley, Boston University School of Medicine, USA
Copyright: © 2015 Zan and Casali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Zan, emFuQHV0aHNjc2EuZWR1;
Paolo Casali, cGNhc2FsaUB1dGhzY3NhLmVkdQ==