 Ana Luisa Perdigoto
Ana Luisa Perdigoto Lucienne Chatenoud
Lucienne Chatenoud Jeffrey A. Bluestone
Jeffrey A. Bluestone Kevan C. Herold
Kevan C. Herold- 1Department of Immunobiology, Yale University, New Haven, CT, USA
- 2Department of Internal Medicine, Yale University, New Haven, CT, USA
- 3Université Paris Descartes, Sorbonne Paris Cité, F-75475, Paris, France
- 4INSERM U1151, CNRS UMR 8253, Hôpital Necker-Enfants Malades, Paris, France
- 5Diabetes Center, University of California San Francisco, San Francisco, CA, USA
Regulatory T cells (Tregs) control unwanted immune responses, including those that mediate tolerance to self as well as to foreign antigens. Their mechanisms of action include direct and indirect effects on effector T cells and important functions in tissue repair and homeostasis. Tregs express a number of cell surface markers and transcriptional factors that have been instrumental in defining their origins and potentially their function. A number of immune therapies, such as rapamycin, IL-2, and anti-T cell antibodies, are able to induce Tregs and are being tested for their efficacy in diverse clinical settings with exciting preliminary results. However, a balance exists with the use of some, such as IL-2, that may have effects on unwanted populations as well as promoting expansion and survival of Tregs requiring careful selection of dose for clinical use. The use of cell surface markers has enabled investigators to isolate and expand ex vivo Tregs more than 500-fold routinely. Clinical trials have begun, administering these expanded Tregs to patients as a means of suppressing autoimmune and alloimmune responses and potentially inducing immune tolerance. Studies in the future are likely to build on these initial technical achievements and use combinations of agents to improve the survival and functional capacity of Tregs.
Introduction
In order to maintain sufficient diversity needed to eliminate foreign antigens, the immune system needs mechanisms to avoid responses to self and to maintain tolerance. Inadequate immune responses can result in life-threatening infections and tumor growth but left unchecked, activation of the immune system can result in autoimmunity, allergy, and organ transplant rejection. T cell-mediated self-tolerance is sustained via a number of checkpoints in the thymus and the periphery (1). Immune regulation mediated by dedicated subsets of T lymphocytes, termed regulatory T cells (Tregs), is one major mechanisms of peripheral tolerance. Autoimmune diseases, including insulin-dependent type 1 diabetes (T1DM), systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, result from a breakdown in self-tolerance. The development of autoimmunity in patients with malignancies treated with agents blocking costimulation highlights the critical role of the balance between Treg and effector T cell responses to prevent or halt the progression of autoimmune diseases (2–4). The major goal of immune therapies in autoimmune disease is to re-establish this balance.
This explains the ever growing interest over the last decade for strategies potentiating the functional capacity of Tregs and for T cell therapy approaches using ex vivo expanded Tregs.
Regulatory T cells include distinct subsets of T lymphocytes derived as a distinct lineage from the thymus, initially termed natural Treg, expressing the FOXP3 transcription factor and also from the periphery, initially termed adaptive Treg and encompassing both FOXP3+ and FOXP3− cells. One may, thus, distinguish FOXP3+ Tregs, FOXP3− IL-10-dependent Tr1, and LAP+TGF-β-dependent Th3 cells (1). Th3 cells play an important role in oral tolerance primarily through secretion of TGF-β and suppression of Th1 and Th2 cells (5). T regulatory type 1 (Tr1) cells develop from conventional T cells when exposed to regulatory dendritic cells (DCs) and have been shown to suppress T cell and antigen-presenting cell (APC) responses mainly via an IL-10 and TGF-β dependent mechanism (6). However, the bulk of critical Treg data has been generated based on the activity and specificity of the FOXP3+ Treg subset that develops within the thymic environment and in some circumstances, following peripheral exposure to self-antigens. This review will focus on the biology of FOXP3+ Tregs, therapeutic efforts to enhance their function in vivo and cell therapy strategies using ex vivo expanded FOXP3+ Tregs.
Biology of Regulatory T Cells
Regulatory T cells play a critical role in immune homeostasis and self-tolerance. They modulate immune responses by inhibiting effector T cells but they also serve an important function in the development and regulation of other lymphocyte and APC subsets. Tregs are found in both primary and secondary lymphoid organs as well as non-lymphoid tissues where they are thought to play a role in protection against immune damage as well as non-immune functions, such as tissue homeostasis and repair (7). Conventional Tregs are characterized by expression of the forkhead family transcription factor, Forkhead box P3 (FOXP3). FOXP3 was originally identified as playing a role in the development and maintenance of Tregs from observations in Scurfy mice, which develop a fatal lymphoproliferative disorder with CD4 T cell hyperactivation and production of proinflammatory cytokines (8). In humans, mutations in FOXP3 lead to a lack of functional Tregs and results in immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome that manifests as multi-organ autoimmunity, including diabetes, thyroiditis and allergy (i.e., eczema); in the absence of a bone marrow transplantation, death occurs within a year of birth (9–13). FOXP3, the lineage determinant of thymus-derived Tregs is a transcriptional repressor and inhibits cytoplasmic and calcineurin-dependent NFATc2, as well as other transcriptional factors, such as NFκB, and AML1/RUNX1 (Figure 1). Interaction of NFAT with FOXP3 is needed for suppressor function of Tregs. FOXP3 also facilitates Treg development by amplifying and stabilizing its own expression and inhibiting transcription factors required for other cell lineages, such as Tbet, GATA3, and RORγt (14).
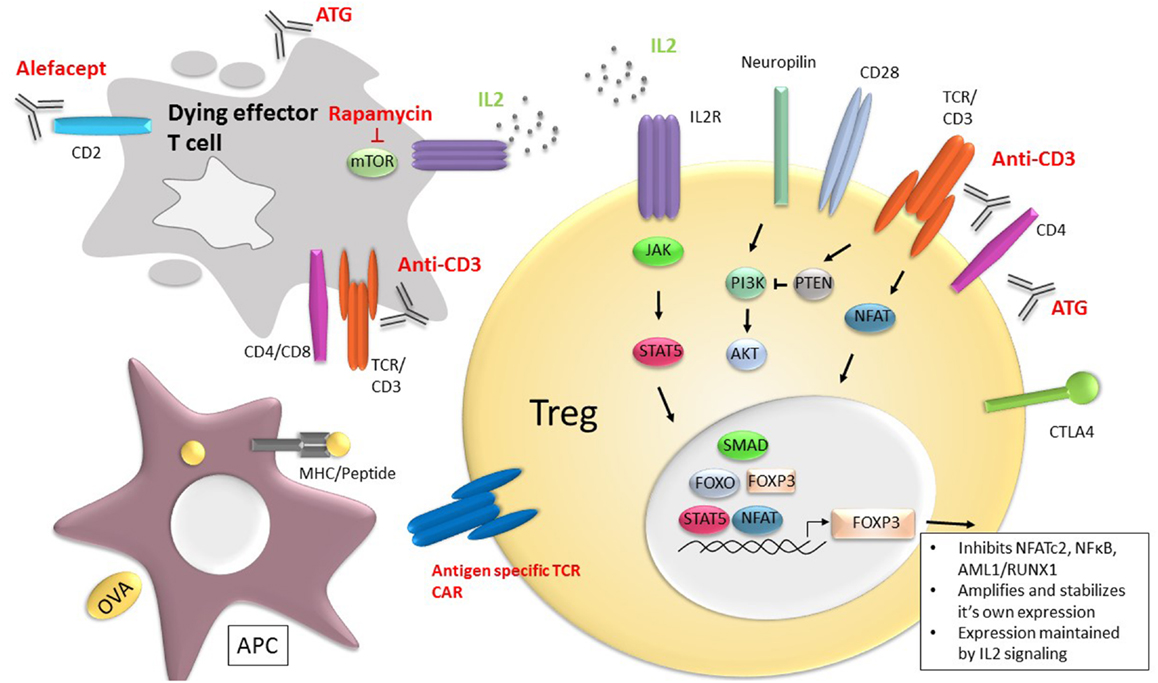
Figure 1. Current and potential future therapies to promote Tregs and immune tolerance. Therapies, such as rapamycin, anti-CD3 mAb, anti-thymocyte globulin (ATG), and Alefacept, a CD2 binding fusion molecule that eradicates CD2 expressing cells, exert their immune suppressive effect by eliminating effector T cells (gray cell) and tipping the balance in favor of Treg function and/or frequency. In addition, the anti-T cell receptor therapies, such as anti-CD3 mAbs may enhance survival and function of Tregs (yellow). IL-2, signaling through the ILR receptor (purple) and pSTAT5 (orange), is central to Treg survival and FOXP3 maintenance through the signaling cascades shown. FOXP3 inhibits cytokine gene expression by inhibiting NFATc2, as well as other transcriptional factors, such as NFκB and AML1/RUNX1. FOXP3 also facilitates Treg development by amplifying and stabilizing its own expression and inhibiting transcription factors required for other cell lineages, such as Tbet, GATA3, and RORγt. Cell-based therapies include use of Tregs engineered to express TCRs directed against specific antigens, including chimeric antigen receptors (CAR) (blue).
The potency of Tregs lies in their ability to deploy various immunosuppressive mechanisms depending on the immunological context as well as extending their influence through the process of infectious tolerance (15, 16). Through contact-dependent mechanisms, Tregs have been shown to cause reduced T cell receptor (TCR)-induced calcium flux, NFAT, and NF-κB signaling and IL-2 production by effector T cells (17). In addition, by virtue of expression of CD25, they have been shown to consume IL-2, needed by effector T cells, and induce effector cell death by granzyme and perforin (18, 19). Tregs can inhibit T cell costimulation by either regulating CD80/86 expression on APCs through CTLA-4 or competing for CD28 binding (20–22). Finally, Tregs can produce regulatory cytokines, such as TGFβ, IL-35, and IL-10, which facilitate a key functional consequence of Tregs, namely bystander suppression (23–27).
The Treg TCR repertoire is highly skewed toward self-reactivity, which may be important in ensuring their ability to prevent the activation of autoreactive effectors and to avoid regulation of effector T cells needed for responses to pathogens and tumors. Work from our lab has shown that disease susceptibility loci, such as CTLA-4 and IL-2, in Type 1 diabetes lead to Treg instability (28). Immune effector T cells generated from destabilized FOXP3+ Tregs can mediate autoimmune reactivity, suggesting that some autoreactive effector T cells may have their origins in the Treg lineage. In addition, data suggest that a subset of effector T cells can become resistant to Tregs enhancing the potential activity of the autoreactive T cells in the type 1 diabetes setting (29).
Definition and Role of Treg Subsets
Thymus-derived cells, tTregs [previously termed natural or nTreg (30)] are needed for general homeostasis and tissue repair. Tregs generated in the periphery, pTregs (previously termed adaptive or inducible), develop from conventional T cells (Tconv) and may be more important in controlling local auto-inflammatory responses (31). These subsets have distinct TCR repertoires where autoantigens may be presented differently in local tissues than during thymic development. In instances where the origin of Tregs is unclear, the general term “FOXP3+ Treg cell” is used (30). Although many markers have been identified as important in the immunobiology of Tregs, most are not unique to Tregs (Table 1). Signaling by IL-2 through the IL-2R has been shown to be particularly critical for Treg homeostasis and the maintenance of FOXP3 expression (32). CD127 expression is low on Tregs when compared to activated, conventional T cells, thus, the combination of CD4, CD25, and CD127 has been used to select T cells for functional studies as well as for expansion and adoptive immune therapy (33, 34). Other cell surface markers have been identified on Tregs that have functional significance. CD39 and CD73 are ectoenzymes that are involved in generation of adenosine, which inhibits effector T cells through interactions with the adenosine receptors A2AR/A2BR and in recruiting Tregs (35–37). Neuropilin-1 is a membrane-bound receptor on Tregs whose ligation by Semaphorin-4a restrains Akt phosphorylation and maintains Treg stability (38, 39). Helios, a transcription factor of the Ikaros family, was originally proposed as a marker of tTregs, distinguishing them from pTregs (40). However, a subsequent study showed that upon induction by APCs, pTregs can also express Helios, preventing its use as a tTreg marker (41). The cell surface receptors TIGIT and FCRL3 have also been recently identified as useful markers for Helios+ Tregs. TIGIT is associated with lineage stability and suppressive capacity. TIGIT+ Tregs were reported to selectively inhibit Th1 and Th17 but not Th2 T cell responses through an Fgl2-dependent mechanism (42–44). In addition, Smigiel et al. have discriminated Treg subsets into tissue-resident (eTregs: CD44hiCD62lo/−), which are dominant in non-lymphoid tissues and highly proliferative or central Tregs (CD44loCD62L+), which are quiescent and recirculate through the secondary lymphoid tissues (45). The latter are dependent on IL-2 for survival and function, whereas the former are largely controlled by signaling through the TCR and IL-33, hence, their role in the control of antigen-specific responses that occur in peripheral tissues.
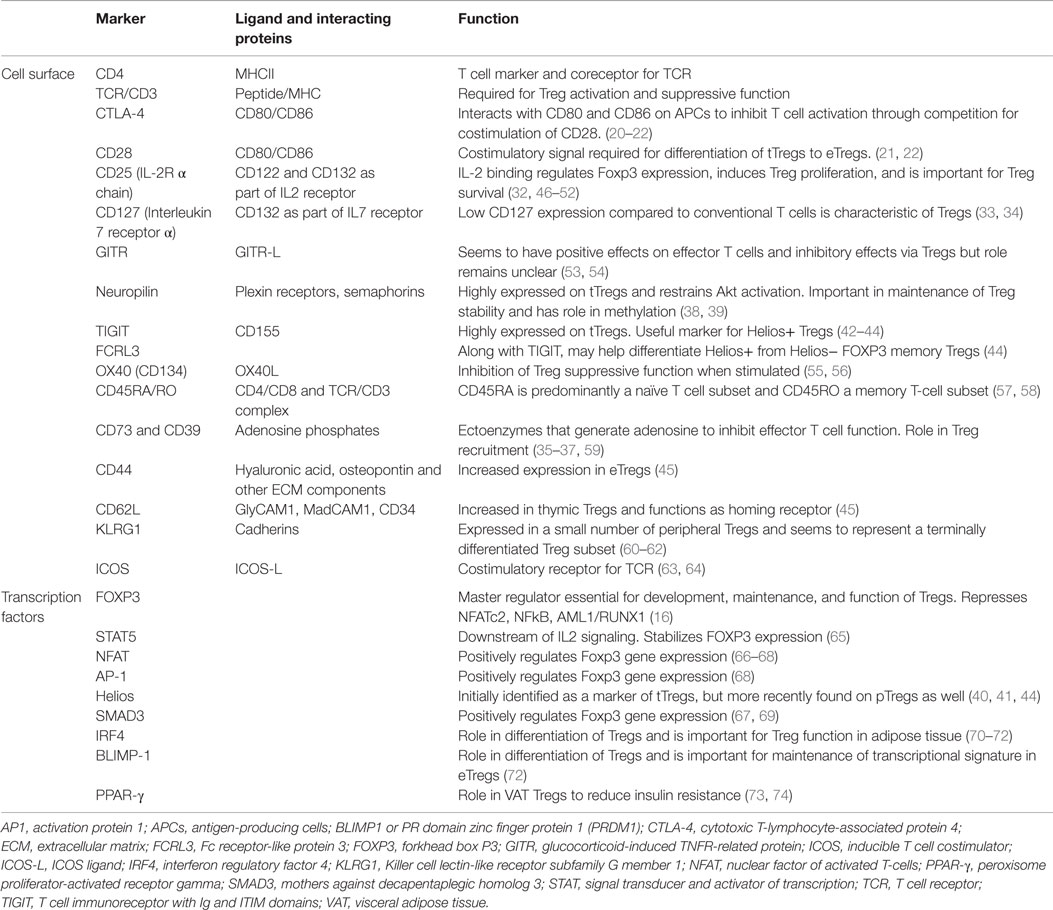
Table 1. Markers of Tregs.
PD1, implicated in negative regulation as well as a marker of T cell activation, is also expressed on Tregs. As in conventional cells, activation of PD1 on Tregs inhibited responses to anti-CD3 mAb but induced a different cluster of genes in Tregs compared to those activated in conventional T cells (75, 76). Finally, some investigators have subdivided FOXP3+ Tregs into effector/memory phenotypes in much the same manner as conventional T cells (57, 58, 77).
Studies in recent years have identified Treg populations in non-lymphoid tissues, including the skin, visceral adipose tissue (VAT), liver, intestine, skeletal muscle, bone, lungs, and placenta. Tissue-specific Tregs have unique phenotypes and roles depending on their location, varying with regard to frequency, TCR repertoire, cytokine production, chemokine receptor expression, and mechanism of action (15, 78–80). Immune-related functions in non-lymphoid tissues include suppression of T cell responses as well as limiting inflammation through control of myeloid populations (70, 73, 74, 78–80).Tregs are recruited by unique homing receptors to the skin and intestinal mucosa where they are needed to regulate the immune response against infectious pathogens as well as tolerance of commensal organisms (78). TGF-β and IL-10, produced by Tregs, play an important role in Treg function in the intestine and the intestinal microbiome also seems to influence Treg development and induction (81). Tregs in VAT express peroxisome proliferator-activated receptor gamma (PPAR-γ), a transcription factor that is required for the accumulation of Tregs at this location where they have a positive effect on insulin resistance and glucose metabolism (73, 74). VAT Tregs express IL-10 and TGF-β and depend on IRF4, BATF, and IL-33 (70).
There is growing evidence that Treg expansion occurs at sites of tissue injury and plays an important role in tissue repair and regeneration beyond immune regulation by influencing tissue resident non-immune cells. In injured skeletal muscle, a Treg population was identified that can promote muscle repair through expression of amphiregulin, an epidermal growth factor, in vitro and in vivo in mice (82). Tregs have been shown to have an amphiregulin-dependent role in tissue repair in the setting of infectious lung injury independent of their immune suppressive function (83). A role for Tregs in bone repair and regeneration has also been proposed to occur not only through the effect of Tregs on other immune cells to maintain bone homeostasis but also potentially through direct interactions with osteoblasts (15).
Various tumors contain Tregs, where their accumulation may suppress the immune response that would otherwise keep tumor cells at bay (84). CCL22 on tumors recruits CCR4+ Treg cells to the site and has been associated with tumor progression through their suppression of effector T cells that target tumor antigens (85, 86). Tregs constitutively express CTLA-4 and the benefit seen with CTLA-4 antibody therapy in cancer may in part be attributed to Treg depletion. Furthermore, CCL28 positive tumors can recruit Tregs where they have a proangiogenic role (87).
Unstable Tregs
Several years ago, we demonstrated that Tregs were unstable in inflammatory tissues as they can lose FOXP3 and begin to turn on a broad array of potentially pathogenic pathways, such as the production of interferon-gamma and IL-17 (88). In part, this instability is due to decreased IL-2 signaling but may also reflect Treg inactivation through cytokines, such as IL-6. Purified IFNγ+ Tregs were suppressive in vitro but lacked Helios expression and were methylated at the Treg-specific demethylated region (TSDR) of FOXP3, characteristic of in vitro-induced Tregs. Thus, efforts to target Tregs in clinical settings will rely on efforts to repair and replace Tregs in autoimmunity and organ transplantation or destabilize and eliminate Tregs in the cancer and infectious disease settings.
Human Therapies that Promote Tregs
Rapamycin
While calcineurin inhibitors (such as Tacrolimus and Cyclosporine A) appear to inhibit Treg generation, rapamycin, which inhibits PI3K/AKT signaling through its direct interaction with the mTORC1 complex enhances Treg expansion and survival at the time it inhibits proliferation of Th1 and Th17 cells (89–102) (Figure 1). This is in part because PI3K/AKT signaling is a principal signaling pathway in Teff cells but less so in Tregs. In mice, rapamycin treatment leads to expansion of Tregs with increased suppressive activity in vitro and an enhanced ability to prevent pancreatic β cell transplant rejection in vivo (96). Administration of rapamycin in NOD mice, a model of T1DM, resulted in prevention of diabetes and restored tolerance to self-antigens due to expansion of Tregs (103). In patients with T1DM, rapamycin promoted expansion of Tregs (97) and enhanced their suppressive capacity (104). Again, unlike calcineurin inhibitors, rapamycin treatment maintained the proportion of Tregs in peripheral blood in renal transplant recipients (105) or even enhanced their frequency and reduced production of inflammatory cytokines, such as IL-1β, IL-6, IL-17, and IFNγ, in patients switched from tacrolimus to rapamycin (106). Similar effects are seen with everolimus, a synthetic derivative of rapamycin (107).
Interleukin-2
The cytokine IL-2 plays a central role in Treg function and the balance between immunity and tolerance (46, 108, 109). Tregs express high-affinity CD25 and require IL-2 for survival. IL-2 interacts with the trimeric IL-2 receptor complex (CD122, CD132, and CD25) and signals primarily through the JAK/STAT pathway in Tregs to maintain FOXP3 expression and the development, proliferation, and suppressive function of Tregs (32, 47). IL-2 can expand Tregs in vivo and enhance their immune suppressive function (7, 110). The role of IL-2 in maintenance of self-tolerance is clear from studies of IL-2 or IL-2R-deficient mice that developed severe multi-organ autoimmune disease and early death (48–51). The levels of IL-2 and expression of CD25 on target cells influence the balance between immunity and tolerance. Tregs have a 10- to 20-fold lower activation threshold for IL-2 than effector T cells as assessed by the level of phosphorylated STAT5 (pSTAT5). The sensitivity to IL-2 in Tregs may be due to the function of the IL-2R signaling specificity: the MAPK, PI3K-AKT, and STAT5 pathways are all activated in effector T cells. In Tregs, the high PTEN expression may inhibit the PI3K signaling pathways and, therefore, activation relies on pSTAT5 signaling (65, 111, 112).
In autoimmune diseases, impaired IL-2 signaling is thought to affect the number and function of Tregs. The IL-2RA gene is one of the T1DM susceptibility genes (51, 52, 113–116). NOD mice treated with low-dose IL-2 showed increase in Tregs and reversal or prevention of diabetes (115, 117). In an EAE mouse model of multiple sclerosis, IL-2 treatment resulted in restoration of FOXP3 expression, Treg stability, and prevention of autoimmunity (118). However, depending on the dose, IL-2 can increase other potentially damaging leukocytes, including NK cells, activated CD8+ T cells and eosinophils [as a direct consequence of the activation of an innate lymphoid cell subset (ILC2)] (76).
Interleukin-2 was first used clinically to augment the immune response against tumor self-antigens in metastatic malignancies, such as melanoma and renal cell carcinomas, but variable clinical responses were seen with significant side effects (119, 120). Thus, in order to maximize the ability of IL-2 to selectively enhance Treg function, a series of mouse and human studies have been undertaken to examine IL-2 dosing as a means to shift the balance between immunity and tolerance in autoimmune diseases (111, 121, 122). IL-2 has been tested in clinical trials to treat graft versus host disease (GVHD), hepatitis C virus-induced vasculitis, and T1DM (108, 123–126) through a Treg-dependent pathway. A clinical response was shown in patients with GVHD enrolled in a Phase I, dose escalation, 8-week trial (123). Subjects exhibited not only an increased number of Tregs but also an increase in eosinophils and NK cells, which also express CD25. These investigators reported that IL-2 therapy increased Treg proliferation, thymic export, and enhanced resistance to apoptosis with minimal effects on conventional T cells (127). There was a selective effect of low concentrations of IL-2 on phosphorylation of STAT5, whereas IL-7 induced similar phosphorylation of STAT5 in conventional and Tregs at low concentrations. Patients with HCV vasculitis were treated with IL-2 in a Phase I/II clinical trial with the objective of increasing Tregs, which had been shown previously to correlate with successful treatment of this disease process. The majority of patients showed clinical improvement and there was an accompanying increase in the number of Tregs (124).
Preclinical studies by Rabinovitch et al. had demonstrated reversal of diabetes with combination of IL-2 and rapamycin (128). Thus, it was postulated that combining rapamycin with IL-2 would optimize inhibition of Teff signaling and concurrent augmentation of Treg signaling. Long et al. found that T1DM patients treated with rapamycin and IL-2 achieved increased frequency of circulating Tregs and sustained IL-2 signaling (125) but had transient worsening of β cell function as assessed by C-peptide following a mixed meal tolerance test. The trial was stopped after treatment of nine subjects. These investigators observed increased eosinophilia and natural killer cells. The relatively high doses of IL-2 (12 doses of 4.5 × 106 IU) administered to the patients in that study was thought to account for the expansion of these potentially β cell toxic cells, although effects of the rapamycin could not be ruled out.
Thus, more recent efforts have been devoted to administering low-doses of IL-2 in patients with new onset diabetes. In a Phase I/II randomized double-blind placebo-controlled study, Hartemann et al. studied treatment with IL-2 at doses of 0.33, 1, or 3 million IU/day for a 5-day course. IL-2 induced a dose-dependent increase in the proportion of Tregs at all doses but at a dose of 1 × 106 IU × 5, approximately one-tenth of the dose used in the trial of Long et al. Effects on Tregs predominated and there were insignificant changes in the proportion of NK cells in the peripheral blood. Adverse events also showed a dose–response with the most common AEs injection-site reactions and influenza-like syndrome (126). These studies were followed by Rosenzwajg et al. to determine the effects of IL-2 on induction of Tregs and NK cells in patients with T1DM (129, 130). They observed an increase in CD4+FOXP3+ and CD8+FOXP3+ Tregs, the proportion and duration of which was dose dependent. The Tregs expressed enhanced levels of activation markers and basal pSTAT5 and had a 20-fold higher sensitivity to IL-2 than Teff and NK cells. Global transcriptome analyses showed a dose-dependent decrease in immune response signatures. However, although they were able to induce a dose-dependent increase in Tregs, they did not observe a change in glucose metabolism (126).
Low-dose IL-2 is being trialed in other clinical settings, including rheumatoid arthritis, ankylosing spondylitis, systemic lupus erythematosus, psoriasis, Behcet’s disease, Wegener’s granulomatosis, Takayasu’s disease, Crohn’s disease, ulcerative colitis, autoimmune hepatitis, and sclerosing cholangitis (TRANSREG, ClinicalTrials.gov NCT01988506).
CD3 Monoclonal Antibodies
In the 1990s, we established that CD3 monoclonal antibodies (mAbs) were shown to cause reversal of disease and induce immunologic tolerance in hyperglycemic NOD mice (131–135). To overcome the adverse events due to cytokine release associated with clinical use of Fc receptor (FcR)-binding CD3 mAb, molecules with mutations in the Fc region of the immunoglobulin were developed and further mechanistic studies in mice used either similarly modified molecules or F(ab’)2 fragments of the hamster anti-mouse CD3 mAb 145-2C11. Similar to the induction of Tregs following engagement of TCR with self-antigens, a relatively weak cognate signal may result in development of a regulatory phenotype rather than effector cells or depletion. The CD3 mAbs depleted effector T cells and caused a transient systemic rise in the percentage of CD4+FOXP3+ Tregs (136, 137). Expression of Helios was increased after anti-CD3 mAb treatment, suggesting that it increased the relative proportion of Tregs and stabilized their function (136). Belghith et al. described induction of adaptive TGFβ-dependent Tregs with anti-CD3 mAb even in CD28−/− mice that lacked naturally occurring Tregs (137). These studies suggested that CD3 mAb induced pTregs from Tconv cells. Expanding on this notion, Esplugues et al. and Waldron-Lynch et al. showed in mice and in humanized mice that teplizumab, a non-FcR binding anti-CD3 mAb, caused migration of peripheral T cells to the lamina propria of the gut. At that location, there was induction of FOXP3 on T cells and Tregs. In the murine studies, this occurred following an inflammatory response heralded by the production of IL-17 and resolution with the formation of TGFβ and IL-10-producing cells. In the humanized mice treated with teplizumab, the gut migrating T cells expressed FOXP3 and produced IL-10. In the humanized mice, treatment with teplizumab prevented rejection of xenogeneic skin grafts (138, 139). Both CD4+ and CD8+ T cells were involved and the effects of the mAb required gut migration because it was blocked with the anti-α4 mAb, natalizumab. Interestingly, more recent studies by You et al. showed, in the setting of islet transplantation, that the effects of the anti-CD3 mAb were greatest after initiation of the graft-specific immune response (140). These findings were consistent with previous work in NOD mice and an experimental animal model of Multiple Sclerosis (EAE) showing that the efficacy was greatest at the time of peak immune response (131). This was confirmed in humans with T1DM where the greatest efficacy was observed at the time of clinical diagnosis (137).
Human trials with CD3 mAbs have shown effects and mechanisms consistent with induction of immune regulation (141–146). In general, an increase in the number of circulating CD4+CD25+FOXP3+ Tregs in the peripheral blood has not been seen in drug-treated patients with new onset T1DM. However, an increase in the number of circulating CD8+ T cells that have been suggested to have regulatory function has been consistently observed (147). These cells produce IL-10 family members and can suppress effector T cells ex vivo. Some evidence suggests that among these cells are CD8+ cells reactive with antigens from EBV, which may be reactivated with high doses of CD3 mAb (148). More recent studies have suggested that these cells show reduced expression of genes associated with T cell activation (149).
Mucosal, oral or nasal, administration of CD3 mAb has been shown to suppress autoimmunity in animal models of encephalomyelitis, collagen-induced arthritis, systemic lupus erythematosus, and diabetes (150–155). These studies demonstrated suppression of autoimmunity via induction of a Th3 type CD4+CD25−LAP (latency-associated peptide)+ Treg population. These Tregs are proposed to be a unique population given lack of CD25 expression and lower expression of FOXP3 that is not induced by mucosal anti-CD3 administration. Mucosal anti-CD3 seems to act primarily locally by inducing Tregs and not by reduction of effector T cells. Oral anti-CD3 has also been used in human studies to promote Tregs (156–158). Patients with non-alcoholic steatohepatitis treated with oral anti-CD3 in a Phase IIa trial showed that treatment was well tolerated and resulted in positive effects on hepatic and metabolic factors. Depending on treatment dose and time of analysis, they observed increased CD4+LAP+, CD4+CD25+LAP+, and CD4+CD25+FOXP3+ cells as well as increased TGF-β, supporting that oral anti-CD3 could induce Tregs in humans (156).
Other Anti-T Cell Modulators
The major effect of anti-thymocyte globulin (ATG) and polyclonal anti-T cell antibodies was thought to involve broad elimination of T cells (159–164). However, subsequent in vitro and in vivo data suggest that ATG may actually selectively deplete Teff cells while sparing or in some cases even promoting the generation of Tregs (160, 165–171). The mechanisms that lead to depletion versus induction of regulatory cells have not been clearly defined.
Alefacept is a LFA3 fusion molecule that binds CD2 and results in eradication of CD2 expressing cells and has primarily been used in the treatment of psoriasis where it resulted in clinical benefit and sustained disease remission long after drug termination, suggesting lasting immune tolerance (172–174). In a randomized placebo-controlled, Phase II trial in T1DM preservation of C-peptide was improved with Alefacept vs. placebo treatment at 1 and 2 years. Phenotype studies of peripheral blood cells showed a decreased frequency of central memory and effector memory T cells and preserved Tregs resulting in increased Treg:Teff ratio similar to what was seen in mouse models with CD3 mAb (175–177).
Cellular Therapy with Tregs
The ability to identify Tregs based on the expression of surface markers enabled investigators to isolate and potentially expand them ex vivo for cell therapy treatment. Moreover, the evidence that defects in Treg signaling that had been observed in patients might be repaired/reversed during culture provided even more support for adoptive cellular therapy with Tregs for treatment of autoimmune diseases. Methods to expand Tregs for clinical use were reported by our group using a 14-day expansion protocol with anti-CD3/28 and IL-2 (178). The expanded cells retained their immune suppressive function and had features consistent with nTregs, including high expression of CD25 and FOXP3 and demethylation of the TSDR. The Tregs could be expanded 500- to 2000-fold to as many as 3 × 109 cells from 400 ml of peripheral blood. This represents more than 20% of the total estimated number of Tregs in humans (179).
Autoantigen-specific Tregs have had superior efficacy to polyclonal Tregs in preclinical studies but difficulties in expanding these cells and maintaining their phenotype and function led to the initial development of polyclonal Tregs for adoptive immune therapy (180, 181). An important consideration was that although Tregs require TCR-mediated activation to develop regulatory activity, their suppressive activity can spread within the affected tissues through bystander suppression and they can regulate local inflammatory responses through a combination of cell–cell contact and suppressive cytokine production. Thus, initial studies have been performed with polyclonal Tregs. In one study of 12 patients with new onset T1DM, aged 5–18 years, Marek-Trzonkowska et al. administered autologous Tregs that had been expanded with CD3/CD28 antibodies and IL-2. FOXP3 expression was found on >90% of the infused cells. A dose escalation of 10–30 × 106 Tregs/kg in 1 or 2 doses was studied. After 1 year, 8/12 met the criteria of clinical remission (<0.5 U/kg/d of insulin) and two were insulin independent. They reported a significant improvement in glucagon-stimulated C-peptide responses and reduced insulin usage at 4 months and 1 year compared to untreated subjects. The infusions were well tolerated and antibody responses to a hepatitis B vaccine and rubella were apparently not affected by the treatment (182).
In a recently completed Phase I trial (NCT01210664), we observed that polyclonal FOXP3+CD4+CD25+CD127lo Tregs could be efficiently isolated and expanded from patients with T1DM (183). This trial involved isolation and expansion of autologous Tregs over a 2-week culture period. The cells could then be shipped to a collaborating institution for infusion into patients, suggesting that development of this strategy for adoptive transfer of cells is feasible and need not be limited only to sites that are capable of performing the expansion on site. There were no significant safety signals from the study and the cells maintained their phenotype in vivo: there was no evidence of differentiation of the infused Tregs into other, potentially pathogenic phenotypes. Importantly, using an in vitro non-radioactive labeling technique (184), a subset of the adoptively transferred Tregs were observed to survive >1 year post infusion. A similar strategy is being used for treatment of patients with cutaneous lupus erythematosus (NCT02428309).
Tregs in Organ Transplantation
Adoptive Treg therapy is being tested in transplantation settings. Tregs, partially matched for MHC, isolated from umbilical cord blood, have been used to treat graft-versus-host disease, associated with double umbilical cord blood transplantation. The Tregs were enriched from cryopreserved umbilical cord blood followed by an 18-day expansion culture with anti-CD3/CD28 beads and IL-2. Patients received a dose of 0.1–30 × 105 Treg/kg after double umbilical cord blood transplantation. After infusion, the Tregs could be detected for 14 days. The Treg infusion was associated with reduced incidence of grades II–IV GVHD compared to historical controls with no deleterious effects on risk of infection, relapse, or early mortality (185). However, the Tregs were short lived, suggesting that either the third party cells were immunogenic or the absence of IL-2 in vivo in the BMT setting reduced Treg survival.
Solid organ transplants offer the opportunity to isolate, expand, and infuse antigen-specific Tregs from the graft recipient. In an ongoing trial (NCT02091232), Tregs are being isolated from mixed lymphocyte cultures in the setting of CTLA4Ig and infusing sorted Tregs from these cultures (The “ONE” Study). In patients with evidence of renal allograft rejection, another trial is expanding polyclonal Tregs for adoptive immune therapy (NCT02088931).
Future Directions
Studies being conducted at UCSF and elsewhere have been initiated that utilize either polyclonal Tregs in other kidney and liver transplant settings with a goal of the discontinuation of immunosuppressive drugs (e.g., NCT02088931). These and other efforts will be essential in determining the potential use of ex vivo expanded cells in patients with these immune dysregulation diseases. Maximizing the functional activity, persistence, and migration of infused Tregs will be essential by either altering the Treg genetically or combining the treatment with growth factors, such as IL-2. Trials of Tregs plus low-dose IL-2 are ongoing (NCT1937468) and others are planned in T1DM. In an effort to increase the persistence of the cells in vivo, Parmer et al. showed that ex vivo fucosylation, which forms the sialyl Lewis X moiety on P-selectin glycoprotein ligand-1, improved in vivo persistence and had improved effects on a murine model of GVHD (186). We expect that additional biologics and antigen-based therapies will be introduced to protect and expand Tregs.
As mentioned above, antigen-specific Tregs are likely to be the best able to block unwanted immunity. In this regard, we have developed alloantigen-specific Tregs using a modified protocol that can selectively expand Tregs specific for MHC molecules expressed by donor tissue in kidney and liver transplant recipients. Donor-alloantigen-reactive Tregs (darTregs), isolated after leukapheresis, are being tested in patients undergoing liver transplantation at doses up to 800 × 106 together with thymoglobulin, everolimus, mycophenylate mofetil (MMF), and solumedrol (NCT02188719). Future studies are likely to include Tregs, engineered to express TCRs, that are specific for auto or alloantigens or even antigens that are on or in the region of target organs to cause localization of the cells to the site of pathology and activation. Strategies used to introduce chimeric antigen receptors (CAR) for tumor antigens in effector T cells may be considered. As such, Ag-specific TCRs or chimeric antigen receptor-based genomic manipulation is now feasible with the use of genetic manipulation techniques, such as CRISPRCas9 to modify and enhance Treg specificity and function (187).
Conclusion
Because of their central role in regulating immune tolerance and modulating immune responses, manipulation of Tregs represents a clear target for treatment of unwanted immune responses. Contrary to the strategy of immune suppression or cell deletion as a means to curtail responses, treatment with agents that enhance or cellular therapies with Tregs represent a strategy to restore immune tolerance. Trials that have enhanced the number and function of Tregs have shown success in treatment of a variety of conditions, and the development of technologies to expand Tregs and even select for antigen specific cells has created a new era for adoptive cellular immune therapies. Further refinements of the ways Tregs are selected and administered, including methods to improve their survival and maintain their efficacy, are likely to occur in the next several years.
Author Contributions
AP, LC, JB, and KH reviewed the data and wrote the manuscript.
Conflict of Interest Statement
Dr. Bluestone maintains agreements with MacroGenics Inc., Caladrius Biosciences, and Juno Therapeutics for development and testing of anti-CD3 mAb and regulatory T cells. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Supported by R01AI046643 (JAB) and R01DK057846 and U01AI02011 (KCH) from the National Institutes of Health and 17-2013-549 and 2-SRA-2014-150 and 17-2011-661 (JAB) and 2-SRA-2014-158 from the Juvenile Diabetes Research Foundation.
References
1. Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. T cells in the control of organ-specific autoimmunity. J Clin Invest (2015) 125(6):2250–60. doi:10.1172/JCI78089
2. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6(4):295–307. doi:10.1038/nri1806
3. Voskens CJ, Goldinger SM, Loquai C, Robert C, Kaehler KC, Berking C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One (2013) 8(1):e53745. doi:10.1371/journal.pone.0053745
4. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
5. Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev (2001) 182:207–14. doi:10.1034/j.1600-065X.2001.1820117.x
6. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi:10.1007/978-3-662-43492-5_3
7. Rothstein DM, Camirand G. New insights into the mechanisms of Treg function. Curr Opin Organ Transplant (2015) 20(4):376–84. doi:10.1097/MOT.0000000000000212
8. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi:10.1038/83784
9. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest (2006) 116(6):1713–22. doi:10.1172/JCI25112
10. Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res (2007) 38(1–3):112–21. doi:10.1007/s12026-007-0022-2
11. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27(1):18–20. doi:10.1038/83707
12. Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445(7129):766–70. doi:10.1038/nature05479
13. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445(7129):771–5. doi:10.1038/nature05543
14. Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol (2007) 8(5):457–62. doi:10.1038/ni1455
15. Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk HD. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front Pharmacol (2015) 6:184. doi:10.3389/fphar.2015.00184
16. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10(7):490–500. doi:10.1038/nri2785
17. Safinia N, Scotta C, Vaikunthanathan T, Lechler RI, Lombardi G. Regulatory T cells: serious contenders in the promise for immunological tolerance in transplantation. Front Immunol (2015) 6:438. doi:10.3389/fimmu.2015.00438
18. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi:10.1038/ni1536
19. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27(4):635–46. doi:10.1016/j.immuni.2007.08.014
20. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi:10.1126/science.1160062
21. Paust S, Lu L, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci U S A (2004) 101(28):10398–403. doi:10.1073/pnas.0403342101
22. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332(6029):600–3. doi:10.1126/science.1202947
23. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450(7169):566–9. doi:10.1038/nature06306
24. Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev (2006) 212:28–50.
25. Shevach EM, Davidson TS, Huter EN, Dipaolo RA, Andersson J. Role of TGF-Beta in the induction of Foxp3 expression and T regulatory cell function. J Clin Immunol (2008) 28(6):640–6. doi:10.1007/s10875-008-9240-1
26. Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J Exp Med (1996) 183(6):2669–74. doi:10.1084/jem.183.6.2669
27. Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR, et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol (2001) 166(6):3789–96. doi:10.4049/jimmunol.166.6.3789
28. Bailey-Bucktrout SL, Bluestone JA. Regulatory T cells: stability revisited. Trends Immunol (2011) 32(7):301–6. doi:10.1016/j.it.2011.04.002
29. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol (2008) 181(10):7350–5. doi:10.4049/jimmunol.181.10.7350
30. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14(4):307–8. doi:10.1038/ni.2554
31. Yadav M, Stephan S, Bluestone JA. Peripherally induced tregs – role in immune homeostasis and autoimmunity. Front Immunol (2013) 4:232. doi:10.3389/fimmu.2013.00232
32. Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol Rev (2011) 241(1):63–76. doi:10.1111/j.1600-065X.2011.01004.x
33. Simonetta F, Chiali A, Cordier C, Urrutia A, Girault I, Bloquet S, et al. Increased CD127 expression on activated FOXP3+CD4+ regulatory T cells. Eur J Immunol (2010) 40(9):2528–38. doi:10.1002/eji.201040531
34. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203(7):1701–11. doi:10.1084/jem.20060772
35. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, et al. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem (2010) 285(36):27571–80. doi:10.1074/jbc.M110.127100
36. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5:304. doi:10.3389/fimmu.2014.00304
37. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol (2006) 177(10):6780–6. doi:10.4049/jimmunol.177.10.6780
38. Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature (2013) 501(7466):252–6. doi:10.1038/nature12428
39. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209(10):S1–19. doi:10.1084/jem.20120822
40. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184(7):3433–41. doi:10.4049/jimmunol.0904028
41. Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol (2012) 188(3):976–80. doi:10.4049/jimmunol.1102964
42. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi:10.1016/j.immuni.2014.02.012
43. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195(1):145–55. doi:10.4049/jimmunol.1402381
44. Bin Dhuban K, d’Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, et al. Coexpression of TIGIT and FCRL3 identifies Helios+ human memory regulatory T cells. J Immunol (2015) 194(8):3687–96. doi:10.4049/jimmunol.1401803
45. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med (2014) 211(1):121–36. doi:10.1084/jem.20131142
46. Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity (2009) 30(2):204–17. doi:10.1016/j.immuni.2008.11.014
47. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, et al. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol (2010) 185(11):6426–30. doi:10.4049/jimmunol.0903940
48. Sadlack B, Löhler J, Schorle H, Klebb G, Haber H, Sickel E, et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol (1995) 25(11):3053–9. doi:10.1002/eji.1830251111
49. Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science (1995) 268(5216):1472–6. doi:10.1126/science.7770771
50. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol (2007) 119(2):482–7. doi:10.1016/j.jaci.2006.10.007
51. Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet (2007) 39(3):329–37. doi:10.1038/ng1958
52. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes (2010) 59(2):407–15. doi:10.2337/db09-0694
53. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi:10.1038/ni759
54. Shevach EM, Stephens GL. The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol (2006) 6(8):613–8. doi:10.1038/nri1867
55. Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood (2005) 105(7):2845–51. doi:10.1182/blood-2004-07-2959
56. Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci U S A (2006) 103(35):13138–43. doi:10.1073/pnas.0603107103
57. Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood (2006) 108(13):4260–7. doi:10.1182/blood-2006-06-027409
58. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi:10.1016/j.immuni.2009.03.019
59. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem (2010) 285(10):7176–86. doi:10.1074/jbc.M109.047423
60. Beyersdorf N, Ding X, Tietze JK, Hanke T. Characterization of mouse CD4 T cell subsets defined by expression of KLRG1. Eur J Immunol (2007) 37(12):3445–54. doi:10.1002/eji.200737126
61. Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol (2009) 10(7):689–95. doi:10.1038/ni.1760
62. Stephens GL, Andersson J, Shevach EM. Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. J Immunol (2007) 178(11):6901–11. doi:10.4049/jimmunol.178.11.6901
63. Kornete M, Sgouroudis E, Piccirillo CA. ICOS-dependent homeostasis and function of Foxp3+ regulatory T cells in islets of nonobese diabetic mice. J Immunol (2012) 188(3):1064–74. doi:10.4049/jimmunol.1101303
64. Nurieva RI, Duong J, Kishikawa H, Dianzani U, Rojo JM, Ho IC, et al. Transcriptional regulation of th2 differentiation by inducible costimulator. Immunity (2003) 18(6):801–11. doi:10.1016/S1074-7613(03)00144-4
65. Bensinger SJ, Walsh PT, Zhang J, Carroll M, Parsons R, Rathmell JC, et al. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J Immunol (2004) 172(9):5287–96. doi:10.4049/jimmunol.172.9.5287
66. Rudensky AY, Gavin M, Zheng Y. FOXP3 and NFAT: partners in tolerance. Cell (2006) 126(2):253–6. doi:10.1016/j.cell.2006.07.005
67. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol (2008) 9(2):194–202. doi:10.1038/ni1549
68. Hu H, Djuretic I, Sundrud MS, Rao A. Transcriptional partners in regulatory T cells: Foxp3, Runx and NFAT. Trends Immunol (2007) 28(8):329–32. doi:10.1016/j.it.2007.06.006
69. Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity (2010) 33(3):313–25. doi:10.1016/j.immuni.2010.09.001
70. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol (2015) 16(3):276–85. doi:10.1038/ni0515-544d
71. Ohkura N, Sakaguchi S. Maturation of effector regulatory T cells. Nat Immunol (2011) 12(4):283–4. doi:10.1038/ni0411-283
72. Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol (2011) 12(4):304–11. doi:10.1038/ni.2006
73. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature (2012) 486(7404):549–53. doi:10.1038/nature11132
74. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15(8):930–9. doi:10.1038/nm.2002
75. Wakamatsu E, Mathis D, Benoist C. Convergent and divergent effects of costimulatory molecules in conventional and regulatory CD4+ T cells. Proc Natl Acad Sci U S A (2013) 110(3):1023–8. doi:10.1073/pnas.1220688110
76. Van Gool F, Molofsky AB, Morar MM, Rosenzwajg M, Liang HE, Klatzmann D, et al. Interleukin-5-producing group 2 innate lymphoid cells control eosinophilia induced by interleukin-2 therapy. Blood (2014) 124(24):3572–6. doi:10.1182/blood-2014-07-587493
77. Beyer M, Schultze JL. CD4+CD25highFOXP3+ regulatory T cells in peripheral blood are primarily of effector memory phenotype. J Clin Oncol (2007) 25(18):2628–30. doi:10.1200/JCO.2006.08.0192 author reply 2630-2,
78. Campbell DJ. Control of regulatory T cell migration, function, and homeostasis. J Immunol (2015) 195(6):2507–13. doi:10.4049/jimmunol.1500801
79. Zhou X, Tang J, Cao H, Fan H, Li B. Tissue resident regulatory T cells: novel therapeutic targets for human disease. Cell Mol Immunol (2015) 12(5):543–52. doi:10.1038/cmi.2015.23
80. Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol (2013) 14(10):1007–13. doi:10.1038/ni.2683
81. Bollrath J, Powrie FM. Controlling the frontier: regulatory T-cells and intestinal homeostasis. Semin Immunol (2013) 25(5):352–7. doi:10.1016/j.smim.2013.09.002
82. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell (2013) 155(6):1282–95. doi:10.1016/j.cell.2013.10.054
83. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell (2015) 162(5):1078–89. doi:10.1016/j.cell.2015.08.021
84. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol (2014) 27:1–7. doi:10.1016/j.coi.2013.12.005
85. Shevach EM. Fatal attraction: tumors beckon regulatory T cells. Nat Med (2004) 10(9):900–1. doi:10.1038/nm0904-900
86. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
87. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T-reg cells. Nature (2011) 475(7355):226–U141. doi:10.1038/nature10169
88. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186(7):3918–26. doi:10.4049/jimmunol.1003099
89. Coenen JJ, Koenen HJ, van Rijssen E, Hilbrands LB, Joosten I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood (2006) 107(3):1018–23. doi:10.1182/blood-2005-07-3032
90. Ruggenenti P, Perico N, Gotti E, Cravedi P, D’Agati V, Gagliardini E, et al. Sirolimus versus cyclosporine therapy increases circulating regulatory T cells, but does not protect renal transplant patients given alemtuzumab induction from chronic allograft injury. Transplantation (2007) 84(8):956–64. doi:10.1097/01.tp.0000284808.28353.2c
91. Segundo DS, Ruiz JC, Izquierdo M, Fernández-Fresnedo G, Gómez-Alamillo C, Merino R, et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4+CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation (2006) 82(4):550–7. doi:10.1097/01.tp.0000229473.95202.50
92. Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, Strom TB. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant (2007) 7(7):1722–32. doi:10.1111/j.1600-6143.2007.01842.x
93. Zeiser R, Nguyen VH, Beilhack A, Buess M, Schulz S, Baker J, et al. Inhibition of CD4+CD25+ regulatory T-cell function by calcineurin-dependent interleukin-2 production. Blood (2006) 108(1):390–9. doi:10.1182/blood-2006-01-0329
94. Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity (2010) 33(3):301–11. doi:10.1016/j.immuni.2010.09.002
95. Ohkura N, Hamaguchi M, Sakaguchi S. FOXP3+ regulatory T cells: control of FOXP3 expression by pharmacological agents. Trends Pharmacol Sci (2011) 32(3):158–66. doi:10.1016/j.tips.2010.12.004
96. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105(12):4743–8. doi:10.1182/blood-2004-10-3932
97. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol (2006) 177(12):8338–47. doi:10.4049/jimmunol.177.12.8338
98. Valmori D, Tosello V, Souleimanian NE, Godefroy E, Scotto L, Wang Y, et al. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4(+) T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4(+) T cells. J Immunol (2006) 177(2):944–9. doi:10.4049/jimmunol.177.2.944
99. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi:10.1016/j.immuni.2009.04.014
100. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12(4):295–303. doi:10.1038/ni.2005
101. Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol (2007) 7(13):1819–24. doi:10.1016/j.intimp.2007.08.027
102. Yuan LF, Li GD, Ren XJ, Nian H, Li XR, Zhang XM. Rapamycin ameliorates experimental autoimmune uveoretinitis by inhibiting Th1/Th2/Th17 cells and upregulating CD4+CD25+ Foxp3 regulatory T cells. Int J Ophthalmol (2015) 8(4):659–64. doi:10.3980/j.issn.2222-3959.2015.04.03
103. Battaglia M, Stabilini A, Draghici E, Migliavacca B, Gregori S, Bonifacio E, et al. Induction of tolerance in type 1 diabetes via both CD4+CD25+ T regulatory cells and T regulatory type 1 cells. Diabetes (2006) 55(6):1571–80. doi:10.2337/db05-1576
104. Monti P, Scirpoli M, Maffi P, Piemonti L, Secchi A, Bonifacio E, et al. Rapamycin monotherapy in patients with type 1 diabetes modifies CD4+CD25+FOXP3+ regulatory T-cells. Diabetes (2008) 57(9):2341–7. doi:10.2337/db08-0138
105. Gong N, Chen Z, Wang J, Fang A, Li Y, Xiang Y, et al. Immunoregulatory effects of sirolimus vs. tacrolimus treatment in kidney allograft recipients. Cell Immunol (2015) 297(2):87–93. doi:10.1016/j.cellimm.2015.07.002
106. Li Y, Shi Y, Liao Y, Yan L, Zhang Q, Wang L. Differential regulation of Tregs and Th17/Th1 cells by a sirolimus-based regimen might be dependent on STAT-signaling in renal transplant recipients. Int Immunopharmacol (2015) 28(1):435–43. doi:10.1016/j.intimp.2015.07.006
107. Sabbatini M, Ruggiero G, Palatucci AT, Rubino V, Federico S, Giovazzino A, et al. Oscillatory mTOR inhibition and T increase in kidney transplantation. Clin Exp Immunol (2015) 182(2):230–40. doi:10.1111/cei.12669
108. Long SA, Buckner JH, Greenbaum CJ. IL-2 therapy in type 1 diabetes: “Trials” and tribulations. Clin Immunol (2013) 149(3):324–31. doi:10.1016/j.clim.2013.02.005
109. Malek TR. The biology of interleukin-2. Annu Rev Immunol (2008) 26:453–79. doi:10.1146/annurev.immunol.26.021607.090357
110. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med (2009) 206(4):751–60. doi:10.1084/jem.20082824
111. Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol (2015) 15(5):283–94. doi:10.1038/nri3823
112. Walsh PT, Buckler JL, Zhang J, Gelman AE, Dalton NM, Taylor DK, et al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Invest (2006) 116(9):2521–31. doi:10.1172/JCI28057
113. Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet (2007) 39(9):1074–82. doi:10.1038/ng2102
114. Dendrou CA, Wicker LS. The IL-2/CD25 pathway determines susceptibility to T1D in humans and NOD mice. J Clin Immunol (2008) 28(6):685–96. doi:10.1007/s10875-008-9237-9
115. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28(5):687–97. doi:10.1016/j.immuni.2008.03.016
116. Garg G, Tyler JR, Yang JH, Cutler AJ, Downes K, Pekalski M, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol (2012) 188(9):4644–53. doi:10.4049/jimmunol.1100272
117. Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med (2010) 207(9):1871–8. doi:10.1084/jem.20100209
118. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity (2013) 39(5):949–62. doi:10.1016/j.immuni.2013.10.016
119. Lotze MT, Chang AE, Seipp CA, Simpson C, Vetto JT, Rosenberg SA. High-dose recombinant interleukin 2 in the treatment of patients with disseminated cancer. Responses, treatment-related morbidity, and histologic findings. JAMA (1986) 256(22):3117–24. doi:10.1001/jama.256.22.3117
120. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med (1985) 313(23):1485–92. doi:10.1056/NEJM198512053132327
121. Bayer AL, Pugliese A, Malek TR. The IL-2/IL-2R system: from basic science to therapeutic applications to enhance immune regulation. Immunol Res (2013) 57(1–3):197–209. doi:10.1007/s12026-013-8452-5
122. Bluestone JA. The yin and yang of interleukin-2-mediated immunotherapy. N Engl J Med (2011) 365(22):2129–31. doi:10.1056/NEJMe1110900
123. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365(22):2055–66. doi:10.1056/NEJMoa1108188
124. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med (2011) 365(22):2067–77. doi:10.1056/NEJMoa1105143
125. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes (2012) 61(9):2340–8. doi:10.2337/db12-0049
126. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1(4):295–305. doi:10.1016/S2213-8587(13)70113-X
127. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5(179):179ra43. doi:10.1126/scitranslmed.3005265
128. Rabinovitch A, Suarez-Pinzon WL, Shapiro AM, Rajotte RV, Power R. Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes (2002) 51(3):638–45. doi:10.2337/diabetes.51.3.638
129. Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes (2015) 64(6):2172–83. doi:10.2337/db14-1322
130. Rosenzwajg M, Churlaud G, Mallone R, Six A, Dérian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun (2015) 58:48–58. doi:10.1016/j.jaut.2015.01.001
131. Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol (1997) 158(6):2947–54.
132. Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A (1994) 91(1):123–7. doi:10.1073/pnas.91.1.123
133. Chatenoud L, Waldmann H. CD3 monoclonal antibodies: a first step towards operational immune tolerance in the clinic. Rev Diabet Stud (2012) 9(4):372–81. doi:10.1900/RDS.2012.9.372
134. Chatenoud L. Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat Rev Endocrinol (2010) 6(3):149–57. doi:10.1038/nrendo.2009.275
135. Kuhn C, You S, Valette F, Hale G, van Endert P, Bach JF, et al. Human CD3 transgenic mice: preclinical testing of antibodies promoting immune tolerance. Sci Transl Med (2011) 3(68):68ra10. doi:10.1126/scitranslmed.3001830
136. Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol (2011) 187(4):2015–22. doi:10.4049/jimmunol.1100713
137. Belghith M, Bluestone JA, Barriot S, Mégret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med (2003) 9(9):1202–8. doi:10.1038/nm924
138. Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, et al. Control of TH17 cells occurs in the small intestine. Nature (2011) 475(7357):514–8. doi:10.1038/nature10228
139. Waldron-Lynch F, Henegariu O, Deng S, Preston-Hurlburt P, Tooley J, Flavell R, et al. Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci Transl Med (2012) 4(118):118ra12. doi:10.1126/scitranslmed.3003401
140. You S, Zuber J, Kuhn C, Baas M, Valette F, Sauvaget V, et al. Induction of allograft tolerance by monoclonal CD3 antibodies: a matter of timing. Am J Transplant (2012) 12(11):2909–19. doi:10.1111/j.1600-6143.2012.04213.x
141. Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes (2005) 54(6):1763–9. doi:10.2337/diabetes.54.6.1763
142. Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med (2002) 346(22):1692–8. doi:10.1056/NEJMoa012864
143. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med (2005) 352(25):2598–608. doi:10.1056/NEJMoa043980
144. Keymeulen B, Walter M, Mathieu C, Kaufman L, Gorus F, Hilbrands R, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia (2010) 53(4):614–23. doi:10.1007/s00125-009-1644-9
145. Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr, et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet (2011) 378(9790):487–97. doi:10.1016/S0140-6736(11)60931-8
146. Daifotis AG, Koenig S, Chatenoud L, Herold KC. Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol (2013) 149(3):268–78. doi:10.1016/j.clim.2013.05.001
147. Ablamunits V, Bisikirska B, Herold KC. Acquisition of regulatory function by human CD8(+) T cells treated with anti-CD3 antibody requires TNF. Eur J Immunol (2010) 40(10):2891–901. doi:10.1002/eji.201040485
148. Keymeulen B, Candon S, Fafi-Kremer S, Ziegler A, Leruez-Ville M, Mathieu C, et al. Transient Epstein-Barr virus reactivation in CD3 monoclonal antibody-treated patients. Blood (2010) 115(6):1145–55. doi:10.1182/blood-2009-02-204875
149. Tooley JE, Vudattu N, Choi J, Cotsapas C, Devine L, Raddassi K, et al. Changes in T-cell subsets identify responders to FcR non-binding anti-CD3 mAb (teplizumab) in patients with Type 1 diabetes. Eur J Immunol (2015). doi:10.1002/eji.201545708
150. Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+ CD25- LAP+ T cells. Nat Med (2006) 12(6):627–35. doi:10.1038/nm1408
151. Wu HY, Maron R, Tukpah AM, Weiner HL. Mucosal anti-CD3 monoclonal antibody attenuates collagen-induced arthritis that is associated with induction of LAP+ regulatory T cells and is enhanced by administration of an emulsome-based Th2-skewing adjuvant. J Immunol (2010) 185(6):3401–7. doi:10.4049/jimmunol.1000836
152. Wu HY, Quintana FJ, Weiner HL. Nasal anti-CD3 antibody ameliorates lupus by inducing an IL-10-secreting CD4+ CD25- LAP+ regulatory T cell and is associated with down-regulation of IL-17+ CD4+ ICOS+ CXCR5+ follicular helper T cells. J Immunol (2008) 181(9):6038–50. doi:10.4049/jimmunol.181.9.6038
153. Ishikawa H, Ochi H, Chen ML, Frenkel D, Maron R, Weiner HL. Inhibition of autoimmune diabetes by oral administration of anti-CD3 monoclonal antibody. Diabetes (2007) 56(8):2103–9. doi:10.2337/db06-1632
154. Wu HY, Center EM, Tsokos GC, Weiner HL. Suppression of murine SLE by oral anti-CD3: inducible CD4+CD25-LAP+ regulatory T cells control the expansion of IL-17+ follicular helper T cells. Lupus (2009) 18(7):586–96. doi:10.1177/0961203308100511
155. Hu C, Ding H, Zhang X, Wong FS, Wen L. Combination treatment with anti-CD20 and oral anti-CD3 prevents and reverses autoimmune diabetes. Diabetes (2013) 62(8):2849–58. doi:10.2337/db12-1175
156. Lalazar G, Mizrahi M, Turgeman I, Adar T, Ben Ya’acov A, Shabat Y, et al. Oral administration of OKT3 MAb to patients with NASH, promotes regulatory T-cell induction, and alleviates insulin resistance: results of a phase IIa blinded placebo-controlled trial. J Clin Immunol (2015) 35(4):399–407. doi:10.1007/s10875-015-0160-6
157. Ilan Y, Zigmond E, Lalazar G, Dembinsky A, Ben Ya’acov A, Hemed N, et al. Oral administration of OKT3 monoclonal antibody to human subjects induces a dose-dependent immunologic effect in T cells and dendritic cells. J Clin Immunol (2010) 30(1):167–77. doi:10.1007/s10875-009-9323-7
158. Halota W, Ferenci P, Kozielewicz D, Dybowska D, Lisovoder N, Samira S, et al. Oral anti-CD3 immunotherapy for HCV-nonresponders is safe, promotes regulatory T cells and decreases viral load and liver enzyme levels: results of a phase-2a placebo-controlled trial. J Viral Hepat (2015) 22(8):651–7. doi:10.1111/jvh.12369
159. Like AA, Rossini AA, Guberski DL, Appel MC, Williams RM. Spontaneous diabetes mellitus: reversal and prevention in the BB/W rat with antiserum to rat lymphocytes. Science (1979) 206(4425):1421–3. doi:10.1126/science.388619
160. Chung DT, Korn T, Richard J, Ruzek M, Kohm AP, Miller S, et al. Anti-thymocyte globulin (ATG) prevents autoimmune encephalomyelitis by expanding myelin antigen-specific Foxp3+ regulatory T cells. Int Immunol (2007) 19(8):1003–10. doi:10.1093/intimm/dxm078
161. Ogawa N, Minamimura K, Kodaka T, Maki T. Short administration of polyclonal anti-T cell antibody (ALS) in NOD mice with extensive insulitis prevents subsequent development of autoimmune diabetes. J Autoimmun (2006) 26(4):225–31. doi:10.1016/j.jaut.2006.03.001
162. Simon G, Parker M, Ramiya V, Wasserfall C, Huang Y, Bresson D, et al. Murine antithymocyte globulin therapy alters disease progression in NOD mice by a time-dependent induction of immunoregulation. Diabetes (2008) 57(2):405–14. doi:10.2337/db06-1384
163. Bevans MF, Shalabi RA. Management of patients receiving antithymocyte globulin for aplastic anemia and myelodysplastic syndrome. Clin J Oncol Nurs (2004) 8(4):377–82. doi:10.1188/04.CJON.377-382
164. Bacigalupo A. Antithymocyte globulin for prevention of graft-versus-host disease. Curr Opin Hematol (2005) 12(6):457–62. doi:10.1097/01.moh.0000183726.42063.ec
165. Minamimura K, Gao W, Maki T. CD4+ regulatory T cells are spared from deletion by antilymphocyte serum, a polyclonal anti-T cell antibody. J Immunol (2006) 176(7):4125–32. doi:10.4049/jimmunol.176.7.4125
166. Lopez M, Clarkson MR, Albin M, Sayegh MH, Najafian N. A novel mechanism of action for anti-thymocyte globulin: induction of CD4+CD25+Foxp3+ regulatory T cells. J Am Soc Nephrol (2006) 17(10):2844–53. doi:10.1681/ASN.2006050422
167. Feng X, Kajigaya S, Solomou EE, Keyvanfar K, Xu X, Raghavachari N, et al. Rabbit ATG but not horse ATG promotes expansion of functional CD4+CD25highFOXP3+ regulatory T cells in vitro. Blood (2008) 111(7):3675–83. doi:10.1182/blood-2008-01-130146
168. Valdez-Ortiz R, Bestard O, Llaudó I, Franquesa M, Cerezo G, Torras J, et al. Induction of suppressive allogeneic regulatory T cells via rabbit antithymocyte polyclonal globulin during homeostatic proliferation in rat kidney transplantation. Transpl Int (2015) 28(1):108–19. doi:10.1111/tri.12448
169. Gurkan S, Luan Y, Dhillon N, Allam SR, Montague T, Bromberg JS, et al. Immune reconstitution following rabbit antithymocyte globulin. Am J Transplant (2010) 10(9):2132–41. doi:10.1111/j.1600-6143.2010.03210.x
170. Crepin T, Carron C, Roubiou C, Gaugler B, Gaiffe E, Simula-Faivre D, et al. ATG-induced accelerated immune senescence: clinical implications in renal transplant recipients. Am J Transplant (2015) 15(4):1028–38. doi:10.1111/ajt.13092
171. Lu Y, Suzuki J, Guillioli M, Umland O, Chen Z. Induction of self-antigen-specific Foxp3+ regulatory T cells in the periphery by lymphodepletion treatment with anti-mouse thymocyte globulin in mice. Immunology (2011) 134(1):50–9. doi:10.1111/j.1365-2567.2011.03466.x
172. Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE, et al. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol (2003) 139(6):719–27. doi:10.1001/archderm.139.6.719
173. Krueger GG, Ellis CN. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol (2003) 148(4):784–8. doi:10.1046/j.1365-2133.2003.05239.x
174. Gottlieb AB, Casale TB, Frankel E, Goffe B, Lowe N, Ochs HD, et al. CD4+ T-cell-directed antibody responses are maintained in patients with psoriasis receiving alefacept: results of a randomized study. J Am Acad Dermatol (2003) 49(5):816–25. doi:10.1016/S0190-9622(03)01836-X
175. Chamian F, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, Sullivan-Whalen M, et al. Alefacept (anti-CD2) causes a selective reduction in circulating effector memory T cells (Tem) and relative preservation of central memory T cells (Tcm) in psoriasis. J Transl Med (2007) 5:27. doi:10.1186/1479-5876-5-27
176. Ellis CN, Krueger GG, Alefacept Clinical Study Group. Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N Engl J Med (2001) 345(4):248–55. doi:10.1056/NEJM200107263450403
177. Rigby MR, Harris KM, Pinckney A, DiMeglio LA, Rendell MS, Felner EI, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest (2015) 125(8):3285–96. doi:10.1172/JCI81722
178. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes (2009) 58(3):652–62. doi:10.2337/db08-1168
179. Tang Q, Lee K. Regulatory T-cell therapy for transplantation: how many cells do we need? Curr Opin Organ Transplant (2012) 17(4):349–54. doi:10.1097/MOT.0b013e328355a992
180. Tang Q, Bluestone JA. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med (2013) 3(11):a015552. doi:10.1101/cshperspect.a015552
181. Tang Q, Bluestone JA, Kang SM. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. J Mol Cell Biol (2012) 4(1):11–21. doi:10.1093/jmcb/mjr047
182. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets – results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi:10.1016/j.clim.2014.03.016
183. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Trans Med (2015) 7:315ra. doi:10.1126/scitranslmed.aad4134
184. Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc (2007) 2(12):3045–57. doi:10.1038/nprot.2007.420
185. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi:10.1182/blood-2010-07-293795
186. Parmar S, Liu X, Najjar A, Shah N, Yang H, Yvon E, et al. Ex vivo fucosylation of third-party human regulatory T cells enhances anti-graft-versus-host disease potency in vivo. Blood (2015) 125(9):1502–6. doi:10.1182/blood-2014-10-603449
Keywords: T regulatory cells, autoimmunity, immune tolerance, immune therapy, cellular therapy
Citation: Perdigoto AL, Chatenoud L, Bluestone JA and Herold KC (2016) Inducing and Administering Tregs to Treat Human Disease. Front. Immunol. 6:654. doi: 10.3389/fimmu.2015.00654
Received: 20 October 2015; Accepted: 21 December 2015;
Published: 22 January 2016
Edited by:
Helmut Jonuleit, Johannes Gutenberg-University Mainz, GermanyReviewed by:
Dan Frenkel, Tel Aviv University, IsraelEdgar Schmitt, Johannes Gutenberg-Universität Mainz, Germany
Copyright: © 2016 Perdigoto, Chatenoud, Bluestone and Herold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevan C. Herold, S2V2YW4uaGVyb2xkQHlhbGUuZWR1