 Claire Hivroz1,2,3*
Claire Hivroz1,2,3* Michael Saitakis1,2,3†
Michael Saitakis1,2,3†
- 1Institut Curie Section Recherche, Paris, France
- 2INSERM U932, Paris, France
- 3PSL Research University, Paris, France
T lymphocyte activation is a pivotal step of the adaptive immune response. It requires the recognition by T-cell receptors (TCR) of peptides presented in the context of major histocompatibility complex molecules (pMHC) present at the surface of antigen-presenting cells (APCs). T lymphocyte activation also involves engagement of costimulatory receptors and adhesion molecules recognizing ligands on the APC. Integration of these different signals requires the formation of a specialized dynamic structure: the immune synapse. While the biochemical and molecular aspects of this cell–cell communication have been extensively studied, its mechanical features have only recently been addressed. Yet, the immune synapse is also the place of exchange of mechanical signals. Receptors engaged on the T lymphocyte surface are submitted to many tensile and traction forces. These forces are generated by various phenomena: membrane undulation/protrusion/retraction, cell mobility or spreading, and dynamic remodeling of the actomyosin cytoskeleton inside the T lymphocyte. Moreover, the TCR can both induce force development, following triggering, and sense and convert forces into biochemical signals, as a bona fide mechanotransducer. Other costimulatory molecules, such as LFA-1, engaged during immune synapse formation, also display these features. Moreover, T lymphocytes themselves are mechanosensitive, since substrate stiffness can modulate their response. In this review, we will summarize recent studies from a biophysical perspective to explain how mechanical cues can affect T lymphocyte activation. We will particularly discuss how forces are generated during immune synapse formation; how these forces affect various aspects of T lymphocyte biology; and what are the key features of T lymphocyte response to stiffness.
Introduction
T lymphocytes are motile small cells, which play a key role in adaptive immune responses against pathogens and tumor cells. T lymphocyte activation is triggered by the recognition via the T-cell receptor (TCR) expressed at the surface of T lymphocytes, of antigenic peptides, derived from pathogens or tumors and associated with major histocompatibility complex (MHC) molecules exposed at the surface of antigen-presenting cells (APCs). Numerous costimulatory or co-inhibitory receptor/ligand pairs present at the plasma membrane of both cells can also modulate T lymphocyte activation (1). Thus, T lymphocyte activation is crucially dependent on the close interaction between both plasma membranes. This interaction is organized in time and space by the formation of structures, termed immune synapses, in which molecules are unevenly distributed and segregated while remaining mobile (2–4).
Thanks to increasingly sophisticated visualization techniques, more and more information is accumulated on the organization of both plasma membrane receptors and signaling molecules at the immune synapses. Visualization of T lymphocyte interactions with APCs showed that these cellular partners were submitted to pulling, pushing, and shearing forces due to cell motility relative to each other (5); continuous spontaneous motion of plasma membrane (6); and cytoskeletal remodeling (7–9). A specific function of mechanical forces in T lymphocyte activation was even proposed in the first study showing the dynamic formation of immune synapse (10). Forces exerted by T lymphocytes during these contacts have only been quantified recently (11–13). The TCR itself was shown to be a mechanosensor, i.e., able to convert the mechanical forces exerted during TCR binding to peptide–MHC complexes into a biochemical signal (14–16). Finally, at the resting T lymphocyte membrane, there are organized complexes of receptors and signaling molecules, maintained in a state of basal activity where the membrane receptors are readily available to interact with their ligands on an APC surface and to induce a signaling cascade. This dynamic organization resembles a buffer condition that is able to respond to a minute amount of agonist pMHC in a sea of endogenous pMHCs and is optimized not only for the identification of antigen but also for the initiation and amplification of signals following successful antigen recognition (17–19).
Although formation of the immune synapse has been extensively studied, information on the mechanical properties of the microenvironment and on how these properties affect T lymphocyte functions has only recently become available. We will thus review herein recent advances on the knowledge of how T lymphocytes generate or respond to forces during antigen recognition and immune synapse formation.
Forces in T Cells
When interacting with an APC, T lymphocyte morphology changes drastically: the cell moves on the APC surface, develops invadosome-like structures which push into the cortex of the APC (20–22), spreads on the APC, and eventually stops. During each of these steps, T lymphocytes exert and/or are submitted to forces, which can affect receptor/ligand bonds. We will discuss the pathways involved in the generation of these forces.
Spontaneous Membrane Oscillations and Formation of Protrusions
Lymphocytes, such as other cell types, display membrane undulations with amplitude of several tens of nanometer and frequency ranging between 0.2 and 30 Hz (23). Moreover, when interacting with a surface, T lymphocytes rapidly develop protrusions and retractions that are organized in lateral waves along the cell membrane (24). Filopodia or microvilli are protrusive structures with a length between 0.1 and several micrometers that display receptors at their tips and present cycles of protrusions/retractions, which allow them to sense both the mechanical and biochemical environments (25). In order to grow, filopodia have to develop protrusion forces against the membrane that are mainly produced by actin polymerization at the filopodial tip (26–28). Filopodial diameter is in the same range as the diameter of membrane tubes, the generation of which requires forces ranging from 5 to 30 pN (29). Filopodia not only exert protrusive/pushing forces but also retracting/pulling ones, which have been measured using traction force microscopy, i.e., by recording the local deformation of a soft substrate of known stiffness in which fluorescent beads are embedded (29, 30). In neuron cells, the pulling forces developed by filopodia have been shown to be in the order of 1 nN (31). During their migration on endothelial cells, T cells can form F-actin-based protrusions, termed invadosome-like protrusions (ILPs) (32). These structures are small (diameter of ~0.2 μm) and transient (half-life of ~20 s) and physically push against the endothelial cell surface (20, 33). It has been postulated that ILPs can sense the stiffness of endothelial cells by “tiptoing” their surface (32, 34, 35). More recently, Yang et al. (36) described the forces developed by chemotactic T lymphocytes. A laser trap was used to position two beads, one as source of chemokine gradient and the other to measure the forces exerted by the migrating T lymphocytes (in their case, the Jurkat leukemic T cell line). The protrusion forces measured at the leading edge of Jurkat cells migrating in a gradient of SDF-1 were as high as 1000 pN and increased in parallel to the chemoattractant gradient. Moreover, the forces required to stop the migrating cells ranged from 100 to 300 pN. Finally, tensile forces may also be present at the membrane when short molecule bridges at the T lymphocyte/APC interface (i.e., TCR/pMHC or CD2/CD58 pairs) exclude other larger molecules (LFA-1/ICAM-1 pairs or CD45) (37). These results (see Figure 1 for summary) demonstrate that spontaneous undulations of the T lymphocyte membrane and formation/retraction of filopodia and other cellular protrusions can generate forces that facilitate probing of the biomechanical microenvironment (38). Meanwhile, receptor/ligand bonds are submitted to a wide range of forces during cell migration. This will result in the modulation of signaling cascades induced by mechanosensitive molecules.
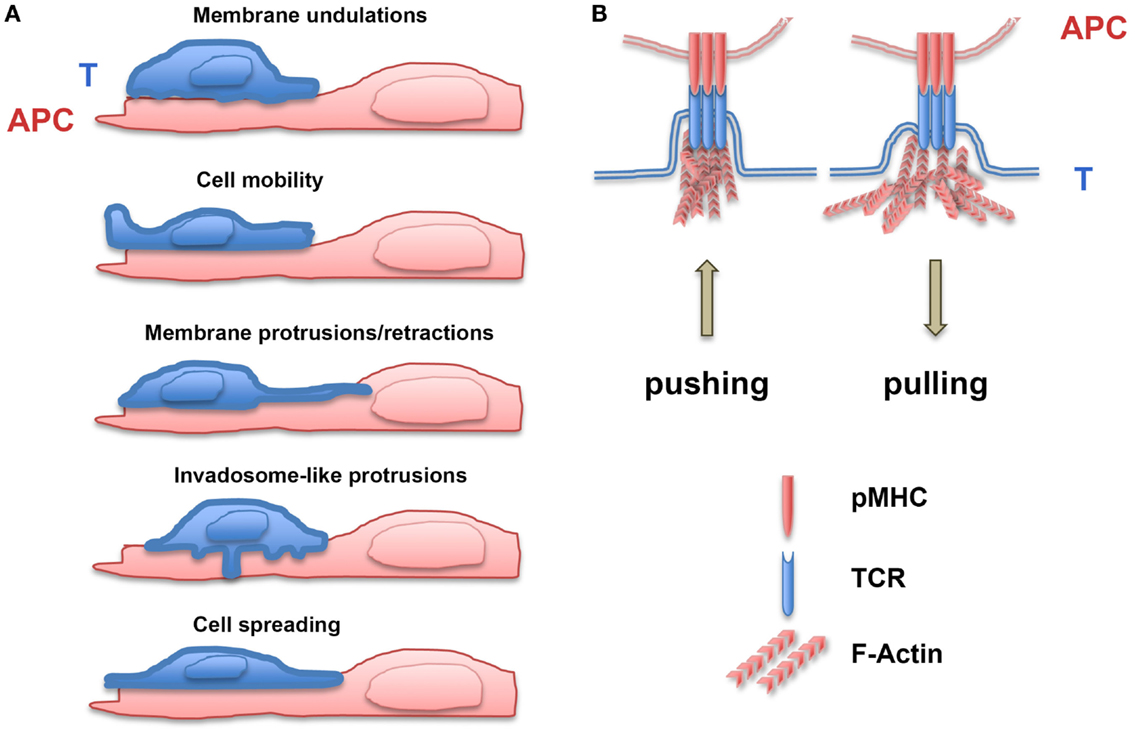
Figure 1. Generation of forces during T lymphocyte/APC contacts. (A) Forces are exerted on receptor/ligand bonds by membrane T lymphocyte undulations, cell mobility, membrane protrusions/retractions, invadosome-like protrusions, and cell spreading on antigen-presenting cells (APC). (B) Upon TCR triggering, T lymphocytes develop pushing and pulling forces on TCR/pMHC bonds, which depend on polymerization of F-actin.
TCR Engagement Induces Force Generation
Despite the increasing knowledge of signaling pathways engaged after recognition of pMHC by the TCR, the triggering mechanism of the signaling cascade still remains controversial (39). Several mechanisms have been proposed, which involve aggregation, conformational changes, and segregation (40). This matter has been reviewed extensively and will not be addressed further. Yet, studies aiming to investigate if and how receptor engagement generates forces that might then be converted in biochemical signal are sparse.
In one study, Hosseini et al. used atomic force microscopy (AFM) to measure the adhesion forces between a T cell hybridoma and a B cell line used as APC (41). Results showed that in the presence of antigen, adhesion forces built up with time of conjugate formation, starting from 1 to 2 nN at the beginning of the interaction to 14 nN after 30 min. The adhesion forces were mainly due to lymphocyte function-associated antigen-1 (LFA-1)-mediated adhesion, since the integrin inhibitor BIRT377 almost completely abolished forces in the conjugates (41). Similar experiments were performed on conjugates formed between mouse primary T lymphocytes expressing the OT1 transgenic TCR and a mouse dendritic cell line presenting OVA peptides of different affinities (42). In this experimental model, adhesion forces between cellular partners were smaller (up to 1.5 nN) and correlated to the ability of the different peptides to activate the T lymphocytes; better agonist peptides induced stronger adhesion forces (42).
Even though the above studies provided values for interaction forces between T lymphocytes and APCs, they could neither address the question of the relative contribution of different molecules to the forces measured nor the question of the contribution of each cell partner in force generation. Therefore, we adapted the biomembrane force probe (BFP) technique, which was developed to probe molecular adhesion (43), in order to assay the generation of forces by T lymphocytes. The BFP consisted of a red blood cell (RBC), which was on one side coupled to a bead coated with antibodies and held on the other side by a pipette. A human primary CD4+ T lymphocyte held by a second pipette was brought into contact with the BFP. Activation of the T lymphocyte was monitored by imaging increases in the intracellular Ca2+ concentration, [Ca2+]i, and forces exerted by the T lymphocyte on the BFP were measured on time lapse stacks of images by determining the elongation of the RBC with respect to the position of the fixed micropipette (11). When the bead was coated with anti-CD3 antibodies, three consecutive phases were observed following T lymphocyte contact with the BFP: a latency phase, which lasted less than a minute during which no force and no [Ca2+]i increase were observed; a pushing phase consisting of the growth of a directional cell protrusion characterized by an initial axial compression of the RBC and a peak in [Ca2+]i increase; and, in most cases, a pulling phase characterized by protrusion/retraction and generation of pulling forces, as witnessed by the elongation of the BFP. The initial forces exerted by T lymphocytes on the RBC were around 25 pN for a probe stiffness of 50 pN/μm. Measurement of elongations showed that CD3 engagement on T lymphocytes triggered a constant pulling loading rate of ~2 pN/s. These characteristic three phases were not observed when the bead was coated with anti-MHC-I antibodies, showing that the mere binding of the bead to the T lymphocyte membrane is not sufficient to induce forces (11). Engagement of LFA-1 together with CD3 modified the forces exerted compared to CD3 alone: when the bead coupled to the BFP was coated with an anti-CD18 antibody (specific against the β2 chain of LFA-1), a clear decrease in growth velocity and protrusion length during the pushing phase was observed. Moreover, the pulling phase started earlier and the protrusion morphology was changed from a “tube-like” to a “cup-like” structure resembling the phagocytic cup. Engagement of LFA-1 alone on resting primary T lymphocytes did not generate any pushing phase. It also generated 100-fold lower pulling loading rates (0.2 pN/s for a probe stiffness of 50 pN/μm) than the pulling loading rates induced by CD3 engagement alone (25 pN/s for the same probe stiffness). Absence of force generation in response to just LFA-1 triggering can be attributed to the fact that T lymphocytes were not pretreated for inside-out signaling induction (i.e., pretreatment with chemokines, anti-CD3, or phorbol-ester). Thus, integrins can generate traction forces and modify the forces induced upon CD3/TCR triggering. Indeed, force measurements performed on human neutrophils submitted to chemotactic gradients on hydrogel substrates revealed that neutrophils also generated traction forces, which were dependent on β2 integrin engagement and signaling (44). This was not specific to LFA-1 engagement since binding of α5β1 integrins to fibronectin and activation of these integrins by addition of Mn2+ were also been shown to induce traction forces (45).
Two more studies confirmed that T lymphocytes generate significant forces upon CD3 engagement. In the first one, Bashour et al. used elastomer pillar arrays of known spring constant coated with activating antibodies (12). In this experimental setting, each pillar tip deflection caused by cell attachment and spreading is monitored using live cell videomicroscopy (46, 47). Human primary CD4+ T lymphocytes were put on micropillars coated with anti-CD3 antibodies and several phases were observed (12). In the first phase, cell spreading generated only minor forces. After this phase, cells ceased to spread and started to exert significant traction forces, which were essentially centripetal and exerted mostly at the cell periphery. The forces generated per pillar were around 50 pN. In the same study, forces exerted by mouse primary CD4+ T lymphocytes on the same pillars were fourfold higher (200 pN/pillar). No forces were measured when pillars were coated with an antibody against the costimulatory molecule CD28 alone. However, the dual presence of anti-CD3 and anti-CD28 antibodies resulted in doubling the traction forces exerted by T lymphocytes on the micropillars. This was observed when the anti-CD28 Ab was present on the pillar together with anti-CD3 or when added in solution (12). These results suggest that the traction forces induced by CD28 engagement are not directly generated through the CD28 receptor. They are rather due to signaling-dependent amplification of the forces triggered by TCR engagement.
In another study, Hui et al. used traction force microscopy to measure the forces exerted by Jurkat cells during TCR activation (13). Jurkat cells were put on polyacrylamide gels coated with anti-CD3 antibodies and embedded with fluorescent beads at the top surface. The traction forces exerted by the cells were measured by tracking fluorescent bead displacement. In the presence of anti-CD3 and for a substrate stiffness rigidity of 1–2 kPa, traction forces were in the order of 2 nN, whereas forces exerted on substrates coated with a non-activating antibody were below 1 nN (13).
From the above results, it is evident that TCR–CD3 engagement can generate forces in T lymphocytes. These forces can be modified by the engagement of costimulators, such as LFA-1 and CD28. We will now discuss the potential outcomes of forces on T cell activation.
Effect of Forces on T Cell Activation
We have seen in the previous paragraphs that membrane undulations, protrusions and retractions, cell migration, and TCR triggering can generate forces that can be exerted on receptor/ligand bonds. In the next section, we will discuss the effect of these forces on specific receptor/ligand pairs at the T lymphocyte/APC interface, i.e., TCR/pMHC and LFA-1/intercellular adhesion molecule-1 (ICAM-1), and on overall T lymphocyte activation.
Forces Exerted on TCR/pMHC Bonds
T lymphocytes typically recognize peptides of 8–11 amino acids presented by MHC molecules. The TCR can “sense” a single amino acid substitution and translate it in a different functional response. Moreover, T lymphocytes can precisely discriminate a small number (2–10) of pMHC complexes for which they are specific within a sea of self or foreign peptide-MHC molecules (17–19). How this exquisite specificity and sensitivity is achieved is still a matter of investigation. Forces exerted on the TCR–pMHC bonds may have a key role in these processes. Indeed, it has been shown that the TCR functions as a mechanosensor, i.e., it can convert mechanical cues into biochemical signals (16). The first direct evidence was obtained by E. Reinherz’s group, who used optically trapped beads coated with non-activating anti-CD3ϵ antibodies or pMHC to apply forces on the TCR and monitored T lymphocyte activation by measuring [Ca2+]i increase (14). They showed that T cells were triggered mechanically, since application of a tangential force (50 pN) to the coated bead induced calcium signaling. Force application on beads coated with pMHC complexes that did not bind TCR had no effect on calcium flux. In another study, Li et al. used a fibroblast cell line expressing a single chain Fv anti-CD3ϵ antibody elongated by a tether (15). Binding to CD3 did not induce calcium signaling unless a mild perpendicular shear stress or a normal pulling force on the T lymphocyte bound to the surrogate APC was applied (15). By contrast, pulling forces applied on CD28 or CD62L did not increase intracellular calcium levels. These studies demonstrated that the TCR could transform a mechanical signal (force) into a biochemical one ([Ca2+]i increase). Yet, several questions still remain unresolved and particularly whether forces applied on TCR/pMHC bonds can affect T cell antigen recognition and discrimination. Work from the group of C. Zhu elegantly demonstrated that mechanical forces applied using a BFP on TCR/pMHC-I (48) and TCR/pMHC-II (49, 50) affected dissociation kinetics in a peptide-specific way. Forces applied to the bonds prolonged the lifetimes of single TCR–pMHC bonds for agonists (catch bonds) but shortened those for antagonists (slip bonds). When forces of 10 pN were applied by BFP on OT1 TCR/pMHC bonds, the ratio of OT1 TCR–pMHC bond lifetimes for the agonist peptide versus a weaker altered peptide grew 57-fold compared to when no force was applied (49, 50), demonstrating that forces can increase the power of antigen discrimination. The functional outcome of different peptides recognized by the same TCR was also shown to be coupled with the cumulative lifetime of the TCR–pMHC bonds (49).
A TCR deformation model where mechanical stress could induce conformational changes that would unmask sites of phosphorylation and allow TCR signaling was also proposed (51). Application of forces on the TCR would expose the immunoreceptor tyrosine-based activation motifs present in the CD3ϵ and ζ chains, otherwise buried into the hydrophobic core of the membrane lipid bilayer (52, 53). More recently, it was proposed that the structural features of TCR–CD3 complexes are adapted to permit sensing and discrimination of the forces to which TCR/pMHC bonds are submitted (54). Das et al. used optical tweezers and DNA tether spacer technology, which allow for application of forces in the order of piconewton with a spatial precision of nanometer, in order to address the mechanisms involved in the control of strength and lifetime of the TCR–pMHC-I bonds (55). They confirmed that forces applied on TCR–pMHC-I bonds increased the lifetime of the bond and showed that the state of the CβFG loop region, a 12-amino acid peptide present in the constant region of the β chain of the TCR of all mammalian αβTCRs (56), is involved in the increased lifetime of TCR–pMHC-I bonds submitted to tensile forces (55). This study suggests that forces physically modify the αβTCR by switching it from an “extended form” that binds weakly to a “compact form” that binds more robustly. The conformational changes of the TCR would then be transmitted to the CD3 signaling complexes associated with the TCR through mechanisms that have yet to be discovered. Finally, a recent study showed that the juxtamembrane region of ζ–ζ homodimers are divaricated within the TCR–CD3ζ complexes and that TCR engagement drives the intra-complex juxtaposition of the ζ–ζ juxtamembrane regions (57). This mechanical switch might thus couple TCR engagement with CD3ζ-dependent signaling.
Forces Exerted on Integrin/Ligand Bonds
Integrins are heterodimeric transmembrane proteins that mediate interactions in-between cells and interactions between cells and the extracellular matrix. Avidity of integrins is regulated by changing their valency, i.e., by changing their density at the cell/cell interface and/or changing their affinity for ligands (58). The LFA-1 integrin plays an essential role for T lymphocyte trafficking, immune synapse formation, and T lymphocyte activation (59). In resting T lymphocytes, LFA-1 is in an inactive bent conformation state, which binds with low avidity to its ligand ICAM-1. TCR stimulation induces a change in LFA-1 conformation, resulting in a more extended conformation of the integrin with an intermediate affinity (60). Finally, binding of LFA-1 to its ligand modifies further its conformation with further increase in its affinity (61, 62). Forces have been shown to play a role in affinity maturation of integrins. Indeed, application of tensile forces on integrin/ligand bonds increases bond strength and longevity (63). This was also reported for LFA-1/ICAM-1 interactions, indicating that, as for TCR/pMHC, these molecules form catch bonds (64). Moreover, it has been shown that the integrin bonds “remember” the history of the forces they have been submitted to. This phenomenon was called “cyclic mechanical reinforcement,” as the bond strength accumulates over repeated cycles of forces and is maintained after force removal (65). For instance, fibronectin/α5β1 integrin bonds dissociate within 1 s at a force of 5 pN, while upon cyclic mechanical reinforcement, the bond lifetimes can be extended to 14 s. Similar mechanisms apply to LFA-1/ICAM-1-specific bonds (65). Although head rearrangements of integrins are induced by ligand binding, this might take seconds to happen in the absence of force (66). Application of forces on the bonds would thus shorten the time required for conformational change. Moreover, cyclic mechanical reinforcement would strengthen the bonds by easing and accumulating the reversible conformational change of integrins with multiple force cycles. Therefore, during immune synapse formation, dynamic cyclic traction forces are exerted on LFA-1/ICAM-1 bonds by cycles of membrane undulations, protrusions, and retractions or by direct LFA-1 engagement, since, as described above, this can lead to force generation in T lymphocytes. By inducing conformational changes of integrins, forces during immune synapse formation can facilitate adhesion between T lymphocytes and APCs and probably participate to the costimulatory activity of LFA-1, although this remains to be tested.
The Actomyosin Cytoskeleton: A Force Generator at the Immune Synapse
Forces experienced by T lymphocytes during synapse formation can come from the exterior but can also come from the interior generated by the cell’s own cytoskeleton. Many reviews have described and discussed remodeling of the cytoskeleton at the immune synapse and its potential role. We will herein concentrate on the role of the actomyosin cytoskeleton on the generation of forces. In the first dynamic study of immune synapse formation on artificial lipid bilayer, Grakoui et al. proposed a model of synapse formation in three stages (10): in the first stage, LFA-1 binding in the center of nascent synapse would provide “a fulcrum for cytoskeletal protrusive mechanisms that force an outermost ring of T cell membrane into close apposition with the substrate”; in the second stage, the transport of TCR–pMHC pairs to the center of the synapse would be actin driven; and in the last stage, the forces exerted would equilibrate, leading to stabilization (10). This model already proposed that forces generated by the T lymphocyte cytoskeleton would play a key role in immune synapse formation. It is remarkable to note that this model fitted so well to the experimental data obtained later on. Actin cytoskeleton has long been known to control T lymphocyte activation at different levels, such as adhesion to APC, early signaling through the TCR, and release of cytolytic granules or cytokines (67–71). T lymphocytes, when activated by the TCR–CD3, spread rapidly (in 2–4 min) on the activating substrate or cell they interact with, they stabilize (for 15–20 min), and then retract (10, 21, 72–74). These phases are reminiscent of the phases observed when adherent cells spread on their substrate (75). Indeed, the different zones of the immune synapse or supramolecular activation clusters (SMACs) have been compared to the lamellipodium (for the distal SMAC), the adhesive lamella (for the peripheral SMAC), and the uropod (for the non-adhesive central SMAC) of a mobile adherent cell (76). During synapse formation, microclusters of receptors form in the periphery and then move toward the center of the synapse (77). LFA-1 clusters stop in the pSMAC lamella zone, whereas TCR microclusters follow their path toward the cSMAC where they are endocytosed (78, 79) or secreted (80).
In the context of spreading described earlier, the centripetal movement of receptor clusters has been proposed to be driven by a combination of pushing forces originating from actin retrograde flow in the lamellipodium and pulling forces generated in the lamella by myosin-based contraction. Indeed, the inward flow of cortical F-actin at the immune synapse has been shown to be the major driving force behind microcluster movement (67, 81–84). The role of myosin II-based contractions at the lamella in microcluster movement, although more controversial (85), has also been shown to control the centripetal movement of both TCR and LFA-1 microclusters (86–88). One can speculate that the resistance of TCR, LFA-1, and other receptors to this mobilization would generate traction forces on the receptor/ligand bonds. Thus, coupling of receptors with the actin cytoskeleton together with mobility of the ligands at the membrane of the APC would be key elements in force generation on receptors. Adaptor molecules, such as talin, mediate interaction of LFA-1 with the actin cytoskeleton (89). The generation of localized traction forces by actin retrograde flow has indeed been shown to regulate adhesion (90, 91) in many cell types, including T lymphocytes forming immune synapses (92). In contrast, coupling of TCR to the actin cytoskeleton remains elusive. Yet, interactions of TCR clusters with actin have been revealed in experiments that introduced selective barriers, which altered TCR microcluster transport to the central SMAC (82, 93). Association of signalosomes with tyrosine-phosphorylated CD3 complexes may contribute to dynamic coupling of TCR–CD3 complexes with actin flow. The mobility of ligands on the surface of APC is another parameter to take into account into the generation of forces on receptor/ligand bonds (92, 94, 95) (Figure 2 and see later discussion in Section “T Lymphocytes Interact with Cells That Have Different Mechanical Properties”). More studies and modeling analysis are required to address these specific aspects.
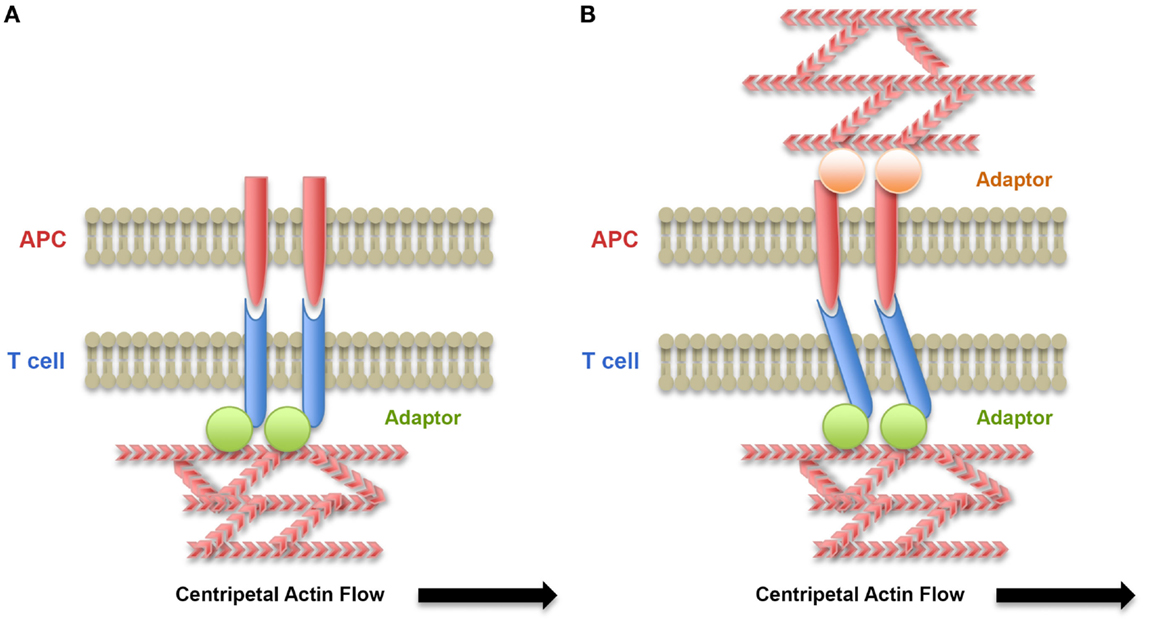
Figure 2. Role of the cytoskeleton of the T lymphocyte and the APC on force exertion on receptor/ligand bonds. (A) Centripetal flow of actin exerts forces on receptor/ligand bonds when receptors are coupled by an adaptor to the cytoskeleton. These forces may lead to conformational changes of the receptors and signaling. (B) When ligands are associated with the APC cytoskeleton, forces on receptor/ligand bonds are submitted to resistance due to reduced mobility of the ligands on the APC surface and the forces exerted on bonds are increased.
Effect of Substrate Stiffness on Force Development and T Cell Activation
The mechanical behavior of solid materials, such as plastic and glass, can be described as purely elastic. This means that their stiffness can be expressed as the ratio of the applied stress and the resulting deformation, which is termed elastic (or Young’s) modulus. On the other hand, cells and tissues display viscous properties in addition to their elastic ones and are, hence, viscoelastic materials. Two components can describe the mechanical properties of viscoelastic materials, one elastic and the other viscous, referred to as storage and loss moduli. In viscoelastic materials, the duration of force/stress application is also important, resulting in time-dependent deformations. Storage and loss moduli are different from the elastic or Young’s modulus that is more often reported in literature, since calculation of the latter is not taking into account the duration of force application. Henceforth, we refer to elastic modulus as a measure of stiffness, unless otherwise mentioned.
The effects of substrate stiffness can be as diverse as growth, differentiation, migration, and survival (96–99). Particularly, it was demonstrated that cells display differential spreading (100), velocities (101), traction forces (102), and physiological behavior (103) in response to variations in stiffness. In a seminal study (104), Discher and co-workers showed that stem cell fate could be influenced just by the stiffness of culture substrates. Another important observation was that cells match their stiffness to that of the environment by regulating their actin cytoskeleton (105). Moreover, several cell types have been reported to display durotaxis, i.e., migration from soft toward stiff substrates (101, 106).
In this section, we will report on recent studies that have begun to shed light on the mechanical properties of T lymphocyte environment and on T cell responses to these properties.
T Lymphocytes Interact with Cells That Have Different Mechanical Properties
T lymphocytes are mobile cells that are exposed to different chemical and mechanical environments. Inside lymph nodes, T lymphocytes interact transiently with a number of different APCs, each of them potentially activated by different stimuli and presenting a varying repertoire of agonist/non-agonist peptides on MHC molecules (5, 107). In blood vessels, T lymphocytes interact with endothelial cells, and inside tissues, effector T lymphocytes interact with their targets, i.e., infected or tumor cells. Much of the studies on immune synapse formation and T cell activation have been performed on plastic or glass surfaces or on planar lipid bilayers supported on glass. Even though these surfaces provide an ideal substrate to follow receptor/ligand interactions and rearrangements, they are flat and rigid with no topological variation. Moreover, plastic and glass display stiffness in the ranges of gigapascal. In contrast, cells in the body generally display stiffness in the range of 50 Pa–40 kPa (97) with primary human T lymphocytes and Jurkat cells being at the soft end of this range (108, 109) with their stiffness ranging from 50 to 90 Pa. Therefore, in order to really study the effect of mechanical properties on T lymphocyte biology, it is vital to know the mechanical landscape that the cells encounter in vivo and use substrates with stiffness values inside this physiological range. Using a single-cell rheometer (110), we recently showed that different human myeloid APCs have different viscoelastic properties and that their Young’s modulus values vary from 500 Pa for monocytes and DCs to 900 Pa for macrophages (109). Moreover, inflammatory conditions modified the viscoelastic properties of myeloid cells, which were halved or doubled when cells were treated with a TNFα/PGE2 cocktail or IFNγ, respectively (109). These results suggest that viscoelastic properties of myeloid cells are additional parameters of inflammation that can be integrated with biochemical factors to generate an adapted T lymphocyte response. Other studies have also reported variations in myeloid cell mechanical properties (111–113). Finally, it is worth noting that endothelial cells have also been shown to change their viscoelastic properties in response to inflammation (35), suggesting that this might be a more general process.
In our study, the viscoelastic properties of human myeloid cells were dependent on myosin IIA activity and correlated to the F-actin content in each type of cells (109). These results suggest that the actomyosin cytoskeleton of myeloid cells is responsible for their mechanical properties. Interestingly, older reports have shown that DC cytoskeleton was indispensable for priming of T cells since following DC treatment with actin depolymerizing drugs, naïve CD4+ T cells were unable to proliferate (114). DCs were shown to polarize their cytoskeleton toward the immune synapse only upon successful antigen recognition by the T cell, and this was critical for TCR triggering and IL-2 production (115, 116). Maturation of DCs has also been associated with remodeling of their cytoskeleton, leading to development of projections directed toward T lymphocytes to optimize cell/cell interactions (117, 118). More recently, it has been shown that the cortical actin network of DCs regulated ICAM-1 lateral mobility at the cell surface and that DC maturation regulated mobility and clustering of ICAM-1 (95). The constrained ICAM-1 mobility associated with DC maturation was shown to promote formation of T lymphocyte/DC conjugates as well as T lymphocyte proliferation. On the T lymphocyte side, it was shown that LFA-1 affinity maturation correlated to ICAM-1 lateral mobility on the DC surface, i.e., low mobility of ICAM-1 induced high-affinity conformational changes of LFA-1 (95). The same group showed that actin flow in T lymphocytes was indispensable to maintain LFA-1 in the high-affinity conformation at the immune synapse and that ICAM-1 mobility directly affected distribution of high-affinity LFA-1 on the surface of engaged T lymphocytes (92). These results suggest a model in which ICAM-1 mobility on APC surface modulates resistance to tensile forces applied by the T lymphocyte actin cytoskeleton on LFA-1/ICAM-1 bonds, highlighting the role of mechanotransduction in cell conjugate formation and T lymphocyte activation (Figure 2). These mechanisms may apply to other receptor/ligand pairs, such as CD28/CD80-CD86, which are also coupled to the actin cytoskeleton (119–122). They may also apply to other cell types, such as endothelial cells. Therefore, these studies show that APCs can contribute both biochemical and mechanical cues to T cell activation. Overall, the above findings are (1) highlighting the requirements for APC/T lymphocyte crosstalk (123) for immune synapse formation and T lymphocyte activation and the need for more studies focusing on the mechanical properties of both sides of the immune synapse and (2) stressing the importance for T lymphocytes to sense the mechanical and topological properties of their environment in order to locate a specific target and respond.
It is worth noting that the mechanical properties of tissues and organs can be also modified in normal and pathological conditions. For example, it was recently shown that the contractility of fibroblastic reticular cells is regulated upon inflammation by the expression of CLEC-2 on mature dendritic cells (124, 125). CLEC-2, by interacting with podoplanin expressed on fibroblastic reticular cells, induces the relaxation of these cells that leads to a decrease of the lymph node stiffness that is probably important for its expansion (125). Moreover, tumor mechanics, and in particular the rigidity of tumoral tissues, has been shown to play a role in tumor development (126). These changes in mechanical properties of tissues and organs might also affect overall T lymphocyte activity.
Finally, it is possible that viscoelastic properties of T lymphocytes themselves are also modified by activation. The strength of TCR signaling may induce changes in T lymphocyte stiffness, which in turn may affect their interactions with APC and target cells as well as their migratory properties. Along this line, it is worth noting that T lymphocytes can adopt two types of migratory behavior (5, 127). Strong TCR stimulation can lead to complete arrest of T lymphocyte migration and stable conjugation with an APC, which can last several hours (128), while when interacting with TCR ligands of low potency or low affinity, T lymphocytes do not completely stop migrating and establish brief dynamic contacts with the APC (129), termed kinapses (127). TCR signaling strength modifies the actomyosin cytoskeleton of T lymphocytes (130, 131), which may lead to an alteration of their mechanical properties. It would thus be interesting to measure the effect of TCR signaling strength and also cytokine environment on T lymphocyte viscoelastic properties.
T Lymphocytes Sense and Adapt to Substrate Stiffness
As discussed above, forces exerted by T lymphocytes may be important to probe their environment and particularly to test the stiffness, as we do when exerting pressure with our finger on a substrate. For example, it was proposed that “T lymphocytes are guided by the mechanical ‘path of least resistance’ as they transverse the endothelium” (34). In fact, T lymphocytes develop ILPs that physically push against the endothelial cell surface (20, 33, 34), suggesting that the role of these protrusions is to test the stiffness of endothelial cells in order to find “soft” areas to cross through (35). It is worth noting that these protrusions have also been proposed to facilitate the activation of memory/effector T cells to pMHC exposed on endothelial cells (22). Thus, T lymphocytes can sense the stiffness of the substrate they interact with. We have shown that not only T lymphocytes sense stiffness but also adapt to it. The pulling forces exerted by T lymphocytes upon TCR–CD3 triggering increased with the stiffness of the BFP used (11). This adaptation of forces to stiffness was not found in another study (12). Yet, the stiffness range used in each study might be very different.
Recent studies have addressed the effect of substrate stiffness on T lymphocyte activation. Using polyacrylamide gels with varying stiffness (range from 2 to 200 kPa) coated with an activating anti-CD3ϵ antibody, it was shown that mouse naïve CD4+ T lymphocytes modulated their response according to the stiffness of gel substrates (132). Production of IL-2 and early phosphorylation of Zap70 and Src family kinases was higher on “stiff” (100–200 kPa) substrates. This response to substrate stiffness was observed only when the anti-CD3ϵ antibody was attached to the gel and was abrogated in the presence of the myosin inhibitor blebbistatin (132). These results suggest that the mechanotransduction involved in T cell activation requires coupling of the TCR–CD3 to the substrate and intact myosin II activity. In another study, human naïve CD4+ and CD8+ T lymphocytes were cultured on poly-dimethoxysilane (PDMS) substrates with stiffness ranging from 100 to 10,000 kPa. The “soft” substrates (~100 kPa) induced higher IL-2 and IFNγ production as well as more T lymphocyte proliferation (133). These results seem inconsistent with the previous study (132). Yet, for poly-acrylamide gel substrates, immobilization was performed by coupling of biotinylated activating antibodies on acrylamide-conjugated streptavidin. In contrast, coating of PDMS substrates was performed by passive adsorption of antibodies on the hydrophobic surface (133), possibly resulting in both loss of immobilized material over time and passive adsorption of proteins from the culture medium. In a third study, human CD4+ T lymphoblasts were activated on PDMS substrates of varying stiffness, which presented anti-CD3 antibodies either alone or together with ICAM-1 molecules (134). In this study, “soft” (5 kPa) substrates induced less tyrosine phosphorylation than the “rigid” (2000 kPa) ones, and ICAM-1 increased the response to “stiff” substrates (134).
Even though the aforementioned reports provide very interesting results, their focus is on a stiffness range (2–10,000 kPa) that is non-physiological for T cells in the body, since APCs were shown to display stiffness ranging from 0.19 to 1.45 kPa (109). A recent study (13) looked at the response of Jurkat cells to substrates of a more physiologically relevant stiffness range (0.2–6 kPa). The authors used polyacrylamide gels, treated with hydrazine hydrate and coated with poly-l-lysine and an activating anti-CD3 antibody. They quantified the effect of substrate stiffness on CD3-induced signaling by following tyrosine phosphorylation by immunoblotting and microscopy (13). They showed that tyrosine phosphorylation peaked higher and more rapidly on “stiff” gels (5 kPa) but decreased more rapidly than on “soft” gels (1 kPa).
Although these studies (summarized in Table 1) are difficult to directly compare because they use different cell types, substrate chemistry, antibody immobilization, and stiffness ranges, they overall reveal that T lymphocytes are indeed mechanosensitive. It is not entirely clear what is the mechanosensing mechanism, yet, as summarized above, it requires TCR-dependent actomyosin remodeling. One explanation for the effects on TCR triggering and subsequent activation could be the local spreading and deformation of the T cell membrane in contact with substrate of different stiffness. In the kinetic-segregation model (37), local membrane deformation and segregation of large glycoproteins have to occur before TCR and pMHC can come close to one another and interact (40). It was proposed that this process required cytoskeleton-derived force (135). One could postulate that this could occur at the tips of ILPs or other small protrusions of the cell, i.e., short filopodia. Regarding the deformability of the substrate, “stiff” substrates would allow more deformation of the T lymphocyte membrane and better molecular segregation at the T cell protrusions compared to “soft” substrates and this, in turn, would result in an increased number of successful interactions between TCR and ligands (anti-CD3 antibodies or pMHC complexes). The increased number of TCR engagements would induce increased intracellular signaling that would then activate the actin cytoskeleton to produce larger cell protrusions and generate forces (11–13). This process can eventually result in increasing bond lifetimes of TCR and LFA-1 for their ligands. By inducing more conformational changes, it would lead to increased T lymphocyte activation. Thus, “stiff” substrates would display a kind of mechanical signal amplification. This mechanism has already been proposed for fibroblast adhesion on substrates of varying stiffness (100). Further work will be required to test this hypothesis for T lymphocytes.
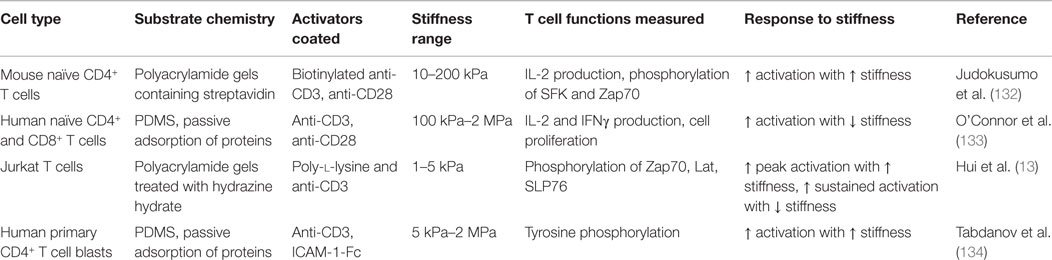
Table 1. T lymphocyte response to substrate stiffness.
Conclusion and Perspectives
Recent evidence has shown that TCR signaling and T lymphocyte activation are not solely regulated by chemical signals of the environment but also by mechanical cues. Forces exerted by the exterior or the T lymphocyte itself regulate the lifetime of receptor/ligand bonds. This, in turn, increases adhesion of T lymphocytes to APCs and allows for better discrimination of agonist pMHC. Forces exerted by T lymphocytes also help the cells probe the substrates they interact with by testing their stiffness, which might be a key parameter of T cell activation. We now need to explore further the viscoelastic properties of cells and tissues in physiological and pathological conditions in order to develop experimental models that better mimic the mechanical landscape of T lymphocytes. At the molecular level, we need to study the role of known costimulators or co-inhibitors of T lymphocyte activation in force development and force sensing and find out potential mechanical crosstalk between receptors. Finally, at the cell level, it would be interesting to study if and how mechanical cues can modulate the functions of different T lymphocyte subsets. It would be particularly important to see if mechanics can modulate naïve T lymphocyte priming or effector T lymphocyte functions (cytotoxicity and cytokine secretion). Elucidating these issues will provide further insight into T lymphocyte activation under normal and pathological conditions that could be translated in novel therapeutic strategies.
Author Contributions
CH wrote the review, edited the manuscript, and designed table and figures. MS wrote part of the review and edited the manuscript and figures and table.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Supported by funds from ANR (ANR-12-BSV5-0007-01, ImmunoMeca; ANR-13-BSV2-0018 “NeuroImmunoSynapse”), DC-Biol Labex, and Fondation pour la Recherche Médicale (FRM, FRM DEQ20140329513).
References
1. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13:227–42. doi:10.1038/nri3405
2. Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, et al. The immunological synapse. Annu Rev Immunol (2001) 19:375–96. doi:10.1146/annurev.immunol.19.1.375
3. Dustin ML. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr Opin Cell Biol (2007) 19:529–33. doi:10.1016/j.ceb.2007.08.003
4. Yokosuka T, Saito T. The immunological synapse, TCR microclusters, and T cell activation. Curr Top Microbiol Immunol (2010) 340:81–107. doi:10.1007/978-3-642-03858-7_5
5. Moreau HD, Bousso P. Visualizing how T cells collect activation signals in vivo. Curr Opin Immunol (2014) 26:56–62. doi:10.1016/j.coi.2013.10.013
6. He HT, Bongrand P. Membrane dynamics shape TCR-generated signaling. Front Immunol (2012) 3:90. doi:10.3389/fimmu.2012.00090
7. Beemiller P, Krummel MF. Regulation of T-cell receptor signaling by the actin cytoskeleton and poroelastic cytoplasm. Immunol Rev (2013) 256:148–59. doi:10.1111/imr.12120
8. Roybal KT, Sinai P, Verkade P, Murphy RF, Wulfing C. The actin-driven spatiotemporal organization of T-cell signaling at the system scale. Immunol Rev (2013) 256:133–47. doi:10.1111/imr.12103
9. Kumari S, Curado S, Mayya V, Dustin ML. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta (2014) 1838:546–56. doi:10.1016/j.bbamem.2013.05.004
10. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285:221–7. doi:10.1126/science.285.5425.221
11. Husson J, Chemin K, Bohineust A, Hivroz C, Henry N. Force generation upon T cell receptor engagement. PLoS One (2011) 6:e19680. doi:10.1371/journal.pone.0019680
12. Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, et al. CD28 and CD3 have complementary roles in T-cell traction forces. Proc Natl Acad Sci U S A (2014) 111:2241–6. doi:10.1073/pnas.1315606111
13. Hui KL, Balagopalan L, Samelson LE, Upadhyaya A. Cytoskeletal forces during signaling activation in Jurkat T-cells. Mol Biol Cell (2015) 26:685–95. doi:10.1091/mbc.E14-03-0830
14. Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, et al. The alphabeta T cell receptor is an anisotropic mechanosensor. J Biol Chem (2009) 284:31028–37. doi:10.1074/jbc.M109.052712
15. Li YC, Chen BM, Wu PC, Cheng TL, Kao LS, Tao MH, et al. Cutting edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J Immunol (2010) 184:5959–63. doi:10.4049/jimmunol.0900775
16. Wang JH, Reinherz EL. The structural basis of alphabeta T-lineage immune recognition: TCR docking topologies, mechanotransduction, and co-receptor function. Immunol Rev (2012) 250:102–19. doi:10.1111/j.1600-065X.2012.01161.x
17. Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature (2002) 419:845–9. doi:10.1038/nature01076nature01076
18. Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol (2004) 5:524–30. doi:10.1038/ni1058
19. Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity (2013) 39:846–57. doi:10.1016/j.immuni.2013.08.036
20. Gerard A, van der Kammen RA, Janssen H, Ellenbroek SI, Collard JG. The Rac activator Tiam1 controls efficient T-cell trafficking and route of transendothelial migration. Blood (2009) 113:6138–47. doi:10.1182/blood-2008-07-167668
21. Ueda H, Morphew MK, McIntosh JR, Davis MM. CD4+ T-cell synapses involve multiple distinct stages. Proc Natl Acad Sci U S A (2011) 108:17099–104. doi:10.1073/pnas.1113703108
22. Sage PT, Varghese LM, Martinelli R, Sciuto TE, Kamei M, Dvorak AM, et al. Antigen recognition is facilitated by invadosome-like protrusions formed by memory/effector T cells. J Immunol (2012) 188:3686–99. doi:10.4049/jimmunol.1102594
23. Krol A, Grinfeldt MG, Levin SV, Smilgavichus AD. Local mechanical oscillations of the cell surface within the range 0.2-30 Hz. Eur Biophys J (1990) 19:93–9. doi:10.1007/BF00185092
24. Dobereiner HG, Dubin-Thaler BJ, Hofman JM, Xenias HS, Sims TN, Giannone G, et al. Lateral membrane waves constitute a universal dynamic pattern of motile cells. Phys Rev Lett (2006) 97:038102. doi:10.1103/PhysRevLett.97.038102
25. Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol (2008) 9:446–54. doi:10.1038/nrm2406
26. Mogilner A, Rubinstein B. The physics of filopodial protrusion. Biophys J (2005) 89:782–95. doi:10.1529/biophysj.104.056515
27. Atilgan E, Wirtz D, Sun SX. Mechanics and dynamics of actin-driven thin membrane protrusions. Biophys J (2006) 90:65–76. doi:10.1529/biophysj.105.071480
28. Lan Y, Papoian GA. The stochastic dynamics of filopodial growth. Biophys J (2008) 94:3839–52. doi:10.1529/biophysj.107.123778
29. Dai J, Sheetz MP. Mechanical properties of neuronal growth cone membranes studied by tether formation with laser optical tweezers. Biophys J (1995) 68:988–96. doi:10.1016/S0006-3495(95)80274-2
30. Schwarz US, Gardel ML. United we stand: integrating the actin cytoskeleton and cell-matrix adhesions in cellular mechanotransduction. J Cell Sci (2012) 125:3051–60. doi:10.1242/jcs.093716
31. Bridgman PC, Dave S, Asnes CF, Tullio AN, Adelstein RS. Myosin IIB is required for growth cone motility. J Neurosci (2001) 21:6159–69.
32. Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J Cell Sci (2009) 122:3025–35. doi:10.1242/jcs.047522
33. Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity (2007) 26:784–97. doi:10.1016/j.immuni.2007.04.015
34. Martinelli R, Zeiger AS, Whitfield M, Sciuto TE, Dvorak A, Van Vliet KJ, et al. Probing the biomechanical contribution of the endothelium to lymphocyte migration: diapedesis by the path of least resistance. J Cell Sci (2014) 127:3720–34. doi:10.1242/jcs.148619
35. Schaefer A, Hordijk PL. Cell-stiffness-induced mechanosignaling – a key driver of leukocyte transendothelial migration. J Cell Sci (2015) 128:2221–30. doi:10.1242/jcs.163055
36. Yang H, Gou X, Wang Y, Fahmy TM, Leung AY, Lu J, et al. A dynamic model of chemoattractant-induced cell migration. Biophys J (2015) 108:1645–51. doi:10.1016/j.bpj.2014.12.060
37. Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol (2006) 7:803–9. doi:10.1038/ni1369
38. Pierres A, Monnet-Corti V, Benoliel AM, Bongrand P. Do membrane undulations help cells probe the world? Trends Cell Biol (2009) 19:428–33. doi:10.1016/j.tcb.2009.05.009
39. Malissen B, Bongrand P. Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu Rev Immunol (2015) 33:539–61. doi:10.1146/annurev-immunol-032414-112158
40. van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol (2011) 11:47–55. doi:10.1038/nri2887
41. Hosseini BH, Louban I, Djandji D, Wabnitz GH, Deeg J, Bulbuc N, et al. Immune synapse formation determines interaction forces between T cells and antigen-presenting cells measured by atomic force microscopy. Proc Natl Acad Sci U S A (2009) 106:17852–7. doi:10.1073/pnas.0905384106
42. Lim TS, Mortellaro A, Lim CT, Hammerling GJ, Ricciardi-Castagnoli P. Mechanical interactions between dendritic cells and T cells correlate with T cell responsiveness. J Immunol (2011) 187:258–65. doi:10.4049/jimmunol.1100267
43. Evans E, Ritchie K, Merkel R. Sensitive force technique to probe molecular adhesion and structural linkages at biological interfaces. Biophys J (1995) 68:2580–7. doi:10.1016/S0006-3495(95)80441-8
44. Jannat RA, Dembo M, Hammer DA. Traction forces of neutrophils migrating on compliant substrates. Biophys J (2011) 101:575–84. doi:10.1016/j.bpj.2011.05.040
45. Lin GL, Cohen DM, Desai RA, Breckenridge MT, Gao L, Humphries MJ, et al. Activation of beta 1 but not beta 3 integrin increases cell traction forces. FEBS Lett (2013) 587:763–9. doi:10.1016/j.febslet.2013.01.068
46. Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A (2003) 100:1484–9. doi:10.1073/pnas.0235407100
47. Schoen I, Hu W, Klotzsch E, Vogel V. Probing cellular traction forces by micropillar arrays: contribution of substrate warping to pillar deflection. Nano Lett (2010) 10:1823–30. doi:10.1021/nl100533c
48. Pryshchep S, Zarnitsyna VI, Hong J, Evavold BD, Zhu C. Accumulation of serial forces on TCR and CD8 frequently applied by agonist antigenic peptides embedded in MHC molecules triggers calcium in T cells. J Immunol (2014) 193:68–76. doi:10.4049/jimmunol.1303436
49. Liu B, Chen W, Evavold BD, Zhu C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell (2014) 157:357–68. doi:10.1016/j.cell.2014.02.053
50. Hong J, Persaud SP, Horvath S, Allen PM, Evavold BD, Zhu C. Force-regulated in situ TCR-peptide-bound MHC class II kinetics determine functions of CD4+ T Cells. J Immunol (2015) 195:3557–64. doi:10.4049/jimmunol.1501407
51. Ma Z, Janmey PA, Finkel TH. The receptor deformation model of TCR triggering. FASEB J (2008) 22:1002–8. doi:10.1096/fj.07-9331hyp
52. Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol (2000) 7:1023–6. doi:10.1038/80930
53. Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell (2008) 135:702–13. doi:10.1016/j.cell.2008.09.044
54. Brazin KN, Mallis RJ, Das DK, Feng Y, Hwang W, Wang JH, et al. Structural features of the alphabetaTCR mechanotransduction apparatus that promote pMHC discrimination. Front Immunol (2015) 6:441. doi:10.3389/fimmu.2015.00441
55. Das DK, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, et al. Force-dependent transition in the T-cell receptor beta-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc Natl Acad Sci U S A (2015) 112:1517–22. doi:10.1073/pnas.1424829112
56. Kim ST, Touma M, Takeuchi K, Sun ZY, Dave VP, Kappes DJ, et al. Distinctive CD3 heterodimeric ectodomain topologies maximize antigen-triggered activation of alpha beta T cell receptors. J Immunol (2010) 185:2951–9. doi:10.4049/jimmunol.1000732
57. Lee MS, Glassman CR, Deshpande NR, Badgandi HB, Parrish HL, Uttamapinant C, et al. A mechanical switch couples T cell receptor triggering to the cytoplasmic juxtamembrane regions of CD3zetazeta. Immunity (2015) 43:227–39. doi:10.1016/j.immuni.2015.06.018
58. Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol (2005) 5:546–59. doi:10.1038/nri1646
59. Hogg N, Patzak I, Willenbrock F. The insider’s guide to leukocyte integrin signalling and function. Nat Rev Immunol (2011) 11:416–26. doi:10.1038/nri2986
60. Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity (2006) 25:583–94. doi:10.1016/j.immuni.2006.07.016
61. Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct (2002) 31:485–516. doi:10.1146/annurev.biophys.31.101101.140922
62. Takagi J, Springer TA. Integrin activation and structural rearrangement. Immunol Rev (2002) 186:141–63. doi:10.1034/j.1600-065X.2002.18613.x
63. Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol (2009) 185:1275–84. doi:10.1083/jcb.200810002
64. Chen W, Lou J, Zhu C. Forcing switch from short- to intermediate- and long-lived states of the alphaA domain generates LFA-1/ICAM-1 catch bonds. J Biol Chem (2010) 285:35967–78. doi:10.1074/jbc.M110.155770
65. Kong F, Li Z, Parks WM, Dumbauld DW, Garcia AJ, Mould AP, et al. Cyclic mechanical reinforcement of integrin-ligand interactions. Mol Cell (2013) 49:1060–8. doi:10.1016/j.molcel.2013.01.015
66. Bednar B, Cunningham ME, McQueney PA, Egbertson MS, Askew BC, Bednar RA, et al. Flow cytometric measurement of kinetic and equilibrium binding parameters of arginine-glycine-aspartic acid ligands in binding to glycoprotein IIb/IIIa on platelets. Cytometry (1997) 28:58–65. doi:10.1002/(SICI)1097-0320(19970501)28:1<58::AID-CYTO7>3.0.CO;2-D
67. Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol (2007) 7:131–43. doi:10.1038/nri2021
68. Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol (2008) 26:233–59. doi:10.1146/annurev.immunol.26.021607.090347
69. Chemin K, Bohineust A, Dogniaux S, Tourret M, Guegan S, Miro F, et al. Cytokine secretion by CD4+ T cells at the immunological synapse requires Cdc42-dependent local actin remodeling but not microtubule organizing center polarity. J Immunol (2012) 189:2159–68. doi:10.4049/jimmunol.1200156
70. Babich A, Burkhardt JK. Coordinate control of cytoskeletal remodeling and calcium mobilization during T-cell activation. Immunol Rev (2013) 256:80–94. doi:10.1111/imr.12123
71. Ritter AT, Asano Y, Stinchcombe JC, Dieckmann NM, Chen BC, Gawden-Bone C, et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity (2015) 42:864–76. doi:10.1016/j.immuni.2015.04.013
72. Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity (2001) 14:315–29. doi:10.1016/S1074-7613(01)00112-1
73. Tskvitaria-Fuller I, Rozelle AL, Yin HL, Wulfing C. Regulation of sustained actin dynamics by the TCR and costimulation as a mechanism of receptor localization. J Immunol (2003) 171:2287–95. doi:10.4049/jimmunol.171.5.2287
74. Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, et al. HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity (2006) 24:741–52. doi:10.1016/j.immuni.2006.03.022
75. Cuvelier D, Thery M, Chu YS, Dufour S, Thiery JP, Bornens M, et al. The universal dynamics of cell spreading. Curr Biol (2007) 17:694–9. doi:10.1016/j.cub.2007.02.058
76. Dustin ML. Visualization of cell-cell interaction contacts: synapses and kinapses. Self Nonself (2011) 2:85–97. doi:10.4161/self.2.2.17931
77. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity (2006) 25:117–27. doi:10.1016/j.immuni.2006.04.010
78. Balagopalan L, Barr VA, Samelson LE. Endocytic events in TCR signaling: focus on adapters in microclusters. Immunol Rev (2009) 232:84–98. doi:10.1111/j.1600-065X.2009.00840.x
79. Griffiths GM, Tsun A, Stinchcombe JC. The immunological synapse: a focal point for endocytosis and exocytosis. J Cell Biol (2010) 189:399–406. doi:10.1083/jcb.201002027
80. Choudhuri K, Llodra J, Roth EW, Tsai J, Gordo S, Wucherpfennig KW, et al. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature (2014) 507:118–23. doi:10.1038/nature12951
81. Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci U S A (2007) 104:20296–301. doi:10.1073/pnas.0710258105
82. DeMond AL, Mossman KD, Starr T, Dustin ML, Groves JT. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys J (2008) 94:3286–92. doi:10.1529/biophysj.107.119099
83. Hartman NC, Nye JA, Groves JT. Cluster size regulates protein sorting in the immunological synapse. Proc Natl Acad Sci U S A (2009) 106:12729–34. doi:10.1073/pnas.0902621106
84. Yu CH, Wu HJ, Kaizuka Y, Vale RD, Groves JT. Altered actin centripetal retrograde flow in physically restricted immunological synapses. PLoS One (2010) 5:e11878. doi:10.1371/journal.pone.0011878
85. Hammer JA III, Burkhardt JK. Controversy and consensus regarding myosin II function at the immunological synapse. Curr Opin Immunol (2013) 25:300–6. doi:10.1016/j.coi.2013.03.010
86. Babich A, Li S, O’Connor RS, Milone MC, Freedman BD, Burkhardt JK. F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation. J Cell Biol (2012) 197:775–87. doi:10.1083/jcb.201201018
87. Kumari S, Vardhana S, Cammer M, Curado S, Santos L, Sheetz MP, et al. T lymphocyte myosin IIA is required for maturation of the immunological synapse. Front Immunol (2012) 3:230. doi:10.3389/fimmu.2012.00230
88. Yi J, Wu XS, Crites T, Hammer JA III. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol Biol Cell (2012) 23:834–52. doi:10.1091/mbc.E11-08-0731
89. Smith A, Carrasco YR, Stanley P, Kieffer N, Batista FD, Hogg N. A talin-dependent LFA-1 focal zone is formed by rapidly migrating T lymphocytes. J Cell Biol (2005) 170:141–51. doi:10.1083/jcb.200412032
90. Giannone G, Dubin-Thaler BJ, Rossier O, Cai Y, Chaga O, Jiang G, et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell (2007) 128:561–75. doi:10.1016/j.cell.2006.12.039
91. Craig EM, Van Goor D, Forscher P, Mogilner A. Membrane tension, myosin force, and actin turnover maintain actin treadmill in the nerve growth cone. Biophys J (2012) 102:1503–13. doi:10.1016/j.bpj.2012.03.003
92. Comrie WA, Babich A, Burkhardt JK. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J Cell Biol (2015) 208:475–91. doi:10.1083/jcb.201406121
93. Manz BN, Groves JT. Spatial organization and signal transduction at intercellular junctions. Nat Rev Mol Cell Biol (2010) 11:342–52. doi:10.1038/nrm2883
94. Dillard P, Varma R, Sengupta K, Limozin L. Ligand-mediated friction determines morphodynamics of spreading T cells. Biophys J (2014) 107:2629–38. doi:10.1016/j.bpj.2014.10.044
95. Comrie WA, Li S, Boyle S, Burkhardt JK. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J Cell Biol (2015) 208:457–73. doi:10.1083/jcb.201406120
96. Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science (2005) 310:1139–43. doi:10.1126/science.1116995
97. Janmey PA, McCulloch CA. Cell mechanics: integrating cell responses to mechanical stimuli. Annu Rev Biomed Eng (2007) 9:1–34. doi:10.1146/annurev.bioeng.9.060906.151927
98. Tee SY, Bausch AR, Janmey PA. The mechanical cell. Curr Biol (2009) 19:R745–8. doi:10.1016/j.cub.2009.06.034
99. Gasiorowski JZ, Murphy CJ, Nealey PF. Biophysical cues and cell behavior: the big impact of little things. Annu Rev Biomed Eng (2013) 15:155–76. doi:10.1146/annurev-bioeng-071811-150021
100. Pelham RJ Jr, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci U S A (1997) 94:13661–5. doi:10.1073/pnas.94.25.13661
101. Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J (2000) 79:144–52. doi:10.1016/S0006-3495(00)76279-5
102. Oakes PW, Patel DC, Morin NA, Zitterbart DP, Fabry B, Reichner JS, et al. Neutrophil morphology and migration are affected by substrate elasticity. Blood (2009) 114:1387–95. doi:10.1182/blood-2008-11-191445
103. Majkut S, Idema T, Swift J, Krieger C, Liu A, Discher DE. Heart-specific stiffening in early embryos parallels matrix and myosin expression to optimize beating. Curr Biol (2013) 23:2434–9. doi:10.1016/j.cub.2013.10.057
104. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell (2006) 126:677–89. doi:10.1016/j.cell.2006.06.044
105. Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J (2007) 93:4453–61. doi:10.1529/biophysj.106.101386
106. Plotnikov SV, Waterman CM. Guiding cell migration by tugging. Curr Opin Cell Biol (2013) 25:619–26. doi:10.1016/j.ceb.2013.06.003
107. Pace L, Tempez A, Arnold-Schrauf C, Lemaitre F, Bousso P, Fetler L, et al. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science (2012) 338:532–6. doi:10.1126/science.1227049
108. Rosenbluth MJ, Lam WA, Fletcher DA. Force microscopy of nonadherent cells: a comparison of leukemia cell deformability. Biophys J (2006) 90:2994–3003. doi:10.1529/biophysj.105.067496
109. Bufi N, Saitakis M, Dogniaux S, Buschinger O, Bohineust A, Richert A, et al. Human primary immune cells exhibit distinct mechanical properties that are modified by inflammation. Biophys J (2015) 108:2181–90. doi:10.1016/j.bpj.2015.03.047
110. Desprat N, Guiroy A, Asnacios A. Microplates-based rheometer for a single living cell. Rev Sci Instrum (2006) 77:055111. doi:10.1063/1.2202921
111. Lautenschlager F, Paschke S, Schinkinger S, Bruel A, Beil M, Guck J. The regulatory role of cell mechanics for migration of differentiating myeloid cells. Proc Natl Acad Sci U S A (2009) 106:15696–701. doi:10.1073/pnas.0811261106
112. Ekpenyong AE, Whyte G, Chalut K, Pagliara S, Lautenschlager F, Fiddler C, et al. Viscoelastic properties of differentiating blood cells are fate- and function-dependent. PLoS One (2012) 7:e45237. doi:10.1371/journal.pone.0045237
113. Patel NR, Bole M, Chen C, Hardin CC, Kho AT, Mih J, et al. Cell elasticity determines macrophage function. PLoS One (2012) 7:e41024. doi:10.1371/journal.pone.0041024
114. Al-Alwan MM, Rowden G, Lee TD, West KA. The dendritic cell cytoskeleton is critical for the formation of the immunological synapse. J Immunol (2001) 166:1452–6. doi:10.4049/jimmunol.166.3.1452
115. Al-Alwan MM, Liwski RS, Haeryfar SM, Baldridge WH, Hoskin DW, Rowden G, et al. Cutting edge: dendritic cell actin cytoskeletal polarization during immunological synapse formation is highly antigen-dependent. J Immunol (2003) 171:4479–83. doi:10.4049/jimmunol.171.9.4479
116. Bouma G, Mendoza-Naranjo A, Blundell MP, de Falco E, Parsley KL, Burns SO, et al. Cytoskeletal remodeling mediated by WASp in dendritic cells is necessary for normal immune synapse formation and T-cell priming. Blood (2011) 118:2492–501. doi:10.1182/blood-2011-03-340265
117. Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, Popoff M, et al. Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science (2004) 305:1150–3. doi:10.1126/science.1099159
118. Fisher PJ, Bulur PA, Vuk-Pavlovic S, Prendergast FG, Dietz AB. Dendritic cell microvilli: a novel membrane structure associated with the multifocal synapse and T-cell clustering. Blood (2008) 112:5037–45. doi:10.1182/blood-2008-04-149526
119. Doty RT, Clark EA. Subcellular localization of CD80 receptors is dependent on an intact cytoplasmic tail and is required for CD28-dependent T cell costimulation. J Immunol (1996) 157:3270–9.
120. Tavano R, Contento RL, Baranda SJ, Soligo M, Tuosto L, Manes S, et al. CD28 interaction with filamin-A controls lipid raft accumulation at the T-cell immunological synapse. Nat Cell Biol (2006) 8:1270–6. doi:10.1038/ncb1492
121. Liang Y, Cucchetti M, Roncagalli R, Yokosuka T, Malzac A, Bertosio E, et al. The lymphoid lineage-specific actin-uncapping protein Rltpr is essential for costimulation via CD28 and the development of regulatory T cells. Nat Immunol (2013) 14:858–66. doi:10.1038/ni.2634
122. Bashour KT, Tsai J, Shen K, Lee JH, Sun E, Milone MC, et al. Cross talk between CD3 and CD28 is spatially modulated by protein lateral mobility. Mol Cell Biol (2014) 34:955–64. doi:10.1128/MCB.00842-13
123. Hivroz C, Chemin K, Tourret M, Bohineust A. Crosstalk between T lymphocytes and dendritic cells. Crit Rev Immunol (2012) 32:139–55. doi:10.1615/CritRevImmunol.v32.i2.30
124. Acton SE, Farrugia AJ, Astarita JL, Mourao-Sa D, Jenkins RP, Nye E, et al. Dendritic cells control fibroblastic reticular network tension and lymph node expansion. Nature (2014) 514:498–502. doi:10.1038/nature13814
125. Astarita JL, Cremasco V, Fu J, Darnell MC, Peck JR, Nieves-Bonilla JM, et al. The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat Immunol (2015) 16:75–84. doi:10.1038/ni.3035
126. Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med (2011) 17:320–9. doi:10.1038/nm.2328
127. Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol Rev (2008) 221:77–89. doi:10.1111/j.1600-065X.2008.00589.x
128. Skokos D, Shakhar G, Varma R, Waite JC, Cameron TO, Lindquist RL, et al. Peptide-MHC potency governs dynamic interactions between T cells and dendritic cells in lymph nodes. Nat Immunol (2007) 8:835–44. doi:10.1038/ni1490
129. Moreau HD, Lemaitre F, Terriac E, Azar G, Piel M, Lennon-Dumenil AM, et al. Dynamic in situ cytometry uncovers T cell receptor signaling during immunological synapses and kinapses in vivo. Immunity (2012) 37:351–63. doi:10.1016/j.immuni.2012.05.014
130. Jacobelli J, Friedman RS, Conti MA, Lennon-Dumenil AM, Piel M, Sorensen CM, et al. Confinement-optimized three-dimensional T cell amoeboid motility is modulated via myosin IIA-regulated adhesions. Nat Immunol (2010) 11:953–61. doi:10.1038/ni.1936
131. Moreau HD, Lemaitre F, Garrod KR, Garcia Z, Lennon-Dumenil AM, Bousso P. Signal strength regulates antigen-mediated T-cell deceleration by distinct mechanisms to promote local exploration or arrest. Proc Natl Acad Sci U S A (2015) 112:12151–6. doi:10.1073/pnas.1506654112
132. Judokusumo E, Tabdanov E, Kumari S, Dustin ML, Kam LC. Mechanosensing in T lymphocyte activation. Biophys J (2012) 102:L5–7. doi:10.1016/j.bpj.2011.12.011
133. O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, et al. Substrate rigidity regulates human T cell activation and proliferation. J Immunol (2012) 189:1330–9. doi:10.4049/jimmunol.1102757
134. Tabdanov E, Gondarenko S, Kumari S, Liapis A, Dustin ML, Sheetz MP, et al. Micropatterning of TCR and LFA-1 ligands reveals complementary effects on cytoskeleton mechanics in T cells. Integr Biol (Camb) (2015) 7:1272–84. doi:10.1039/c5ib00032g
Keywords: T lymphocytes, immune synapse, force control, TCR, LFA-1, biomechanics, stiffness
Citation: Hivroz C and Saitakis M (2016) Biophysical Aspects of T Lymphocyte Activation at the Immune Synapse. Front. Immunol. 7:46. doi: 10.3389/fimmu.2016.00046
Received: 02 November 2015; Accepted: 31 January 2016;
Published: 15 February 2016
Edited by:
Vincenzo Di Bartolo, Institut Pasteur, FranceCopyright: © 2016 Hivroz and Saitakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire Hivroz, Y2xhaXJlLmhpdnJvekBjdXJpZS5mcg==
†Michael Saitakis is the recipient of a grant from the Fondation pour la Recherche Médicale.