 Ana Catarina Lunz Macedo
Ana Catarina Lunz Macedo Lourdes Isaac
Lourdes Isaac- 1Department of Immunology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil
- 2Faculty of Medicine, Children’s Hospital, Clinics Hospital, University of São Paulo, São Paulo, Brazil
The complement system plays an important role in the innate and acquired immune response against pathogens. It consists of more than 30 proteins found in soluble form or attached to cell membranes. Most complement proteins circulate in inactive forms and can be sequentially activated by the classical, alternative, or lectin pathways. Biological functions, such as opsonization, removal of apoptotic cells, adjuvant function, activation of B lymphocytes, degranulation of mast cells and basophils, and solubilization and clearance of immune complex and cell lysis, are dependent on complement activation. Although the activation of the complement system is important to avoid infections, it also can contribute to the inflammatory response triggered by immune complex deposition in tissues in autoimmune diseases. Paradoxically, the deficiency of early complement proteins from the classical pathway (CP) is strongly associated with development of systemic lupus erythematous (SLE) – mainly C1q deficiency (93%) and C4 deficiency (75%). The aim of this review is to focus on the deficiencies of early components of the CP (C1q, C1r, C1s, C4, and C2) proteins in SLE patients.
Systemic Lupus Erythematosus
Systemic lupus erythematous (SLE) is a systemic autoimmune disorder in which multiple autoantibodies against cell nuclear constituents – DNA, histones, and ribonucleoproteins – are produced. The deposition of immune complexes in several organs, mainly kidney, skin, and joints, causes inflammation and tissue damage, producing a broad spectrum of clinical manifestations (1–3) (Table 1) (4). SLE classification criteria was defined by the American College of Rheumatology (ACR) in 1982 (5) and revised in 1997 (6). In 2012, the Systemic Lupus Collaborating Clinics (SLICC) validated the new classification criteria (4). SLE is classified by 4 out of 17 criteria (including at least one clinical and one immunological criterion) or by biopsy-proven lupus nephritis in presence of ANA or anti-dsDNA antibodies (Table 1) (4). Criteria are cumulative and do not need be present concurrently.

Table 1. Clinical and immunological criteria for SLE diagnostic [modified from Ref. (4)].
The etiopathogenesis of SLE depends on complement system activation triggered by the presence of immune complexes, leading to inflammation and complement proteins consumption. The acute or continuous low-grade chronic inflammation of lupus often predisposes patients to infections and cardiovascular diseases. Interaction of genetic background with environmental factors makes SLE one of the most complex diseases (7–11). SLE incidence ranges from 1 to 10 per 100,000 person-years and prevalence ranges from 20 to 70 per 100,000. SLE affects both sexes of every age, but predominates in women of child-bearing age with the highest incidence observed in African-Americans. The prevalence for men is approximately 10% of that observed in women (10–12). The disease is extremely rare in children <5 years of age, which demands a more accurate investigation for genetic and immunodeficiency’s causes of SLE. Juvenile systemic lupus erythematous (JSLE – onset before 16 years old) represents 15–20% of all SLE cases and differ in multiple aspects. JSLE is more severe, with higher incidence of nephritis, hematological disorders, photosensitivity, butterfly rash, and mucosal ulceration (13–17).
To study influence of genetic factors, Deapen et al. compared 107 pairs of SLE twins. They found 24% of concordance in 45 pairs of monozygotic while only 6% of concordance in dizygotic twins. This reinforces the importance of genetic predisposition to SLE, though less than first expected, indicating the influence of environmental factors as well (18).
One of the most remarkable genetic associations in SLE is the high frequency of deficiency of the early components of the complement system classical pathway (CP), mainly C1q (90–93%), C1r/C1s (50–57%), C4 (75%), and C2 (10%) (19–22).
Complement System Activation
The complement system participates in innate and acquired immunity. The proper activation of this system against microorganisms becomes clearly evident in complement immunodeficient patients, who are highly susceptible to recurrent infections.
The complement system is formed by more than 30 proteins present in a soluble form or on the surface of cell membranes. Some proteins act in sequential activation by different pathways: alternative pathway (AP), lectin pathway (LP), and CP. All pathways can lead to activation of a common terminal pathway, generating a membrane attack complex (MAC). This activation is tightly regulated to avoid excessive consumption of complement and tissue injury. Complement proteins may also act as cell surface receptors mediating important interactions with immune cells.
The AP is initiated by hydrolysis of an intramolecular thioester bond located in the C3 alpha chain in the native molecule. Once this bond is broken, the native structural conformation of C3 is modified generating (C3H2O), which now exhibits previously hidden binding sites for Factor B leading to the formation of the C3H2OB complex. Factor D cleaves Factor B, releasing the fragment Ba generating C3H2OBb. This complex has a catalytic site that cleaves C3 into C3a and C3b. C3b has a structural conformation that resembles C3H2O and therefore also binds to Factor B (23–27). C3bB is cleaved by Factor D, generating C3bBb (the second C3-convertase) and releasing Ba in the fluid phase. Properdin binds to C3b in this complex and stabilizes C3-convertases of the AP, increasing their half-lives (26, 28–32). Properdin can also bind directly to the surface of certain pathogens (33) and initiates a new platform for deposition of C3-convertase (34–36).
The LP is initiated once lectins, such as mannose-binding lectin (MBL) or ficolins (ficolin-1, ficolin-2, and ficolin-3), bind to microorganisms carbohydrates. MBL-associated serine proteases (MASP 1–3) (37–39) are complexed to MBL or ficolins and they cleave C4 and C2 (40, 41), generating the complex C4b2a (C3-convertase).
The CP is activated mainly by the presence of immune complexes formed by IgM or IgG. However, other proteins, such as C1q, pentraxins, and C-reactive protein, can bind directly to pathogens or other particles and directly activate the CP even in the absence of specific antibodies. Once the immune complex is formed, C1q binds to the Fc portion of the immunoglobulin and immediately activates C1r, which activates C1s. Activated C1s cleaves C4 into C4a and C4b and later C2 already complexed to C4 (C4bC2) generating the C3-convertase (C4b2a), which cleaves C3 into fragments C3a and C3b (42, 43).
All pathways converge to the activation of C3 protein, which can be cleaved by all C3-convertases. C3b fragments can bind to several acceptor molecules including C3-convertases (C3bBbC3bn or C4b2aC3bn) generating C5-convertases, which cleave C5 into C5a and C5b. The next component, C6, binds to C5b, generating C5b6, which now promotes the incorporation of C7 to form C5b67. This complex binds firmly to the cell membrane and later C8 binds to C7 generating C5b678. This complex has lytic activity, which is incremented when several C9 molecules bind to form the MAC – a transmembrane pore (42, 44–46).
Functions of the Complement System
The activation of the complement system generates active proteins and fragments that play important roles for the innate and acquired immune response (47–49): (a) production of opsonins: fragments C3b, iC3b, and C3d bind covalently to acceptor surfaces and molecules and may act as a bridge between pathogens or host cells and complement receptors (CR1 and CR3) present on phagocytic cells. This property may facilitate pathogen killing and antigen presentation complexed to MHC products for T lymphocytes; (b) production of anaphylatoxins, chemotactic factors, and inflammatory modulators: fragments C3a and C5a released during complement activation can bind to C3aR or C5aR1 and C5aR2 receptors triggering mast cells or basophil degranulation (50, 51), leading to production and release of important inflammatory mediators. C3a and C5a recruit leukocytes to the inflammatory area. Besides its proinflammatory effects, C3a could act too as inflammatory modulator on neutrophils (52); (c) increase antibody production: antigen-bound C3d interacts with CR2 and BCR on B lymphocytes that are activated, proliferates, and produces higher levels of specific antibodies (53). These coated antigens also bind to follicular dendritic cells (DCs), that trap them into germinal centers of lymphoid organs, and display them to B cells to selection of high-affinity B cells (54); (d) clearance of immune complexes: fragments C4b and C3b covalently bind to immune complexes. Erythrocyte-CR1 binds to C3b/C4b-coated immune complexes, which are removed from circulation in the spleen and liver; (e) cell lysis: the assembly of MAC (C5b6789n) forms a pore in the membrane surface, which leads to osmotic cell lysis; and (f) removal of apoptotic cells and debris: C1q (55), C4b, and C3b (56) enhance the ingestion of dead cells by phagocytes (57).
Pathogeneses of SLE: Inadequate Removal of Debris and Reduction of Self-Tolerance
Apoptosis is a fundamental event for embryogenesis, negative selection of T and B lymphocytes, and maturation of organs. This type of cell death is a coordinated process in which cells undergo cytoskeletal disruption, shrinkage, DNA fragmentation, and plasma membrane blebbing under active control so as not to trigger local inflammation. Production of anti-inflammatory cytokines, such as TGF-beta, after apoptosis is observed (58). Consequently to apoptosis, the cell membrane is modified, and internal phospholipids are exposed on the surface. Opsonization of apoptotic cells is mediated by C1q, C4b, C3b, pentraxins, and collectins. Phagocytic macrophage and DCs remove the opsonized apoptotic debris (58). While apoptosis takes place over a few hours, the removal of apoptotic debris is a very quick process, so as to avoid inflammatory response and activation of T lymphocytes (8). However, apoptotic cells may represent an important source of auto-antigens, breaking self-tolerance and triggering autoimmune diseases.
NETosis is a fast process of cell death in which neutrophils undergoes self-disruption, extruding fibrillary networks composed of DNA, citrullinated histones, and antimicrobial granule peptides [neutrophil extracellular traps (NETs)] that entrap and may kill bacteria, virus, fungi, and protozoan pathogens. In pathological conditions, NETs are associated with tissue damage and autoimmune diseases (7, 59, 60). In lupus, apoptosis and NETosis are deregulated and overactive, providing an overload of self-antigens that under normal circumstances would not be available to be targeted by the immune system. In SLE patients, the clearance of apoptotic bodies is diminished, as is the capability to degrade NETs, related to the reduced DNase I activity (61). The increased load of nuclear auto-antigens could amplify the autoimmune response in SLE patients. Histone post-translational modifications in incompletely cleared apoptotic cells or NETs may generate neoantigens and danger signals with an increased antigenic and immunogenic potential (7, 59, 60).
Systemic lupus erythematous patients commonly present skin photosensitivity. Keratinocytes upon sun exposure undergo apoptosis and expel subcellular blebs, which trigger the production of specific autoantibodies against DNA and nucleosomes. SLE patients also present impairment in the clearance of apototic debris by phagocytic cells (58).
C1q and other proteins, such as immunoglobulins, MBL, serum amyloid P, and C-reactive protein, can influence the adequate removal of apoptotic material, which may be dysfunctional in some SLE patients due to gene polymorphism (7, 59, 60). Macrophages of SLE patients may have defective uptake of apoptotic material (62). The expression of FcγRII and FcγRIII receptors on the monocyte plasma membrane is diminished in monocytes from SLE patients, which may contribute to the impaired clearance of apoptotic debris (63). Complement receptor 1 (CR1 and CD35) present in erythrocytes and other leukocytes is important for the clearance of circulating immune complexes. In SLE patients, the expression of CR1 on erythrocytes is also lower when compared to blood cells from normal individuals. This could be a factor that contributes to a defective clearance of C3b- or C4b-coated immune complexes from the circulation (64).
C1q plays an important role since it binds to apoptotic debris and accelerates the removal of auto-antigens from the immune system (55). Moreover, C1q-deficient and C4-deficient mice present impaired uptake of apoptotic bodies by elicited peritoneal macrophages when compared to wild type control (56). In addition, monocytes not only secrete C1q, C1r, and C1s, as it has at its membrane a functional anchored C1q with globular heads outward that could assemble C1r/C1s, serving to capture apoptotic cells, immune complexes, and pathogen- and danger-associated molecular patterns (PAMPs and DAMPs). Monocytes can also recognize these patterns by cC1qR (receptor for collagen domain of C1q) and gC1qR (receptor for globular domain of C1q) triggering different intracellular signaling pathways (65). Membrane C1q and C1qR on monocytes have been suggested to function as sensing molecules, switching them to DCs or macrophages, depending on the stimuli (66).
Dendritic cells have an important role in preventing autoimmune reactions. The tolerance induction is not fully understood but depends on DC phenotype, type and dose of antigens, cytokine microenvironment, and local synthesis of C1q and its receptors (66, 67). C1q could provide active protection by regulating autoreactive immune cells. Experiments in vitro have shown that C1q inhibits T cell proliferation through gC1q/C1q interactions (66, 68, 69).
Immunodeficiencies of Complement and SLE
According to the European Society for Immunodeficiencies (ESID) registry, deficiencies of complement proteins were responsible for 4.9% (946 out of 19,355) of all primary immunodeficiencies (PID) between 2004 and 2014 (http://esid.org/Working-Parties/Registry/ESID-Database-Statistics). Turley et al. (70), studying 77 complement-deficient patients from the ESID registry in 18 European cities observed that 43% presented defects in the CP, 31% in the AP, and 26% presented defects in terminal complement components. C2 deficiency was the most common of the observed deficiencies (29% of the total). In this series, 37% of patients with defects in the CP have SLE-like disease. These immunodeficiencies were implicated in higher susceptibility to infections: mainly pneumococcal in patients with the CP defects and meningococcal disease in patients with terminal component defects (70).
Since complement-activation products lead to an accentuated inflammatory response in SLE, the disruption of complement activity associated with pathological changes in autoimmune diseases is considered a paradox. Deficiency of early complement components frequently leads to the development of autoimmunity or autoimmune-like manifestations (93% of individuals with C1q deficiency, 60–66% of individuals with C1s–C1r deficiency, 75% of individuals with C4 deficiency, and 10% of individuals with C2 deficiency) (71). Inflammatory and autoimmune diseases were not usually seen in patients deficient in proteins from the terminal pathway. On the on hand, late complement factor deficiencies are preferentially linked to infections and not to autoimmunity (72, 73). On the other hand, homozygous complement deficiency occurs in approximately 1% of SLE patients (74), while 8% of Brazilian and 20% of Indian JSLE subgroup patients have deficiencies of early complement components (75, 76).
C1q, C1r, or C1s Deficiencies
C1q can recognize a broad range of ligands, from PAMPs to DAMPs, and have been explored as a possible major bridge between innate and acquired immunity. Patients with C1 deficiencies usually present SLE at an early age, in similar female:male proportions, with severe symptoms and prominent cutaneous manifestations (77). Defects in C1 complex proteins are related to point mutations, gene polymorphism, and partial gene deletion (19, 78, 79). Combined deficiencies of C1s/C1r are commonly inherited together. More than 50% of these patients develop SLE. Sixty-seven cases of complete C1q deficiency have been reported (80). More than 90% of patients with homozygous deficiency of C1q are reported to have SLE or lupus-like syndrome. Rash (95%), glomerulonephritis (42%), and alterations in the central nervous system are observed in 18% of C1q-deficient patients. High titers of autoantibodies are observed in more than 70% of these patients (19–21, 78). Successful cure of C1q deficiency was reported with hematopoietic stem cells transplantation in three patients, but one died in consequence of graft-versus-host disease and multi-organ failure (81–83).
C1q Autoantibodies and SLE
C1q autoantibodies are present in 2–8% of the healthy population, but in SLE, they are present in 30–48% of patients (84). The presence of anti-C1q autoantibodies is accompanied by intense activation of the CP, with very low titers of C1q, C4, and C2 (85). These autoantibodies target a neoepitope of bound C1q that is not expressed in the intact C1 complex (8). This helps to explain the strong association with C1q autoantibodies and nephritis. Their titer correlates to active renal disease with a sensitivity of 44–100% and a specificity of 70–92% (86).
C2 Deficiency
Homozygous C2 deficiency is more frequent in Western European populations with a prevalence of 1:10,000–20,000, in which the majority (>60%) of these individuals are asymptomatic. Heterozygous C2 deficiency has a frequency of 1–2% in Caucasian populations (19, 87). About 10–30% of homozygous C2-deficient patients develop SLE (19, 88, 89). C2-deficient patients present SLE with a female:male proportion of 7:1. Arthritis, malar rash, discoid rash, and photosensitivity are seen in the majority of C2-deficient patients with SLE (19, 20, 22, 78, 89). The human C2 gene is located in chromosome 6p21.3. Type I C2 deficiency is caused by a 28-bp deletion in the C2 gene, which results in the deletion of exon 6 and no translation of the C2 protein. Type II C2 deficiency is usually caused by a point mutation (Ser189Phe and Gly144Arg), which leads to an impairment in C2 secretion (87, 90), reducing the plasma levels of this protein.
C4 Deficiency
Complete homozygous deficiency of C4 is rare but is strongly related to SLE. More than 75% of these patients develop this disease. C4 genes are located in chromosome 6p21.3 in the MHC Class III cluster, with two genes encoding C4 protein (C4A and C4B) (91). The gene copy number (CNV) of C4 ranges from 2 to 8. The more common CNV in the healthy population is 2 of each isotype codominantly expressed. SLE is related to reduction of total C4 copy number, but increased number of copies is a protective factor (91, 92). Approximately 50% of SLE-C4-deficient patients develop glomerulonephritis and more than 70% carry ANA and anti-Rho autoantibodies in their serum (19, 20, 78).
The clinical characteristics, prevalence, and presence of autoantibodies in patients with deficiency of early components of the CP are summarized in Table 2 (8, 19–22).
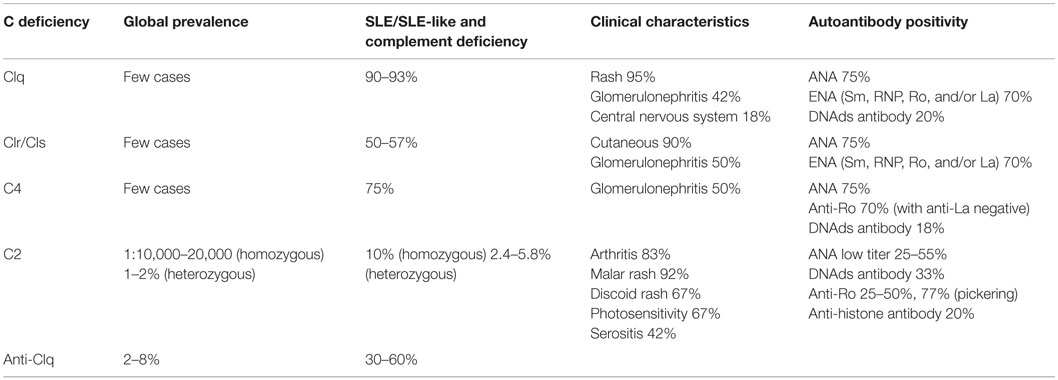
Table 2. Clinical characteristics and prevalence of patients with early complement component deficiencies (8, 19–21).
SLE, Classical Complement Component Deficiencies, and Mortality
Systemic lupus erythematous patients with deficiencies in classical complement components present disease at an early age, with more aggressive symptoms and worse prognosis. Walport et al. (21) reviewed a series of 41 C1q deficiency cases reported in the literature, 38 of which were SLE patients. In these SLE cases, eight individuals younger than 10 years of age died and four of them due of sepsis. Kallel-Sellami et al. (22) reported a 3-year-old girl with SLE and C1q deficiency who died of digestive hemorrhage during treatment of infection. Schaarenburg et al. described 20% fatality in C1q-deficient patients before age of 20 years (93). Jesus et al. (75) studied a total of 72 JLES patients of which six were complement-deficient patients. Three of the six deaths occurred in patients deficient in CP proteins, compared with 1 out of 59 (1.7%) in the group without PID. Two deaths were caused by sepsis, and the third was caused by pneumococcal meningitis. In the same study, disease activity and severity over time, measured by SLICC–ACR damage index, were significantly greater in PID patients (including deficiency of early components of complement and/or immunoglobulins). These descriptions of severe infections in reported cases emphasize the role of the CP in early childhood, when protective antibodies and the anamnestic response have not yet developed. Taken together, these reports reinforce the severity of disease in young SLE patients with complement immunodeficiencies of the CP.
Final Considerations
Systemic lupus erythematous is more severe when associated with deficiencies in components of the CP mainly in young patients. We still do not fully comprehend the real prevalence of complement deficiencies and their association with SLE.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Pesquisa e Desenvolvimento (CNPq), Brazil.
References
1. Liu C-C, Ahearn JM. Complement and systemic lupus erythematosus. 7th ed. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus. (Chap. 13), Philadelphia: Lippincott Williams & Wilkins (2007). p. 214–35.
2. Grammatikos AP, Tsokos GC. Immunodeficiency and autoimmunity: lessons from systemic lupus erythematosus. Trends Mol Med (2012) 18:101–8. doi: 10.1016/j.molmed.2011.10.005
3. Ippolito A, Wallace DJ, Gladman D, Fortin PR, Urowitz M, Werth V, et al. Auto-antibodies in systemic lupus erythematosus: comparison of historical and current assessment of seropositivity. Lupus (2011) 20:250–5. doi:10.1177/0961203310385738
4. Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum (2012) 64(8):2677–86. doi:10.1002/art.34473
5. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1982) 25(11):1271–7. doi:10.1002/art.1780251101
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1997) 40(9):1725. doi:10.1002/art.1780400928
7. Pieterse E, van der Vlag J. Breaking immunological tolerance in systemic lupus erythematosus. Front Immunol (2014) 5:164. doi:10.3389/fimmu.2014.00164
8. Botto M, Walport MJ. C1q autoimmunity and apotosis. Immunobiology (2002) 205:396–406. doi:10.1078/0171-2985-00141
9. Crispín JC, Liossis SN, Kis-Toth K, Lieberman LA, Kyttaris VC, Juang YT, et al. Pathogenesis of human systemic lupus erythematosus: recent advances. Trends Mol Med (2010) 16(2):47–57. doi:10.1016/j.molmed.2009.12.005
10. Pons-Estel GJ, Alarcón GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum (2010) 39(4):257. doi:10.1016/j.semarthrit.2008.10.007
11. Fernandez M, Alarcón GS, Calvo-Alen J, Andrade R, McGwin G Jr, Vilá LM, et al. A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum (2007) 57:576–84. doi:10.1002/art.22672
12. McCarty DJ, Manzi S, Medsger TA Jr, Ramsey-Goldman R, LaPorte RE, Kwoh CK. Incidence of systemic lupus erythematosus. Race and gender differences. Arthritis Rheum (1995) 38:1260–70. doi:10.1002/art.1780380914
13. Malattia C, Martini A. Pediatric-onset systemic lupus erythematosus. Best Pract Res Clin Rheumatol (2013) 27:351–62. doi:10.1016/j.berh.2013.07.007
14. Morgan TA, Watson L, McCann LJ, Beresford MW. Children and adolescents with SLE: not just little adults. Lupus (2013) 22:1309–19. doi:10.1177/0961203313502863
15. Tarr T, Dérfalvi B, Győri N, Szántó A, Siminszky Z, Malik A, et al. Similarities and differences between pediatric and adult patients with systemic lupus erythematosus. Lupus (2015) 24:796–803. doi:10.1177/0961203314563817
16. Choi JH, Park DJ, Kang JH, Yim YR, Lee KE, Lee JW, et al. Comparison of clinical and serological differences among juvenile-, adult-, and late-onset systemic lupus erythematosus in Korean patients. Lupus (2015) 24:1342–9. doi:10.1177/0961203315591024
17. Papadimitraki ED, Isenberg DA. Childhood- and adult-onset lupus: an update of similarities and differences. Expert Rev Clin Immunol (2009) 5:391–403. doi:10.1586/eci.09.29
18. Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum (1992) 35(3):311–8. doi:10.1002/art.1780350310
19. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus complement deficiency and apoptosis. Adv Immunol (2000) 76:227–324. doi:10.1016/S0065-2776(01)76021-X
20. Barilla-LaBarca ML, Atkinson JP. Rheumatic syndromes associated with complement deficiency. Curr Opin Rheumatol (2003) 15:55–60. doi:10.1097/00002281-200301000-00010
21. Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology (1998) 199:265–85. doi:10.1016/S0171-2985(98)80032-6
22. Kallel-Sellami M, Baili-Klila L, Zerzeri Y, Laadhar L, Blouin J, Abdelmalek R, et al. Hereditary complement deficiency and lupus: report of four Tunisian cases. Ann N Y Acad Sci (2007) 1108:197–202. doi:10.1196/annals.1422.022
23. Isenman DE, Kells DI, Cooper NR, Muller-Eberhard HJ, Pangburn MK. Nucleophilic modification of human complement protein C3: correlation of conformational changes with acquisition of C3b-like functional properties. Biochemistry (1981) 20(15):4458–67. doi:10.1021/bi00518a034
24. Isenman DE, Kells DI. Conformational and functional changes in the fourth component of human complement producer by nucleophilic modification and by proteolysis with C1s-. Biochemistry (1982) 21(6):1109–17. doi:10.1021/bi00535a001
25. Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med (1981) 145(3):856–67. doi:10.1084/jem.154.3.856
26. Pangburn MK, Müller-Eberhard HJ. Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of C3. Ann N Y Acad Sci (1983) 421:291–8. doi:10.1111/j.1749-6632.1983.tb18116.x
27. Tack BF, Harrison RA, Janatova J, Thomas ML, Prahl JW. Evidence for presence of an internal thiolester bond in third component of human complement. Proc Natl Acad Sci U S A (1980) 77(10):5764–8. doi:10.1073/pnas.77.10.5764
28. Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med (1975) 142:856–63. doi:10.1084/jem.142.4.856
29. Fearon DT. Regulation of the amplification C3 convertase of human complement by an inhibitory protein isolated from human erythrocyte membrane. Proc Natl Acad Sci U S A (1979) 76(11):5867–71. doi:10.1073/pnas.76.11.5867
30. Lepow IH. Presidential address to American Association of Immunologists in Anaheim, California, April 16, 1980. Louis Pillemer, properdin, and scientific controversy. J Immunol (1980) 125(2):471–5.
31. Müller-Eberhard HJ, Schreiber RD. Molecular biology and chemistry of the alternative pathway of complement. Adv Immunol (1980) 29:1–53. doi:10.1016/S0065-2776(08)60042-5
32. Pangburn MK, Müller-Eberhard HJ. The alternative pathway of complement. Springer Semin Immunopathol (1984) 7(2–3):163–92. doi:10.1007/BF01893019
33. Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC. The properdin system and immunity: I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science (1954) 120:279–85. doi:10.1126/science.120.3112.279
34. Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol (2007) 179:2600–8. doi:10.4049/jimmunol.179.4.2600
35. Kemper C, Hourcade DE. Properdin: new roles in pattern recognition and target clearance. Mol Immunol (2008) 45(16):4048–56. doi:10.1016/j.molimm.2008.06.034
36. Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol (2010) 28:131–55. doi:10.1146/annurev-immunol-030409-101250
37. Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, et al. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity (2001) 15(1):127–35. doi:10.1016/S1074-7613(01)00161-3
38. Fujita T, Matsushita M, Endo Y. The lectin – complement pathway – its role in innate immunity and evolution. Immunol Rev (2004) 198:185–202. doi:10.1111/j.0105-2896.2004.0123.x
39. Matsushita M, Fujita T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J Exp Med (1992) 176(6):1497–502. doi:10.1084/jem.176.6.1497
40. Matsushita M, Endo Y, Fujita T. Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J Immunol (2000) 164(5):2281–4. doi:10.4049/jimmunol.164.5.2281
41. Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature (1997) 386:506–10. doi:10.1038/386506a0
42. Walport MJ. First of two parts. N Engl J Med (2001) 344(14):1058–66. doi:10.1056/NEJM200104053441406
43. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi:10.1038/ni.1923
44. Mayer MM. Mechanism of cytolysis by complement. Proc Natl Acad Sci U S A (1972) 69(10):2954–8. doi:10.1073/pnas.69.10.2954
45. Morgan BP. Regulation of the complement membrane attack pathway. Crit Rev Immunol (1999) 19(3):173–98. doi:10.1615/CritRevImmunol.v19.i3.10
46. Whaley K, Lemercier C. The complement system. In: Sim E, editor. Humoral Factors. (Chap. 5), Oxford: Oxford University Press (1993). p. 121–50.
47. Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol (2007) 7(1):9–18. doi:10.1038/nri1994
48. Rooijakkers SH, van Strijp JA. Bacterial complement evasión. Mol Immunol (2007) 44:23–32. doi:10.1016/j.molimm.2006.06.011
49. Zipfel PF, Würzner R, Skerka C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol (2007) 44(16):3850–7. doi:10.1016/j.molimm.2007.06.149
50. da Silva WD, Eisele JW, Lepow IH. Complement as a mediator of inflammation. 3. Purification of the activity with anaphylatoxin properties generated by interaction of the first four components of complement and its identification as a cleavage product of C3. J Exp Med (1967) 126(6):1027–48. doi:10.1084/jem.126.6.1027
51. Osler AG, Randall HG, Hill BM, Ovary Z. Studies on the mechanism of hypersensitivity phenomena. III. The participation of complement in the formation of anaphylatoxin. J Exp Med (1959) 110(2):311–39. doi:10.1084/jem.110.2.311
52. Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflamatory molecule? Re-evaluating the evidence and the myth. J Immunol (2015) 194:3542–8. doi:10.4049/jimmunol.1403068
53. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science (1996) 271(5247):348–50. doi:10.1126/science.271.5247.348
54. Carroll MC. A protective role for innate immunity in systemic lupus erythematosus. Nat Rev Immunol (2004) 4(10):825–31. doi:10.1038/nri1456
55. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol (1997) 158(10):4525–8.
56. Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med (2000) 192(3):359–66. doi:10.1084/jem.192.3.359
57. Navratil JS, Korb LC, Ahearn JM. Systemic lupus erythematosus and complement deficiency: clues to a novel role for the classical complement pathway in the maintenance of immune tolerance. Immunopharmacology (1999) 42:47–52. doi:10.1016/S0162-3109(99)00018-1
58. Flierman R, Daha MR. The clearance of apoptotic cells by complement. Immunobiology (2007) 212(4–5):363–70. doi:10.1016/j.imbio.2006.11.005
59. Bouts YM, Wolthuis DF, Dirkx MF, Pieterse E, Simons EM, Van Boekel AM, et al. Apoptosis and NET formation in the pathogenesis of SLE. Autoimmunity (2012) 45:597–601. doi:10.3109/08916934.2012.719953
60. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extra cellular trap degradation is associated whit lupus nephritis. Proc Natl Acad Sci U S A (2010) 107:9813–8. doi:10.1073/pnas.0909927107
61. Skiljevic D, Jeremic I, Nikolic M, Andrejevic S, Sefik-Bukilica M, Stojimirovic B, et al. Serum DNase I activity in systemic lupus erythematosus: correlation with immunoserological markers, the disease activity and organ involvement. Clin Chem Lab Med (2013) 51:1083–91. doi:10.1515/cclm-2012-0521
62. Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum (1998) 41:1241–50. doi:10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H
63. Katsiari CG, Liossis SN, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum (2010) 39:491–503. doi:10.1016/j.semarthrit.2008.11.002
64. Ross GD, Yount WJ, Walport MJ, Winfield JB, Parker CJ, Fuller CR, et al. Disease-associated loss of erytrocyte complement receptors (CR1, C3b receptors) in patients with systemic lupus erythematosus and other diseases involving auto-antibodies and/or complement activation. J Immunol (1985) 135:2005–14.
65. Hosszu KK, Valentino A, Ji Y, Matkovic M, Pednekar L, Rehage N, et al. Cell surface expression and function of the macromolecular C1 complex on the surface of human monocytes. Front Immunol (2012) 3:38. doi:10.3389/fimmu.2012.00038
66. Ghebrehiwet B, Hosszu KK, Valentino A, Ji Y, Peerschke EIB. Monocyte expressed macromolecular C1 and C1q receptors as molecular sensors of danger: implications in SLE. Front Immunol (2014) 5:278. doi:10.3389/fimmu.2014.00278
67. Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med (2000) 191:411–6. doi:10.1084/jem.191.3.411
68. Chen A, Gaddipati S, Volkman DJ, Peerschke EIB, Ghebrehiwet B. Human T cells possess specific receptors for C1q: role in activation and proliferation. J Immunol (1994) 153:1430–40.
69. Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest (2000) 106(10):1239–49. doi:10.1172/JCI10323
70. Turley AJ, Gathmann B, Bangs C, Bradbury M, Seneviratne S, Gonzalez-Granado LI, et al. Spectrum and management of complement immunodeficiencies (excluding hereditary angioedema) across Europe. J Clin Immunol (2015) 35(2):199–205. doi:10.1007/s10875-015-0137-5
71. Arason GJ, Jorgensen GH, Ludviksson BR. Primary immunodeficiency and autoimmunity: lessons from human diseases. Scand J Immunol (2010) 71:317–28. doi:10.1111/j.1365-3083.2010.02386.x
72. Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol (2014) 61:110–7. doi:10.1016/j.molimm.2014.06.030
73. Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun (2010) 34:276–86. doi:10.1016/j.jaut.2009.11.014
74. Walport MJ, Davies KA, Morley BJ, Botto M. Complement deficiency and autoimmunity. Ann N Y Acad Sci (1997) 815:267–81. doi:10.1111/j.1749-6632.1997.tb52069.x
75. Jesus AA, Liphaus BL, Silva CA, Bando SY, Andrade LEC, Coutinho A, et al. Complement and antibody primary immunodeficiency in juvenile systemic lupus erythematosus patients. Lupus (2011) 20:1275–84. doi:10.1177/0961203311411598
76. Bhattad S, Rawat A, Gupta A, Suri D, Garg R, de Boer M, et al. Early complement component deficiency in a single-centre cohort of pediatric onset lupus. J Clin Immunol (2015) 35(8):777–85. doi:10.1007/s10875-015-0212-y
77. Stegert M, Bock M, Trendelenburg M. Clinical presentation of human C1q deficiency: how much of a lupus? Mol Immunol (2015) 67:3–11. doi:10.1016/j.molimm.2015.03.007
78. Mayilyan KR. Complement genetics, deficiencies, and disease associations. Protein Cell (2012) 3:487–96. doi:10.1007/s13238-012-2924-6
79. Amano MT, Ferriani VP, Florido MP, Reis ES, Delcolli MI, Azzolini AE, et al. Genetic analysis of complement C1s deficiency associated with systemic lupus erythematosus highlights alternative splicing of normal C1s gene. Mol Immunol (2008) 45(6):1693–702. doi:10.1016/j.molimm.2007.09.034
80. Jlajla H, Sellami MK, Sfar I, Laadhar L, Zerzeri Y, Abdelmoula MS, et al. New C1q mutation in a Tunisian family. Immunobiology (2014) 219:241–6. doi:10.1016/j.imbio.2013.10.010
81. Olsson R, Hagelberg S, Ringden O, Truedsson L, Åhlin A. Allogeneic haematopoietic stem cell transplantation restores complement function in human hereditary C1q deficiency. Bone Marrow Transplant (2013) 217(2012):1129–222. doi:10.1016/j.imbio.2012.08.022
82. Arkwright PD, Riley P, Hughes SM, Alachkar H, Wynn RF. Successful cure of C1q deficiency in human subjects treated with hematopoietic stem cell transplantation. J Allergy Clin Immunol (2014) 133:265–7. doi:10.1016/j.jaci.2013.07.035
83. Olsson R, Hagelberg S, Schiller B, Ringden O, Truedsson L, Åhlin A. Allogeneic haematopoietic stem cell transplantation in the treatment for human C1q deficiency – the Karolinska experience. Bone Marrow Transpl (2015) 50:S489. doi:10.1097/TP.0000000000000975
84. Beurskens FJ, van Schaarenburg RA, Trouw LA. C1q, antibodies and anti-C1q autoantibodies. Mol Immunol (2015) 68(1):6–13. doi:10.1016/j.molimm.2015.05.010
85. Fremeaux-Bacchi V, Weiss L, Demouchy C, Blouin J, Kazatchkine MD. Auto-antibodies to the collagen-like region of C1q are strongly associated with classical pathway mediated hypocomplementemia in systemic lupus erythematosus. Lupus (1996) 5:216. doi:10.1177/096120339600500309
86. Sinico RA, Rimoldi L, Radice A, Bianchi L, Gallelli B, Moroni G. Anti-C1q auto-antibodies in lupus nephritis. Ann N Y Acad Sci (2009) 1173:47–51. doi:10.1111/j.1749-6632.2009.04746.x
87. O’Neil KM. Complement deficiency. Clin Rev Allergy Immunol (2000) 19:83. doi:10.1385/CRIAI:19:2:83
88. Laich A, Sim RB. Cross-talk between the human complement classical and alternative pathways: evidence for a C4bBb “hybrid” C3 convertase. Mol Immunol (2001) 38:105.
89. Jonsson G, Sjoholm AG, Truedsson L, Bengtsson AA, Braconier JH, Sturfelt G. Rheumatological manifestations, organ damage and autoimmunity in hereditary C2 deficiency. Rheumatology (Oxford) (2007) 46:1133–9. doi:10.1093/rheumatology/kem023
90. Wetsel RA, Kulics J, Lokki ML, Kiepiela P, Akama H, Johnson CA, et al. Type II human complement C2 deficiency. Allele-specific amino acid substitutions (Ser189→Phe; Gly444→Arg) cause impaired C2 secretion. J Biol Chem (1996) 271(10):5824–31. doi:10.1074/jbc.271.10.5824
91. Yang Y, Lhotta K, Chung EK, Eder P, Neumair F, Yu CY. Complete complement components C4A and C4B deficiencies in human kidney diseases and systemic lupus erythematosus. J Immunol (2004) 173:2803–14. doi:10.4049/jimmunol.173.4.2803
92. Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene copy-number variation and associated polymorphisms of complement component c4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet (2007) 80:1037–54. doi:10.1086/518257
Keywords: SLE, lupus, complement deficiency, C1q, C2, C4
Citation: Macedo ACL and Isaac L (2016) Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway. Front. Immunol. 7:55. doi: 10.3389/fimmu.2016.00055
Received: 02 October 2015; Accepted: 05 February 2016;
Published: 24 February 2016
Edited by:
Brian Reilly, Texas Tech University, USAReviewed by:
Bin Li, Chinese Academy of Sciences, ChinaMichael Kirschfink, University of Heidelberg, Germany
Copyright: © 2016 Macedo and Isaac. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lourdes Isaac, bG91aXNhYWNAaWNiLnVzcC5icg==