 Marieke Griffioen
Marieke Griffioen Cornelis A. M. van Bergen
Cornelis A. M. van Bergen J. H. Frederik Falkenburg
J. H. Frederik Falkenburg- Department of Hematology, Leiden University Medical Center, Leiden, Netherlands
Allogeneic stem cell transplantation (alloSCT) can be a curative treatment for hematological malignancies. Unfortunately, the desired anti-tumor or graft-versus-leukemia (GvL) effect is often accompanied with undesired side effects against healthy tissues known as graft-versus-host disease (GvHD). After HLA-matched alloSCT, GvL and GvHD are both mediated by donor-derived T-cells recognizing polymorphic peptides presented by HLA surface molecules on patient cells. These polymorphic peptides or minor histocompatibility antigens (MiHA) are produced by genetic differences between patient and donor. Since polymorphic peptides may be useful targets to manipulate the balance between GvL and GvHD, the dominant repertoire of MiHA needs to be discovered. In this review, the diversity of autosomal MiHA characterized thus far as well as the various molecular mechanisms by which genetic variants create immune targets and the role of cryptic transcripts and proteins as antigen sources are described. The tissue distribution of MiHA as important factor in GvL and GvHD is considered as well as possibilities how hematopoietic MiHA can be used for immunotherapy to augment GvL after alloSCT. Although more MiHA are still needed for comprehensive understanding of the biology of GvL and GvHD and manipulation by immunotherapy, this review shows insight into the composition and kinetics of in vivo immune responses with respect to specificity, diversity, and frequency of specific T-cells and surface expression of HLA–peptide complexes and other (accessory) molecules on the target cell. A complex interplay between these factors and their environment ultimately determines the spectrum of clinical manifestations caused by immune responses after alloSCT.
Allogeneic Stem Cell Transplantation
Allogeneic stem cell transplantation (alloSCT) can be a curative treatment for hematological malignancies. In alloSCT, the immune system from a healthy donor is transplanted into the patient to induce an effective response against the leukemic cells (1, 2). Unfortunately, desired anti-tumor or graft-versus-leukemia (GvL) reactivity is often accompanied with graft-versus-host disease (GvHD) affecting predominantly skin, gut, liver, and lungs (3–6). To reduce GvHD, donor T-cells can be (partially) depleted from the stem cell graft (7–9). T-cell depletion decreases the incidence and severity of GvHD, but increases the risk of leukemia relapse and opportunistic infections. Once toxicity of the conditioning has subsided, donor T-cells can be administered after alloSCT as donor lymphocyte infusions (DLI) to reinstall beneficial GvL (10, 11). Although DLI can induce long-lasting clinical remissions, GvHD remains a major cause of morbidity and mortality. Since DLI is applied to patients who do not receive (or only limited) immunosuppression, postponed DLI after alloSCT creates an ideal platform to study specificity, magnitude, and duration of GvL and GvHD.
Minor Histocompatibility Antigens
To minimize GvHD, patients with hematological malignancies are preferably transplanted with HLA-matched donors (12, 13). After HLA-matched alloSCT, donor-derived T-cells can mediate GvL and GvHD by recognizing polymorphic peptides presented on patient cells by shared HLA molecules. In the classical dogma, intracellular proteins are degraded in the cytosol by the proteasome and peptides are presented by HLA class I to CD8 T-cells (14, 15). HLA class II molecules present peptides derived from intra- and extracellular proteins to CD4 T-cells (14, 16). In the autologous situation, peptides from normal cellular proteins cannot be recognized by the immune system due to negative selection and deletion of self-specific T-cells in the thymus (17). After HLA-matched alloSCT, however, donor T-cells recognize polymorphic peptides presented on patient cells by shared HLA as “non-self.” These polymorphic peptides or so-called minor histocompatibility antigens (MiHA) can be encoded by the male-specific Y-chromosome (H-Y antigens) or other chromosomes (autosomal MiHA) and are produced by genetic differences between patient and donor (18–21). This review is focused on discovery strategies and molecular mechanisms behind autosomal MiHA.
T-Cell Isolation
Minor histocompatibility antigen-specific T-cells can be directly isolated from in vivo immune responses after alloSCT or generated in vitro by stimulating antigen-experienced or naive donor T-cells (22, 23). T-cells have been isolated from in vivo immune responses by their capacity to produce IFN-γ upon in vitro stimulation with patient hematopoietic cells (24, 25). A disadvantage is that not all specific T-cells produce IFN-γ during 5–24 h of incubation, resulting in low T-cell isolation efficiencies. HLA-DR is a marker that has successfully been used to isolate in vivo activated T-cells (26, 27). Since HLA-DR is expressed on activated T-cells for a prolonged time, its expression does not require in vitro stimulation and enables analysis of the in vivo immune response without introducing a bias. CD137 is another marker that allows direct isolation of antigen-experienced T-cells (28, 29). This marker is specific for T-cells that are recently activated and requires in vitro stimulation for re-expression on the cell surface.
Discovery Strategies
To develop strategies that allow manipulation of GvL and GvHD, the dominant repertoire of autosomal MiHA needs to be discovered. HA-1 and HA-2 are the first MiHA that have been identified as T-cell targets in a patient with GvHD (Table 1). The antigens have been characterized as peptides eluted from HLA surface molecules that are recognized by specific T-cells by mass spectrometry. Other MiHA characterized by this approach are HA-8, HA-3, PANE1, and LB-ADIR-1F (Table 1). cDNA library screening in which pools of plasmids are tested for T-cell recognition is another technique that has been used for discovery of HB-1H, UGT2B17/A29, UGT2B17/B44, ACC-4, ACC-5, ACC-6, SP110, LB-ECGF-1H, C19ORF48, TRIM22, and LB-TRIP10-1EPC (Table 1). In addition, five HLA class II-restricted MiHA encoded by PI4K2B, PTK2B, LY75, MR1, and MTHFD1 have been characterized by screening a library in which recombinant bacteria are screened for T-cell recognition (Table 1).
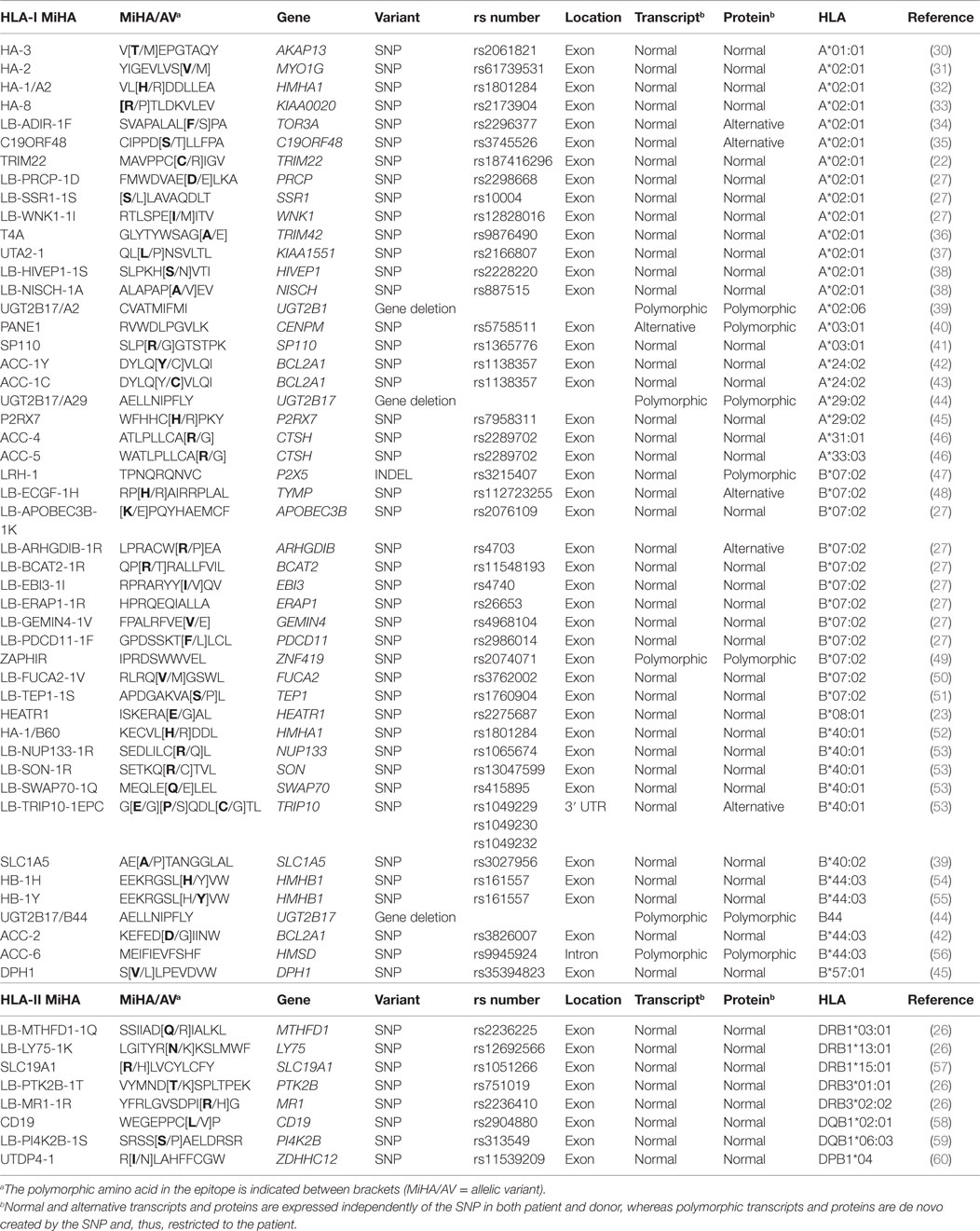
Table 1. HLA class I- and II-restricted autosomal minor histocompatibility antigens.
Due to advanced array techniques to measure single nucleotide polymorphisms (SNPs), whole genome association scanning (WGAs) became available as efficient method for MiHA discovery (39, 43). In this approach, a panel of test cells with known SNP genotypes is used to measure T-cell recognition. T-cell recognition is subsequently investigated for association with individual SNPs to identify the genomic region that encodes the MiHA. Before SNP arrays became commercially available, WGAs was performed with low-resolution genetic markers, leading to identification of large genomic regions of which all genes needed to be investigated for encoding the antigen. MiHA characterized by WGAs with low-resolution markers are ACC-1Y, ACC-2, LRH-1, and HEATR1 (Table 1). When high-resolution SNP data are used, WGAs enables direct identification of the MiHA-producing SNP or identification of small genomic regions with SNP(s) that are in linkage disequilibrium with the MiHA-producing SNP. MiHA identified with high-resolution SNP data are ACC-1C, SLC1A5, UGT2B17/A2, DPH1, P2RX7, LB-PRCP-1D, SSR1-1S, LB-WNK1-1I, LB-EBI3-1I, LB-BCAT2-1R, LB-ARHGDIB-1R, LB-PDCD11-1F, LB-APOBEC3B-1K, LB-GEMIN4-1V, LB-ERAP1-1R, ZAPHIR, LB-SON-1R, LB-NUP133-1R, LB-SWAP70-1Q, UTA2-1, and LB-FUCA2-1V (Table 1). WGAs with high-resolution SNPs also led to discovery of HLA class II-restricted MiHA encoded by CD19, SLC19A1, and ZDHHC12 (Table 1). Nowadays, data for all SNPs as present in the human genome are available in the 1000 Genomes Project and the value of this dataset has recently been illustrated by discovery of UTDP4-1 (Table 1).
Whereas T-cells are used to identify MiHA by forward strategies; in reverse strategies, peptides are selected to search for specific T-cells. Polymorphic peptides identified in HLA-ligandomes (38, 51), hematopoiesis-restricted genes (52, 55, 61), and peptides identified based on association of SNPs with good clinical outcome after alloSCT (36) have been selected to search for specific T-cells in transplanted patients or healthy individuals.
In total, 48 HLA class I-restricted and 8 HLA class II-restricted autosomal MiHA have thus far been characterized. These numbers are expected to rapidly increase in the near future, in particular, if WGAs is performed with cell panels for which all SNPs are measured by whole genome sequencing.
Molecular Mechanisms
HLA class I-restricted autosomal MiHA are generated by different molecular mechanisms. An overview of the various mechanisms by which genetic variants create MiHA is shown in Figure 1. Of the 48 HLA class I-restricted MiHA, 36 antigens are encoded by SNPs in coding exons, leading to single amino acid changes in proteins that are translated from primary gene transcripts in the normal reading frame (Figure 1A). MiHA can, however, also be translated from normal gene transcripts in an alternative reading frame. These SNPs can be located in coding exons (C19ORF48, LB-ECGF1-1H, LB-ADIR-1F, LB-ARHGDIB-1R) (Figure 1B) or in 5’ or 3’ UTR regions (LB-TRIP10-1EPC) (Figure 1C). Though not yet discovered, it is expected that MiHA can also be encoded by SNP in intron regions that are retained in alternative transcripts (Figure 1D). Proteins translated in alternative reading frames are considered as aberrant proteins that lack any cellular function, the so-called defective ribosomal products (DRiPs). DRiPs are rapidly degraded during or shortly after translation and evidence has been found that they may be a main source of peptide precursors for T-cell immunosurveillance (62). Degradation is an important factor for HLA presentation (63), but relative abundance of normal functional proteins in the cell may counteract the actual contribution of DRiPs to the HLA-ligandome.
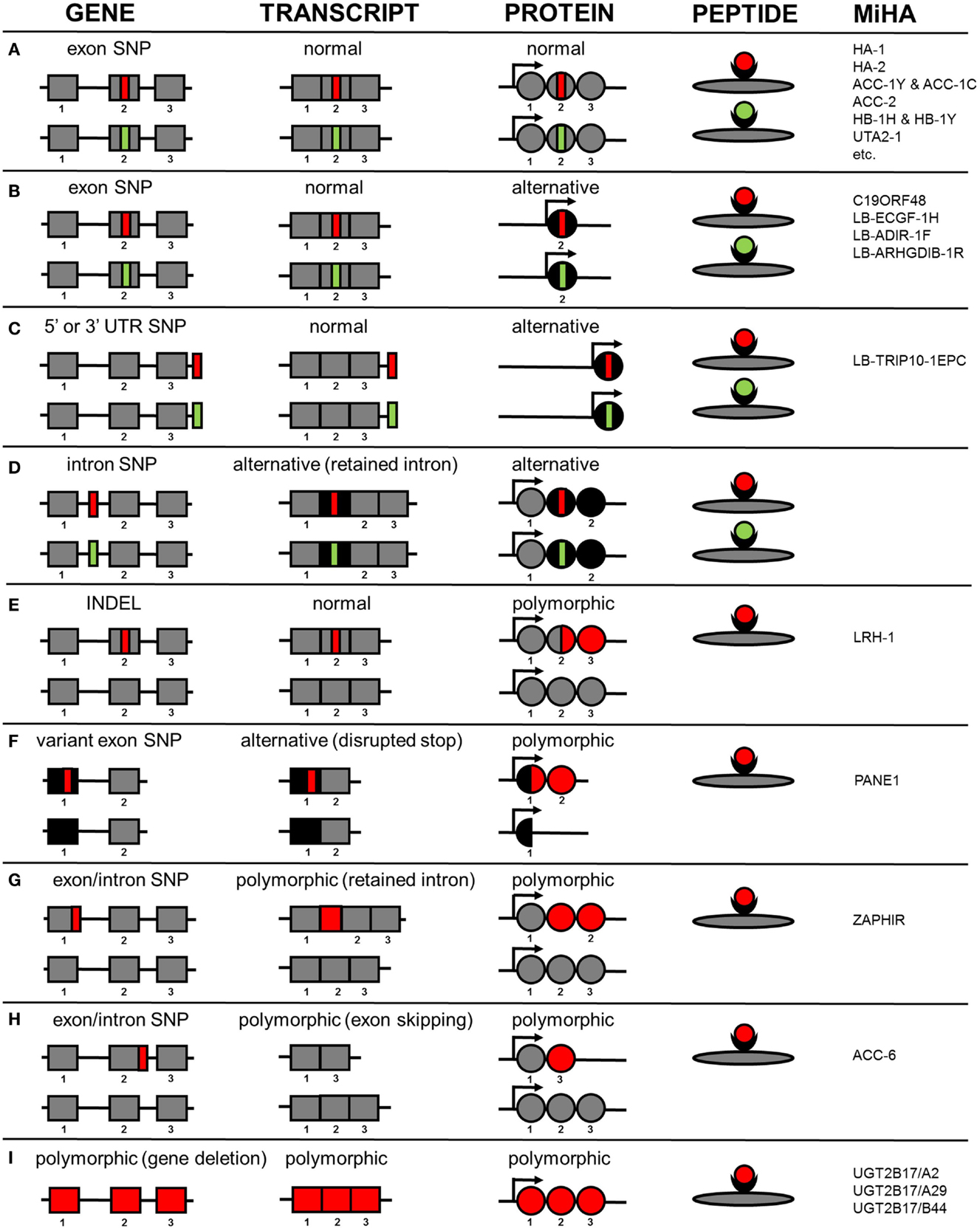
Figure 1. Molecular mechanisms by which genetic variants create autosomal MiHA. Normal non-polymorphic sequences are indicated in gray, whereas alternative non-polymorphic sequences are shown in black. Polymorphic patient-specific sequences are shown in red and donor-specific sequences are indicated in green (if allelic variants exist). Whether the allelic variants are actually presented on the cell surface is also dependent on intracellular processing and presentation mechanisms, which are not taken into consideration in this figure. (A) MiHA created by SNPs in primary gene transcripts in the normal reading frame. (B) MiHA created by SNPs in primary gene transcripts in an alternative reading frame. (C) MiHA created by SNPs in 5´ or 3´ UTR of primary gene transcripts. (D) MiHA created by intron SNPs as retained in alternative gene transcripts. (E) MiHA derived from polymorphic proteins as created by frameshift insertions or deletions in primary gene transcripts. (F) MiHA derived from polymorphic proteins as created by SNP in alternative gene transcripts. (G) MiHA translated from intron sequences in polymorphic gene transcripts as created by exon or intron SNPs. (H) MiHA translated from polymorphic gene transcripts in which exon sequences are skipped as created by exon or intron SNPs. (I) MiHA encoded by polymorphic genes.
In addition to the proteins described above that are expressed independently of the SNP in both patient and donor, MiHA can also be derived from proteins or protein products that are de novo created by SNPs. Expression of these polymorphic proteins is restricted to the patient and allelic variants in the donor do not exist. As a result, the epitope that is recognized by the T-cell can be derived from another protein region than the amino acids that are directly encoded by the SNP. Examples of antigens from polymorphic proteins that are de novo created are LRH-1, an antigen that is produced by an insertion/deletion variant (INDEL) that induces a frameshift in protein translation (Figure 1E) and PANE1, which is an antigen from an elongated protein created by a SNP that disrupts the stop codon (Figure 1F). PANE1 and LRH-1 are both polymorphic proteins translated from transcripts that are expressed independently of the SNP in both patient and donor. However, MiHA can also be encoded by transcripts that are newly created by the SNP. These polymorphic transcripts are expressed in the patient and do not exist in the donor. Antigens encoded by polymorphic transcripts that are newly created by SNPs are ZAPHIR, which is translated from a ZNF419 transcript in which an intron is retained (Figure 1G), and ACC-6, which is encoded by an HMSD transcript that is generated by exon skipping (Figure 1H). Finally, MiHA can be encoded by polymorphic genes as illustrated by UGT2B17, which is present in the patient but absent in the donor genome (Figure 1I).
The numbers of MiHA that have been characterized for each molecular mechanism as shown in Figure 1 probably do not reflect the actual contribution of the various mechanisms to the entire repertoire of MiHA that are recognized by specific T-cells after alloSCT. This is suggested by the finding that for various T-cell clones, associating SNPs have successfully been identified by WGAs in genomic regions outside known exons, whereas epitope discovery failed due to absence of SNP disparities in the normal gene transcript. MiHA recognized by these T-cells are probably encoded by cryptic transcripts. RNA-sequence data can be used to search for these cryptic transcripts in the genomic region that contains the associating SNPs and single RNA-sequence reads can be analyzed to determine the exact sequence composition of the transcripts, thereby facilitating discovery of these MiHA. As such, implementation of RNA-sequence analysis in a combined approach of whole genome and transcriptome analysis may increase the efficiency of MiHA discovery. The various molecular mechanisms how genetic variants create MiHA as shown in Figure 1 are probably similar for neoantigens. Neoantigens are peptides created by tumor-specific mutations that are presented by HLA and recognized by specific T-cells (64). In cancer neoantigen discovery, research is focused on selecting peptides encoded by mutations in coding exons with single amino acid changes in the normal protein reading frame (Figure 1A), whereas other molecular mechanisms (Figures 1B–H) are often not taken into consideration. RNA-sequence analysis may, therefore, also be relevant to elucidate transcript variants for neoantigens in particular since splicing defects often occur in cancer (65).
Tissue Distribution
The tissue distribution of MiHA is an important factor in clinical manifestations caused by immune responses after alloSCT. Various T-cells recognize leukemic cells in vitro with no or minimal reactivity against non-hematopoietic cells. These T-cells are expected to mediate beneficial GvL after alloSCT without GvHD. Other T-cells are reactive with both hematopoietic and non-hematopoietic cells, suggesting a role in GvHD. Since in alloSCT, patient hematopoiesis is replaced by a blood-forming system from a healthy donor, donor T-cells for hematopoiesis-restricted MiHA eliminate the malignant cells of the patient, while sparing healthy hematopoietic cells of donor origin. Therefore, discovery of hematopoietic MiHA is an explicit research goal.
Various methods are available to investigate the tissue distribution of MiHA to estimate their efficacy and toxicity as T-cell targets. Toxicity can be analyzed by measuring T-cell reactivity against non-hematopoietic cells from organs that are targeted in GvHD. This analysis, however, requires collection of a variety of tissues expressing the relevant MiHA and HLA restriction allele. Skin fibroblasts are frequently used to estimate toxicity and have also been cultured with cytokines to mimic the inflammatory environment of the early post-transplantation period. T-cells often recognize skin fibroblasts when cultured under inflammatory conditions, what may be explained by efficient antigen processing and presentation and enhanced surface expression of HLA, costimulatory, and adhesion molecules. Other non-hematopoietic cells, however, are more difficult to culture and often not available in quantities that allow in depth T-cell analysis. Therefore, as second best option, the tissue distribution can be investigated by gene expression analysis. Thus far, only a limited number of MiHA are encoded by genes with restricted or predominant expression in (malignant) hematopoietic cells, i.e., HMHB1 (54), MYO1G (66), HMHA1 (67), BCL2A1 (42), P2×5 (47), CENPM (40), HMSD (56), KIAA1551 (37), and ARHGDIB (68). Although gene expression analysis allows rapid selection of hematopoietic antigens, the therapeutic value of MiHA needs to be validated by demonstrating the capacity of specific T-cells to kill leukemic cells and confirming their failure to react with non-hematopoietic cells.
In Vivo Immune Responses
Minor histocompatibility antigens characterization enabled ex vivo quantification of specific T-cells by pMHC multimers in individual patients after alloSCT. Staining with pMHC multimers demonstrated a peak in the immune response in patients who responded to DLI after HLA-matched alloSCT (27, 37, 47, 69). In these patients, high frequencies of circulating T-cells coincided with development of GvL. Detailed analysis of peak responses between 4 and 12 weeks after DLI demonstrated that a diversity of HLA class I- and II-restricted MiHA are targeted by CD8 and CD4 T-cells (26, 27, 53). These T-cells expand and retract with similar kinetics, although frequencies and timing of the peak may differ between MiHA (27, 53).
Although GvL after alloSCT is often accompanied with GvHD, strong anti-tumor responses without severe side effects are occasionally observed (69), illustrating that GvL can be separated from GvHD. In the pathophysiology of GvHD, the tissue distribution of MiHA is important as well as the frequencies of circulating T-cells, their homing behavior and capacity to destroy non-hematopoietic cells in situ (70). Although tissue distribution is relevant, occurrence of GvHD cannot entirely be explained by induction of T-cells targeting MiHA on non-hematopoietic tissues. This became clear when T-cells for hematopoietic and ubiquitous MiHA were simultaneously detected in patients with severe GvHD (71) and patients without GvHD (53). Since immune responses in patients with GvHD are generally strong, it can be speculated that T-cell reactivity against non-hematopoietic tissues needs to exceed a certain threshold in GvHD (72). Since T-cells for ubiquitous MiHA may stimulate development of GvL by releasing cytokines, strategies that retain reactivity against healthy tissues below the threshold may effectively separate GvL from GvHD.
Therapeutic Use
As the number of characterized MiHA increases, T-cells from different patients more often recognize MiHA that are already known, suggesting that the repertoire of MiHA that are presented by HLA and recognized by specific T-cells is limited and follow rules for immunodominance that cannot be predicted by measuring only SNP disparities (73). If true, a large proportion of all MiHA with balanced population frequencies will be characterized in the coming years. Discovery of these MiHA is needed to analyze and compare in vivo immune responses in GvL and GvHD with respect to specificity, diversity, frequency, and dynamics of specific T-cells. Moreover, it enables to follow GvL and GvHD in large patient groups, which is essential to investigate and compare efficacy and toxicity of different alloSCT (and DLI) transplantation protocols.
With the discovery of a large proportion of common MiHA, a variety of targets become available for therapy to augment GvL after alloSCT (74, 75). One strategy is in vitro production and adoptive transfer of donor T-cells for hematopoietic MiHA (75, 76). Patients with leukemia who relapsed after alloSCT have been treated with in vitro expanded T-cells for leukemic cells (77, 78), T-cells for HA-1 (79) or MiHA-specific T-cells that lacked reactivity against fibroblasts (45). Other strategies for adoptive transfer are isolation of MiHA-specific T-cells from the DLI by pMHC multimers and T-cell receptor (TCR) gene transfer. In the latter study, patients are treated with virus-specific donor T-cells that are genetically engineered with the TCR for HA-1 (80). Besides adoptive transfer, patients with hematological malignancies can be in vivo vaccinated with donor (or patient) dendritic cells loaded with peptides or mRNA (81–83). In conclusion, hematopoietic MiHA (and their specific TCRs) may be easily implemented in ongoing clinical trials to increase efficacy, reduce toxicity, and broaden applicability of immunotherapy after alloSCT.
Concluding Remarks
In this review, the various molecular mechanisms how genetic variants create autosomal MiHA are described as well as the relevance of these antigens as tools to understand the biology of GvL and GvHD and as targets for immunotherapy to treat hematological cancers after alloSCT. Although more MiHA are needed for comprehensive understanding and manipulation by immunotherapy, this review shows insight into the composition and kinetics of in vivo immune responses with respect to specificity, diversity, and frequency of specific T-cells and surface expression of HLA-peptide complexes and other (accessory) molecules on the target cell. A complex interplay between these factors and their environment (84) ultimately determines the spectrum of clinical manifestations that are caused by immune responses after alloSCT.
Author Contributions
All authors listed, have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
alloSCT, allogeneic stem cell transplantation; DLI, donor lymphocyte infusion; DRiP, defective ribosomal product; GvHD, graft-versus-host disease; GvL, graft-versus-leukemia reactivity; MiHA, minor histocompatibility antigen; SNP, single nucleotide polymorphism; WGAs, whole genome association scanning.
References
1. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood (1990) 75:555–62.
2. Falkenburg JHF, Warren EH. Graft versus leukemia reactivity after allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2011) 17:S33–8. doi: 10.1016/j.bbmt.2010.11.009
3. Gratwohl A, Brand R, Apperley J, Biezen AA, Bandini G, Devergie A, et al. Graft-versus-host disease and outcome in HLA-identical sibling transplantations for chronic myeloid leukemia. Blood (2002) 100:3877–86. doi:10.1182/blood.V100.12.3877
5. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet (2009) 373:1550–61. doi:10.1016/S0140-6736(09)60237-3
6. Stern M, de Wreede LC, Brand R, van Biezen A, Dreger P, Mohty M, et al. Sensitivity of hematological malignancies to graft-versus-host effects: an EBMT megafile analysis. Leukemia (2014) 28:2235–40. doi:10.1038/leu.2014.145
7. Marmont AM, Horowitz MM, Gale RP, Sobocinski K, Ash RC, van Bekkum DW, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood (1991) 78:2120–30.
8. Barge RM, Starrenburg CW, Falkenburg JHF, Fibbe WE, Marijt EW, Willemze R. Long-term follow-up of myeloablative allogeneic stem cell transplantation using Campath “in the bag” as T-cell depletion: the Leiden experience. Bone Marrow Transplant (2006) 37:1129–34. doi:10.1038/sj.bmt.1705385
9. Hobbs GS, Perales MA. Effects of T-cell depletion on allogeneic hematopoietic stem cell transplantation outcomes in AML patients. J Clin Med (2015) 4:488–503. doi:10.3390/jcm4030488
10. Kolb HJ. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood (2008) 112:4371–83. doi:10.1182/blood-2008-03-077974
11. Deol A, Lum LG. Role of donor lymphocyte infusions in relapsed hematological malignancies after stem cell transplantation revisited. Cancer Treat Rev (2010) 36:528–38. doi:10.1016/j.ctrv.2010.03.004
12. Hansen JA, Petersdorf EW, Lin MT, Wang S, Chien JW, Storer B, et al. Genetics of allogeneic hematopoietic cell transplantation. Role of HLA matching, functional variation in immune response genes. Immunol Res (2008) 41:56–78. doi:10.1007/s12026-007-0043-x
13. Petersdorf EW. The major histocompatibility complex: a model for understanding graft-versus-host disease. Blood (2013) 122:1863–72. doi:10.1182/blood-2013-05-355982
14. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol (2011) 11:823–36. doi:10.1038/nri3084
15. Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst (2013) 105:1172–87. doi:10.1093/jnci/djt184
16. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol (2015) 15:203–16. doi:10.1038/nri3818
17. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi:10.1038/nri3667
18. Falkenburg JHF, van de Corput L, Marijt EW, Willemze R. Minor histocompatibility antigens in human stem cell transplantation. Exp Hematol (2003) 31:743–51. doi:10.1016/S0301-472X(03)00190-5
19. Warren EH, Zhang XC, Li S, Fan W, Storer BE, Chien JW, et al. Effect of MHC and non-MHC donor/recipient genetic disparity on the outcome of allogeneic HCT. Blood (2012) 120:2796–806. doi:10.1182/blood-2012-04-347286
20. Spierings E. Minor histocompatibility antigens: past, present, and future. Tissue Antigens (2014) 84:374–360. doi:10.1111/tan.12445
21. Zilberberg J, Feinman R, Korngold R. Strategies for the identification of T cell-recognized tumor antigens in hematological malignancies for improved graft-versus-tumor responses after allogeneic blood and marrow transplantation. Biol Blood Marrow Transplant (2015) 21:1000–7. doi:10.1016/j.bbmt.2014.11.001
22. Wölfel C, Lennerz V, Lindemann E, Hess G, Derigs HG, Huber C, et al. Dissection and molecular analysis of alloreactive CD8+ T cell responses in allogeneic haematopoietic stem cell transplantation. Cancer Immunol Immunother (2008) 57:849–57. doi:10.1007/s00262-007-0421-1
23. Bleakley M, Otterud BE, Richardt JL, Mollerup AD, Hudecek M, Nishida T, et al. Leukemia-associated minor histocompatibility antigen discovery using T-cell clones isolated by in vitro stimulation of naive CD8+ T cells. Blood (2010) 115:4923–33. doi:10.1182/blood-2009-12-260539
24. Kloosterboer FM, Luxemburg-Heijs SAP, van Soest RA, van Egmond HM, Barbui AM, Strijbosch MP, et al. Minor histocompatibility antigen-specific T cells with multiple distinct specificities can be isolated by direct cloning of IFNgamma-secreting T cells from patients with relapsed leukemia responding to donor lymphocyte infusion. Leukemia (2005) 19:83–90. doi:10.1038/sj.leu.2403572
25. Campbell JD, Foerster A, Lasmanowicz V, Niemoller M, Scheffold A, Fahrendorff M, et al. Rapid detection, enrichment and propagation of specific T cell subsets based on cytokine secretion. Clin Exp Immunol (2011) 163:1–10. doi:10.1111/j.1365-2249.2010.04261.x
26. Stumpf AN, van der Meijden ED, van Bergen CAM, Willemze R, Falkenburg JHF, Griffioen M. Identification of 4 new HLA-DR-restricted minor histocompatibility antigens as hematopoietic targets in antitumor immunity. Blood (2009) 114:3684–92. doi:10.1182/blood-2009-03-208017
27. van Bergen CAM, Rutten CE, van der Meijden ED, van Luxemburg-Heijs SAP, Lurvink EGA, Houwing-Duistermaat JJ, et al. High-throughput characterization of 10 new minor histocompatibility antigens by whole genome association scanning. Cancer Res (2010) 70:9073–83. doi:10.1158/0008-5472.CAN-10-1832
28. Wehler TC, Nonn M, Brandt B, Britten CM, Grone M, Todorova M, et al. Targeting the activation-induced antigen CD137 can selectively deplete alloreactive T cells from antileukemic and antitumor donor T-cell lines. Blood (2007) 109:365–73. doi:10.1182/blood-2006-04-014100
29. Wolfl M, Kuball J, Eyrich M, Schlegel PG, Greenberg PD. Use of CD137 to study the full repertoire of CD8+ T cells without the need to know epitope specificities. Cytometry A (2008) 73:1043–9. doi:10.1002/cyto.a.20594
30. Spierings E, Brickner AG, Caldwell JA, Zegveld S, Tatsis N, Blokland E, et al. The minor histocompatibility antigen HA-3 arises from differential proteasome-mediated cleavage of the lymphoid blast crisis (Lbc) oncoprotein. Blood (2003) 102:621–9. doi:10.1182/blood-2003-01-0260
31. den Haan JM, Sherman NE, Blokland E, Huczko E, Koning F, Drijfhout JW, et al. Identification of a graft versus host disease-associated human minor histocompatibility antigen. Science (1995) 268:1476–80. doi:10.1126/science.7539551
32. den Haan JM, Meadows LM, Wang W, Pool J, Blokland E, Bishop TL, et al. The minor histocompatibility antigen HA-1: a diallelic gene with a single amino acid polymorphism. Science (1998) 279:1054–7. doi:10.1126/science.279.5353.1054
33. Brickner AG, Warren EH, Caldwell JA, Akatsuka Y, Golovina TN, Zarling AL, et al. The immunogenicity of a new human minor histocompatibility antigen results from differential antigen processing. J Exp Med (2001) 193:195–206. doi:10.1084/jem.193.2.195
34. van Bergen CAM, Kester MGD, Jedema I, Heemskerk MHM, van Luxemburg-Heijs SAP, Kloosterboer FM, et al. Multiple myeloma-reactive T cells recognize an activation-induced minor histocompatibility antigen encoded by the ATP-dependent interferon-responsive (ADIR) gene. Blood (2007) 109:4089–96. doi:10.1182/blood-2006-08-043935
35. Tykodi SS, Fujii N, Vigneron N, Lu SM, Mito JK, Miranda MX, et al. C19orf48 encodes a minor histocompatibility antigen recognized by CD8+ cytotoxic T cells from renal cell carcinoma patients. Clin Cancer Res (2008) 14:5260–9. doi:10.1158/1078-0432.CCR-08-0028
36. Armistead PM, Liang S, Li H, Lu S, van Bergen CAM, Alatrash G, et al. Common minor histocompatibility antigen discovery based upon patient clinical outcomes and genomic data. PLoS One (2011) 6:e23217. doi:10.1371/journal.pone.0023217
37. Oostvogels R, Minnema MC, van Elk M, Spaapen RM, te Raa GD, Giovannone B, et al. Towards effective and safe immunotherapy after allogeneic stem cell transplantation: identification of hematopoietic-specific minor histocompatibility antigen UTA2-1. Leukemia (2013) 27:642–9. doi:10.1038/leu.2012.277
38. Hombrink P, Hassan C, Kester MGD, de Ru AH, van Bergen CAM, Nijveen H, et al. Discovery of T cell epitopes implementing HLA-peptidomics into a reverse immunology approach. J Immunol (2013) 190:3869–77. doi:10.4049/jimmunol.1202351
39. Kamei M, Nannya Y, Torikai H, Kawase T, Taura K, Inamoto Y, et al. HapMap scanning of novel human minor histocompatibility antigens. Blood (2009) 113:5041–8. doi:10.1182/blood-2008-07-171678
40. Brickner AG, Evans AM, Mito JK, Xuereb SM, Feng X, Nishida T, et al. The PANE1 gene encodes a novel human minor histocompatibility antigen that is selectively expressed in B-lymphoid cells and B-CLL. Blood (2006) 107:3779–86. doi:10.1182/blood-2005-08-3501
41. Warren EH, Vigneron NJ, Gavin MA, Coulie PG, Stroobant V, Dalet A, et al. An antigen produced by splicing of noncontiguous peptides in the reverse order. Science (2006) 313:1444–7. doi:10.1126/science.1130660
42. Akatsuka Y, Nishida T, Kondo E, Miyazaki M, Taji H, Iida H, et al. Identification of a polymorphic gene, BCL2A1, encoding two novel hematopoietic lineage-specific minor histocompatibility antigens. J Exp Med (2003) 197:1489–500. doi:10.1084/jem.20021925
43. Kawase T, Nannya Y, Torikai H, Yamamoto G, Onizuka M, Morishima S, et al. Identification of human minor histocompatibility antigens based on genetic association with highly parallel genotyping of pooled DNA. Blood (2008) 111:3286–94. doi:10.1182/blood-2007-10-118950
44. Murata M, Warren EH, Riddell SR. A human minor histocompatibility antigen resulting from differential expression due to a gene deletion. J Exp Med (2003) 197:1279–89. doi:10.1084/jem.20030044
45. Warren EH, Fujii N, Akatsuka Y, Chaney CN, Mito JK, Loeb KR, et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood (2010) 115:3869–78. doi:10.1182/blood-2009-10-248997
46. Torikai H, Akatsuka Y, Miyazaki M, Tsujimura A, Yatabe Y, Kawase T, et al. The human cathepsin H gene encodes two novel minor histocompatibility antigen epitopes restricted by HLA-A*3101 and -A*3303. Br J Haematol (2006) 134:406–16. doi:10.1111/j.1365-2141.2006.06205.x
47. de Rijke B, Horssen-Zoetbrood A, Beekman JM, Otterud B, Maas F, Woestenenk R, et al. A frameshift polymorphism in P2X5 elicits an allogeneic cytotoxic T lymphocyte response associated with remission of chronic myeloid leukemia. J Clin Invest (2005) 115:3506–16. doi:10.1172/JCI24832
48. Slager EH, Honders MW, van der Meijden ED, Luxemburg-Heijs SAP, Kloosterboer FM, Kester MGD, et al. Identification of the angiogenic endothelial-cell growth factor-1/thymidine phosphorylase as a potential target for immunotherapy of cancer. Blood (2006) 107:4954–60. doi:10.1182/blood-2005-09-3883
49. Broen K, Levenga H, Vos J, van Bergen CAM, Fredrix H, Greupink-Draaisma A, et al. A polymorphism in the splice donor site of ZNF419 results in the novel renal cell carcinoma-associated minor histocompatibility antigen ZAPHIR. PLoS One (2011) 6:e21699. doi:10.1371/journal.pone.0021699
50. van Bergen CAM, Verdegaal EM, Honders MW, Hoogstraten C, Steijn-van Tol AQ, de Quartel L, et al. Durable remission of renal cell carcinoma in conjuncture with graft versus host disease following allogeneic stem cell transplantation and donor lymphocyte infusion: rule or exception? PLoS One (2014) 9:e85198. doi:10.1371/journal.pone.0085198
51. Hombrink P, Hassan C, Kester MGD, Jahn L, Pont MJ, de Ru AH, et al. Identification of biological relevant minor histocompatibility antigens within the B-lymphocyte-derived HLA-ligandome using a reverse immunology approach. Clin Cancer Res (2015) 21:2177–86. doi:10.1158/1078-0432.CCR-14-2188
52. Mommaas B, Kamp J, Drijfhout JW, Beekman N, Ossendorp F, van Veelen P, et al. Identification of a novel HLA-B60-restricted T cell epitope of the minor histocompatibility antigen HA-1 locus. J Immunol (2002) 169:3131–6. doi:10.4049/jimmunol.169.6.3131
53. Griffioen M, Honders MW, van der Meijden ED, van Luxemburg-Heijs SAP, Lurvink EGA, Kester MGD, et al. Identification of 4 novel HLA-B*40:01 restricted minor histocompatibility antigens and their potential as targets for graft-versus-leukemia reactivity. Haematologica (2012) 97:1196–204. doi:10.3324/haematol.2011.049478
54. Dolstra H, Fredrix H, Maas F, Coulie PG, Brasseur F, Mensink E, et al. A human minor histocompatibility antigen specific for B cell acute lymphoblastic leukemia. J Exp Med (1999) 189:301–8. doi:10.1084/jem.189.2.301
55. Dolstra H, de Rijke B, Fredrix H, Balas A, Maas F, Scherpen F, et al. Bi-directional allelic recognition of the human minor histocompatibility antigen HB-1 by cytotoxic T lymphocytes. Eur J Immunol (2002) 32:2748–58. doi:10.1002/1521-4141(2002010)32:10<2748::AID-IMMU2748>3.0.CO;2-T
56. Kawase T, Akatsuka Y, Torikai H, Morishima S, Oka A, Tsujimura A, et al. Alternative splicing due to an intronic SNP in HMSD generates a novel minor histocompatibility antigen. Blood (2007) 110:1055–63. doi:10.1182/blood-2007-02-075911
57. Spaapen RM, de Kort RA, van den Oudenalder K, van Elk M, Bloem AC, Lokhorst HM, et al. Rapid identification of clinical relevant minor histocompatibility antigens via genome-wide zygosity-genotype correlation analysis. Clin Cancer Res (2009) 15:7137–43. doi:10.1158/1078-0432.CCR-09-1914
58. Spaapen RM, Lokhorst HM, van den Oudenalder K, Otterud BE, Dolstra H, Leppert MF, et al. Toward targeting B cell cancers with CD4+ CTLs: identification of a CD19-encoded minor histocompatibility antigen using a novel genome-wide analysis. J Exp Med (2008) 205:2863–72. doi:10.1084/jem.20080713
59. Griffioen M, van der Meijden ED, Slager EH, Honders MW, Rutten CE, van Luxemburg-Heijs SAP, et al. Identification of phosphatidylinositol 4-kinase type II beta as HLA class II-restricted target in graft versus leukemia reactivity. Proc Natl Acad Sci U S A (2008) 105:3837–42. doi:10.1073/pnas.0712250105
60. Oostvogels R, Lokhorst HM, Minnema MC, van Elk M, van den Oudenalder K, Spierings E, et al. Identification of minor histocompatibility antigens based on the 1000 Genomes Project. Haematologica (2014) 99:1854–9. doi:10.3324/haematol.2014.109801
61. Broen K, Greupink-Draaisma A, Woestenenk R, Schaap N, Brickner AG, Dolstra H. Concurrent detection of circulating minor histocompatibility antigen-specific CD8+ T cells in SCT recipients by combinatorial encoding MHC multimers. PLoS One (2011) 6:e21266. doi:10.1371/journal.pone.0021266
62. Anton LC, Yewdell JW. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J Leukoc Biol (2014) 95:551–62. doi:10.1189/jlb.1113599
63. Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics (2015) 14:658–73. doi:10.1074/mcp.M114.042812
64. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi:10.1126/science.aaa4971
65. Daguenet E, Dujardin G, Valcárcel J. The pathogenicity of splicing defects: mechanistic insights into pre-mRNA processing inform novel therapeutic approaches. EMBO Rep (2015) 16:1640–55. doi:10.15252/embr.201541116
66. Pierce RA, Field ED, Mutis T, Golovina TN, Kap-Herr C, Wilke M, et al. The HA-2 minor histocompatibility antigen is derived from a diallelic gene encoding a novel human class I myosin protein. J Immunol (2001) 167:3223–30. doi:10.4049/jimmunol.167.6.3223
67. Wilke M, Dolstra H, Maas F, Pool J, Brouwer R, Falkenburg JHF, et al. Quantification of the HA-1 gene product at the RNA level; relevance for immunotherapy of hematological malignancies. Hematol J (2003) 4:315–20. doi:10.1038/sj.thj.6200318
68. Pont MJ, Hobo W, Honders MW, van Luxemburg-Heijs SAP, Kester MGD, van Oeveren-Rietdijk AM, et al. LB-ARHGDIB-1R as novel minor histocompatibility antigen for therapeutic application. Haematologica (2015) 100(10):e419–22. doi:10.3324/haematol.2015.125021
69. Marijt WAF, Heemskerk MHM, Kloosterboer FM, Goulmy E, Kester MGD, van der Hoorn MAWG, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci U S A (2003) 100:2742–7. doi:10.1073/pnas.0530192100
70. Dickinson AM, Wang XN, Sviland L, Vyth-Dreese FA, Jackson GH, Schumacher TN, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med (2002) 8:410–4. doi:10.1038/nm0402-410
71. Mutis T, Gillespie G, Schrama E, Falkenburg JHF, Moss P, Goulmy E. Tetrameric HLA class I-minor histocompatibility antigen peptide complexes demonstrate minor histocompatibility antigen-specific cytotoxic T lymphocytes in patients with graft-versus-host disease. Nat Med (1999) 5:839–42. doi:10.1038/10563
72. van der Zouwen B, Kruisselbrink AB, Jordanova ES, Rutten CE, von dem Borne PA, Falkenburg JHF, et al. Alloreactive effector T cells require the local formation of a proinflammatory environment to allow crosstalk and high avidity interaction with nonhematopoietic tissues to induce GVHD reactivity. Biol Blood Marrow Transplant (2012) 18:1353–67. doi:10.1016/j.bbmt.2012.06.017
73. Jameson-Lee M, Koparde V, Griffith P, Scalora AF, Sampson JK, Khalid H, et al. In silico derivation of HLA-specific alloreactivity potential from whole exome sequencing of stem-cell transplant donors and recipients: understanding the quantitative immunobiology of allogeneic transplantation. Front Immunol (2014) 5:529. doi:10.3389/fimmu.2014.00529
74. Spaapen R, Mutis T. Targeting haematopoietic-specific minor histocompatibility antigens to distinguish graft-versus-tumour effects from graft-versus-host disease. Best Pract Res Clin Haematol (2008) 21:543–57. doi:10.1016/j.beha.2008.06.001
75. Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol (2011) 89:396–407. doi:10.1038/icb.2010.124
76. Tsirigotis P, Shimoni A, Nagler A. The expanding horizon of immunotherapy in the treatment of malignant disorders: allogeneic hematopoietic stem cell transplantation and beyond. Ann Med (2014) 46:384–96. doi:10.3109/07853890.2014.918463
77. Falkenburg JHF, Wafelman AR, Joosten P, Smit WM, van Bergen CAM, Bongaerts R, et al. Complete remission of accelerated phase chronic myeloid leukemia by treatment with leukemia-reactive cytotoxic T lymphocytes. Blood (1999) 94:1201–8.
78. Marijt EW, Wafelman AR, van der Hoorn MAWG, van Bergen CAM, Bongaerts R, van Luxemburg-Heijs SAP, et al. Phase I/II feasibility study evaluating the generation of leukemia-reactive cytotoxic T lymphocyte lines for treatment of patients with relapsed leukemia after allogeneic stem cell transplantation. Haematologica (2007) 92:72–80. doi:10.3324/haematol.10433
79. Meij P, Jedema I, van der Hoorn MAWG, Bongaerts R, Cox L, Wafelman AR, et al. Generation and administration of HA-1-specific T-cell lines for the treatment of patients with relapsed leukemia after allogeneic stem cell transplantation: a pilot study. Haematologica (2012) 97:1205–8. doi:10.3324/haematol.2011.053371
80. Heemskerk MHM, Griffioen M, Falkenburg JHF. T-cell receptor gene transfer for treatment of leukemia. Cytotherapy (2008) 10:108–15. doi:10.1080/14653240701883087
81. Overes IM, Fredrix H, Kester MGD, Falkenburg JHF, van der Voort R, de Witte TM, et al. Efficient activation of LRH-1-specific CD8+ T-cell responses from transplanted leukemia patients by stimulation with P2X5 mRNA-electroporated dendritic cells. J Immunother (2009) 32:539–51. doi:10.1097/CJI.0b013e3181987c22
82. Rezvani K. Posttransplantation vaccination: concepts today and on the horizon. Hematology Am Soc Hematol Educ Program (2011) 2011:299–304. doi:10.1182/asheducation-2011.1.299
83. Hosen N, Maeda T, Hashii Y, Tsuboi A, Nishida S, Nakata J, et al. Vaccination strategies to improve outcome of hematopoietic stem cell transplant in leukemia patients: early evidence and future prospects. Expert Rev Hematol (2014) 7:671–81. doi:10.1586/17474086.2014.953925
84. Stevanovic S, van Bergen CAM, van Luxemburg-Heijs SAP, van der Zouwen B, Jordanova ES, Kruisselbrink AB, et al. HLA class II upregulation during viral infection leads to HLA-DP-directed graft-versus-host disease after CD4+ donor lymphocyte infusion. Blood (2013) 122:1963–73. doi:10.1182/blood-2012-12-470872
Keywords: allogeneic stem cell transplantation, hematological malignancy, graft-versus-leukemia reactivity, graft-versus-host disease, donor lymphocyte infusion, T-lymphocytes, minor histocompatibility antigens, immunotherapy
Citation: Griffioen M, van Bergen CAM and Falkenburg JHF (2016) Autosomal Minor Histocompatibility Antigens: How Genetic Variants Create Diversity in Immune Targets. Front. Immunol. 7:100. doi: 10.3389/fimmu.2016.00100
Received: 02 October 2015; Accepted: 01 March 2016;
Published: 15 March 2016
Edited by:
Gilles Benichou, Massachusetts General Hospital and Harvard Medical School, USAReviewed by:
Ralf Dressel, University Medical Center Göttingen, GermanyEmmanuel Zorn, Columbia University Medical Center, USA
Copyright: © 2016 Griffioen, van Bergen and Falkenburg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marieke Griffioen, bS5ncmlmZmlvZW5AbHVtYy5ubA==