 Akio Ohta
Akio Ohta- Center for Drug Discovery, Northeastern University, Boston, MA, USA
Within tumors, some areas are less oxygenated than others. Since their home ground is under chronic hypoxia, tumor cells adapt to this condition by activating aerobic glycolysis; however, this hypoxic environment is very harsh for incoming immune cells. Deprivation of oxygen limits availability of energy sources and induces accumulation of extracellular adenosine in tumors. Extracellular adenosine, upon binding with adenosine receptors on the surface of various immune cells, suppresses pro-inflammatory activities. In addition, signaling through adenosine receptors upregulates a number of anti-inflammatory molecules and immunoregulatory cells, leading to the establishment of a long-lasting immunosuppressive environment. Thus, due to hypoxia and adenosine, tumors can discourage antitumor immune responses no matter how the response was induced, whether it was spontaneous or artificially introduced with a therapeutic intention. Preclinical studies have shown the significance of adenosine in tumor survival strategy by demonstrating tumor regression after inactivation of adenosine receptors, inhibition of adenosine-producing enzymes, or reversal of tissue hypoxia. These promising results indicate a potential use of the inhibitors of the hypoxia–adenosine pathway for cancer immunotherapy.
Immune Checkpoint
The discovery of tumor-associated antigens and tumor-reactive immune cells led to a promise of a new cancer therapy based on tumor antigen-specific immune responses. Preventing successful immunotherapy of cancer is the immunosuppressive nature of the tumor microenvironment (1–3). In animals expressing the same antigen in both tumors and normal tissue, the intensity of antigen-specific T cell response was diminished in tumors, while the same T cells were vigorously activated in normal tissue (4, 5). T cells infiltrated in tumor tissue are often capable of recognizing tumor-associated antigen, but they coexist with their target, tumor cells, without significant antitumor activity. However, when isolated from tumor tissue, those tumor-infiltrated T cells could kill the tumor cells efficiently in vitro (6). These studies indicate that tumors establish a stern environment for antitumor immune cells, cells that can be active effector cells otherwise.
Many of the immunosuppressive mechanisms in tumors are common to physiological immunoregulation in normal tissues. Such immunoregulation is very important in keeping the immune system under control in order to block a self-reactive immune response and to prevent an ongoing immune response from causing critical tissue damage. The lack of physiological immunoregulation often results in overwhelming immune activation that accompanies autoimmunity. For example, CTLA-4 is a physiological mechanism that negatively regulates T cell activity by blocking a costimulatory signal through CD28–B7 interaction. The lack of CTLA-4 causes non-specific T cell activation, and CTLA-4-deficient mice die in several weeks with massive lymphocytic tissue infiltration (7, 8). In humans, heterozygous mutation in the CTLA-4 gene is enough to cause immune dysregulation similar to homozygous CTLA-4-knockout mice (9). PD-1 also provides a T cell inhibitory signal upon interaction with its ligands, PD-L1 and PD-L2. Deficiency of PD-1 in mice is known to cause various types of autoimmune disorders depending on the genetic strains (10).
Besides cell surface transducers of immunosuppressive signal, e.g., CTLA-4 and PD-1, immunosuppression in the tumor microenvironment involves anti-inflammatory cytokines (IL-10, TGF-β), enzymes (indoleamine-2,3-dioxygenase), and professional immunoregulatory cells [regulatory T cells, myeloid-derived suppressor cells (MDSCs)] (1, 2). These immunosuppressive mechanisms play an important role in controlling immune response in normal tissues, not tumor tissue specific. Since tumors take advantage of such physiological immunoregulatory mechanisms to protect their tissue from immune attack, these mechanisms intended to prevent inflammatory complication, now turn out to be major obstacles hampering spontaneous cancer regression and immunological cancer treatment. The identification of immunosuppressive mechanisms in tumors pointed out molecular targets to restore the antitumor immune response. Thus, these negative immunoregulatory mechanisms, so-called immune checkpoints, became a focus in drug discovery. The effort resulted in FDA approval of anti-CTLA-4 and anti-PD-1 antibodies for cancer treatment. This achievement finally convinced people that immunotherapy of cancer is realistic, and it further encouraged the development of inhibitors of other immune checkpoint molecules (10–12).
Extracellular adenosine has been known as an inhibitor of immune functions. While intracellular adenosine is involved in energy metabolism, nucleic acid metabolism, and the methionine cycle, extracellular adenosine plays an important role in intercellular signaling. Its signal is transmitted by G protein-coupled adenosine receptors on the cell surface, and it affects diverse physiological functions including neurological, cardiovascular, and immunological systems (13). Extracellular concentration of adenosine can increase in response to metabolic change. When cells are deprived of nutrients or oxygen, insufficient ATP biosynthesis tends to lower the ATP/adenosine ratio. To reduce ATP expenditure, cells may suspend energy-consuming activities such as cell proliferation, which requires biosynthesis of a large amount of cellular components (14, 15). Indeed, tissue hypoxia strongly represses proliferation of activated T cells (16). Interestingly, extracellular adenosine is known to accumulate under hypoxic conditions. Adenosine signaling may play a role in the improvement of energy status by promoting catabolism of stored metabolic energy. Correspondingly, extracellular adenosine increases energy expenditure through the induction of lipolysis (17). Tumors contain high levels of extracellular adenosine (18, 19), suggesting that tumor cells may benefit from its immunosuppressive effect and catabolic energy production. The current review focuses on the pro-cancer roles of extracellular adenosine and discusses application of the inhibitors of this metabolic immune checkpoint to cancer immunotherapy.
Adenosine Receptors and Suppression of Antitumor Immunity
Of the four known types of adenosine receptors, A2A adenosine receptor (A2AR) is the predominantly expressed subtype in most immune cells (13). Stimulation of A2AR generally provides an immunosuppressive signal that inhibits activities of T cells (proliferation, cytokine production, cytotoxicity), NK cells (cytotoxicity), NKT cells (cytokine production, CD40L upregulation), macrophages/dendritic cells (antigen presentation, cytokine production), and neutrophils (oxidative burst) (20, 21). Administration of A2AR agonist strongly inhibits induction of various inflammatory diseases (22, 23). Endogenous adenosine shows similar immunoregulatory activity via A2AR, and this mechanism is critical to the control of the immune response. Exaggeration of inflammatory tissue injury in A2AR-deficient mice indicated that no other immunoregulatory mechanism could compensate for the lack of the adenosine-dependent immunoregulation (20, 24–28).
In 2006, the presence of high levels of extracellular adenosine in tumors was found to play a significant role in the evasion of antitumor immune response (19). The study showed that A2AR-deficient mice could spontaneously regress the inoculated tumor, whereas no wild-type mice showed similar tumor regression. A2AR antagonists were also beneficial in tumor-bearing wild-type animals. Importantly, depletion of T cells and NK cells impaired the retardation of tumor growth by A2AR antagonists, suggesting improvement of antitumor cellular immune response (19, 29). Effector functions of T cells and NK cells are susceptible to A2AR stimulation. In addition, when activated in the presence of A2AR agonist, the effector function of T cells is persistently impaired even after removal of A2AR agonist (30, 31). This result suggests that the adenosine-rich environment in tumors may induce T cells that are anergic to the tumor cells. Consistent with this change, A2AR stimulation induces immunoregulatory molecules such as CTLA-4 and PD-1 on T cells (32–34). Antigen-presenting cells (APCs) are also targets of adenosine. A2AR agonists suppress IL-12 and induce IL-10 production from APCs, discouraging cellular immune response (35, 36). Conversely, myeloid-specific deletion of A2AR enhanced tumor infiltration of effector T cells and NK cells (37). These studies show A2AR-dependent suppression of antitumor T cell activity. However, a recent study reported an interesting finding that A2AR-deficient antitumor T cells might have a shorter half-life in vivo (38). A2AR-deficient T cells effectively elicit antitumor activities in the adenosine-rich tumor microenvironment, but if these cells were to disappear prematurely, they may not complete tumor regression and may actually result in tumor regrowth. Further studies with this issue may result in an improved treatment in which adenosine-resistant antitumor effector cells can offer persistent immune response.
Furthermore, adenosine recruits immunoregulatory activity of regulatory T (Treg) cells. FoxP3, a key transcriptional factor for the immunosuppressive activity of Treg cells, is inducible by A2AR stimulation (30). T cell activation in the presence of A2AR stimulation largely increases CD4+ FoxP3+ cells (33, 39–41). Interestingly, in addition to the numerical increase of Treg cells, A2AR stimulation augmented the immunoregulatory activity of Treg cells (33). Consistent with the increase in regulatory activity in vitro, Treg cell-dependent prevention of in vivo inflammatory disease induction could be enhanced by pretreating Treg cells with A2AR agonist before transfer (39). In contrast, transfer of A2AR-deficient Treg cells failed to save the tissue from inflammatory damage (42), suggesting that endogenous adenosine may be important in the modulation of Treg activity in vivo.
A2B adenosine receptor (A2BR) can also potentially mediate immunosuppressive activity of extracellular adenosine, but the mechanisms of A2BR-dependent immunosuppression are different from those of A2AR. A2BR-dependent immunoregulation is notable in myeloid cells. A2BR plays a predominant role in the adenosine-dependent differentiation of macrophages into M2-type (43–45). Those macrophages activated in the presence of A2BR stimulation express arginase, indoleamine-2,3-dioxygenase, and TGF-β, and have limited T cell stimulatory activity. In addition to the induction of such tolerogenic APCs, the adenosine–A2BR interaction results in the expansion of MDSCs (46). Indeed, treatment of tumor-bearing mice with A2BR agonist increased MDSC in tumors and accelerated tumor growth (47).
Immunoregulatory activity of A2BR is also involved in the suppression of antitumor immunity by adenosine present in the tumor microenvironment. Slower tumor growth was observed in A2BR-deficient mice and A2BR antagonist-treated wild-type mice (48, 49). Recruitment of T cell immunity was necessary to provoke antitumor effect of A2BR antagonist because the treatment was not effective in T cell-deficient animals (49). Diminished tumor growth by A2BR antagonist accompanied a greater number of T cell infiltration but reduced numbers of Treg cells and MDSC (47, 49, 50).
Thus, extracellular adenosine downregulates antitumor immune response in various ways. The mechanisms of immunosuppression involve not only direct effects on antitumor effector cells but also indirect effects on APCs and professional immunoregulatory cells such as Treg cells and MDSC. A2AR and/or A2BR mediate immunosuppressive effects of adenosine depending on cell types. Interestingly, A2AR and A2BR seem to mainly target lymphoid cells and myeloid cells, respectively. The impact of adenosine-mediated immunosuppression seems to be persistent rather than transient because the outcome of adenosine exposure can induce M2-type tumor-associated macrophages, Treg cells, MDSC, and “anergic” effector T cells. These facts support an idea that extracellular adenosine is a negative immune checkpoint molecule that plays a significant role in establishing an immunosuppressive tumor microenvironment. Therefore, it is reasonable to target the adenosine-dependent pathway in order to improve cancer therapy. The blockade of A2AR and A2BR using antagonists has demonstrated promise for a new therapeutic approach (Table 1).

Table 1. Representative A2AR/A2BR antagonists (majority of A2AR antagonists were developed for Parkinson’s disease; *under cancer clinical trial).
Cancer Immunotherapy by Targeting Adenosine-Producing Mechanism
To inactivate the adenosine-dependent immunosuppression, it would be beneficial to think about the metabolism of extracellular adenosine. The source of extracellular adenosine is believed to be ATP in the extracellular compartment. There are nucleotidases on the cell surface called CD39 and CD73. CD39 catalyzes hydrolysis of ATP/ADP to AMP, and CD73 converts AMP to adenosine. Very low extracellular adenosine levels in CD73-deficient mice suggest that degradation of ATP is a major source of extracellular adenosine (51, 52). Adenosine is metabolized to inosine by adenosine deaminase or converted to AMP by the function of adenosine kinase. Tissue hypoxia can induce accumulation of extracellular adenosine by increasing adenosine production and by decreasing adenosine removal. Hypoxia induces the mRNA levels and enzymatic activities of CD39 and CD73 leading to an increase in adenosine production (53, 54). The rate of adenosine removal can be reduced by hypoxia-dependent inhibition of adenosine kinase, resulting in further accumulation of adenosine (55, 56).
Inflammation consequently induces focal hypoxia in the inflamed tissue. Since inflammatory tissue damage involves non-specific injury to vasculature, disruption of blood circulation in the tissue causes deprivation of nutrients and oxygen downstream, and the resulting tissue hypoxia may increase extracellular adenosine production (57, 58). In addition, cellular damage, another consequence of proinflammatory activities, can result in the release of a large amount of adenine nucleotides, such as ATP, ADP, AMP, and adenosine, into the extracellular space. A combination of adenine nucleotide release and CD39/CD73 upregulation induces a robust accumulation of adenosine. Indeed, inflammation has been found to accompany an increase in extracellular adenosine levels (59–61). This hypoxia-induced metabolic switch favoring adenosine accumulation changes the course of inflammation. When inflammatory damage is excessive, the tissue will face an imminent danger, the loss of function. Adenosine produced from the hypoxic and damaged tissue can stop proinflammatory activities and, instead, facilitate anti-inflammatory activities (58). Moreover, adenosine promotes angiogenesis, which is an ultimate solution to tissue hypoxia, and tissue remodeling (62, 63). Therefore, adenosine upregulation in the hypoxic tissue can serve as a negative feedback mechanism of inflammation, promoting a switch from propagation of proinflammatory response to resolution of inflammation.
An important factor in keeping inflammation under control is the induction of a metabolic switch favoring adenosine accumulation. Just as A2AR deficiency caused exaggeration of inflammation, CD39- or CD73-deficient mice provided further evidence for the importance of endogenous adenosine in controlling inflammation (64–69). Although the release of intracellular adenosine from damaged cells can also increase extracellular adenosine levels, exaggerated inflammation in the absence of CD39 or CD73 indicates the pathophysiological significance of the metabolic adenosine production at the outer membrane. The importance of CD73 enzymatic activity was confirmed using an inhibitor of nucleotidase activity. It is also interesting that Treg cells express both CD39 and CD73, and the produced adenosine serves as an immunoregulatory mechanism of Treg cells (70–72).
Such biological significance of ectonucleotidases implied the role in the suppression of antitumor immunity. Interestingly, CD73 expression has been observed in mouse tumor cell lines (73–75) and tumor cells from various cancer patients (76–83). Those tumor cells are likely to generate adenosine by themselves for protection from immune cells. Inhibition of CD73 can reduce tumor growth (75), and indeed the suppression of tumor growth was found to be immune response dependent (84). Neutralization of CD73 by antibodies reduced tumor growth and metastasis from the primary tumor (84, 85). The reduction in tumor growth was not observed in immunocompromised mice, suggesting an involvement of antitumor immunity. Since anti-CD73 antibody did not block tumor growth in A2AR-deficient mice (84), it is likely that adenosine produced from CD73-expressing tumor cells suppresses antitumor immune response in an A2AR-dependent fashion. Supporting this speculation, knockdown of CD73 in tumor cells reduced their growth in vivo and increased their susceptibility to antitumor immune cells (84, 86). Tumor growth is also retarded in CD73-deficient mice, suggesting that adenosine production from non-tumor host cells also contributes to the establishment of the immunosuppressive environment (67, 68).
CD39, in conjunction with CD73, can promote adenosine accumulation and thereby inhibit immune activities in tumors. Deletion of CD39 from vasculature or bone marrow-derived cells as well as administration of a CD39 inhibitor blocked tumor growth in a model of hepatic metastasis (87). In human cancer, CD39 expression was observed in tumor cells, tumor stroma, and infiltrated lymphocytes (88). Inhibition of CD39 on tumor cells can relieve coexisting immune effector cells from immunosuppression (88, 89). Taken together, tumors often upregulate the extracellular adenosine-producing mechanism. Therefore, CD39 and CD73 are potential targets to block adenosine-dependent immunosuppression in tumors.
When mutated cells arise, immune cells may be able to recognize newly developed nascent tumor cells as abnormal and eliminate them (90). This immunosurveillance is considered to prevent cancer formation as shown by increased cancer incidence in immunocompromised mice that are lacking IFN-γ or perforin (91). Although most immune cells can exert a tumor-eliminating response, emergence of immune cells that produce anti-inflammatory cytokines or have immunoregulatory functions will change the microenvironment into tumor-permissive one. Since adenosine suppresses cellular immunity and promotes the switch to a tolerant immune profile, an abundance of extracellular adenosine may be detrimental to the prevention of cancer development. Consistent with this view, double transgenic mice expressing human papilloma virus type 16 E7 oncogene (E7) and A2AR were prone to spontaneous cancer development compared to E7-single transgenic mice. Thyroid specific coexpression of E7 and A2AR resulted in rapid development of thyroid carcinoma and lung metastasis of malignant thyroid cancer cells (92). Conversely, A2AR-deficient mice showed a reduced cancer incidence in 3-methylcholanthrene-induced chemical carcinogenesis (93). CD73-deficient mice were also resistant to 3-methylcholanthrene-induced carcinogenesis (68). CD73 deficiency is preventive to spontaneous prostate cancer development in TRAMP-transgenic mice (68). These studies suggest the significance of the adenosine system in cancer development, although the mechanism of cancer prevention needs further investigation.
Non-Immune Targets of Adenosine in Cancer
As discussed above, immunosuppressive effects of extracellular adenosine provide tumors a protection from antitumor immunity. However, immune cells are not the only targets of the pro-tumor effect of adenosine. Some tumor cells have been found to express adenosine receptors, and adenosine may support tumor progression directly. In tumor cell culture in vitro, the addition of adenosine to the culture enhanced tumor cell proliferation. Cotreatment with antagonists suggested that the effect was mediated by A2AR or other adenosine receptors (94, 95). Adenosine can also prevent apoptosis of tumor cells, and A2AR antagonist reversed this effect (95). As discussed above, some tumor cells express CD73 and are able to increase extracellular adenosine levels. Tumor cell-derived adenosine not only protects the tumor from immune cells but also autonomously enhances tumor growth. Downregulation of CD73 in siRNA-transduced tumor cells reduced cell proliferation in vitro as well as tumor growth in vivo (75, 96). APCP, an inhibitor of enzymatic activity of CD73, also inhibited CD73-expressing tumor cell proliferation and increased apoptosis (94, 96). In contrast, CD73 overexpression increased tumor cell proliferation and viability (96).
CD73 expression on human cancer cells may induce resistance to chemotherapy. In human breast cancer cells, higher CD73 expression was associated with lower therapeutic response to anthracyclines (79). CD73 inhibitor sensitized the tumors to doxorubicin. In chronic lymphocytic leukemia, cancer cells constitutively express A2AR, and autonomous adenosine production with their own CD39/CD73 can result in A2AR-dependent prevention of drug-induced apoptosis (78). The inhibition of CD73 activity in glioblastoma cells decreased tumor cell viability to vincristine by downregulating multidrug transporter protein 1 (MRP1) (80).
Moreover, adenosine and CD73 expression may play a role in metastasis of cancer cells. CD73 downregulation by antibody or siRNA reduced adhesion, invasion, and migration of CD73-expressing tumor cells (75, 84). Conversely, CD73 overexpression increased adhesion and migration (97). When tumor cells were pretreated with anti-CD73 mAb before injection into mice, their extravasation and metastasis in vivo were decreased compared to control tumor cells (85). Addition of adenosine to the tumor cell culture increases tumor cell adhesion and migration (97, 98). Since adenosine does not increase tumor cell migration in the absence of A2AR (98), the adenosine-A2AR system may promote tumor metastasis by, in addition to its suppressive effect on antitumor immune response (29, 84, 99), directly activating tumor cell adhesion and migration. Although the pro-metastatic effect of CD73 may be mediated by adenosine production through its enzymatic activity, the non-enzymatic role of CD73 is also suggested in tumor cell migration (85).
Tissue Hypoxia and Immunosuppression in Tumors
Thus, adenosine plays important roles in the establishment of an immunosuppressive tumor microenvironment and cancer progression. Adenosine is present in tumors at much higher levels than in normal tissues (18, 19). Tissue hypoxia seems to be crucial to the increase in intratumoral adenosine levels because reversal of tissue hypoxia by exposing tumor-bearing mice to hyperoxic atmosphere (60% oxygen) largely reduced tumor adenosine levels (100). Tumors are found to be hypoxic (101, 102) probably because of high oxygen demand by proliferating cells and insufficient oxygen supply due to sluggish blood flow in the disorganized tumor vasculature. Tissues under hypoxic stress shift adenosine metabolism toward its accumulation in the extracellular space. On one hand, hypoxia induces CD39 and CD73 and increases catabolic adenosine production (53, 103). On the other hand, hypoxia downregulates adenosine kinase and thereby inhibits conversion of adenosine (55, 56).
Corresponding to the increase in adenosine levels, hypoxia is known to be immunosuppressive. When cultured at various degrees of hypoxic atmosphere (1–5% oxygen), T cells undergo an impaired T cell receptor-mediated activation process (104) and reduce proliferation, cytokine production, and cytotoxicity (16, 105–108). T cell stimulation in mice breathing hypoxic atmosphere (10% oxygen) confirmed the impairment of T cell activation in vivo (16, 109). Consistent with the effects on T cells, hypoxia impairs the antigen-presenting function of dendritic cells and macrophages. Exposure of APCs to hypoxia downregulated both antigen uptake (110) and expression of MHC and costimulatory molecules (111, 112), resulting in the inability to achieve full activation of T cells. Immunosuppressive effects of hypoxia further include inhibition of NK cell activity (113) and induction of immunosuppressive activity of MDSCs (114). Although hypoxia conducts various functional changes through adenosine receptor-dependent mechanisms (115–117), an unidentified adenosine receptor-independent mechanism may also contribute to T cell suppression under hypoxia (16). While T cell proliferating activity is relatively resistant to A2AR-mediated T cell suppression, hypoxia strongly reduces T cell proliferation in an A2AR-independent manner (16, 31).
Nonetheless, tissue hypoxia, at least in part, contributes to immunosuppression in the tumor microenvironment. Many anti-inflammatory molecules are present in tumors; hypoxia may be responsible for the upregulation of some, if not all, of them. Reversal of hypoxia by whole body exposure to hyperoxic atmosphere (60% oxygen) reduced intratumoral expression of TGF-β, CTLA-4, COX-2, and FoxP3 (100, 118). In the same study, levels of CD39, CD73, A2AR, and A2BR in tumors were also reduced in response to hyperoxia, suggesting oxygen-dependent regulation of adenosine signaling pathway. Consistent with these changes indicating a switch to a more immuno-permissive tumor microenvironment, hyperoxia treatment inhibited tumor progression with an enhanced cellular immune response.
When deprived of oxygen, cells urgently need to prepare for the inevitable loss of oxidative energy production. To adapt to this dangerously stressful condition, cells quickly promote anaerobic energy production by glycolysis. A crucial molecule conducting this metabolic switch is HIF-1α, a transcriptional factor regulating glycolytic enzymes together with angiogenesis and erythropoiesis (119, 120). Tumor cells are known to utilize glycolysis even under aerobic conditions and satisfy their high energy demand with this all-out energy production. Therefore, it seems reasonable that increased HIF-1α levels in human cancers are positively correlated with the increased risk of mortality (121). While HIF-1α expression in tumors may indicate the presence of potentially immunosuppressive tissue hypoxia, HIF-1α may actively regulate immunosuppressive mechanisms. HIF-1α can mediate CD73 induction by hypoxia and promote adenosine-dependent immunosuppression (53). HIF-1α may be inhibitory to T cell activation as indicated by attenuated T cell receptor signaling (104). Indeed, T cells lacking HIF-1α showed stronger effector functions (122, 123) and caused severe inflammation (124). Contrary to the T cell suppressive role of HIF-1α, however, deletion of HIF-1α in myeloid cells diminished inflammation (125, 126). Bactericidal activities of macrophages and neutrophils are impaired when they lack HIF-1α (127). Interestingly, HIF-1α also positively regulates activity of MDSC, supporting their immunosuppressive activity, PD-L1 expression and conversion into tumor-associated macrophages (114, 128). These studies suggest that myeloid cells need HIF-1α for their activities, while HIF-1α may be inhibitory to T cell effector functions.
HIF-1α is also vital in controlling Treg cells, but its role is complex. Initially, hypoxia was reported to upregulate FoxP3 in mouse and human T cells (129). The induction of Treg cells in hypoxic conditions was confirmed (130) and extended in the demonstration of decreased immunoregulatory activity of HIF-1α-deficient Treg cells (131). Contrary to the positive role of HIF-1α in Treg cell induction, functional differentiation of HIF-1α-deficient CD4+ T cells demonstrated an increase in FoxP3+ Treg cells and a reciprocal decrease of Th17 cells (132, 133). The preferential differentiation into Treg cells in the absence of HIF-1 α was consistent with the reduced magnitude of inflammation in these animals. Inflammation was rather enhanced when Treg cells overexpress HIF-1α due to the lack of von Hippel–Lindau tumor suppressor (VHL), which degrades HIF-1α protein in normoxic conditions (134). Such a proinflammatory phenotype of these mice corresponded well with the reduction of FoxP3 and immunoregulatory activity. Similar changes were observed in Deltex1-deficient mice in which the lack of HIF-1α degradation led to its accumulation (135). In these studies, abundance of HIF-1α is considered to promote FoxP3 degradation leading to downregulation of Treg cells (133, 135). Nonetheless, even if roles of hypoxia and HIF-1α on Treg cell induction and immunregulatory activity are not clear, tumors contain a number of Treg cells. Another possibility for the increase of intratumoral Treg cell population is the active recruitment of Treg cells from the outside of tumors. Indeed, hypoxia triggers induction of a chemokine, CCL28, which attracts Treg cells into tumor tissues (136).
Targeting the Hypoxia–Adenosine Pathway for Immune Checkpoint Therapy
For immune cells, the battle against tumors is a war that has to be fought in the enemy’s home ground. Although hypoxic tissue is a harsh environment for every cell, it may be a more challenging condition for immune cells than for tumor cells, which are ready to utilize various kinds of energy sources very aggressively. Since tumor cells drain energy sources as much as possible, it is hard for immune cells to keep on fighting in this scorched battleground. In addition, the inside of tumor tissue is a very hostile environment filled with a number of traps that will try to discourage the antitumor activity of immune cells. Many of such immunosuppressive activities are the use (abuse) of physiological immunoregulatory mechanisms that were originally intended to save vital organs from substantial inflammatory damage. Deceived by this false order, immune cells stop fighting tumor cells. As a result, immune cells, even highly activated immune effector cells that can recognize tumor-associated antigens, struggle mightily in the tumor. Hypoxia-inducible extracellular adenosine represents the physiological immunoregulatory molecules that are used to fortify tumors against immune system.
After adenosine-dependent tumor protection was first reported in 2006, extensive research on this pathway has revealed its significance in cancer immunology and cancer biology. Hypoxia and adenosine not only directly inhibits immune cell functions but also triggers induction of various anti-inflammatory molecules as well as emergence of immunoregulatory cells such as Treg cells, MDSC, and M2-type macrophages. The latter may contribute to establishing a long-lasting immunosuppressive environment in tumors. The pro-tumor effect of adenosine is not limited to immunosuppression, and it involves non-immunological pro-tumor effects, which promote tumor cell proliferation, tumor cell survival, metastasis, and angiogenesis. Clinical data support the pro-tumor effects of the hypoxia–adenosine pathway. Many cancer cells express CD73, and CD73 expression is associated with poor prognosis of various cancers such as breast (79), ovarian (77, 83), prostate (81, 82), brain cancers (80), and leukemia (76, 78). Resistance to chemotherapy has been reported in CD73-expressing cancers (78–80). An association with increased mortality has also been observed in cancers with poorly oxygenated areas (102, 137, 138) and in cancers expressing HIF-1α at high levels (139, 140). Corresponding to the poor prognosis, hypoxic cancers are found to be refractory to radiotherapy and chemotherapy.
Inactivation of the hypoxia–adenosine pathway may be able to relieve antitumor immune effector cells from immunosuppressive signaling, convert an immunosuppressive tumor microenvironment to a more immuno-permissive one, and reduce metastatic activity and resistance to chemotherapy and radiotherapy (Figure 1). The pathway can be targeted in different steps. Antagonists of adenosine receptors block adenosine signaling and improve tumor regression (19, 21, 41). Reduction of extracellular adenosine levels by CD73 inhibitor also improves tumor regression (141, 142). Improvement of tissue oxygenation status reduces adenosine levels and enhances immune cell activities as observed in mice breathing hyperoxic atmosphere (100, 118). When combined with therapeutic approaches that increase the number of antitumor cells, the anti-hypoxia–adenosine strategy may synergistically enhance efficacy by preventing inactivation of effector cells. Preclinical studies have shown further retardation of tumor growth and reduction of metastatic tumor nodules in combined treatments with adoptive immunotherapy (19, 86) or cancer vaccines (143). Interestingly, anti-hypoxia–adenosine therapy cooperates well with other immune checkpoint inhibitors such as ani-CTLA-4 and anti-PD-1 mAbs (29, 34, 47, 144). The complexity of regulating antitumor immunity may limit success in treatments targeting a single mechanism. However, combined treatment with the anti-hypoxia–adenosine strategy may improve the clinical response to other immunotherapy as well as chemotherapy and radiotherapy.
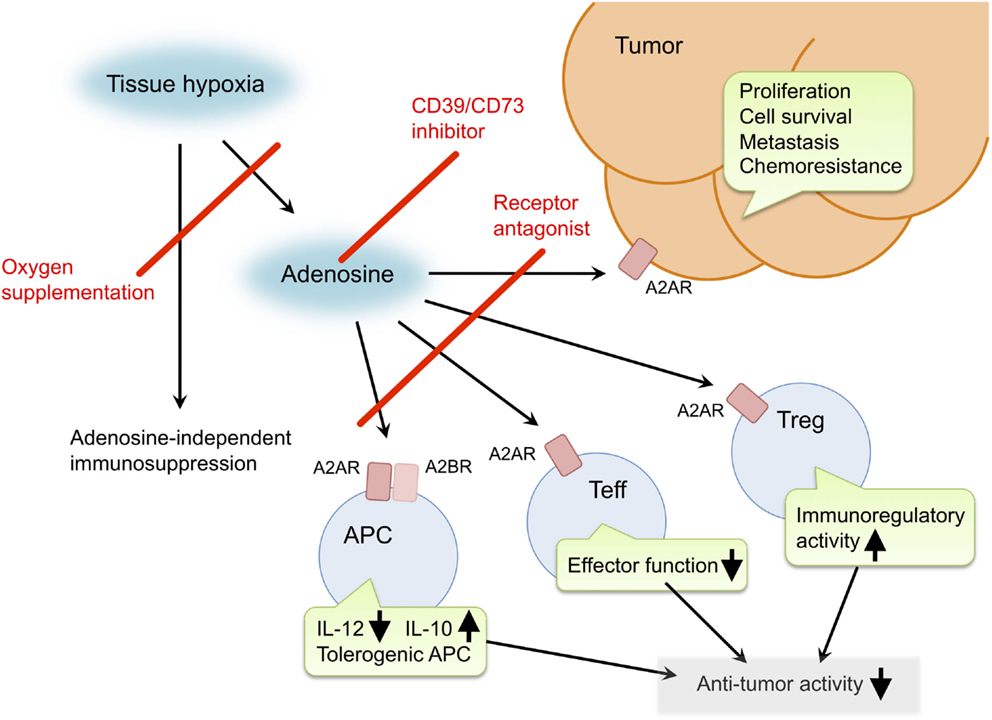
Figure 1. Pro-cancer activities of extracellular adenosine and therapeutic targets for improvement of cancer immunotherapy. Tissue hypoxia in tumors increases extracellular adenosine production through induction of CD39 and CD73. Produced adenosine transmits immunosuppressive signals through adenosine receptors on various immune cells. A2AR stimulation in effector T cells (Teff) blocks T cell receptor signaling and impairs effector functions including IFN-γ production and cytotoxicity. In antigen-presenting cells (APC), signals through A2AR and A2BR reduce Th1-type cytokine milieu and induce tolerogenic APC. Interaction of Teff with these APC will impair activation of cellular immune response against cancer cells. Adenosine enhances immunoregulatory activity of regulatory T cells (Treg). The qualitative and quantitative increase of Treg results in stronger inactivation of antitumor immune response. In addition, adenosine can promote proliferation, survival and metastatic activity of cancer cells. Suppression of adenosine pathway will be able to weaken the intensity of immunosuppression in tumor microenvironment and direct effect on cancer cells. Potential target molecules are adenosine receptors (A2AR and A2BR) and adenosine-producing enzymes (CD39 and CD73). Oxygen supplementation can also decrease pro-cancer effects of adenosine as well as adenosine-independent immunosuppression by hypoxia.
Author Contributions
AO conceived, wrote, and edited the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Susan Ohman for her careful reading that improved the manuscript tremendously. The author is thankful to Dr. Michail Sitkovsky (Northeastern University) for a kind suggestion and Dr. Robert Abbott (La Jolla Institute for Allergy & Immunology) for helpful discussions.
References
1. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
2. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol (2008) 8:74–80. doi:10.1038/nri2233
3. Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res (2012) 72:3125–30. doi:10.1158/0008-5472.CAN-11-4094
4. Drake CG, Doody AD, Mihalyo MA, Huang CT, Kelleher E, Ravi S, et al. Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell (2005) 7:239–49. doi:10.1016/j.ccr.2005.01.027
5. Bai A, Higham E, Eisen HN, Wittrup KD, Chen J. Rapid tolerization of virus-activated tumor-specific CD8+ T cells in prostate tumors of TRAMP mice. Proc Natl Acad Sci U S A (2008) 105:13003–8. doi:10.1073/pnas.0805599105
6. Itoh K, Tilden AB, Balch CM. Interleukin 2 activation of cytotoxic T-lymphocytes infiltrating into human metastatic melanomas. Cancer Res (1986) 46:3011–7.
7. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270:985–8. doi:10.1126/science.270.5238.985
8. Gozalo-Sanmillan S, McNally JM, Lin MY, Chambers CA, Berg LJ. Cutting edge: two distinct mechanisms lead to impaired T cell homeostasis in Janus kinase 3- and CTLA-4-deficient mice. J Immunol (2001) 166:727–30. doi:10.4049/jimmunol.166.2.727
9. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345:1623–7. doi:10.1126/science.1255904
10. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol (2013) 14:1212–8. doi:10.1038/ni.2762
11. Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev (2008) 224:166–82. doi:10.1111/j.1600-065X.2008.00662.x
12. Azoury SC, Straughan DM, Shukla V. Immune checkpoint inhibitors for cancer therapy: clinical efficacy and safety. Curr Cancer Drug Targets (2015) 15:452–62. doi:10.2174/156800961506150805145120
13. Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev (2001) 53:527–52.
14. Tripmacher R, Gaber T, Dziurla R, Haupl T, Erekul K, Grutzkau A, et al. Human CD4(+) T cells maintain specific functions even under conditions of extremely restricted ATP production. Eur J Immunol (2008) 38:1631–42. doi:10.1002/eji.200738047
15. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14:435–46. doi:10.1038/nri3701
16. Ohta A, Madasu M, Subramanian M, Kini R, Jones G, Chouker A, et al. Hypoxia-induced and A2A adenosine receptor-independent T-cell suppression is short lived and easily reversible. Int Immunol (2014) 26:83–91. doi:10.1093/intimm/dxt045
17. Gnad T, Scheibler S, von Kugelgen I, Scheele C, Kilic A, Glode A, et al. Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature (2014) 516:395–9. doi:10.1038/nature13816
18. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res (1997) 57:2602–5.
19. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A (2006) 103:13132–7. doi:10.1073/pnas.0605251103
20. Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol (2004) 22:657–82. doi:10.1146/annurev.immunol.22.012703.104731
21. Ohta A, Sitkovsky M. The hypoxia-adenosinergic immunosuppression and redirection of immune response in tumor microenvironment. In: Gabrilovich DI, Hurwitz AA, editors. Tumor-Induced Immune Suppression. New York: Springer (2014). doi:10.1007/978-0-387-69118-3
22. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367:2322–33. doi:10.1056/NEJMra1205750
23. Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets – what are the challenges? Nat Rev Drug Discov (2013) 12:265–86. doi:10.1038/nrd3955
24. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414:916–20. doi:10.1038/414916a
25. Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, et al. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol (2005) 3:e174. doi:10.1371/journal.pbio.0030174
26. Mohsenin A, Mi T, Xia Y, Kellems RE, Chen JF, Blackburn MR. Genetic removal of the A2A adenosine receptor enhances pulmonary inflammation, mucin production, and angiogenesis in adenosine deaminase-deficient mice. Am J Physiol Lung Cell Mol Physiol (2007) 293:L753–61. doi:10.1152/ajplung.00187.2007
27. Nadeem A, Fan M, Ansari HR, Ledent C, Jamal Mustafa S. Enhanced airway reactivity and inflammation in A2A adenosine receptor-deficient allergic mice. Am J Physiol Lung Cell Mol Physiol (2007) 292:L1335–44. doi:10.1152/ajplung.00416.2006
28. Alam MS, Kurtz CC, Wilson JM, Burnette BR, Wiznerowicz EB, Ross WG, et al. A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol (2009) 2:232–42. doi:10.1038/mi.2009.4
29. Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, et al. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res (2014) 74:3652–8. doi:10.1158/0008-5472.CAN-14-0957
30. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood (2008) 111:251–9. doi:10.1182/blood-2007-03-081646
31. Ohta A, Ohta A, Madasu M, Kini R, Subramanian M, Goel N, et al. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol (2009) 183:5487–93. doi:10.4049/jimmunol.0901247
32. Sevigny CP, Li L, Awad AS, Huang L, McDuffie M, Linden J, et al. Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J Immunol (2007) 178:4240–9. doi:10.4049/jimmunol.178.7.4240
33. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi:10.3389/fimmu.2012.00190
34. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res (2013) 19:5626–35. doi:10.1158/1078-0432.CCR-13-0545
35. Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, et al. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J (2000) 14:2065–74. doi:10.1096/fj.99-0508com
36. Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, et al. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood (2003) 101:3985–90. doi:10.1182/blood-2002-07-2113
37. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res (2014) 74:7250–9. doi:10.1158/0008-5472.CAN-13-3583
38. Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res (2014) 74:7239–49. doi:10.1158/0008-5472.CAN-13-3581
39. Han KL, Thomas SV, Koontz SM, Changpriroa CM, Ha SK, Malech HL, et al. Adenosine A(2)A receptor agonist-mediated increase in donor-derived regulatory T cells suppresses development of graft-versus-host disease. J Immunol (2013) 190:458–68. doi:10.4049/jimmunol.1201325
40. Lee DJ, Taylor AW. Both MC5r and A2Ar are required for protective regulatory immunity in the spleen of post-experimental autoimmune uveitis in mice. J Immunol (2013) 191:4103–11. doi:10.4049/jimmunol.1300182
41. Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol (2014) 5:304. doi:10.3389/fimmu.2014.00304
42. Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, et al. Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J Am Soc Nephrol (2012) 23:1528–37. doi:10.1681/ASN.2012010070
43. Ben Addi A, Lefort A, Hua X, Libert F, Communi D, Ledent C, et al. Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A(2B) receptor. Eur J Immunol (2008) 38:1610–20. doi:10.1002/eji.200737781
44. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood (2008) 112:1822–31. doi:10.1182/blood-2008-02-136325
45. Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26:376–86. doi:10.1096/fj.11-190934
46. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol (2011) 187:6120–9. doi:10.4049/jimmunol.1101225
47. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia (2013) 15:1400–9. doi:10.1593/neo.131748
48. Ryzhov S, Novitskiy SV, Zaynagetdinov R, Goldstein AE, Carbone DP, Biaggioni I, et al. Host A(2B) adenosine receptors promote carcinoma growth. Neoplasia (2008) 10:987–95. doi:10.1593/neo.08478
49. Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol (2012) 188:198–205. doi:10.4049/jimmunol.1101845
50. Kaji W, Tanaka S, Tsukimoto M, Kojima S. Adenosine A(2B) receptor antagonist PSB603 suppresses tumor growth and metastasis by inhibiting induction of regulatory T cells. J Toxicol Sci (2014) 39:191–8. doi:10.2131/jts.39.191
51. Volmer JB, Thompson LF, Blackburn MR. Ecto-5’-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol (2006) 176:4449–58. doi:10.4049/jimmunol.176.7.4449
52. Grenz A, Zhang H, Weingart J, von Wietersheim S, Eckle T, Schnermann J, et al. Lack of effect of extracellular adenosine generation and signaling on renal erythropoietin secretion during hypoxia. Am J Physiol Renal Physiol (2007) 293:F1501–11. doi:10.1152/ajprenal.00243.2007
53. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110:993–1002. doi:10.1172/JCI15337
54. Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood (2004) 104:3986–92. doi:10.1182/blood-2004-06-2066
55. Kobayashi S, Zimmermann H, Millhorn DE. Chronic hypoxia enhances adenosine release in rat PC12 cells by altering adenosine metabolism and membrane transport. J Neurochem (2000) 74:621–32. doi:10.1046/j.1471-4159.2000.740621.x
56. Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood (2008) 111:5571–80. doi:10.1182/blood-2007-11-126763
57. Karhausen J, Haase VH, Colgan SP. Inflammatory hypoxia: role of hypoxia-inducible factor. Cell Cycle (2005) 4:256–8. doi:10.4161/cc.4.2.1407
58. Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol (2005) 26:299–304. doi:10.1016/j.it.2005.04.004
59. Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis (1993) 148:91–7. doi:10.1164/ajrccm/148.1.91
60. Nishiyama A, Miura K, Miyatake A, Fujisawa Y, Yue W, Fukui T, et al. Renal interstitial concentration of adenosine during endotoxin shock. Eur J Pharmacol (1999) 385:209–16. doi:10.1016/S0014-2999(99)00716-5
61. Martin C, Leone M, Viviand X, Ayem ML, Guieu R. High adenosine plasma concentration as a prognostic index for outcome in patients with septic shock. Crit Care Med (2000) 28:3198–202. doi:10.1097/00003246-200009000-00014
62. Montesinos MC, Gadangi P, Longaker M, Sung J, Levine J, Nilsen D, et al. Wound healing is accelerated by agonists of adenosine A2 (G alpha s-linked) receptors. J Exp Med (1997) 186:1615–20. doi:10.1084/jem.186.9.1615
63. Montesinos MC, Desai A, Chen JF, Yee H, Schwarzschild MA, Fink JS, et al. Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A(2A) receptors. Am J Pathol (2002) 160:2009–18. doi:10.1016/S0002-9440(10)61151-0
64. Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol (2007) 178:8127–37. doi:10.4049/jimmunol.178.12.8127
65. Louis NA, Robinson AM, MacManus CF, Karhausen J, Scully M, Colgan SP. Control of IFN-alphaA by CD73: implications for mucosal inflammation. J Immunol (2008) 180:4246–55. doi:10.4049/jimmunol.180.6.4246
66. Ohtsuka T, Changelian PS, Bouis D, Noon K, Harada H, Lama VN, et al. Ecto-5’-nucleotidase (CD73) attenuates allograft airway rejection through adenosine 2A receptor stimulation. J Immunol (2010) 185:1321–9. doi:10.4049/jimmunol.0901847
67. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res (2011) 71:2892–900. doi:10.1158/0008-5472.CAN-10-4246
68. Stagg J, Beavis PA, Divisekera U, Liu MC, Moller A, Darcy PK, et al. CD73-deficient mice are resistant to carcinogenesis. Cancer Res (2012) 72:2190–6. doi:10.1158/0008-5472.CAN-12-0420
69. Tsukamoto H, Chernogorova P, Ayata K, Gerlach UV, Rughani A, Ritchey JW, et al. Deficiency of CD73/ecto-5’-nucleotidase in mice enhances acute graft-versus-host disease. Blood (2012) 119:4554–64. doi:10.1182/blood-2011-09-375899
70. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol (2006) 177:6780–6. doi:10.4049/jimmunol.177.10.6780
71. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
72. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Lenzner DE, Jackson EK, et al. Increased ectonucleotidase expression and activity in regulatory T cells of patients with head and neck cancer. Clin Cancer Res (2009) 15:6348–57. doi:10.1158/1078-0432.CCR-09-1143
73. Ujhazy P, Klobusicka M, Babusikova O, Strausbauch P, Mihich E, Ehrke MJ. Ecto-5’-nucleotidase (CD73) in multidrug-resistant cell lines generated by doxorubicin. Int J Cancer (1994) 59:83–93. doi:10.1002/ijc.2910590117
74. Sadej R, Spychala J, Skladanowski AC. Expression of ecto-5’-nucleotidase (eN, CD73) in cell lines from various stages of human melanoma. Melanoma Res (2006) 16:213–22. doi:10.1097/01.cmr.0000215030.69823.11
75. Zhi X, Chen S, Zhou P, Shao Z, Wang L, Ou Z, et al. RNA interference of ecto-5’-nucleotidase (CD73) inhibits human breast cancer cell growth and invasion. Clin Exp Metastasis (2007) 24:439–48. doi:10.1007/s10585-007-9081-y
76. Coustan-Smith E, Song G, Clark C, Key L, Liu P, Mehrpooya M, et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood (2011) 117:6267–76. doi:10.1182/blood-2010-12-324004
77. Hausler SF, Montalban del Barrio I, Strohschein J, Anoop Chandran P, Engel JB, Honig A, et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol Immunother (2011) 60:1405–18. doi:10.1007/s00262-011-1040-4
78. Serra S, Horenstein AL, Vaisitti T, Brusa D, Rossi D, Laurenti L, et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood (2011) 118:6141–52. doi:10.1182/blood-2011-08-374728
79. Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A (2013) 110:11091–6. doi:10.1073/pnas.1222251110
80. Quezada C, Garrido W, Oyarzun C, Fernandez K, Segura R, Melo R, et al. 5’-ectonucleotidase mediates multiple-drug resistance in glioblastoma multiforme cells. J Cell Physiol (2013) 228:602–8. doi:10.1002/jcp.24168
81. Yang Q, Du J, Zu L. Overexpression of CD73 in prostate cancer is associated with lymph node metastasis. Pathol Oncol Res (2013) 19:811–4. doi:10.1007/s12253-013-9648-7
82. Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F, et al. CD73 expression is an independent prognostic factor in prostate cancer. Clin Cancer Res (2016) 22:158–66. doi:10.1158/1078-0432.CCR-15-1181
83. Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res (2015) 75:4494–503. doi:10.1158/0008-5472.CAN-14-3569
84. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A (2010) 107:1547–52. doi:10.1073/pnas.0908801107
85. Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol (2013) 191:4165–73. doi:10.4049/jimmunol.1301274
86. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res (2010) 70:2245–55. doi:10.1158/0008-5472.CAN-09-3109
87. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology (2010) 139:1030–40. doi:10.1053/j.gastro.2010.05.007
88. Bastid J, Regairaz A, Bonnefoy N, Dejou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res (2015) 3:254–65. doi:10.1158/2326-6066.CIR-14-0018
89. Hausler SF, Del Barrio IM, Diessner J, Stein RG, Strohschein J, Honig A, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am J Transl Res (2014) 6:129–39.
90. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi:10.1126/science.1203486
91. Chow MT, Moller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol (2012) 22:23–32. doi:10.1016/j.semcancer.2011.12.004
92. Coppee F, Gerard AC, Denef JF, Ledent C, Vassart G, Dumont JE, et al. Early occurrence of metastatic differentiated thyroid carcinomas in transgenic mice expressing the A2a adenosine receptor gene and the human papillomavirus type 16 E7 oncogene. Oncogene (1996) 13:1471–82.
93. Eini H, Frishman V, Yulzari R, Kachko L, Lewis EC, Chaimovitz C, et al. Caffeine promotes anti-tumor immune response during tumor initiation: involvement of the adenosine A2A receptor. Biochem Pharmacol (2015) 98:110–8. doi:10.1016/j.bcp.2015.08.092
94. Bavaresco L, Bernardi A, Braganhol E, Cappellari AR, Rockenbach L, Farias PF, et al. The role of ecto-5’-nucleotidase/CD73 in glioma cell line proliferation. Mol Cell Biochem (2008) 319:61–8. doi:10.1007/s11010-008-9877-3
95. Mediavilla-Varela M, Luddy K, Noyes D, Khalil FK, Neuger AM, Soliman H, et al. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol Ther (2013) 14:860–8. doi:10.4161/cbt.25643
96. Zhi X, Wang Y, Zhou X, Yu J, Jian R, Tang S, et al. RNAi-mediated CD73 suppression induces apoptosis and cell-cycle arrest in human breast cancer cells. Cancer Sci (2010) 101:2561–9. doi:10.1111/j.1349-7006.2010.01733.x
97. Wang L, Zhou X, Zhou T, Ma D, Chen S, Zhi X, et al. Ecto-5’-nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J Cancer Res Clin Oncol (2008) 134:365–72. doi:10.1007/s00432-007-0292-z
98. Zhou JZ, Riquelme MA, Gao X, Ellies LG, Sun LZ, Jiang JX. Differential impact of adenosine nucleotides released by osteocytes on breast cancer growth and bone metastasis. Oncogene (2015) 34:1831–42. doi:10.1038/onc.2014.113
99. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci U S A (2013) 110:14711–6. doi:10.1073/pnas.1308209110
100. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med (2015) 7:277ra230. doi:10.1126/scitranslmed.aaa1260
101. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer (2004) 4:437–47. doi:10.1038/nrc1367
102. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev (2007) 26:225–39. doi:10.1007/s10555-007-9055-1
103. Deaglio S, Robson SC. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol (2011) 61:301–32. doi:10.1016/B978-0-12-385526-8.00010-2
104. Neumann AK, Yang J, Biju MP, Joseph SK, Johnson RS, Haase VH, et al. Hypoxia inducible factor 1 alpha regulates T cell receptor signal transduction. Proc Natl Acad Sci U S A (2005) 102:17071–6. doi:10.1073/pnas.0506070102
105. Loeffler DA, Juneau PL, Masserant S. Influence of tumour physico-chemical conditions on interleukin-2-stimulated lymphocyte proliferation. Br J Cancer (1992) 66:619–22. doi:10.1038/bjc.1992.326
106. Naldini A, Carraro F, Silvestri S, Bocci V. Hypoxia affects cytokine production and proliferative responses by human peripheral mononuclear cells. J Cell Physiol (1997) 173:335–42. doi:10.1002/(SICI)1097-4652(199712)173:3<335:AID-JCP5>3.0.CO;2-O
107. Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, et al. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol (2001) 167:6140–9. doi:10.4049/jimmunol.167.11.6140
108. Atkuri KR, Herzenberg LA, Herzenberg LA. Culturing at atmospheric oxygen levels impacts lymphocyte function. Proc Natl Acad Sci U S A (2005) 102:3756–9. doi:10.1073/pnas.0409910102
109. Ohta A, Diwanji R, Kini R, Subramanian M, Ohta A, Sitkovsky M. In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Front Immunol (2011) 2:27. doi:10.3389/fimmu.2011.00027
110. Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W, et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology (2009) 128:e237–49. doi:10.1111/j.1365-2567.2008.02954.x
111. Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, et al. Divergent effects of hypoxia on dendritic cell functions. Blood (2008) 112:3723–34. doi:10.1182/blood-2008-02-142091
112. Wang Q, Liu C, Zhu F, Liu F, Zhang P, Guo C, et al. Reoxygenation of hypoxia-differentiated dentritic cells induces Th1 and Th17 cell differentiation. Mol Immunol (2010) 47:922–31. doi:10.1016/j.molimm.2009.09.038
113. Fink T, Ebbesen P, Koppelhus U, Zachar V. Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand J Immunol (2003) 58:607–12. doi:10.1111/j.1365-3083.2003.01347.x
114. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207:2439–53. doi:10.1084/jem.20100587
115. Takagi H, King GL, Robinson GS, Ferrara N, Aiello LP. Adenosine mediates hypoxic induction of vascular endothelial growth factor in retinal pericytes and endothelial cells. Invest Ophthalmol Vis Sci (1996) 37:2165–76.
116. Kobayashi S, Conforti L, Pun RY, Millhorn DE. Adenosine modulates hypoxia-induced responses in rat PC12 cells via the A2A receptor. J Physiol (1998) 508(Pt 1):95–107. doi:10.1111/j.1469-7793.1998.095br.x
117. Fisher JW, Brookins J. Adenosine A(2A) and A(2B) receptor activation of erythropoietin production. Am J Physiol Renal Physiol (2001) 281:F826–32. doi:10.1152/ajprenal.0083.2001
118. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl) (2014) 92:1283–92. doi:10.1007/s00109-014-1189-3
119. Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J (2007) 405:1–9. doi:10.1042/BJ20070389
120. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell (2010) 40:294–309. doi:10.1016/j.molcel.2010.09.022
121. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123:3664–71. doi:10.1172/JCI67230
122. Lukashev D, Klebanov B, Kojima H, Grinberg A, Ohta A, Berenfeld L, et al. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol (2006) 177:4962–5. doi:10.4049/jimmunol.177.8.4962
123. Guo J, Lu W, Shimoda LA, Semenza GL, Georas SN. Enhanced interferon-gamma gene expression in T Cells and reduced ovalbumin-dependent lung eosinophilia in hypoxia-inducible factor-1-alpha-deficient mice. Int Arch Allergy Immunol (2009) 149:98–102. doi:10.1159/000189191
124. Higashiyama M, Hokari R, Hozumi H, Kurihara C, Ueda T, Watanabe C, et al. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J Leukoc Biol (2012) 91:901–9. doi:10.1189/jlb.1011518
125. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112:645–57. doi:10.1016/S0092-8674(03)00154-5
126. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol (2009) 9:609–17. doi:10.1038/nri2607
127. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 41:518–28. doi:10.1016/j.immuni.2014.09.008
128. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211:781–90. doi:10.1084/jem.20131916
129. Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol (2008) 38:2412–8. doi:10.1002/eji.200838318
130. Wu J, Cui H, Zhu Z, Wang L, Li H, Wang D. Effect of HIF1alpha on Foxp3 expression in CD4+ CD25- T lymphocytes. Microbiol Immunol (2014) 58:409–15. doi:10.1111/1348-0421.12168
131. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A (2012) 109:E2784–93. doi:10.1073/pnas.1202366109
132. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
133. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
134. Lee JH, Elly C, Park Y, Liu YC. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity (2015) 42:1062–74. doi:10.1016/j.immuni.2015.05.016
135. Hsiao HW, Hsu TS, Liu WH, Hsieh WC, Chou TF, Wu YJ, et al. Deltex1 antagonizes HIF-1alpha and sustains the stability of regulatory T cells in vivo. Nat Commun (2015) 6:6353. doi:10.1038/ncomms7353
136. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475:226–30. doi:10.1038/nature10169
137. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer (2006) 6:583–92. doi:10.1038/nrc1893
138. Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer (2008) 8:425–37. doi:10.1038/nrc2397
139. Imamura T, Kikuchi H, Herraiz MT, Park DY, Mizukami Y, Mino-Kenduson M, et al. HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. Int J Cancer (2009) 124:763–71. doi:10.1002/ijc.24032
140. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene (2010) 29:625–34. doi:10.1038/onc.2009.441
141. Zhang B. CD73: a novel target for cancer immunotherapy. Cancer Res (2010) 70:6407–11. doi:10.1158/0008-5472.CAN-10-1544
142. Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol (2012) 33:231–7. doi:10.1016/j.it.2012.02.009
143. Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother (2012) 61:917–26. doi:10.1007/s00262-011-1155-7
Keywords: adenosine, tumor microenvironment, A2A adenosine receptor, CD73, tissue hypoxia, immunosuppression, immune checkpoint, cancer immunotherapy
Citation: Ohta A (2016) A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 7:109. doi: 10.3389/fimmu.2016.00109
Received: 15 January 2016; Accepted: 14 March 2016;
Published: 29 March 2016
Edited by:
Heriberto Prado-Garcia, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosio Villegas”, MexicoReviewed by:
Pedro Berraondo, Centro para la Investigación Médica Aplicada, SpainJohn Stagg, University of Montreal, Canada
Mikhail M. DIkov, The Ohio State University Medical Center, USA
Copyright: © 2016 Ohta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akio Ohta, YS5vaHRhQG5ldS5lZHU=